Back to Journals » OncoTargets and Therapy » Volume 9
JAK2 inhibitor combined with DC-activated AFP-specific T-cells enhances antitumor function in a Fas/FasL signal-independent pathway
Authors Liu Y, Wang Y, Ding G, Yang T, Yao L, Hua J, He Z, Qian M
Received 9 October 2015
Accepted for publication 7 March 2016
Published 20 July 2016 Volume 2016:9 Pages 4425—4433
DOI https://doi.org/10.2147/OTT.S97941
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Geoffrey Pietersz
Yang Liu,1 Yue-ru Wang,2 Guang-hui Ding,1 Ting-song Yang,1 Le Yao,1 Jie Hua,1 Zhi-gang He,1 Ming-ping Qian1
1Department of Hepatobiliary Surgery, Shanghai 10th People’s Hospital, School of Medicine, Tongji University, Shanghai, People’s Republic of China; 2Department of Infection, Shanghai First People’s Hospital Affiliated to Jiaotong University, Shanghai, People’s Republic of China
Objective: Combination therapy for cancer is more effective than using only standard chemo- or radiotherapy. Our previous results showed that dendritic cell-activated α-fetoprotein (AFP)-specific T-cells inhibit tumor in vitro and in vivo. In this study, we focused on antitumor function of CD8+ T-cells combined with or without JAK2 inhibitor.
Methods: Proliferation and cell cycle were analyzed by CCK-8 and flow cytometry. Western blot was used to analyze the expression level of related protein and signaling pathway.
Results: We demonstrated reduced viability and induction of apoptosis of tumor cells with combination treatment. Intriguingly, cell cycle was blocked at the G1 phase by using AFP-specific CD8+ T-cells combined with JAK2 inhibitor (AG490). Furthermore, an enhanced expression of BAX but no influence on Fas/FasL was detected from the tumor cells.
Conclusion: These results indicate a Fas/FasL-independent pathway for cellular apoptosis in cancer therapies with the treatment of AFP-specific CD8+ T-cells combined with JAK2 inhibitor.
Keywords: AFP-specific CD8+ T-cells, JAK2 inhibitor, Fas/FasL signal, antitumor, apoptosis
Introduction
Since the first time bacteria were injected directly into a tumor in 1891, treatment of cancers have been performed using various methods.1 Monoclonal antibodies have been used as vaccines to treat cancer for decades.2–5 Bevacizumab, which was given a trade name Avastin®, was found to be effective and was approved for treating colorectal cancer, non-small-cell lung cancer, breast cancer, and ovarian cancer, and it works by targeting vascular endothelial growth factor.6–13 Recently, a conjugated monoclonal antibody called ADCETRIS® was introduced; it targets CD30 on Hodgkin’s lymphoma and one type of non-Hodgkin’s lymphoma.14,15 In addition, large-scale production of cytokines is used to enhance immune responses against tumors in clinical cases. IL-2 alone and in combination with other cytokines is widely used for treating metastatic melanoma in the United States.16,17 Furthermore, researchers have tried to collect tumor infiltrating lymphocytes for re-expansion in vitro to give patients adoptive T-cell transfer therapies.18 Similarly, the strategy of infusing antigen-stimulated or antigen-specific cytotoxic T lymphocytes into patients was used and a convincing result was obtained.19 Despite considerable progress in this field, combined treatments with specific, efficient, and safe strategies remain poorly understood.
α-Fetoprotein (AFP) is highly expressed in the serum during embryonic development, especially in the fetal liver and gastrointestinal tract. Its expression level is very low in the serum of healthy adults, but an increased expression in serum is reported in up to 60%–70% of patients with hepatocellular carcinoma. In mouse models, AFP-based vaccines that were expressed by plasmid DNA could elicit specific CD8+ T-cell response against tumors expressing AFP.20,21 It was also reported that murine and human CD8+ T-cells specifically recognize AFP peptide epitopes.22,23 These data indicate that AFP is one of the possible targets in treatment of hepatocellular carcinoma (using specific vaccines).
As one of four protein-tyrosine kinases, JAK2 is an essential factor in cellular proliferation, differentiation, survival, and senescence.24–28 It plays an important role in mediating the cell signaling pathway through various membrane receptors binding with specific cytokine or growth factor. In recent studies, researchers demonstrated JAK2 as an ideal target for therapy on a number of cancers. JAK2V617F, which is a somatic mutation recognized in 2005, can be detected in patients with myeloproliferative neoplasms.29,30 Inhibition of JAK2 activity was thought to be a new strategy against its mutation. CYT387, a novel JAK2 inhibitor, was demonstrated to recover normal cytokines profiles in murine myeloproliferative neoplasms.31 However, the combination strategy of JAK2 inhibitor and immunotherapy is seldom reported.
Our previous study showed that dendritic cell (DC)-activated AFP-specific T-cells presented a promising approach for immunotherapy because of its antitumor effect.32 Here, we detected cytokines secreted by the CD8+ T-cells with different treatments in a coculture system. Combined with the JAK2 inhibitor, the production of antitumor cytokines was increased except for IL-10. On the basis of the enhanced antitumor effect, the ability of cell proliferation and livability of target HepG2 cells were reduced by coculturing with AFP-specific CD8+ T-cells and JAK2 inhibitor. It was noted that target cells were blocked in G1 phase in this treatment, which led to cell apoptosis at the end. By detecting key players in cell proliferation and apoptosis pathway, we found that the combined treatment involved a JAK2 inhibitor-induced apoptosis pathway with the high expression of Bax but no variation in Fas/FasL signal.
Materials and methods
Ethics
The study was approved by the Ethics Committee of Shanghai 10th People’s Hospital.
Preparation and identification of DCs and CD8+ T-cells
Peripheral blood mononuclear cells (PBMCs) were prepared from peripheral blood through venous puncture, followed by Ficoll gradient separation. PBMCs were cultured in RPMI 1640 medium supplemented with 10% human AB serum for 2 hours. The suspended cells were removed, and adherent cells in the same medium were recultured along with 800 U/mL GM-CSF and 500 U/mL IL-4 for 7 days. DCs were collected from adherent cells and their morphology and phenotyping were assessed. CD8+ T-cells were isolated from PBMCs through MACS (Miltenyi Biotec, Bergisch Gladbach, Germany) to examine purity through flow cytometry analysis.
Construction, infection, and coculture
AFP cassette was amplified from the expression vector pCMV-sport6-AFP with the following primers: Forward, 5′-CGG GGTACC GTC GAC ATG AAG TGG GTG GAA TCA AT-3′ and reverse: 5′-CCC AAG CTT CTC GAG TTA AAC TCC CAA AGC AGC AC-3′. The recombinant pAd-SV40 vector expressing AFP was digested with restriction enzymes, and then the fragment containing the AFP cassette was harvested by gel extraction following agarose gel electrophoresis. DCs were infected by adenovirus-expressing AFP, which was prepared as previously described.32 Briefly, pAd-SV40-AFP was transfected into 293T cells and cultured for 72 hours. The supernatant containing the recombinant adenovirus was then harvested and used to infect the DCs. DCs were infected with pAd-SV40-AFP at a multiplicity of infection of 10 for 2 hours. Infected DCs were washed and plated at 1×105 cells/mL to serve as stimulators for AFP-specific T-cell generation. Purified autologous CD8+ T-cells were plated with the infected DCs at 2×106 cells/mL in 10% AB serum in the presence of IL-7 (25 ng/mL). Cultures were supplemented with IL-2 at 10 U/mL every 3 days. Cells were harvested after culture for 7 days. For coculture, AFP-specific CD8+ T-cells were cocultured with equal number of HepG2 cells in a 96-well plate in triplicate for 24 hours. After coculturing for 24 hours, cell viability, cell cycle, and apoptosis analysis were performed. Empty pAd-SV40 vector-infected DC was used as a mock.
Cell viability assay
HepG2 cells were seeded in a 96-well plate (1×105 cells/well) and cultured at 37°C for 24 hours. Cell viability was assessed using Cell Counting Kit-8. Measurement was carried out at an absorbance of 450 nm with a microplate reader according to the manufacturer’s instructions.
Cell cycle analysis
HepG2 cells were harvested and fixed with ice-cold ethanol overnight after being cocultured with different groups of CD8+ T-cells. Fixed cells were washed three times with phosphate-buffered solution (PBS) and resuspended in PBS containing 500 ng/μL RNase A and incubated at 37°C for half an hour. All samples were analyzed by flow cytometry after staining with propidum iodide (PI) and 0.1% TritonX-100.
ELISA quantification of cytokines
About 2×105/well AFP-specific CD8+ T-cells with DCs (activated or inactivated) were cocultured with HepG2 cells for 24 hours. The supernatants were collected and analyzed for IL-2, IL-6, IL-10, tumor necrosis factor (TNF)-α, INF-γ, Granzyme B, and pore-forming protein (PFP) with the help of commercially available enzyme-linked immunosorbent assay (ELISA) kits (R&D System, Minneapolis, MN, USA) according to the manufacturer’s instructions.
Apoptosis analysis
HepG2 cells were harvested and washed twice with cold PBS and resuspended in 1× binding buffer at a concentration of 1×106 cells/mL to a final volume of 100 μL. About 5 μL Annexin V-FITC was added to each tube for 20 minutes, and 5 μL PI was added for another 10 minutes, the mixture was then incubated on ice. Finally, each sample was made to a final volume of 500 μL with 1× binding buffer and analyzed through flow cytometry within 30 minutes.
Western blot analysis
Western blotting assay was performed as previously described.33 Total protein was extracted from cells using radioimmunoprecipitation buffer (JRDUN Biotechnology Co., Ltd. Shanghai, People’s Republic of China). The protein concentration was assessed using a bicinchoninic acid protein assay kit (cat number PICPI23223; Thermo Fisher Scientific, Waltham, MA, USA). About 50 μg of protein lysate was separated by 10%–15% sodium dodecyl sulfate–polyacrylamide gel electrophoresis and was transferred to polyvinylidene fluoride membranes (Sigma-Aldrich, St Louis, MO, USA), followed by blocking in fat-free milk overnight at 4°C. The membrane was first incubated with mouse monoclonal antibodies anti-STAT3 (cat number ab119352; 1:5,000; Abcam, Cambridge, MA, USA) and anti-Bcl-2 (cat number ab117115; 1:1,000; Abcam); rabbit monoclonal antibodies anti-p-STAT3 (cat number ab76315; 1:200,000; Abcam) and anti-Bax (cat number ab32503; 1:1,000; Abcam); and rabbit polyclonal antibodies anti-Fas (cat number ab82419; 1:1,000; Abcam), anti-FasL (cat number ab15285; 1:500; Abcam), anti-JAK2 (cat number ab39636; 1:1,000; Abcam), and anti-p-JAK2 (cat number ab68268; 1:1,000; Abcam). Blots were then incubated with goat anti-mouse or anti-rabbit secondary antibody (cat numbers A0208 and A0216; 1:1,000; Beyotime Institute of Biotechnology, Haimen, People’s Republic of China) and visualized using enhanced chemiluminescence (Thermo Fisher Scientific). Glyceraldehyde 3-phosphate dehydrogenase antibody was used as an internal control. The blotting bands were quantified with ImageJ software.
Statistical analysis
Results are shown as mean ± SD; statistical analysis was performed using the GraphPad Prism 5.0 software. All assays were performed in triplicate and repeated at least three times. Different groups were compared using two-tailed Student’s t-test. All statistical analyses were two-sided, and differences below P<0.05 were considered statistically significant.
Results
Detection of cytokine profiles in coculture system
Our previous study32 showed that AFP-specific CD8+ T-cells killed target cells and demonstrated no significant differences between AFP-specific CD8+ T-cells and the ones with CD4+ T-cells included. To further investigate the mechanism of this function, using JAK2 inhibitor (AG490), we incubated CD8+ T-cells stimulated with specific or nonspecific AFP-infected DCs against the target HepG2 cells.
We first detected cytokines in coculture supernatants. From the ELISA results, we found that AFP-specific T-cells combined with JAK2 inhibitor secreted highest levels of IL-2, IL-6, IFN-γ, and TNF-α, as well as cytotoxic effector molecules perforin and granzyme B (Figure 1A–F). It was shown that IL-10 (also known to be produced by HepG2 cells) appeared in a counter-expression pattern and could contribute to the negative effect on the function of AFP-specific CD8+ T-cells to target cells (Figure 1G).
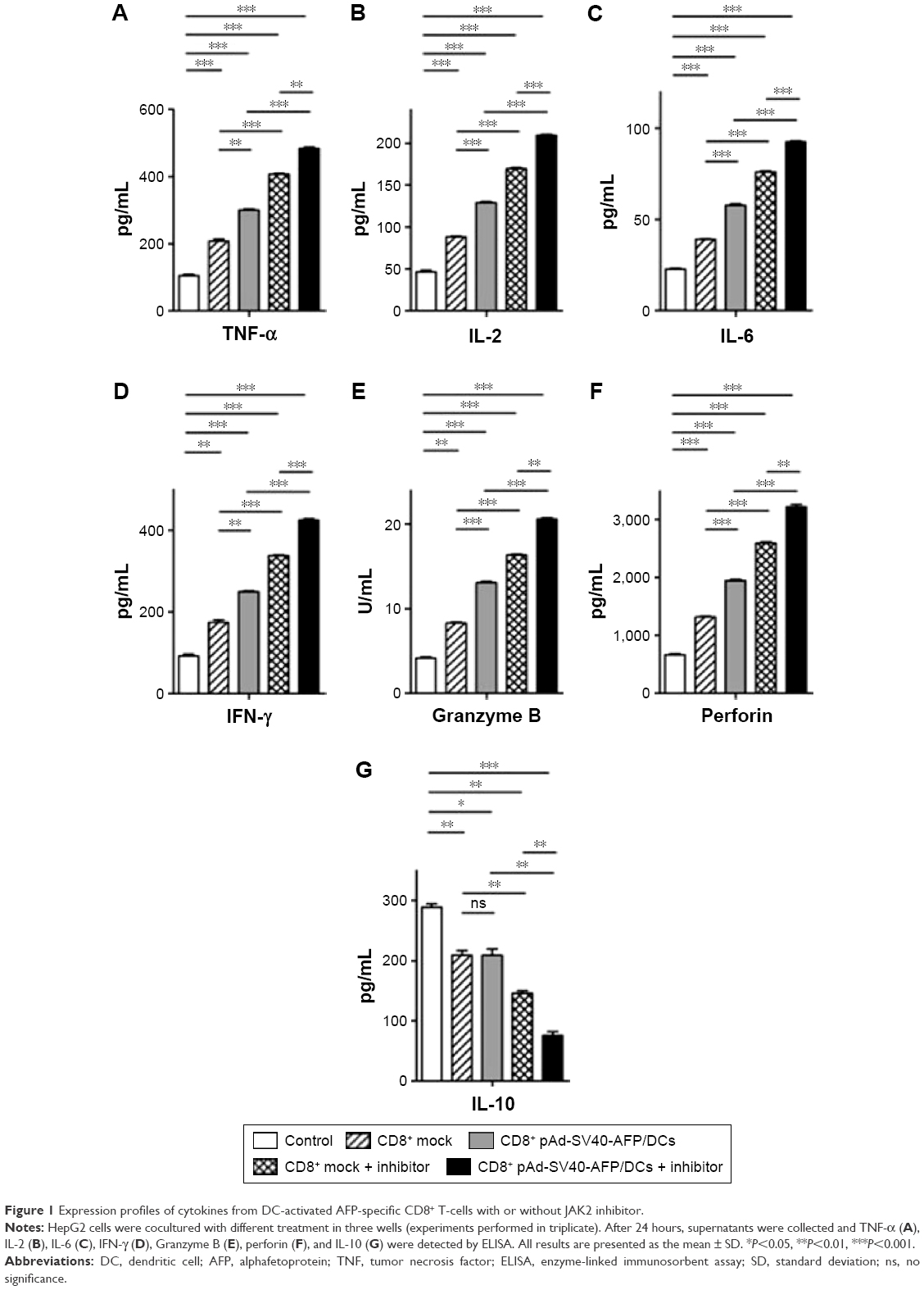 | Figure 1 Expression profiles of cytokines from DC-activated AFP-specific CD8+ T-cells with or without JAK2 inhibitor. |
AFP-specific CD8+ T-cells promote HepG2 cell apoptosis with JAK2 inhibitor
The analysis of cytokine profiles gave us further evidence on assessing functions of AFP-specific CD8+ T-cells against target cells. We next cocultured the effector and target cells, followed by assessing the proliferation ability of cells after CCK-8 incorporation at the indicated time points. The results showed that CD8+ T-cells inhibited HepG2 cell proliferation to some extent irrespective of whether specific AFP-infected DCs and JAK2 inhibitor was used or not. Compared with the mock control (empty pAd-SV40 vector-infected DC), CD8+ T-cells stimulated with specific AFP-infected DCs showed more negative effect on target cell proliferation. Intriguingly, inhibition of proliferation was enhanced by using JAK2 inhibitor combined with specific AFP-infected DCs, but there were no significant differences between two mock controls (Figure 2A).
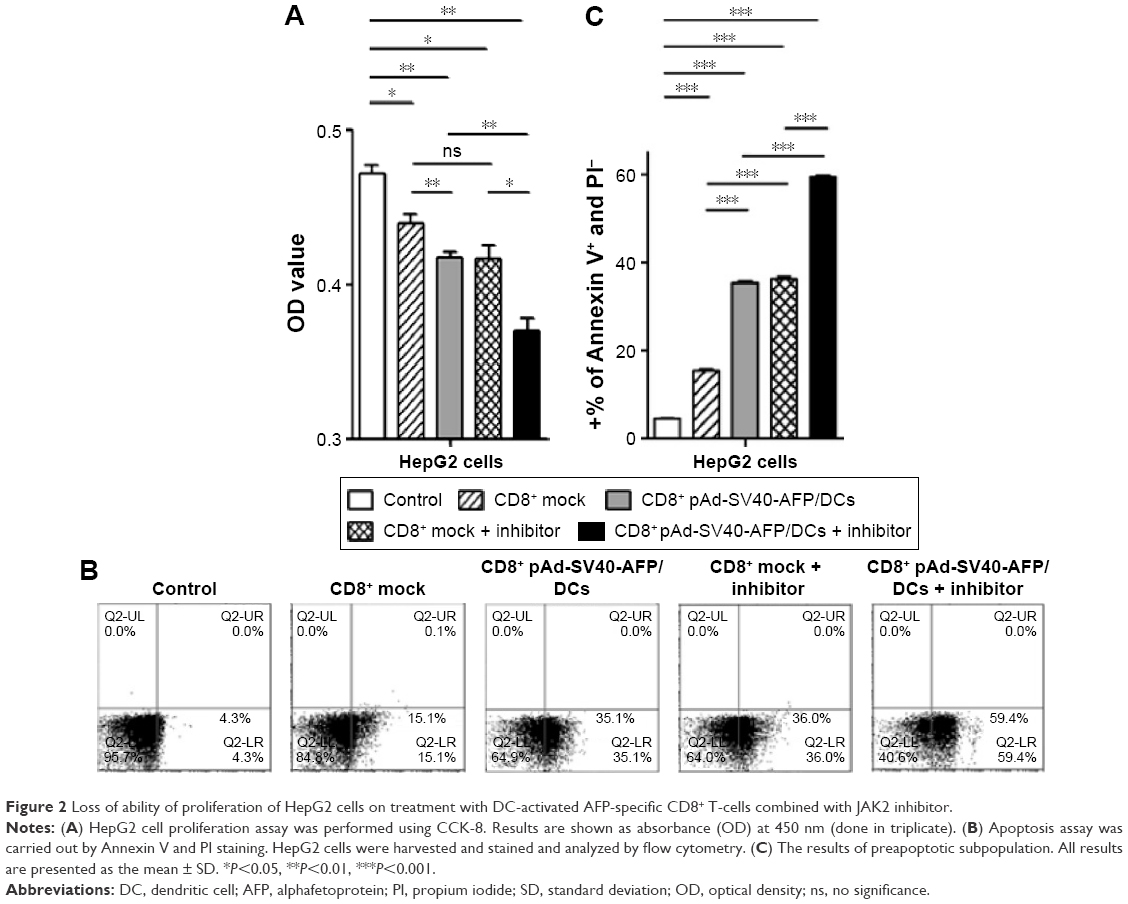 | Figure 2 Loss of ability of proliferation of HepG2 cells on treatment with DC-activated AFP-specific CD8+ T-cells combined with JAK2 inhibitor. |
It was presumed that apoptosis contributed to the negative effect on proliferation assay. For this reason, we detected apoptosis immediately with the same samples in which CCK-8 was incorporated. As expected, more than 59.47%±0.41% HepG2 cells that were cocultured with AFP-specific CD8+ T-cells with JAK2 inhibitor underwent apoptosis and showed reduction in cell proliferation (Figure 2B and C).
HepG2 cells were blocked in G1 phase by coculture of AFP-specific CD8+ T-cells with JAK2 inhibitor
To illustrate the intrinsic reason for apoptosis in our study, we first determined whether cell cycle was significantly changed. HepG2 cells were treated in various ways by using CD8+ T-cells stimulated with specific or nonspecific AFP-infected DCs and JAK2 inhibitor. From the flow cytometry analysis, results demonstrate that more target cells were seen in G1 phase compared to the control without any treatment. In the treatment of JAK2 inhibitor combined with AFP-specific CD8+ T-cells, we found that 64.89%±0.47% HepG2 cells were accumulated in G1 phase, which represented an obvious block in G1 phase and led to inhibition of cell cycle progression (Figure 3A and B). This evidence demonstrated the mechanism that AFP-specific CD8+ T-cells, especially those combined with JAK2 inhibitor, efficiently inhibited cell proliferation and induced apoptosis of HepG2 owing to G1 phase blocking.
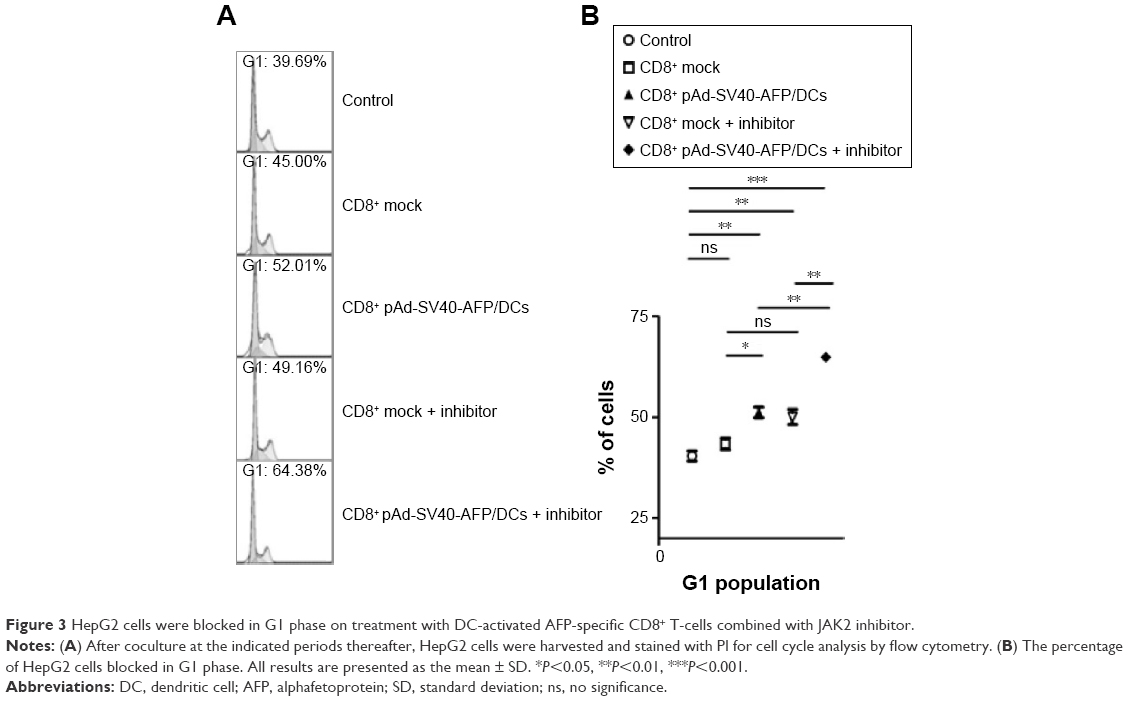 | Figure 3 HepG2 cells were blocked in G1 phase on treatment with DC-activated AFP-specific CD8+ T-cells combined with JAK2 inhibitor. |
JAK2 inhibitor enhanced expression of BAX but had no influence on Fas/FasL in treatment
According to the aforementioned results, we speculated that changeable expression of molecules on regulating apoptosis may occur by using these treatments. From the Western blotting results, we found that the expression of p-STAT3, Bcl-2, and p-JAK2 decreased by using JAK2 inhibitor (Figure 4A). On the contrary, the key factor Bax, which is popularly known as an upstream inducer to apoptosis, was obviously shown to have increased expression. This was direct evidence that apoptosis occurred with treatment using JAK2 inhibitor. Surprisingly, we did not find any changes in the expression of Fas or FasL, which revealed that our treatment involved an independent pathway on Fas/FasL signal (Figure 4B). All these results demonstrated the mechanisms of inhibition on cell proliferation by reduced p-STAT3, thus strengthening cell apoptosis by increasing Bax and reducing Bcl-2, which in turn gave us a new clue on the function of target cells cocultured with AFP-specific CD8+ T-cells with JAK2 inhibitor.
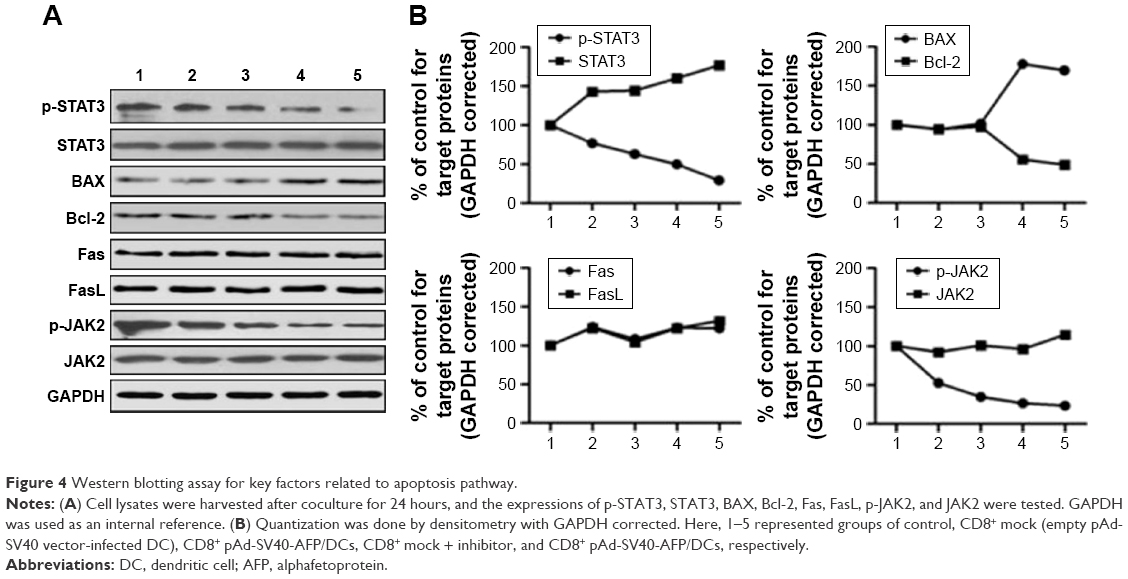 | Figure 4 Western blotting assay for key factors related to apoptosis pathway. |
Discussion
Researchers have taken a long time to find an effective way for fighting cancer by using the patient’s immune system itself. Studies in this field showed that immunotherapy works in various ways. Some of them boost the patients’ own immune system, while others help imitate an early stage of initiating immune reaction. Cancer vaccines are administered to evoke a specific T-cell or B-cell response to cancer.34–36 By using vaccines, DCs are activated from resting state and loaded with antigen.37 They send activation signals to enlarge a specific T-cell population and help T-cells acquire effector function. In this study, we further investigated the mechanism of combined treatment on antitumor activity of AFP-specific CD8+ T-cells, which were used in our previous study. The results showed that the combined treatment inhibited target HepG2 cell proliferation and revealed a mechanism of tumor cell apoptosis in a JAK2 inhibitor inducible pathway.
In addition, we found that cytotoxic antitumor cytokines were significantly increased. Combined with JAK2 inhibitor, the production of cytokines was higher than those by any other treatment. Previous studies32 showed an effective strategy against tumors by using effector cytokines. IL-2 has been applied to solid tumors with acceptable results and few side effects and acts by mediating tumor-associated vasculature destruction.38 However, we also demonstrated the reduced secretion of IL-10 in the AFP-specific CD8+ T-cell combined with JAK2 inhibitor group. Despite the discovery of antitumor function of IL-10, it is still recognized as a negative effector and is usually used as a target in immunotherapies.39 What we observed on IL-10 gave us more belief of its antitumor effect in combined treatment.
By using JAK2 inhibitor, we observed a better treatment response compared to culture with AFP-specific CD8+ T-cells only. JAK inhibitors reduce the activity of the Janus kinase family of enzymes and have demonstrated therapeutic application against cancer and inflammatory diseases. Ruxolitinib, which is the first potent, JAK1/2 inhibitor to enter the clinic, was approved by the US Food and Drug Administration in 2011 for the treatment of intermediate- or high-risk myelofibrosis.40,41 JAK family kinases are well known as one of the most important factors for continuous STAT3 activation in cancer tissues and cells. For the survival of some types of cancer, however, a decrease in the level of p-STAT3 activation is definitely required for the activity/activation of some antitumor proteins and factors.42,43 From the Western blotting results, we did find reduced expression on p-STAT3 by using JAK2 inhibitor, which indicates that the effect is a result of both the inhibitor and AFP-specific CD8+ T-cells.
On the basis of our study, the combination treatment of AFP-specific CD8+ T-cells and JAK2 inhibitor is of high potential value for clinical applications in immunotherapy against various cancers. Meanwhile, we found that more tumor cells underwent apoptosis and were blocked in G1 phase, as assessed by flow cytometry. It was reported that Fas receptor, which is a member of TNF family, is essential to induce apoptosis in tumor cells.44–46 However, the results of Western blotting from cell lysates showed no differences among all these groups, which reveal an independent Fas/FasL apoptosis pathway.
Conclusion
In conclusion, this study clarified an independent Fas/FasL signal pathway on tumor cells apoptosis and demonstrated the effectiveness of combining AFP-specific CD8+ T-cells and JAK2 inhibitor in treating cancers by showing inhibition to tumor cells proliferation and cell cycle blockage in G1 phase.
Acknowledgments
The study was supported by the grant from National Natural Science Foundation of China (NSFC#81372461).
Disclosure
The authors report no conflicts of interest in this work.
References
Terlikowski SJ. Tumour necrosis factor and cancer treatment: a historical review and perspectives. Rocz Akad Med Bialymst. 2001;46:5–18. | ||
Tempero MA, Pour PM, Uchida E, Herlyn D, Steplewski Z. Monoclonal antibody CO17-1A and leukapheresis in immunotherapy of pancreatic cancer. Hybridoma. 1986;5(Suppl 1):S133–S138. | ||
Yu DS, Yeh MY, Chang SY, Ma CP, Han SH. Immunotherapy of xenografted human bladder cancer in nude mice using monoclonal antibody. Eur Urol. 1987;13:198–202. | ||
Shiraiwa H, Sekine T, Tobisu K, Kakizoe T, Koiso K. A new form of specific targeting cancer immunotherapy using anti-tumor monoclonal antibody-conjugated lymphokine-activated killer cells. Jpn J Cancer Res. 1991;82:621–623. | ||
Rader C, Sinha SC, Popkov M, Lerner RA, Barbas CF 3rd. Chemically programmed monoclonal antibodies for cancer therapy: adaptor immunotherapy based on a covalent antibody catalyst. Proc Natl Acad Sci U S A. 2003;100:5396–5400. | ||
Hurwitz H. Integrating the anti-VEGF-A humanized monoclonal antibody bevacizumab with chemotherapy in advanced colorectal cancer. Clin Colorectal Cancer. 2004;4(Suppl 2):S62–S68. | ||
Emmanouilides C, Pegram M, Robinson R, Hecht R, Kabbinavar F, Isacoff W. Anti-VEGF antibody bevacizumab (Avastin) with 5FU/LV as third line treatment for colorectal cancer. Tech Coloproctol. 2004;8(Suppl 1):S50–S52. | ||
An SJ, Huang YS, Chen ZH, et al. Posttreatment plasma VEGF levels may be associated with the overall survival of patients with advanced non-small cell lung cancer treated with bevacizumab plus chemotherapy. Med Oncol. 2012;29:627–632. | ||
Li H, Takayama K, Wang S, et al. Addition of bevacizumab enhances antitumor activity of erlotinib against non-small cell lung cancer xenografts depending on VEGF expression. Cancer Chemother Pharmacol. 2014;74:1297–1305. | ||
Burstein HJ, Chen YH, Parker LM, et al. VEGF as a marker for outcome among advanced breast cancer patients receiving anti-VEGF therapy with bevacizumab and vinorelbine chemotherapy. Clin Cancer Res. 2008;14:7871–7877. | ||
Pentheroudakis G, Kotoula V, Kouvatseas G, et al. Association of VEGF-A splice variant mRNA expression with outcome in bevacizumab-treated patients with metastatic breast cancer. Clin Breast Cancer. 2014;14:330–338. | ||
Madsen CV, Steffensen KD, Olsen DA, et al. Serial measurements of serum PDGF-AA, PDGF-BB, FGF2, and VEGF in multiresistant ovarian cancer patients treated with bevacizumab. J Ovarian Res. 2012;5:23. | ||
van der Bilt AR, Terwisscha van Scheltinga AG, Timmer-Bosscha H, et al. Measurement of tumor VEGF-A levels with 89Zr-bevacizumab PET as an early biomarker for the antiangiogenic effect of everolimus treatment in an ovarian cancer xenograft model. Clin Cancer Res. 2012;18:6306–6314. | ||
Rothe A, Sasse S, Goergen H, et al. Brentuximab vedotin for relapsed or refractory CD30+ hematologic malignancies: the German Hodgkin Study Group experience. Blood. 2012;120:1470–1472. | ||
Yang QM, Hong JY, Ko YH, et al. Brentuximab vedotin for relapsed or refractory CD30+ Hodgkin lymphoma: a multicenter analysis from Asia. Onco Targets Ther. 2014;7:1717–1722. | ||
Tomala J, Chmelova H, Mrkvan T, Rihova B, Kovar M. In vivo expansion of activated naive CD8+ T cells and NK cells driven by complexes of IL-2 and anti-IL-2 monoclonal antibody as novel approach of cancer immunotherapy. J Immunol. 2009;183:4904–4912. | ||
Bruns H, Meinken C, Schauenberg P, et al. Anti-TNF immunotherapy reduces CD8+ T cell-mediated antimicrobial activity against Mycobacterium tuberculosis in humans. J Clin Invest. 2009;119:1167–1177. | ||
Toh U, Fujii T, Seki N, Niiya F, Shirouzu K, Yamana H. Characterization of IL-2-activated TILs and their use in intrapericardial immunotherapy in malignant pericardial effusion. Cancer Immunol Immunother. 2006;55:1219–1227. | ||
Liu K, Caldwell SA, Greeneltch KM, Yang D, Abrams SI. CTL adoptive immunotherapy concurrently mediates tumor regression and tumor escape. J Immunol. 2006;176:3374–3382. | ||
Grimm CF, Ortmann D, Mohr L, et al. Mouse alpha-fetoprotein-specific DNA-based immunotherapy of hepatocellular carcinoma leads to tumor regression in mice. Gastroenterology. 2000;119:1104–1112. | ||
Meng WS, Butterfield LH, Ribas A, et al. Alpha-fetoprotein-specific tumor immunity induced by plasmid prime-adenovirus boost genetic vaccination. Cancer Res. 2001;61:8782–8786. | ||
Vollmer CM Jr, Eilber FC, Butterfield LH, et al. Alpha-fetoprotein-specific genetic immunotherapy for hepatocellular carcinoma. Cancer Res. 1999;59:3064–3067. | ||
Butterfield LH, Meng WS, Koh A, et al. T cell responses to HLA-A*0201-restricted peptides derived from human alpha fetoprotein. J Immunol. 2001;166:5300–5308. | ||
Watanabe S, Itoh T, Arai K. JAK2 is essential for activation of c-fos and c-myc promoters and cell proliferation through the human granulocyte-macrophage colony-stimulating factor receptor in BA/F3 cells. J Biol Chem. 1996;271:12681–12686. | ||
Chung BM, Kang HC, Han SY, et al. Jak2 and Tyk2 are necessary for lineage-specific differentiation, but not for the maintenance of self-renewal of mouse embryonic stem cells. Biochem Biophys Res Commun. 2006;351:682–688. | ||
Campbell PJ, Griesshammer M, Dohner K, et al. V617F mutation in JAK2 is associated with poorer survival in idiopathic myelofibrosis. Blood. 2006;107:2098–2100. | ||
Zhou H, Huang B, Du J, Wang L. Role of the JAK2/STAT pathway and losartan in human glomerular mesangial cell senescence. Mol Med Rep. 2010;3:393–398. | ||
Zhou H, Huang B, Han Y, Jin R, Chen S. Probucol inhibits JAK2-STAT pathway activation and protects human glomerular mesangial cells from tert-butyl hydroperoxide induced premature senescence. Can J Physiol Pharmacol. 2013;91:671–679. | ||
Goerttler PS, Steimle C, Marz E, et al. The Jak2V617F mutation, PRV-1 overexpression, and EEC formation define a similar cohort of MPD patients. Blood. 2005;106:2862–2864. | ||
Tefferi A, Gilliland DG. The JAK2V617F tyrosine kinase mutation in myeloproliferative disorders: status report and immediate implications for disease classification and diagnosis. Mayo Clin Proc. 2005;80:947–958. | ||
Tyner JW, Bumm TG, Deininger J, et al. CYT387, a novel JAK2 inhibitor, induces hematologic responses and normalizes inflammatory cytokines in murine myeloproliferative neoplasms. Blood. 2010;115:5232–5240. | ||
Liu Y, Butterfield LH, Fu X, et al. Lentivirally engineered dendritic cells activate AFP-specific T cells which inhibit hepatocellular carcinoma growth in vitro and in vivo. Int J Oncol. 2011;39:245–253. | ||
Hao J, Song X, Wang J, et al. Cancer-testis antigen MAGE-C2 binds Rbx1 and inhibits ubiquitin ligase-mediated turnover of cyclin E. Oncotarget. 2015;6:42028–42039. | ||
Chen J, Zhang L, Wen W, et al. Induction of HCA587-specific antitumor immunity with HCA587 protein formulated with CpG and ISCOM in mice. PLoS One. 2012;7:e47219. | ||
Batchu RB, Gruzdyn O, Potti RB, Weaver DW, Gruber SA. MAGE-A3 with cell-penetrating domain as an efficient therapeutic cancer vaccine. JAMA Surg. 2014;149:451–457. | ||
Florcken A, Grau M, Wolf A, et al. Gene expression profiling of peripheral blood mononuclear cells during treatment with a gene-modified allogeneic tumor cell vaccine in advanced renal cell cancer: tumor-induced immunosuppression and a possible role for NF-kappaB. Int J Cancer. 2015;136:1814–1826. | ||
Zarnani AH, Torabi-Rahvar M, Bozorgmehr M, Zareie M, Mojtabavi N. Improved efficacy of a dendritic cell-based vaccine against a Murine model of colon cancer: the helper protein effect. Cancer Res Treat. 2015;47:518. | ||
Jackaman C, Bundell CS, Kinnear BF, et al. IL-2 intratumoral immunotherapy enhances CD8+ T cells that mediate destruction of tumor cells and tumor-associated vasculature: a novel mechanism for IL-2. J Immunol. 2003;171:5051–5063. | ||
Bodas M, Jain N, Awasthi A, et al. Inhibition of IL-2 induced IL-10 production as a principle of phase-specific immunotherapy. J Immunol. 2006;177:4636–4643. | ||
Moisan A, Lee YK, Zhang JD, et al. White-to-brown metabolic conversion of human adipocytes by JAK inhibition. Nat Cell Biol. 2015;17:57–67. | ||
Cervantes F, Vannucchi AM, Kiladjian JJ, et al. Three-year efficacy, safety, and survival findings from COMFORT-II, a phase 3 study comparing ruxolitinib with best available therapy for myelofibrosis. Blood. 2013;122:4047–4053. | ||
Grandis JR, Drenning SD, Zeng Q, et al. Constitutive activation of Stat3 signaling abrogates apoptosis in squamous cell carcinogenesis in vivo. Proc Natl Acad Sci U S A. 2000;97:4227–4232. | ||
Bowman T, Garcia R, Turkson J, Jove R. STATs in oncogenesis. Oncogene. 2000;19:2474–2488. | ||
Nagata S, Golstein P. The Fas death factor. Science. 1995;267:1449–1456. | ||
Muzio M. Signalling by proteolysis: death receptors induce apoptosis. Int J Clin Lab Res. 1998;28:141–147. | ||
Magnusson C, Vaux DL. Signalling by CD95 and TNF receptors: not only life and death. Immunol Cell Biol. 1999;77:41–46. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.