Back to Journals » OncoTargets and Therapy » Volume 13
JuBei Oral Liquid Induces Mitochondria-Mediated Apoptosis in NSCLC Cells
Authors Pan Z, Chen Q, Zheng X, Wang K, Duan Y, Xiao K, Jia Z, Ding X
Received 23 March 2020
Accepted for publication 10 July 2020
Published 31 July 2020 Volume 2020:13 Pages 7585—7598
DOI https://doi.org/10.2147/OTT.S254464
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Geoffrey Pietersz
Zhenzhen Pan,1,* Qiufang Chen,2,* Xiulan Zheng,1 Kai Wang,1 Yalei Duan,1 Kang Xiao,1 Zhirong Jia,1 Xuansheng Ding1
1Department of Basic Medicine and Clinical Pharmacy, China Pharmaceutical University, Nanjing 211198, People’s Republic of China; 2Department of Science and Education, Women and Children’s Hospital, School of Medicine, Xiamen University, Xiamen 361003, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Xuansheng Ding Tel +86 13357823738
Email [email protected]
Background: Although gefitinib brings about tremendous advances in the treatment of non-small cell lung cancer (NSCLC) harboring epidermal growth factor receptor (EGFR) mutations, most of patients become incurable due to drug resistance. JuBei oral liquid (JB) has been widely used to treat pneumonia in clinic. Components of JB were reported to induce apoptosis in NSCLC, which indicated that JB could be a potential antitumor agent for NSCLC patients. In this study, we investigated the effect of JB on gefitinib-sensitive PC-9 and gefitinib-resistant PC-9/GR, H1975 cells as well as its underlying molecular mechanisms.
Methods: PC-9, PC-9/GR and H1975 cells were treated with JB, LY294002, SCH772984, gefitinib alone or in combination. Then, cell viability, colony formation, cell death, expression of mitochondria-dependent pathway proteins, expression of EGFR, PI3K/AKT, MAPK signal pathway proteins, Bcl-2 mitochondrial translocation, ROS generation and cell apoptosis were examined by MTT, colony forming, live/dead cell staining, Western blot, immunofluorescence and flow cytometry assay.
Results: Our results showed that JB significantly induced cell growth inhibition and apoptotic cell death in PC-9, PC-9/GR and H1975 cells. JB activated mitochondria-mediated apoptotic pathway through inhibiting Bcl-2 mitochondrial translocation while inducing Bax translocated into mitochondria along with accumulated ROS production, thereby increasing the release of cytochrome c, subsequently cleaving procaspase9 into cleaved-caspase9 and then cleaving procaspase3 into cleaved-caspase3. Furthermore, the employment of protein kinase inhibitors LY294002 and SCH772984 revealed that the induction of mitochondria-mediated apoptosis by JB was reliant on inactivation of PI3K/AKT and MAPK signal pathways. Moreover, JB could synergize with gefitinib to induce apoptosis in PC-9, PC-9/GR and H1975 cells.
Conclusion: These data indicated that JB could be a potential therapeutic agent for NSCLC patients harboring EGFR mutations as well as those under gefitinib resistance.
Keywords: non-small cell lung cancer, gefitinib, resistance, JuBei oral liquid, apoptosis
Introduction
EGFR mutations occur in about 67% of NSCLC patients of East Asian and exon 19 deletion is one of the most common classical mutations.1 NSCLC cells holding these mutations are in the nature of relying on the signal generated by EGFR to survive. Thus, drugs targeting this spot are born. The first-generation EGFR tyrosine kinase inhibitors (EGFR-TKIs), such as gefitinib and erlotinib, prevent ATP form binding to the kinase domain of EGFR in a reversible manner and consequently leading to the loss of activity, are widely used as target therapy in NSCLC harboring EGFR mutations including EGFR exon 19 deletion.2 The application of EGFR-TKIs has provided amazing clinical responses and survival benefits for 80% of NSCLC patients carrying these mutations compared with those receiving standard chemotherapy.3,4 Although treatment with EGFR-TKIs target therapy improves outcomes in NSCLC patients, many patients eventually develop acquired drug resistance after 10–14 months.5 The third-generation EGFR-TKIs, such as osimertinib and rociletinib have been proven to be effective in gefitinib-resistant patients, however progression occurs.6–8 Therefore, new alternative treatment strategies for gefitinib-resistant NSCLC are needed eagerly. Responses of NSCLC to EGFR-TKIs require the induction of apoptosis upon inhibiting EGFR; thus, alternations in cell signal pathway that manipulate apoptosis can change the sensitivity to EGFR-TKIs.9 The Bcl-2 family proteins are critical features of the intrinsic apoptotic pathway.10 It has been reported that Bcl-2 could inhibit mitochondrial permeability transition pore (mPTP) from opening thus leading to pro-apoptotic factors cytochrome c release into the cytosol and subsequently activating caspase cascade.11 Previous studies demonstrated that the transcription of Bcl-2 was regulated by cAMP response element binding protein (CREB) transcription factor, which is activated by two survival signal pathways, PI3K/AKT and MAPK.12,13 Additionally, PI3K/AKT signal pathway was also reported to suppress cell apoptosis and promote cell survival through regulating Bad phosphorylation, a protein of Bcl-2 family.14
JuBei oral liquid (JB) is a traditional Chinese medicine, which is widely used in clinical practice for the treatment of chronic obstructive pulmonary disease and pneumonia. Seven medicinal herbs, Platycodon grandiflorum, Fritillaria thunbergii, Bitter apricot kernel, Ophiopogon japonicas, Scutellaria baicalensis, Eriobotrya japonica leaf and Glycyrrhiza Uralensis make up JB according to the standard of quality control in the Drug Standard of Ministry of Public Health of the Peoples Republic of China. Evidence has shown that JB possessed antipyretic, antibiosis, antiviral and immunomodulatory activities. Yet, to date, direct evidence associated with the antitumor effect of JB remain absent. Previous studies demonstrated that several components of JB exerted outstanding anticancer function. Platycodon grandiflorum was reported to induce cell death and apoptosis in human NSCLC cells through inhibiting AKT/mTOR and MAPK signal pathways plus regulating Bcl-2 family proteins expression.15,16 It has been suggested that Ophiopogon japonicus could active autophagy in NSCLC cells so as to prevent cancer process via inhibiting PI3K/AKT/mTOR signal pathway.17 In addition, Scutellaria baicalensis not only induced NSCLC cell cycle arrest and apoptosis in vitro but also enhanced the therapeutic efficacy of cisplatin in vivo.18,19 Licochalcone A, an active compound extracted from Glycyrrhiza Uralensis was shown to inhibit proliferation and induce apoptosis in NSCLC cells.20 Though all of these works indicated that JB had the potential to be an antitumor agent candidate for NSCLC patients, there has been no attempt to identify this possibility.
In the present study, gefitinib-sensitive PC-9 cells harboring EGFR exon 19 deletion (E746-A750), gefitinib-resistant PC-9/GR cells with no EGFR-T790M mutation and gefitinib-resistant H1975 cells with EGFR-T790M mutation were used as models for detecting the anticancer function of JB.21 Our work aims to investigate the effects of JB on PC-9, PC-9/GR and H1975 cells, as well as demonstrate the possible underlying molecular mechanism.
Materials and Methods
Materials
JuBei oral liquid (JB, Z50020208) was purchased from Taiji Group Chongqing TongJunGe Pharmaceutical Co., Ltd. (Chongqing, China). For cell culture, JB was filtered by 0.22μm filter to remove bacteria and then stored at 4°C. Gefitinib was purchased from Aladdin Industrial Corporation (Shanghai, China). LY294002 and SCH772984 were purchased from AbMole BioScience (Houston, USA) and dissolved in dimethyl sulfoxide (DMSO) at a concentration of 10mmol/L and stored at −20°C. The 3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT) and Penicillin-Streptomycin Solution were purchased from KeyGen (Nanjing, China). The Annexin V-FITC/PI Apoptosis Detection kit was purchased from Vazyme (Nanjing, China). DMSO, Calcein AM/PI Double Stain Kit and MitoTracker® Red CMXRos were purchased from Yeasen (Shanghai, China). The ROS assay kit, DAPI staining solution, BCA Protein Assay kit and goat anti-rabbit IgG H&L (HRP) antibody were purchased from Beyotime Biotechnology (Shanghai, China). RPMI 1640 and fetal bovine serum were purchased from Biological Industries (Kibbutz Beit Haemek, Israel). Anti-Bcl-2 and goat anti-rabbit IgG H&L (FITC) antibodies were purchased from Abcam (New Territories, HK). Mitochondria Isolation Kit, anti-p-EGFR (Tyr1172), anti-EGFR, anti-p-AKT (Ser473), anti-AKT, anti-p-ERK (Thr202/Tyr204), anti-ERK, anti-cleaved-caspase3, anti-cleaved-caspase9, anti-Cytochrome C, anti-Bax, anti-Bak, anti-Bcl-xl, anti-Mcl-1 and anti-COX IV antibodies were purchased from Wanlei Bio. (Shenyang, China). Anti-GAPDH antibody was purchased from Abways Technology (Beijing, China).
Cell Culture
Human lung adenocarcinoma PC-9 cells harboring EGFR exon 19 deletion (E746-A750), gefitinib-resistant PC-9/GR cells with no EGFR-T790M mutation and H1975 cells with EGFR-T790M mutation were provided by Dr. Zhou Caicun (Shanghai pulmonary hospital, Shanghai, China).22 The gifted cells were approved by China Pharmaceutical University ethics committee. All cells were cultured in RPMI 1640 containing 10% fetal bovine serum and 1% penicillin-streptomycin solution at 37°C in an atmosphere of 5% CO2.
Cell Viability Assay
Cell viability was determined by the MTT assay. Briefly, cells in 96-well plates at 80% confluence were treated with indicated concentration of drugs. Then, MTT solution (5 mg/mL dissolved in RPMI 1640) was added to each well and incubated at 37°C. After 4h, the culture medium was removed, and insoluble formazan crystals were dissolved by adding 150μL DMSO. Finally, the absorbance was measured using a microplate reader at 570nm.
Drug Synergy Analysis
The data from cell viability assay were analyzed by CompuSyn software (Biosoft, Cambridge, UK) to investigate the synergistic effects in vitro. The combination index (CI)-isobologram equation was described as previously. Points that fall below the line indicate synergistic relationship between the two drugs.23
Colony Forming Assay
For the measurement of colony formation, a density of 500 cells per well were seeded on 12-well plates and cultured in RPMI 1640 medium for 24h. Next, cells were treated medium with indicated drugs at 37°C in 5% CO2 and allowed to proliferate for another 2 weeks. Then, the cells were washed with PBS and fixed in 4% paraformaldehyde for 15 min. Finally, the cells were stained with crystal violet solution for 20 min and photographed.
Live/Dead Cell Staining
For live/dead cell measurement, cells were stained with Calcein AM and Propidium Iodide (PI) for 15 min. Then, the cells were assessed using fluorescence microscope (Olympus IX53, Japan).
Apoptosis Assay
Apoptosis cells were detected by Annexin V-FITC/PI Apoptosis Detection kit. In brief, cells were treated with indicated drugs for 24 h and collected. The cells were then washed with ice-cold PBS, and 1×106 cells were gathered. Next, the cells were re-suspended with 500 µL binding buffer. After Annexin V-FITC being added and mixed well, PI was added and incubated for 5–15min. Cells were analyzed by flow cytometry (Becton Dickinson FACS Calibur; Becton-Dickinson, USA).
Measurement of Intracellular ROS
Generation of intracellular ROS was measured by flow cytometer using dichlorodihydrofluorescein diacetate (DCFH-DA) fluorescent probe, which can be oxidized by cellular oxidants into the highly fluorescent compound DCF. Therefore, the fluorescence intensity is equivalent to the level of peroxide generated by the cells. Cells were treated by JB for 24h, collected and washed. Then, they were re-suspended in RPMI 1640 and incubated with DCFH-DA for 30 min at 37°C in the dark. The fluorescence intensity was detected by flow cytometry.
Isolation of Mitochondrial Protein
Cells were treated with drugs, then collected and washed using PBS. Mitochondrial fractions were gathered by a Mitochondria isolation kit according to the manufacturer’s instructions.
Western Blot Assay
The proteins were lysed in lysis buffer containing 1% PMSF for 15min on the ice. The lysates were then clarified by centrifugation at 4°C for 10min. Protein concentrations were determined by the BCA Protein Assay kit. Then, 20μg protein was separated by 10–15% SDS-PAGE and electroblotted onto polyvinylidene difluoride (PVDF) membranes. The membranes were blocked using 5% nonfat milk in TBST for 1.5h and then incubated with primary antibodies at 4°C overnight. Next, the membranes were washed and incubated with secondary antibodies for 2h at room temperature. The final detection was performed by ECL reagents. Protein expression was quantified by densitometry using Image J software.
Immunofluorescence Assay
Cells were seeded in 0.17 mm glass bottom dish, being treated with indicated drugs for a given time, and then incubated with MitoTracker® Red CMXRos for 30min according to the manufacturer’s instructions. Next, cells were fixed in 4% paraformaldehyde and incubated with anti-Bcl-2 antibody overnight at 4°C in the dark. After washing cells with PBS for 3 times, goat anti-rabbit IgG H&L (FITC) was added and incubated for 1h at room temperature. For staining of nuclei, the cells were exposed to DAPI for 15 min. Finally, the images were obtained by ZEISS LSM 800 with Airyscan confocal microscope for co-localization analysis.
Statistical Analysis
All experiments were performed for at least 3 times and the data were shown as mean ± S.E.M. Data among groups was analyzed by one-way ANOVA, using GraphPad Prism 6.0 statistical software. P<0.05 was considered as statistically significant.
Results
Effects of JB on Cell Growth and Death
We firstly examined the inhibitory effect of JB in PC-9 cells by MTT assay. As shown in Figure 1A, exposure to increasing concentration of JB (0, 0.78, 1.56, 3.125, 6.25, 12.5 and 25mg/mL) for 24, 48 and 72h caused a dose- and time-dependent decrease in the cell viability. The half maximal inhibitory concentration (IC50) was 7.92±0.08, 4.45±0.26 and 2.39±0.12 mg/mL at 24, 48 and 72h, respectively. We found a similar effect in gefitinib-resistant PC-9/GR and H1975 cells. Gefitinib (Iressa, ZD1839), a first-generation oral EGFR tyrosine kinase inhibitor, blocks the growth of NSCLC cells harboring EGFR mutations and is highly effective in inducing disease remission in NSCLC patients.24 Unfortunately, the pre-existing genetic alterations within a heterogeneous cancer cell population or the acquisition of new alterations under drug pressure ultimately lead to therapy failure.25 JB inhibited the viability of PC-9/GR and H1975 cells in a dose- and time-dependent manner. The IC50 of PC-9/GR cells were 6.61±0.27, 3.95±0.25 and 2.61±0.24 mg/mL, while the IC50 of H1975 cells were 2.78±0.03, 1.27±0.01 and 0.77±0.01 mg/mL at 24, 48 and 72h, respectively.
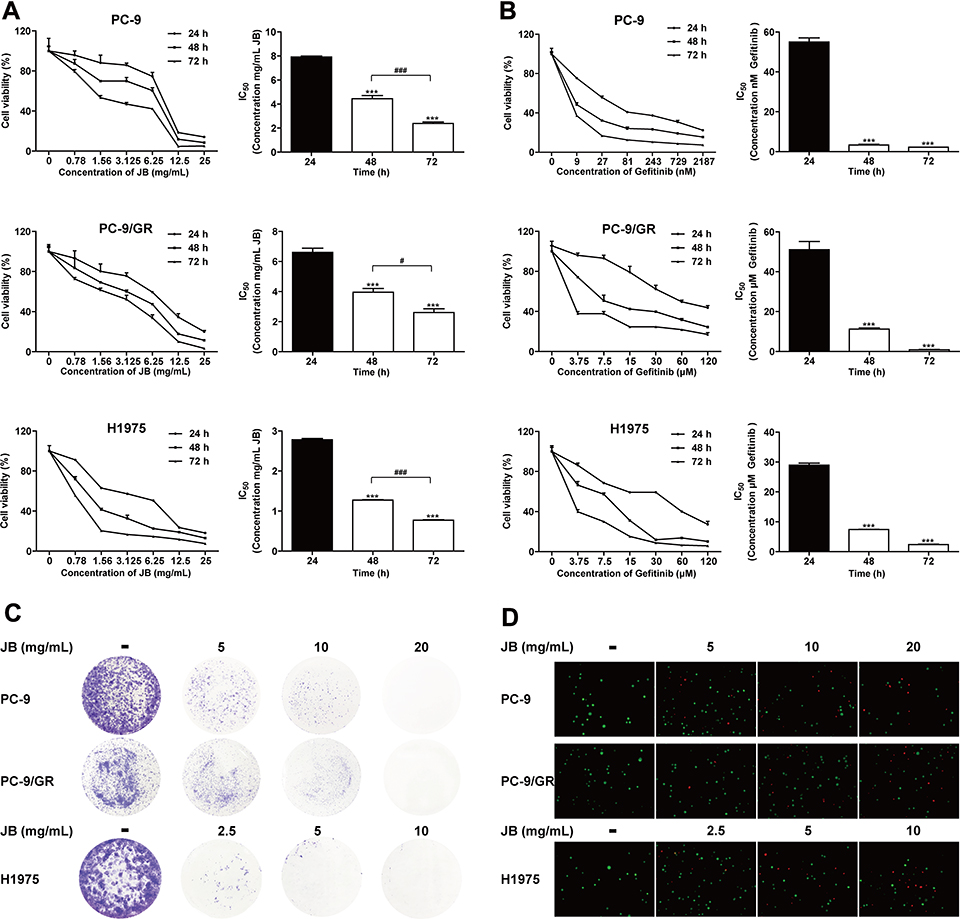 |
Figure 1 JB suppresses cell growth in NSCLC cells. (A) PC-9, PC-9/GR and H1975 cells were treated with JB for 24, 48 or 72h at the doses indicated. Cell viability was detected by MTT assay. ***p<0.001 compared to 24h group. #p<0.05 or ###p<0.001 compared to 48h group. (B) Cells were treated with gefitinib for 24, 48 or 72h at the doses indicated. Cell viability was detected by MTT assay. ***p<0.001 compared to 24h group. (C) Cells were treated with JB at the doses indicated. Representative images of colony forming assay were shown. (D) Cells were treated with JB for 24h, following by staining with Calcein AM (live) and PI (dead). The live (green)/dead (red) cells were observed via the fluorescence microscopy. |
To compare the inhibitory effect of JB with gefitinib, we detected the IC50 of gefitinib. As shown in Figure 1B, gefitinib significantly inhibited the proliferation of PC-9 cells with low doses and the IC50 were 55.02±2.06, 3.33±0.41 and 2.22±0.03 nM at 24, 48 and 72h. However, PC-9/GR and H1975 cells were resistant to gefitinib with the IC50 on PC-9/GR cells were 51.09±4.06, 11.23±0.52 and 0.88±0.16 μM, while H1975 cells were 28.95±0.67, 7.43±0.09 and 2.37±0.18 μM at 24, 48 and 72h.
To further confirm the effect of JB, colony-forming assay was performed. Results showed that JB markedly suppressed the formation of colony in a dose-dependent manner in PC-9, PC-9/GR and H1975 cells (Figure 1C). In addition, increasing concentration of JB treatment caused increasing ratio of dead cells in PC-9, PC-9/GR and H1975 cells (Figure 1D). These results demonstrated that JB induced cell growth inhibition and cell death in NSCLC cells.
Above all, JB induced cell growth inhibition and cell death in both gefitinib-sensitive PC-9 and gefitinib-resistant PC-9/GR, H1975 cells with similar sensitivity.
JB Induced Apoptosis by Activating Mitochondrial Signal Pathway
Previous study demonstrated that JB promoted cell death in PC-9, PC-9/GR and H1975 cells, we endeavored to determine whether the reduction of cell number involved apoptosis. Thus, we measured the effect of JB on apoptosis. PC-9, PC-9/GR and H1975 cells were treated with JB for 24h, following by staining with fluorescent agents Annexin V and PI. Increasing apoptosis cells were detected in JB-treated groups, suggesting that JB induced cell death partially through apoptosis (Figure 2A).
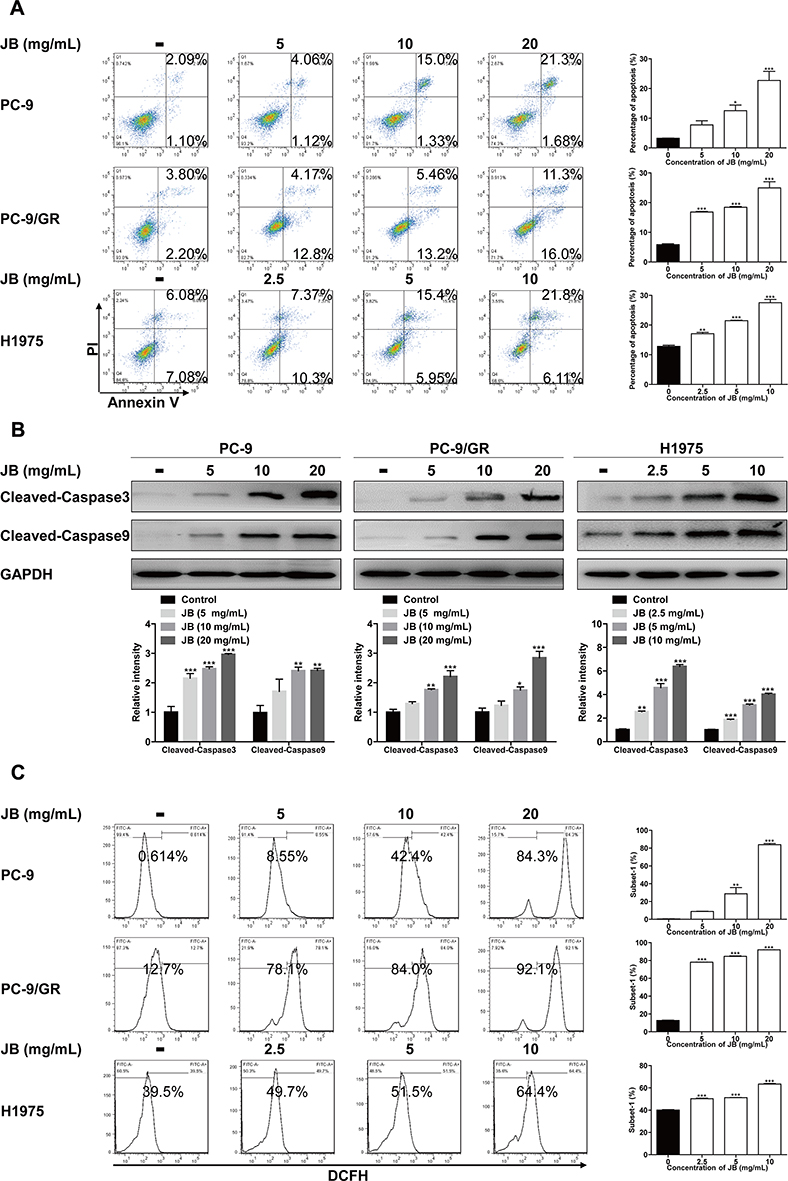 |
Figure 2 JB induces apoptosis and increases ROS production. (A) After being treated with indicated doses JB for 24h, PC-9, PC-9/GR and H1975 cells were stained with Annexin V and PI. Apoptosis cells were detected by flow cytometer. *p<0.05, **p<0.01 or ***p<0.001 compared to control group. (B) Cells were treated with JB at indicated doses for 24h. The protein level of cleaved-caspase3 and cleaved-caspase9 were detected by Western blot assay. GAPDH was used as a loading control. *p<0.05, **p<0.01 or ***p<0.001 compared to control group. (C) Cells were treated with JB for 24h. ROS production was monitored using 10μM DCFH-DA and detected by flow cytometer. **p<0.01 or ***p<0.001 compared to control group. |
The activation of a series of caspase cascade is the final pathway that causes execution of the cell apoptosis.26 To uncover the mechanism of JB induced apoptosis, Western blot assay for caspase cascade was performed. Apoptosis occurs through two major pathways, the extrinsic pathway (or death receptor pathway) and the intrinsic pathway (or mitochondrial pathway), both of which converge to caspase3.27 Results showed that JB treatment dose-dependently increased cleaved-caspase3 protein level in PC-9, PC-9/GR and H1975 cells (Figure 2B). Moreover, we detected the protein expression of cleaved-caspase9 and found similar results with cleaved-caspase3, suggesting that JB induced apoptosis belonged to the mitochondrial pathway. Then we further determined the level of ROS, a production generated along with mitochondrial permeabilization process.28 Cells were exposed to raising concentration of JB for 24h. As shown in Figure 2C, the ROS level was markedly increased in JB treatment groups. Taken together, JB mediated mitochondria-mediated apoptosis in NSCLC cells, applying by increased ROS production.
JB Induced Mitochondria-Mediated Apoptosis Through Altering Mitochondrial Membrane Permeable and Inhibiting Bcl-2 Mitochondrial Translocation
In response to stimuli, the integrity of mitochondria is damaged and become permeable, along with the release of cytochrome c.29 Here, we determined cytochrome c intracellular distribution. As shown in Figure 3A, JB treatment caused the decreasing of cytochrome c protein level in mitochondrial extracts in PC-9, PC-9/GR and H1975 cells. Additionally, the results of Western blot assay also indicated that Bax was accumulated in mitochondria while Bcl-2 mitochondrial translocation was reduced by JB treatment. However, the protein level of Bcl-xl, Mcl-1 and Bak in mitochondria were not changed with significance, indicating that decrease in Bcl-2 and subsequent increase in BAX is important for JB’s activity. It has been reported that Bcl-2 family proteins contributed to mitochondrial permeability transition.30 In further immunofluorescence assay study, we directly found that mitochondria (Red) was significantly decreased in response to JB, especially in high dose group. Meanwhile, Bcl-2 mitochondrial translocation (Green) was seen to be inhibited by JB (Figure 3B). These data demonstrated that the mechanism of JB induced mitochondria-mediated apoptosis was associated with increasing mitochondrial membrane permeable and Bcl-2 mitochondrial translocation inhibition.
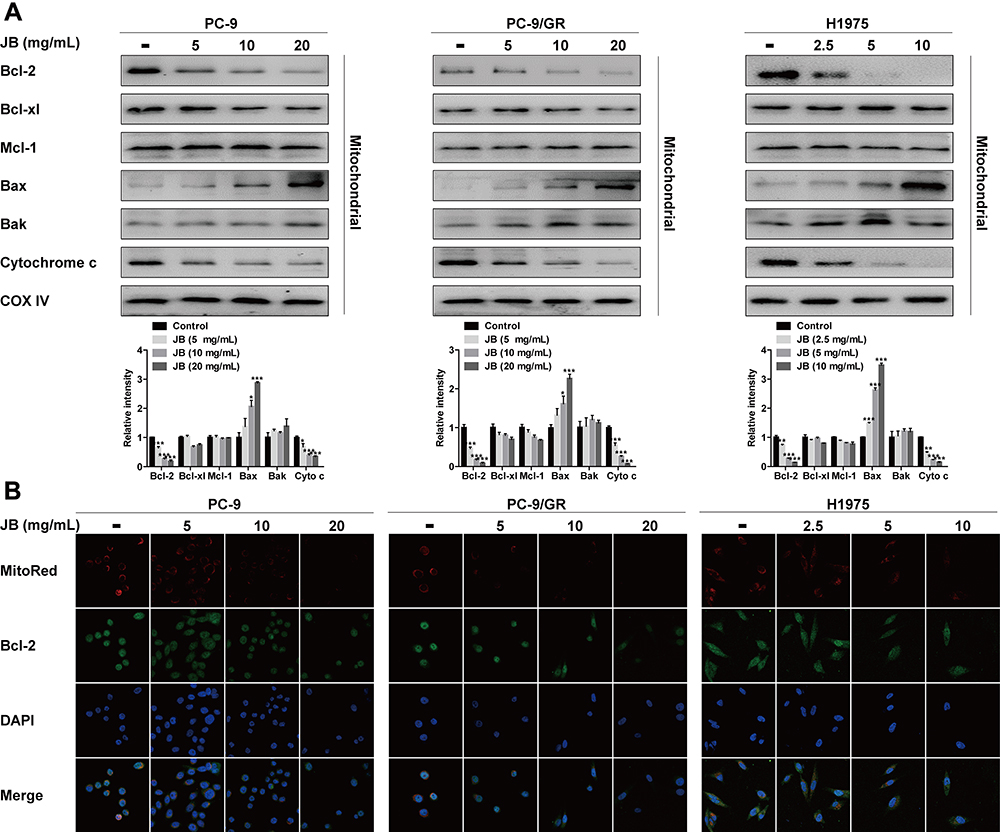 |
Figure 3 JB increases mitochondrial membrane permeable and inhibits Bcl-2 mitochondrial translocation. (A) PC-9, PC-9/GR and H1975 cells were treated with JB at indicated doses for 24h. The distribution of Bcl-2, Bcl-xl, Mcl-1, Bax, Bak and Cytochrome C in mitochondria was detected by Western blot assay. COX IV was used as a mitochondrial protein control to monitor for equal loading. *p<0.05 or ***p<0.001 compared to control group. (B) After treatment of cells with JB for 24h, immunofluorescence assay was conducted to detect Bcl-2 mitochondrial translocation. Confocal images showed the fluorescence of mitochondria in red, Bcl-2 in green, nucleus in blue, and the merged images in the bottom. |
Induction of Mitochondria-Mediated Apoptosis by JB Appeared to Be Reliant on Inactivation of PI3K/AKT and MAPK Signal Pathways
PI3K/AKT and MAPK signal pathways are two vital survival pathways downstream EGFR in tumors. The reactivation of EGFR and two pathways occurs at multiple gefitinib-resistant NSCLC patients.31,32 To investigate whether JB could directly suppress EGFR, AKT and ERK phosphorylation, Western blot assay was conducted. As shown in Figure 4A, JB decreased p-AKT and p-ERK protein expression in PC-9, PC-9/GR and H1975 cells in a dose-dependent manner, while the total EGFR, AKT and ERK protein level remains constant throughout the course of JB treatment. However, JB inhibited EGFR phosphorylation significantly in PC-9 and H1975 cells but slightly in PC-9/GR cells. These data suggested that the inhibition of AKT and ERK phosphorylation might be involved in JB’s antitumor effects on NSCLC cells.
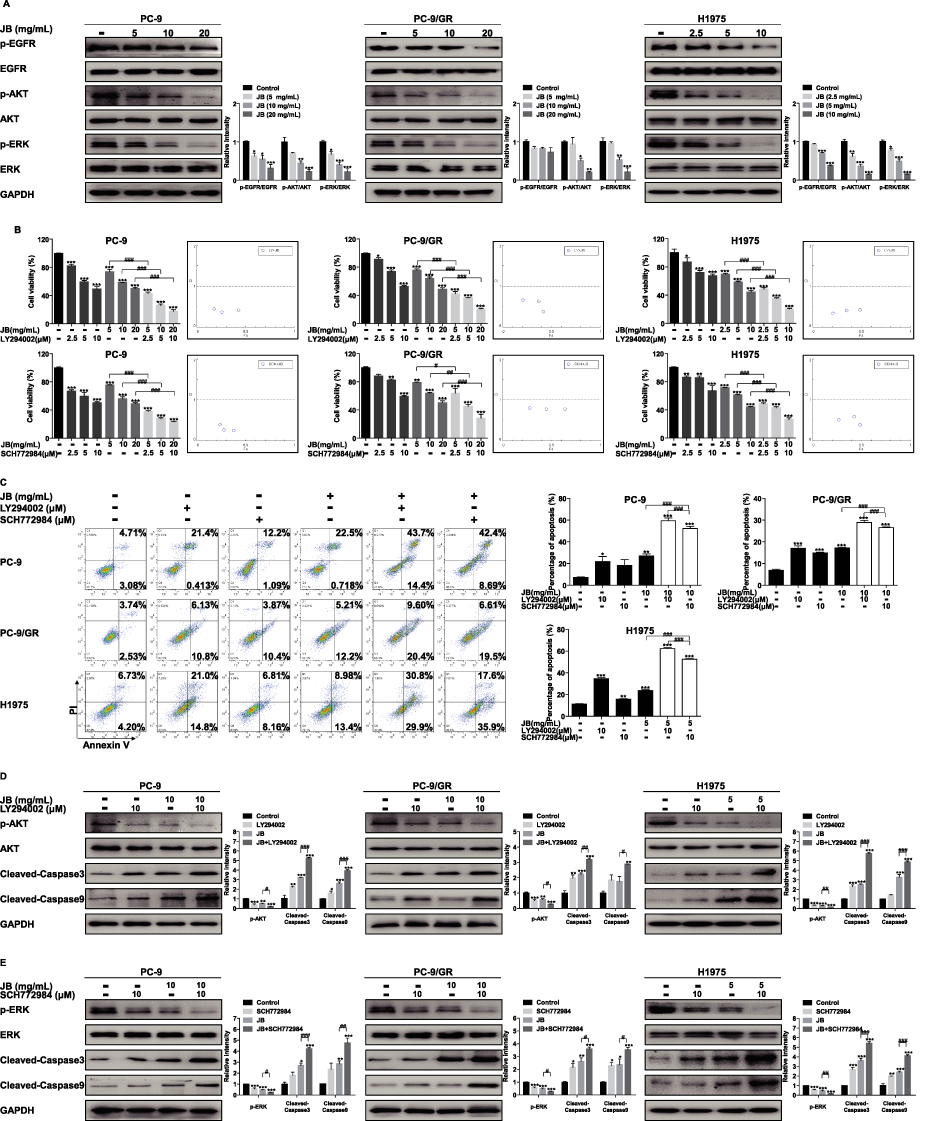 |
Figure 4 JB induces mitochondria-mediated apoptosis through inhibition of PI3K/AKT and MAPK signal pathways. (A) PC-9, PC-9/GR and H1975 cells were treated with JB at indicated doses for 24h. The expression of p-EGFR, EGFR, p-AKT, AKT, p-ERK and ERK protein were assessed by Western blot assay. GAPDH was used as a loading control. *p<0.05, **p<0.01 or ***p<0.001 compared to control group. (B) Cells were treated with JB, LY294002 or SCH772984 at the doses indicated for 24h. Cell viability was detected by MTT assay. *p<0.05, **p<0.01 or ***p<0.001 compared to control group. #p<0.05, ##p<0.01 or ###p<0.001 compared to JB group. The Fa-CI (Combination Index) Plot was generated using CompuSyn software. Points that fall below the line indicate synergistic relationship between the two drugs. (C) After treatment of cells with JB, LY294002 or SCH772984 at the doses indicated, cells were stained with Annexin V and PI, and apoptosis cells were detected by flow cytometer. *p<0.05, **p<0.01 or ***p<0.001 compared to control group. ###p<0.001 compared to JB group. (D) After treatment of cells with JB or LY294002 at the doses indicated for 24h, Western blot assay was performed to determine p-AKT, AKT, cleaved-caspase3 and cleaved-caspase9 protein expression. GAPDH was used as a loading control. *p<0.05, **p<0.01 or ***p<0.001 compared to control group. #p<0.05, ##p<0.01 or ###p<0.001 compared to JB group. (E) Cells were treated with JB or SCH772984 at the doses indicated for 24h. The expression of p-ERK and ERK, cleaved-caspase3 and cleaved-caspase9 proteins was detected by Western blot assay. GAPDH was used as a loading control. *p<0.05, **p<0.01 or ***p<0.001 compared to control group. #p<0.05, ##p<0.01 or ###p<0.001 compared to JB group. |
Evidence showed that PI3K/AKT and MAPK signal pathways are associated with protection against cell apoptosis by regulating Bcl-2 family proteins.33 To further study the role of PI3K/AKT and MAPK pathways on JB induced cell viability inhibition and apoptosis, MTT assay and apoptosis assay were performed. PC-9, PC-9/GR and H1975 cells were pretreated with LY294002 (a PI3K/AKT inhibitor) and SCH772984 (an ERK inhibitor) for 1h before treatment with JB. The results of MTT assay showed that LY294002 and SCH772984 markedly decreased cell viability, not only alone but also in the presence of JB. Additionally, both LY294002 and SCH772984 enhanced the effect of JB on cell growth. Results of the Fa-CI (Combination Index) Plot demonstrated that JB and LY294002 had synergistic effects on PC-9 (CI: 0.365–0.446), PC-9/GR (CI: 0.380–0.654), as well as H1975 (CI: 0.339–0.427) cells. Meanwhile, JB and SCH772984 also showed synergistic effects on PC-9 (CI: 0.249–0.392), PC-9/GR (CI: 0.771–0.799), as well as H1975 (CI: 0.391–0.589) cells (Figure 4B).
According to the results of apoptosis assay, we observed significant raised apoptosis cells in LY294002 and SCH772984 treatment group, similar to JB group. Furthermore, the combined treatment induced more apoptosis in PC-9, PC-9/GR and H1975 cells when compared with JB alone (Figure 4C). Then, the protein level of cleaved-caspase3 and cleaved-caspase9 was detected. As shown in Figure 4D, LY294002 enhanced the effect of JB on the protein expression of p-AKT and caspase cascade. Meanwhile, SCH772984 enhanced the effect of JB on the protein expression of p-ERK and caspase cascade (Figure 4E). These results suggested that PI3K/AKT and MAPK signal pathways were essential upstream targets for JB induced mitochondria-mediated apoptosis.
JB Synergized with Gefitinib to Induce Apoptosis
Previous studies have shown that JB induced cell growth inhibition and apoptosis in both gefitinib-sensitive and gefitinib-resistant cells, we wonder whether JB could enhance gefitinib sensitivity in NSCLC cells. To test the synergistic effect, MTT assay was performed. Cells were treated with gefitinib and JB alone or in combination for 24h. As shown in Figure 5A, the combined treatment significantly inhibited the growth of PC-9, PC-9/GR and H1975 cells when compared with a single drug alone. The results of the Fa-CI Plot showed that JB and gefitinib had synergistic effects on PC-9 (CI: 0.103–0.190), PC-9/GR (CI: 0.405–0.566), and H1975 (CI: 0.677–0.886) cells.
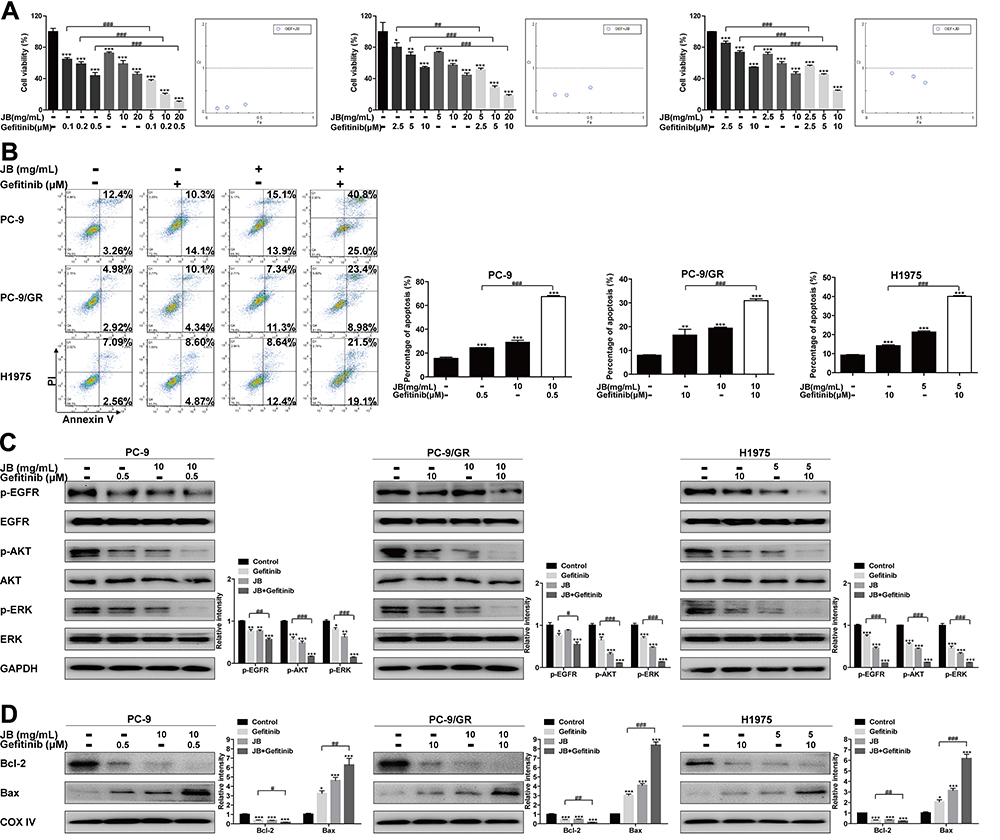 |
Figure 5 JB synergizes with gefitinib to induce cell growth inhibition and apoptosis in NSCLC cells. (A) PC-9, PC-9/GR and H1975 cells were treated with JB or gefitinib at the doses indicated for 24h. Cell viability was detected by MTT assay. *p<0.05, **p<0.01 or ***p<0.001 compared to control group. ##p<0.01 or ###p<0.001 compared to gefitinib group. The Fa-CI Plot was generated using CompuSyn software. (B) Cells were treated with JB or gefitinib at the doses indicated for 24h. Cells were then stained with Annexin V and PI. Apoptosis cells were detected by flow cytometer. **p<0.01 or ***p<0.001 compared to control group. ###p<0.001 compared to gefitinib group. (C) After treatment of cells with JB or gefitinib at indicated doses for 24h. The expression of p-EGFR, EGFR, p-AKT, AKT, p-ERK and ERK proteins was assessed by Western blot assay. GAPDH was used as a loading control. *p<0.05, **p<0.01 or ***p<0.001 compared to control group. #p<0.05, ##p<0.01 or ###p<0.001 compared to gefitinib group. (D) Cells were treated with JB or gefitinib at the doses indicated for 24h. The distribution of Bcl-2 and Bax in mitochondria was detected by Western blot assay. COX IV was used as a mitochondrial protein control to monitor for equal loading. *p<0.05 or ***p<0.001 compared to control group. #p<0.05, ##p<0.01 or ###p<0.001 compared to gefitinib group. |
To further evaluate the synergistic effect, apoptosis assay was performed. As shown in Figure 5B, JB synergized with gefitinib to induce apoptosis in PC-9, PC-9/GR and H1975 cells. Furthermore, the combined treatment decreased the phosphorylation of EGFR, AKT, ERK and Bcl-2 protein level in mitochondria, while increased the Bax level in mitochondria (Figure 5C and D). Altogether, these findings provided evidence that JB not only killed NSCLC cells directly but also could enhance gefitinib sensitivity.
Discussion
Lung cancer is a destructive disease with poor prognosis and the major cause of cancer-related deaths worldwide.34,35 NSCLC represents 85% histological subtype of the disease, which remains a serious health issue and heave economic burden.36,37 The expanding spectrum of oncogenic driver mutations identified in NSCLC, such as EGFR, coupled with the growing number of clinically available target therapeutic drugs, offers an opportunity to improve patient outcome. Although these molecular drugs bring about tremendous advances in the treatment of NSCLC, almost all patients become incurable because of drug resistance.38 The mechanisms of resistance to gefitinib, an FDA-approved first-generation EGFR-TKI for the treatment of NSCLC with EGFR mutations, can be described as two aspects, one is the secondary mutation on EGFR and another is the activation in the downstream and parallel pathways. Upon recognizing these molecular alterations underlying the development of gefitinib resistance, strategies like the development of third-generation EGRF-TKIs and combinatorial therapeutic regimen are applied in clinical practice.39 However, gefitinib resistance has never been completely resolved. The new strategies for the treatment of NSCLC harboring EGFR mutations and those under gefitinib resistance are needed urgently.
Several components of JB, such as Platycodon grandiflorum, Scutellaria baicalensis and Glycyrrhiza Uralensis were recently reported to suppress tumor growth in NSCLC.40–43 Yet, the direct effect of JB has not been confirmed. In this study, we firstly demonstrated that JB effectively inhibited the proliferation of gefitinib-sensitive PC-9 cells and gefitinib-resistant PC-9/GR, H1975 cells with similar sensitivity. Additionally, we observed significant cell death induced by JB in live/dead staining assay and wondered whether the death was due to apoptosis. Apoptosis, also called programmed cell death, is a physiological process that controls cell death when DNA damage is not repaired thus protecting tissue homeostasis.44 Inhibition of apoptosis is regarded as an important step in lung cancer genesis, allowing infinite proliferation of cancer cells.45 Standard platinum-based chemotherapy and EGFR-TKIs target therapy kill lung cancer cells at least partially through activating apoptotic signal pathway. Recently studies suggested that the decreased apoptotic response to EGFR-TKIs occurred in resistant NSCLC cells.46 Thereby, induction of apoptosis is a promising strategy to eradicate NSCLC cells and even to overcome gefitinib resistance. In the present study, JB induced cell apoptosis in a dose-dependent manner with the increased protein expression of active caspase3. Caspase3 is a key executioner of caspase involved in two major apoptotic pathways, the intrinsic pathway (or mitochondrial pathway) and the extrinsic pathway (or death receptor pathway). Apart from caspase3 being cleaved, the mitochondrial pathway is also featured as cytochrome c releasing from mitochondria following by combining with Apaf-1 and procaspase9 to produce active caspase9, thus cleaving procaspase3 into active caspase3 and triggering apoptosis.47 Accumulating evidence has shown that ROS function as promoter of cytochrome c releasing from mitochondria in the Bax-dependent intrinsic apoptotic pathway.48 Our results showed that JB increased the protein level of cleaved-caspase9. Meanwhile, JB decreased the cytochrome c protein level in mitochondria and induced accumulation of ROS production in NSCLC cells, indicating that mitochondrial apoptotic pathway was active.
The Bcl-2 family proteins, including pro-apoptotic proteins, such as Bax, Bad and Bak, and anti-apoptotic proteins, such as Bcl-2, Bcl-xl and Mcl-1, regulate the mitochondrial disruption.49 Pro-apoptotic proteins like Bax and Bak act as promotors of mitochondrial pathway. When receiving stimulation, Bax and Bak translocate into mitochondrial membrane and increase mitochondrial out membrane permeabilization, leading the release of cytochrome c and caspase cascade activation.50 Additionally, anti-apoptotic proteins act as suppressors by blocking the release of cytochrome c. It has been reported that Bcl-2 could directly decrease the mitochondrial membrane permeabilization by binding with the voltage-dependent anion channel 1 (VDAC-1), an outer mitochondrial membrane protein.51 Thereby, induction of Bax translocation into mitochondria and inhibition of Bcl-2 mitochondrial translocation are two strategies to induce mitochondria-mediated apoptosis. To explore the mechanism underlying JB induced apoptosis in PC-9, PC-9/GR and H1975 cells, we investigated the protein level of Bcl-2, Bcl-xl, Mcl-1, Bax and Bak in the mitochondria. The results showed that JB increased the expression of Bax while decreased Bcl-2 expression in the mitochondria. However, Bcl-xl, Mcl-1 and Bak protein level in mitochondria were not changed significantly, indicating that decrease in Bcl-2 and subsequent increase in Bax but not Bcl-xl, Mcl-1, Bak is important for JB’s activity. In addition, confocal microscopy suggested that Bcl-2 translocation to mitochondria increased in response to JB treatment.
EGFR is the most common mutated oncogenic gene in NSCLC patients and is crucial for the survival of tumor. PI3K/AKT and MAPK signal pathways are two important pathways downstream EGFR, regulating cell growth, apoptosis and metabolism by phosphorylating a series of substrates in NSCLC. The reactivation of EGFR and the two pathways was found in EGFR-TKIs-therapy-resistant patients.52–54 Concurrently, our previous studies have found that PI3K/AKT and MAPK signal pathways were reactivated in PC-9/GR and H1975 cells even under the pressure of gefitinib, which suggested that the reactivation of PI3K/AKT and MAPK signal pathways was partially leading cells to become resistant to gefitinib.55,56 Additionally, Integrin beta 1 and miR-200c over-expression were also reported in PC-9/GR cells, which related to the activation of PI3K/AKT and MAPK signal pathways.21,57 So we detected the effect of JB on EGFR, PI3K/AKT and MAPK signal pathways. Results showed that JB could directly inhibit AKT and ERK phosphorylation in a dose-dependent manner while had no effect on the expression of total AKT and ERK in PC-9 and H1975 cells. However, in PC-9/GR cells, JB could only inhibit AKT and ERK phosphorylation significantly while slightly inhibited EGFR phosphorylation. These interesting phenomena revealed that the mechanism of JB’s anticancer effect in PC-9/GR cells was different from gefitinib.
PI3K/AKT signal pathway was reported to block cell apoptosis by phosphorylating Bad.58 Recently, studies showed that MAPK signal pathway affects the Bcl-2 protein expression.12 In order to uncover the role of PI3K/AKT and MAPK signal pathways on JB induced apoptosis, we combined JB with the inhibitor of AKT (LY294002) and ERK (SCH772984) respectively. Results showed that LY294002 and SCH772984 synergized with JB to induce apoptosis in PC-9, PC-9/GR and H1975 cells, as well as inhibiting the level of p-AKT and p-ERK and increasing the level of cleaved-caspase3 and cleaved-caspase9. These findings indicated that PI3K/AKT and MAPK signal pathways were vital upstream targets for JB induced mitochondria-mediated apoptosis.
The mechanisms of gefitinib resistance have been identified, including T790M mutation, Epithelial-mesenchymal transition (EMT) and bypass signal pathways reactivation, but resistant NSCLC cells are largely still dependent on EGFR signaling.38,59 It is a potential therapeutic strategy to develop methods to enhance gefitinib sensitivity in NSCLC cells. In this study, we found that JB synergized with gefitinib to inhibit cell proliferation and induce apoptosis in PC-9, PC-9/GR and H1975 cells. Thus, adjuvant therapy of JB could be a potential strategy to overcome gefitinib resistance in NSCLC.
However, in the present study, we roughly just assessed the overall roles of JB in NSCLC cells, while lacking in vivo work and organoid work. It is important for further work to be done before JB being used in clinical practice to assess this feasibility.
Conclusion
In conclusion, we demonstrated that JB inhibited cell growth both in gefitinib-sensitive and gefitinib-resistant cells. JB could induce mitochondria-mediated apoptosis by inhibiting the activation of PI3K/AKT and MAPK signal pathways. Additionally, JB could also synergize with gefitinib to inhibit NSCLC cell proliferation and induce apoptosis (Figure 6).
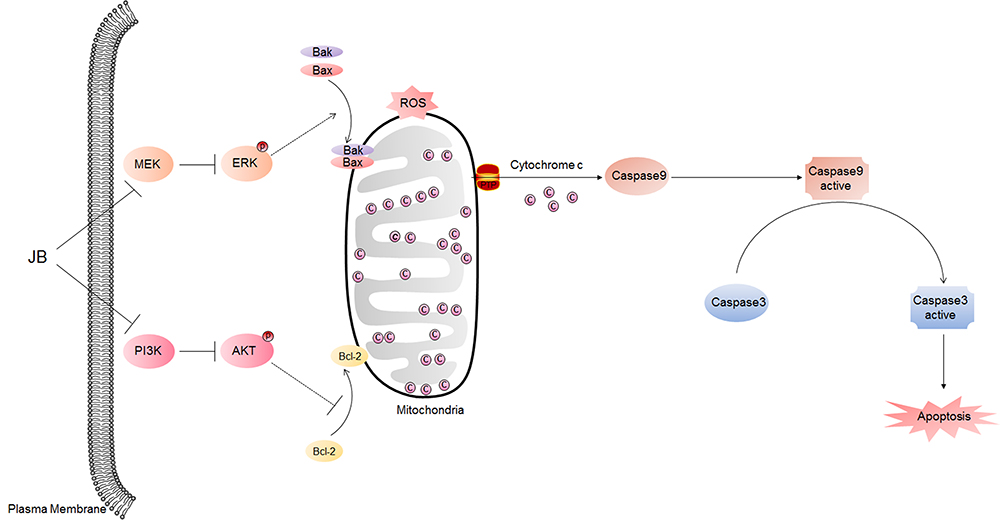 |
Figure 6 Possible molecular mechanisms involved in JB induced apoptosis in NSCLC cells. JB regulates PI3K/AKT and MAPK signal pathways by inhibiting AKT and ERK phosphorylation. Then, the Bcl-2 translocated away from mitochondria while Bax translocated into mitochondria, leading to mitochondria membrane permeable increasing, along with ROS production raising. Next, cytochrome c releases from mitochondria into cytoplasm, where it combines with pro-caspase9 and apoptosis protease activating factor-1 (Apaf-1) to produce active caspase9 (cleaved-caspase9). Active caspase9 cleaves pro-caspase3 to active caspase3 (cleaved-caspase3) and triggers apoptosis in PC-9, PC-9/GR and H1975 cells. |
Abbreviations
AKT, serine/threonine-specific protein kinas; cAMP, cyclic adenosine monophosphate; COX, cytochrome c oxidase; CREB, cAMP response element binding protein; DAPI, 4′:6-diamidino-2-phenylindole; DCFH-DA, dichlorodihydrofluorescein diacetate; DMSO, dissolved in dimethyl sulfoxide; EGFR, epidermal growth factor receptor; EGFR-TKIs, EGFR tyrosine kinase inhibitors; EMT, epithelial-mesenchymal transition; ERK, extracellular signal-regulated kinase; FITC, fluorescein isothiocyanate; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; HRP, horseradish peroxidase; JB, JuBei oral liquid; MAPK, mitogen-activated protein kinase; mPTP, mitochondrial permeability transition pore; mTOR, mammalian target of rapamycin; MTT, 3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide; NSCLC, non-small cell lung cancer; PI, propidium iodide; PI3K, phosphatidylinositol-4, 5-bisphosphate 3-kinase; PVDF, polyvinylidene difluoride; ROS, reactive oxygen species.
Data Sharing Statement
The data used and analyzed during the current study are available from the corresponding author on reasonable request.
Acknowledgments
Thanks to Dr. Zhou Caicun for providing Human lung adenocarcinoma PC-9, PC-9/GR, H1975 cells as kind gifts. Thanks to Dr. Rongpin Tao for helping complete our research work.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Kosaka T, Yatabe Y, Endoh H, Kuwano H, Takahashi T, Mitsudomi T. Mutations of the epidermal growth factor receptor gene in lung cancer: biological and clinical implications. Cancer Res. 2004;64(24):8919–8923. doi:10.1158/0008-5472.CAN-04-2818
2. Pao W, Chmielecki J. Rational, biologically based treatment of EGFR-mutant non-small-cell lung cancer. Nat Rev Cancer. 2010;10(11):760–774. doi:10.1038/nrc2947
3. Maemondo M, Inoue A, Kobayashi K, et al. Gefitinib or chemotherapy for non-small-cell lung cancer with mutated EGFR. N Engl J Med. 2010;362(25):2380–2388. doi:10.1056/NEJMoa0909530
4. Rosell R, Carcereny E, Gervais R, et al. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): a multicentre, open-label, randomised Phase 3 trial. Lancet Oncol. 2012;13(3):239–246. doi:10.1016/S1470-2045(11)70393-X
5. Rosell R, Moran T, Queralt C, et al. Screening for epidermal growth factor receptor mutations in lung cancer. N Engl J Med. 2009;361(10):958–967. doi:10.1056/NEJMoa0904554
6. Mok TS, Wu YL, Ahn MJ, et al. Osimertinib or platinum-pemetrexed in EGFR T790M-positive lung cancer. N Engl J Med. 2017;376(7):629–640. doi:10.1056/NEJMoa1612674
7. Govindan R. Overcoming resistance to targeted therapy for lung cancer. N Engl J Med. 2015;372(18):1760–1761. doi:10.1056/NEJMe1500181
8. Ramalingam SS, Yang JC, Lee CK, et al. Osimertinib as first-line treatment of EGFR mutation-positive advanced non-small-cell lung cancer. J Clin Oncol. 2018;36(9):841–849. doi:10.1200/JCO.2017.74.7576
9. Gong Y, Somwar R, Politi K, et al. Induction of BIM is essential for apoptosis triggered by EGFR kinase inhibitors in mutant EGFR-dependent lung adenocarcinomas. PLoS Med. 2007;4(10):e294. doi:10.1371/journal.pmed.0040294
10. Marsden VS, O’Connor L, O’Reilly LA, et al. Apoptosis initiated by Bcl-2-regulated caspase activation independently of the cytochrome c/Apaf-1/caspase-9 apoptosome. Nature. 2002;419(6907):634–637. doi:10.1038/nature01101
11. van Loo G, Saelens X, van Gurp M, MacFarlane M, Martin SJ, Vandenabeele P. The role of mitochondrial factors in apoptosis: a Russian roulette with more than one bullet. Cell Death Differ. 2002;9(10):1031–1042. doi:10.1038/sj.cdd.4401088
12. Pugazhenthi S, Nesterova A, Sable C, et al. Akt/protein kinase B up-regulates Bcl-2 expression through cAMP-response element-binding protein. J Biol Chem. 2000;275(15):10761–10766. doi:10.1074/jbc.275.15.10761
13. Changchien JJ, Chen YJ, Huang CH, Cheng TL, Lin SR, Chang LS. Quinacrine induces apoptosis in human leukemia K562 cells via p38 MAPK-elicited BCL2 down-regulation and suppression of ERK/c-Jun-mediated BCL2L1 expression. Toxicol Appl Pharmacol. 2015;284(1):33–41. doi:10.1016/j.taap.2015.02.005
14. Zhang X, Tang N, Hadden TJ, Rishi AK. Akt, FoxO and regulation of apoptosis. Biochim Biophys Acta. 2011;1813(11):1978–1986. doi:10.1016/j.bbamcr.2011.03.010
15. Yim NH, Hwang YH, Liang C, Ma JY. A platycoside-rich fraction from the root of Platycodon grandiflorum enhances cell death in A549 human lung carcinoma cells via mainly AMPK/mTOR/AKT signal-mediated autophagy induction. J Ethnopharmacol. 2016;194:1060–1068. doi:10.1016/j.jep.2016.10.078
16. Park DI, Lee JH, Moon SK, et al. Induction of apoptosis and inhibition of telomerase activity by aqueous extract from Platycodon grandiflorum in human lung carcinoma cells. Pharmacol Res. 2005;51(5):437–443. doi:10.1016/j.phrs.2004.11.003
17. Chen J, Yuan J, Zhou L, et al. Regulation of different components from Ophiopogon japonicus on autophagy in human lung adenocarcinoma A549Cells through PI3K/Akt/mTOR signaling pathway. Biomed Pharmacother. 2017;87:118–126. doi:10.1016/j.biopha.2016.12.093
18. Gao J, Morgan WA, Sanchez-Medina A, Corcoran O. The ethanol extract of Scutellaria baicalensis and the active compounds induce cell cycle arrest and apoptosis including upregulation of p53 and Bax in human lung cancer cells. Toxicol Appl Pharmacol. 2011;254(3):221–228. doi:10.1016/j.taap.2011.03.016
19. Huang TH, Wu TH, Guo YH, Li TL, Chan YL, Wu CJ. The concurrent treatment of Scutellaria baicalensis georgi enhances the therapeutic efficacy of cisplatin but also attenuates chemotherapy-induced cachexia and acute kidney injury. J Ethnopharmacol. 2019;243:112075. doi:10.1016/j.jep.2019.112075
20. Qiu C, Zhang T, Zhang W, et al. Licochalcone a inhibits the proliferation of human lung cancer cell lines A549 and H460 by inducing G2/M cell cycle arrest and ER stress. Int J Mol Sci. 2017;18(8):1761. doi:10.3390/ijms18081761
21. Ju L, Zhou C, Li W, Yan L. Integrin beta1 over-expression associates with resistance to tyrosine kinase inhibitor gefitinib in non-small cell lung cancer. J Cell Biochem. 2010;111(6):1565–1574. doi:10.1002/jcb.22888
22. Pan H, Jiang T, Cheng N, et al. Long non-coding RNA BC087858 induces non-T790M mutation acquired resistance to EGFR-TKIs by activating PI3K/AKT and MEK/ERK pathways and EMT in non-small-cell lung cancer. Oncotarget. 2016;7(31):49948–49960. doi:10.18632/oncotarget.10521
23. Chou TC, Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 1984;22:27–55. doi:10.1016/0065-2571(84)90007-4
24. Wheeler DL, Dunn EF, Harari PM. Understanding resistance to EGFR inhibitors-impact on future treatment strategies. Nat Rev Clin Oncol. 2010;7(9):493–507. doi:10.1038/nrclinonc.2010.97
25. Bivona TG, Doebele RC. A framework for understanding and targeting residual disease in oncogene-driven solid cancers. Nat Med. 2016;22(5):472–478. doi:10.1038/nm.4091
26. Soldatenkov VA, Smulson M. Poly(ADP-ribose) polymerase in DNA damage-response pathway: implications for radiation oncology. Int J Cancer. 2000;90(2):59–67. doi:10.1002/(SICI)1097-0215(20000420)90:2<59::AID-IJC1>3.0.CO;2-4
27. Ghobrial IM, Witzig TE, Adjei AA. Targeting apoptosis pathways in cancer therapy. CA Cancer J Clin. 2005;55(3):178–194. doi:10.3322/canjclin.55.3.178
28. Polster BM, Fiskum G. Mitochondrial mechanisms of neural cell apoptosis. J Neurochem. 2004;90(6):1281–1289. doi:10.1111/j.1471-4159.2004.02572.x
29. Li H, Kolluri SK, Gu J, et al. Cytochrome c release and apoptosis induced by mitochondrial targeting of nuclear orphan receptor TR3. Science. 2000;289(5482):1159–1164. doi:10.1126/science.289.5482.1159
30. Marzo I, Brenner C, Zamzami N, et al. The permeability transition pore complex: a target for apoptosis regulation by caspases and Bcl-2–related proteins. J Exp Med. 1998;187(8):1261–1271. doi:10.1084/jem.187.8.1261
31. Ohashi K, Sequist LV, Arcila ME, et al. Lung cancers with acquired resistance to EGFR inhibitors occasionally harbor BRAF gene mutations but lack mutations in KRAS, NRAS, or MEK1. Proc Natl Acad Sci U S A. 2012;109(31):E2127–2133. doi:10.1073/pnas.1203530109
32. Ludovini V, Bianconi F, Pistola L, et al. Phosphoinositide-3-kinase catalytic alpha and KRAS mutations are important predictors of resistance to therapy with epidermal growth factor receptor tyrosine kinase inhibitors in patients with advanced non-small cell lung cancer. J Thorac Oncol. 2011;6(4):707–715. doi:10.1097/JTO.0b013e31820a3a6b
33. Kwon OS, Hong SK, Kwon SJ, Go YH, Oh E, Cha HJ. BCL2 induced by LAMTOR3/MAPK is a druggable target of chemoradioresistance in mesenchymal lung cancer. Cancer Lett. 2017;403:48–58. doi:10.1016/j.canlet.2017.05.019
34. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin. 2020;70(1):7–30. doi:10.3322/caac.21590
35. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424. doi:10.3322/caac.21492
36. Meza R, Meernik C, Jeon J, Cote ML. Lung cancer incidence trends by gender, race and histology in the United States, 1973–2010. PLoS One. 2015;10(3):e0121323. doi:10.1371/journal.pone.0121323
37. Molina JR, Yang P, Cassivi SD, Schild SE, Adjei AA. Non-small cell lung cancer: epidemiology, risk factors, treatment, and survivorship. Mayo Clin Proc. 2008;83(5):584–594. doi:10.1016/S0025-6196(11)60735-0
38. Wu SG, Shih JY. Management of acquired resistance to EGFR TKI-targeted therapy in advanced non-small cell lung cancer. Mol Cancer. 2018;17(1):38. doi:10.1186/s12943-018-0777-1
39. Rotow J, Bivona TG. Understanding and targeting resistance mechanisms in NSCLC. Nat Rev Cancer. 2017;17(11):637–658. doi:10.1038/nrc.2017.84
40. Li Y, Wu Y, Xia Q, Zhao Y, Zhao R, Deng S. Platycodon grandiflorus enhances the effect of DDP against lung cancer by down regulating PI3K/Akt signaling pathway. Biomed Pharmacother. 2019;120:109496. doi:10.1016/j.biopha.2019.109496
41. Seo YS, Kang OH, Kong R, et al. Polygalacin D induces apoptosis and cell cycle arrest via the PI3K/Akt pathway in non-small cell lung cancer. Oncol Rep. 2018;39(4):1702–1710. doi:10.3892/or.2018.6230
42. Cheng CS, Chen J, Tan HY, Wang N, Chen Z, Feng Y. Scutellaria baicalensis and cancer treatment: recent progress and perspectives in biomedical and clinical studies. Am J Chin Med. 2018;46(1):25–54. doi:10.1142/S0192415X18500027
43. Tang ZH, Chen X, Wang ZY, et al. Induction of C/EBP homologous protein-mediated apoptosis and autophagy by licochalcone A in non-small cell lung cancer cells. Sci Rep. 2016;6(1):26241. doi:10.1038/srep26241
44. Wong RS. Apoptosis in cancer: from pathogenesis to treatment. J Exp Clin Cancer Res. 2011;30(1):87. doi:10.1186/1756-9966-30-87
45. Dean EJ, Ranson M, Blackhall F, Holt SV, Dive C. Novel therapeutic targets in lung cancer: inhibitor of apoptosis proteins from laboratory to clinic. Cancer Treat Rev. 2007;33(2):203–212. doi:10.1016/j.ctrv.2006.11.002
46. Hata AN, Niederst MJ, Archibald HL, et al. Tumor cells can follow distinct evolutionary paths to become resistant to epidermal growth factor receptor inhibition. Nat Med. 2016;22(3):262–269. doi:10.1038/nm.4040
47. Liu G, Pei F, Yang F, et al. Role of autophagy and apoptosis in non-small-cell lung cancer. Int J Mol Sci. 2017;18(2):367. doi:10.3390/ijms18020367
48. Kirkland RA, Windelborn JA, Kasprzak JM, Franklin JL. A bax-induced pro-oxidant state is critical for cytochrome c release during programmed neuronal death. J Neurosci. 2002;22(15):6480–6490. doi:10.1523/JNEUROSCI.22-15-06480.2002
49. Korsmeyer SJ. BCL-2 gene family and the regulation of programmed cell death. Cancer Res. 1999;59(7 Suppl):1693s–1700s.
50. Maes ME, Schlamp CL, Nickells RW. BAX to basics: how the BCL2 gene family controls the death of retinal ganglion cells. Prog Retin Eye Res. 2017;57:1–25. doi:10.1016/j.preteyeres.2017.01.002
51. Arbel N, Shoshan-Barmatz V. Voltage-dependent anion channel 1-based peptides interact with Bcl-2 to prevent antiapoptotic activity. J Biol Chem. 2010;285(9):6053–6062. doi:10.1074/jbc.M109.082990
52. Liu Q, Yu S, Zhao W, Qin S, Chu Q, Wu K. EGFR-TKIs resistance via EGFR-independent signaling pathways. Mol Cancer. 2018;17(1):53. doi:10.1186/s12943-018-0793-1
53. Lemmon MA, Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2010;141(7):1117–1134. doi:10.1016/j.cell.2010.06.011
54. Yarden Y, Sliwkowski MX. Untangling the ErbB signalling network. Nat Rev Mol Cell Biol. 2001;2(2):127–137. doi:10.1038/35052073
55. Chen Q, Pan Z, Zhao M, et al. High cholesterol in lipid rafts reduces the sensitivity to EGFR-TKI therapy in non-small cell lung cancer. J Cell Physiol. 2018;233(9):6722–6732. doi:10.1002/jcp.26351
56. Pan Z, Wang K, Chen Q, Zheng X, Song Z, Ding X. SFI enhances therapeutic efficiency of gefitinib: an insight into reversal of resistance to targeted therapy in non-small cell lung cancer cells. J Cancer. 2020;11(2):334–344. doi:10.7150/jca.32989
57. Li J, Li X, Ren S, et al. miR-200c overexpression is associated with better efficacy of EGFR-TKIs in non-small cell lung cancer patients with EGFR wild-type. Oncotarget. 2014;5(17):7902–7916. doi:10.18632/oncotarget.2302
58. Franke TF, Hornik CP, Segev L, Shostak GA, Sugimoto C. PI3K/Akt and apoptosis: size matters. Oncogene. 2003;22(56):8983–8998. doi:10.1038/sj.onc.1207115
59. Jia Y, Yun CH, Park E, et al. Overcoming EGFR(T790M) and EGFR(C797S) resistance with mutant-selective allosteric inhibitors. Nature. 2016;534(7605):129–132. doi:10.1038/nature17960
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.