Back to Journals » Infection and Drug Resistance » Volume 13
iTRAQ-Based Proteomics Reveals Potential Anti-Virulence Targets for ESBL-Producing Klebsiella pneumoniae
Authors Wang Y, Cong S, Zhang Q, Li R, Wang K
Received 2 May 2020
Accepted for publication 28 July 2020
Published 19 August 2020 Volume 2020:13 Pages 2891—2899
DOI https://doi.org/10.2147/IDR.S259894
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Suresh Antony
Yan Wang,1 Shan Cong,1 Qinghua Zhang,1 Ranwei Li,2 Ke Wang1
1Department of Respiratory and Critical Care Medicine., The Second Hospital of Jilin University, Changchun, People’s Republic of China; 2Department of Urology, The Second Hospital of Jilin University, Changchun, People’s Republic of China
Correspondence: Ke Wang
Department of Respiratory and Critical Care Medicine, The Second Hospital of Jilin University, 218 Ziqiang Street, Nanguan District, Changchun, Jilin Province 130041, People’s Republic of China
Tel +86 18643111766
Fax +86-431-81136820
Email [email protected]
Purpose: Treatment of infections with Klebsiella pneumoniae strains producing extended-spectrum beta-lactamases (ESBLs) is challenging due to the coexistence of multiple resistance mechanisms and the hypervirulent variant. Therefore, new targets or more effective treatment options aimed at ESBL-producing Klebsiella pneumoniae are urgently needed.
Materials and Methods: Here, we collected ESBL-producing and non-ESBL Klebsiella pneumoniae isolates and studied their differences from a proteomic point of view.
Results: We revealed treA, wza, gnd, rmlA, rmlC, rmlD, galE, aceE, and sucD as important virulence-related proteins in ESBL-producing Klebsiella pneumoniae, distinct from those in non-ESBL strains.
Conclusion: Our findings provide plausible anti-virulence targets and suggest new therapeutic avenues against ESBL-producing Klebsiella pneumoniae.
Keywords: Klebsiella pneumoniae, extended-spectrum beta-lactamases, proteomics, anti-virulence
Introduction
Antibiotics can kill or inhibit growth of bacteria by disrupting biosynthetic processes or essential cellular pathways. However, the misuse of antibiotics has led to the evolution of antimicrobial resistance (AMR) genes and the selection of multiple antibiotic-resistant bacteria. This has prompted the development of alternative strategies to treat drug-resistant bacterial infections.1 When bacteria invade the host, they activate virulence factors including toxins, adhesins, capsular polysaccharides (CPS), lipopolysaccharide (LPS), and exopolysaccharide (EPS). The activated virulence factors may aid bacterial attachment to the host cells, cloak the subcapsular epitopes on the surface, and escape the host immune response.2,3 Therefore, preventing the expression or activation of bacterial virulence factors could be an alternative strategy, distinct from the classic antibiotic mechanisms, which will not interfere with bacterial survival, allowing the host to fight the infection through immune responses.
Infections due to Gram-negative bacteria are a cause of morbidity and mortality worldwide. Extended-spectrum beta-lactamases (ESBLs) render treatment of these pathogens particularly difficult due to the limited options of effective antibiotics.4 Due to their virulence factors, ESBL-producing Klebsiella pneumoniae strains are significantly more resistant to the antibacterial activity of serum than non-ESBL-producing K. pneumoniae strains.5,6 However, the potential additional factors that render ESBL-producing Klebsiella pneumoniae more virulent remain elusive. In this study, we collected ESBL-producing and non-ESBL-producing K. pneumoniae isolates randomly, and studied their proteomic differences. We identified important virulence-related proteins in ESBL-producing K. pneumoniae distinct from those in the non-ESBL strains. Furthermore, we report novel plausible anti-virulence targets and promising treatment options against infection with ESBL-producing K. pneumoniae.
Materials and Methods
Isobaric tags for relative and absolute quantification (iTRAQ) Reagent‐4/8plex Multiplex Kit (AB Sciex). Antibodies: anti-GALE (Proteintech, 14,414-1-AP), anti-Trehalase (Abcam, 133,808), anti-PGD (Proteintech, 14,718-1-AP), anti-β-actin (ABclonal, AC004), HRP Goat Anti-Mouse IgG (ABclonal, AS003HRP), HRP-conjugated Affinipure Goat Anti-Rabbit IgG (Proteintech, SA00001-2). Chemiluminescent HRP substrate (Millipore, WBKLS0100).
Methods
Sample Collection
ESBL-producing K. pneumoniae (n=60) and sensitive K. pneumoniae (n=60) were collected by the microbiology lab at the Second Hospital of Jilin University. All the selected ESBL-producing isolates carried resistance genes CTX-M, SHV and TEM simultaneously. In addition, ensure that the drug sensitivity results of all ESBL-producing or sensitive isolates are the same. For iTRAQ labeling experiments, 30 ESBL-producing isolates and 30 non-ESBL-producing K. pneumoniae isolates were randomly distributed in six groups. For validation experiments, another 30 isolates from each type were employed. The colonies were incubated in 200 mL LB broth liquid medium (Invitrogen, 10,855,001) and cultured for 16–18 h at 37°C in shaking conical flasks. Thereafter, the medium was centrifuged at 3500 g for 15 min, and the sediment was collected. Subsequently, the sediments were washed three times with sterile PBS.
Protein Extraction and Quantification
For both the iTRAQ labeling experiment and the validation experiment, every 10 isolates of sediments from each type were mixed as one group. Each group of sediments were all added to the SDT pyrolysis solution (4% SDS, 100 mM Tris-HCl, pH 7.6), transferred to lysing matrix A tubes, and broken by MP homogenizer (24 × 2, 6.0 M/s, 60 s, twice). Subsequently, after ultrasonication, the homogenate was boiled for 10 min, followed by centrifugation at 14,000 g for 15 min. The supernatants were then filtered using 0.22 μm filters and collected. Proteins were quantified using BCA (Beyotime, P0012) method. The samples were stored at −80°C.
SDS-PAGE Electrophoresis
For the iTRAQ labeling experiment, 20 μg protein from each sample was mixed with 6X loading buffer solution and boiled for 5 min. Proteins were separated using 12% SDS-PAGE electrophoresis (constant 250 V, 40 min), followed by Coomassie Brilliant blue staining. For Western blot, 50 μg protein from each sample was separated by SDS-PAGE electrophoresis and transferred to polyvinylidene difluoride membranes. The blots were blocked by Tris-buffered saline solution containing 5% non-fat milk before incubation with primary and secondary HRP-conjugated antibodies, prepared in blocking buffer. Bands were visualized using the enhanced chemiluminescence (ECL) detection system (Amersham Biosciences, Pittsburgh, PA, USA).
Protein Digestion and iTRAQ Labeling
Prior to the iTRAQ labeling experiments, equal quality of protein from each group extracted as described above were trypsinized according to the FASP protocol.7 The eluted peptides were quantified using a NanoDrop (Thermo Fisher scientific ND2000).
Subsequently, 100 μg of peptides from each group were iTRAQ-labeled using an 8-plex iTRAQ labeling kit following the manufacturer’s protocol. Three controls (non-ESBL-producing K. pneumoniae) were labelled with iTRAQ reagents 115, 116, and 117, and 3 EBSL+ (ESBL-producing K. pneumoniae) groups were labelled with iTRAQ reagents 118, 119, and 121, respectively.
High pH One-D Reverse Phase Chromatography Fractionation
Fractionation of the peptide mixture was performed on an Agilent 1260 Infinity II HPLC system. Buffer A contained 10 mM HCO2NH4, 5% ACN, pH 10.0; buffer B contained 10 mM HCO2NH4, 85% ACN, pH 10.0. The column was initially equilibrated in buffer A. The samples were separated on the chromatographic column at a flow rate of 1 mL/min. The gradient of liquid phase was the following: buffer B 0%, 0 min-25 min; buffer B 0%-7%, 25 min-30 min; buffer B 7%-40%, 30 min-65 min; buffer B 40%-100%, 65 min-70 min; buffer B 100%, 70 min-85 min. During elution, UV absorbance was monitored at 214 nm. Fractions were collected every minute during the course of the run. Subsequently, all 36 fractions were collected, lyophilized and kept at −80°C prior to mass spectrometry analysis.
Mass Spectrometry Analysis
The collected fractions were examined by Easy nLC system. Buffer A contained 0.1% formic acid aqueous; buffer B contained 0.1% formic acid acetonitrile (acetonitrile concentration 80%). The column was initially equilibrated in 100% buffer A. The samples were separated on the column (Acclaim PepMap RSLC 50 µm × 15 cm, nano viper, P/N164943) at a flow rate of 300 nl/min. The gradient of liquid phase was the following: buffer B 6%, 0 min-5 min; buffer B 6%-28%, 5 min-45 min; buffer B 28%-38%, 45 min-50 min; buffer B 38%-100%, 50 min-55 min; buffer B 100%, 55 min-60 min.
After separation on the chromatography column, the samples were analyzed by Q Exactive mass spectrometer (Thermo Fisher Scientific). Key parameter settings for the Q Exactive mass spectrometer were: (1) First grade MS parameters: Resolution: 70,000, AGC target: 3e6, Maximum IT: 50 ms, Scan range: 350 to 1800 m/z; (2) Second grade MS parameters: Resolution: 17,500, Maximum IT: 45 ms, Isolation window: 2 m/z, Microscans: 1, Maximum IT: 45ms, Normalized Collision Energy 30eV.
Database Search for Protein Identification
The original map file produced by Q Exactive was transformed into. Mgf file by Proteome Discoverer 2.1 (Thermo Fisher Scientific) and submitted to the server MASCOT2.6 for database retrieval through the tools in the software. The database for the search was Uniprot_KlebsiellaPneumoniae_223866_20180301 (http://www.uniprot.org). Then, the library file (.Dat) formed on the MASCOT server was transferred back to the software through Proteome Discoverer 2.1, and the data were filtered based on the standard of false discovery rate (FDR)<0.01. Finally, highly reliable qualitative results were obtained.
In the process of quantitative analysis, the Proteome Discoverer 2.1 software was applied to extract the peak strength of the peptide ion. The search parameters were: (1) peak integration, integration window tolerance: 20 ppm; integration method: most confident centroid; (2) scan event filters, mass analyzer: FTMS; MS order: MS2; activation type: HCD; (3) quantification – general, peptides to use: unique; (4) normalization scaling, normalization mode: total peptide amount; scaling mode: on channels average.
All identified proteins and peptides are listed in S1 and S2 File.
Bioinformatics Analysis
For computing the differential ratio, peptides and proteins having an ion score greater than 99% confidence were included. Peptides with low confidence score of 1%, without peaks corresponding to iTRAQ labels, as well as the peptides found in multiple proteins were excluded. The quantitative estimates for each protein included: protein abundance; average protein abundance of corresponding groups; differential expression fold change ratios; p-value; and FDR. After identification and quantification, proteins identified at 1% FDR with a confidence threshold of 95% and p-values <0.05 were retained. The volcano plot of fold-change ratios was set up, and the proteins with fold change ratio>1.2 and <0.833 were screened out as upregulated and downregulated proteins in ESBL+ groups compared with ESBL— groups, respectively.
The enrichment analysis of GO annotations (www.geneontology.org) was performed to evaluate the significance of a GO term enrichment. Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis (http://www.genome.jp/kegg/) was performed to annotate the functional pathways of all identified proteins or peptides. KEGG pathway enrichment analysis was similar to that of GO. The significance of protein enrichment in each pathway was calculated by Fisher accurate test (Fisher’s Exact Test), taking KEGG pathway as unit and all qualitative proteins as background. The significantly affected metabolic and signal transduction pathways were determined. Certain differentially expressed proteins were mapped by the online Search Tool for the Retrieval of Interacting Genes (STRING) database (https://version-10-5.string-db.org/) to construct the PPI network and disclose the possible connections among proteins. The PPI network was constructed by setting the minimum required interaction score to medium confidence (0.4). The active interaction sources included were “experiments”, “textmining”, and “database”. The setting parameters for maximum number of interactors to show for first shell and second shell were respectively “none/query protein only” and “none”.
Results
Identification of Differentially Expressed Proteins
The Volcano plot generated by Protein pilot in function of the protein ratio obtained by comparison of ESBL+ K. pneumoniae and non-ESBL K. pneumoniae groups is depicted in Figure 1. To identify differentially expressed proteins, 4340 proteins were used as background to screen candidates with ratio >1.2 as upregulated proteins or <0.833 as downregulated proteins. The abundance ratio values of certain differentially expressed protein were similar in three independent experiments. After statistical analysis, in total 934 proteins were considered upregulated and 726 were downregulated in ESBL+ compared to controls. The basic information of differentially expressed proteins, the abundance ratio values as well as the p values are listed in S3 File.
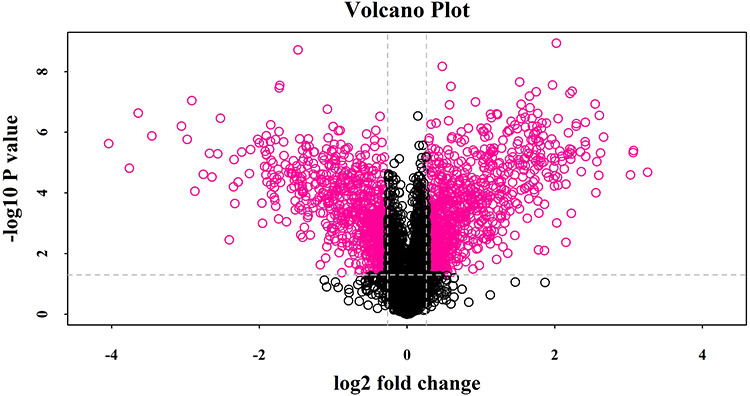 |
Figure 1 Volcano plot showing log2 fold change plotted against -log10 adjusted P value for ESBLs+ Klebsiella pneumoniae samples versus non-ESBL Klebsiella pneumoniae samples. Data points in the upper right (fold change > 1.2) and upper left (fold change < 0.83) sections with P<0.05 represent proteins that are significantly different in ESBLs+ Klebsiella pneumoniae compared with non-ESBL Klebsiella pneumoniae. |
Functional Enrichment Analysis of Differentially Expressed Proteins
Functional enrichment analysis of the 1610 differentially expressed proteins identified 1138 proteins in the category of “biological process”, 767 in “molecular function”, and 144 in “cellular component”. The functional terms in each category, ranked by statistical significance, are summarized in S4(1–3) File. As presented in Figure 2, the differentially expressed proteins were mostly enriched in “cellular process”, “metabolic process”, “single-organism process”, “catalytic activity”, and “binding”.
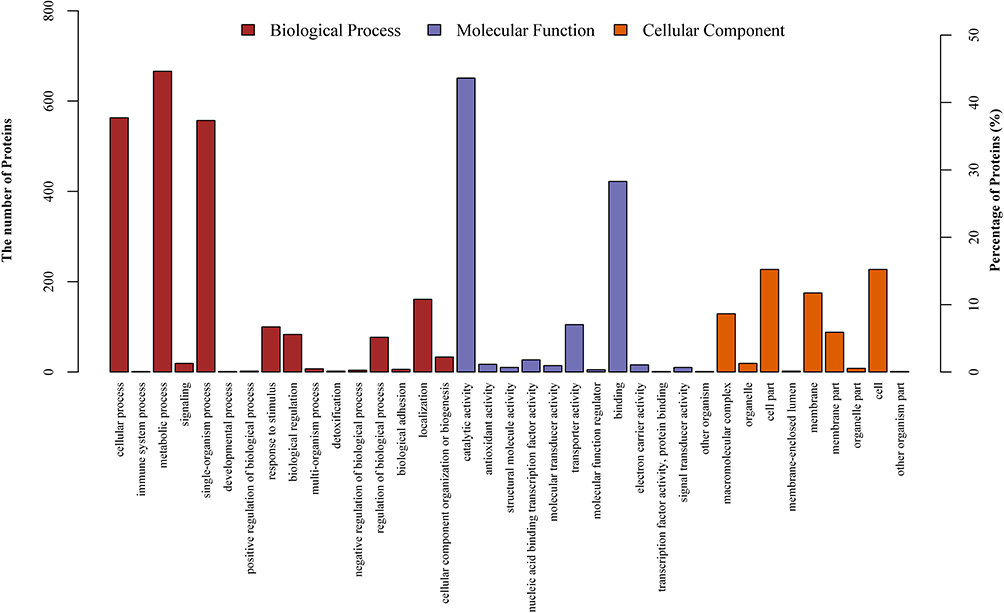 |
Figure 2 Gene Ontology (GO) classification of differentially expressed proteins by iTRAQ-based proteomics experiments between ESBLs+ Klebsiella pneumoniae and non-ESBL Klebsiella pneumoniae. The differentially expressed proteins are grouped into three GO terms: biological process, cellular component, and molecular function. The y-axis indicates the number and percent of proteins in each GO term. |
Construction of Protein–Protein Interaction Network
Subsequently, we further screened out the differentially expressed proteins with fold change ratio ≥2 and ≤0.5. The 264 upregulated and 182 downregulated proteins were combined and mapped by online STRING database to disclose the possible connections among the 446 differentially expressed proteins and visualize the protein-protein interaction (PPI) network (Figure 3). The screened differentially expressed proteins formed a complex regulatory network containing 90 nodes and 115 edges, with an average node degree of 2.56 and a clustering coefficient of 0.421 (Figure 3). The number of expected edges was 71, and the PPI enrichment p-value was 8.58e-07.
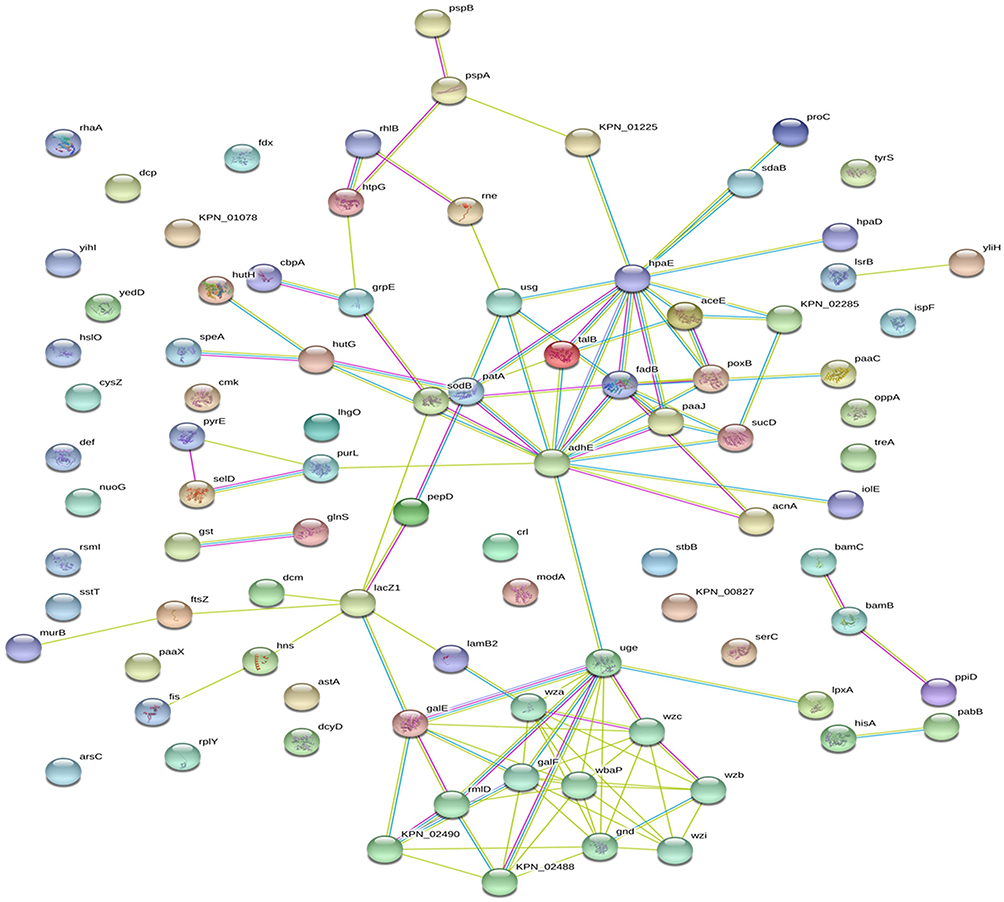 |
Figure 3 Differentially expressed proteins were combined to build a regulatory network by STRING. The differentially expressed proteins are represented as nodes, and the relationship between two nodes is represented as an edge. The empty nodes represent the proteins of unknown 3D structure, while the filled nodes indicate that some 3D structure is known or predicted. The interaction information between proteins from curated databases is represented by an blue edge; experimentally determined: purple edge; predicted interactions: cyan edge; and textming: lawn green edge. The network contained 90 nodes and 115 edges with an average node degree of 2.56 and a clustering coefficient of 0.421. The PPI enrichment p-value was equal to 8.58e-07. |
The Potential Anti-Virulence Targets in ESBL-Producing Klebsiella pneumoniae
Among the screened differentially expressed proteins in ESBL+ K. pneumoniae, and based on the center-nodes in the constructed network (Figure 3) and the ratios in S3 File, we screened out the top 9 significantly different proteins that showed the potential to be anti-virulence targets in ESBL-producing K. pneumoniae. The top 9 candidates included periplasmic trehalase (treA), integral outer membrane lipoprotein (Wza), 6-phosphogluconate dehydrogenase (gnd), dTDP-L-rhamnose synthesis (rmlA, rmlC and rmlD), UDP-galactose-4-epimerase (galE), pyruvate dehydrogenase E1 component (aceE), and succinate-CoA synthetase subunit alpha (sucD) (Table 1).
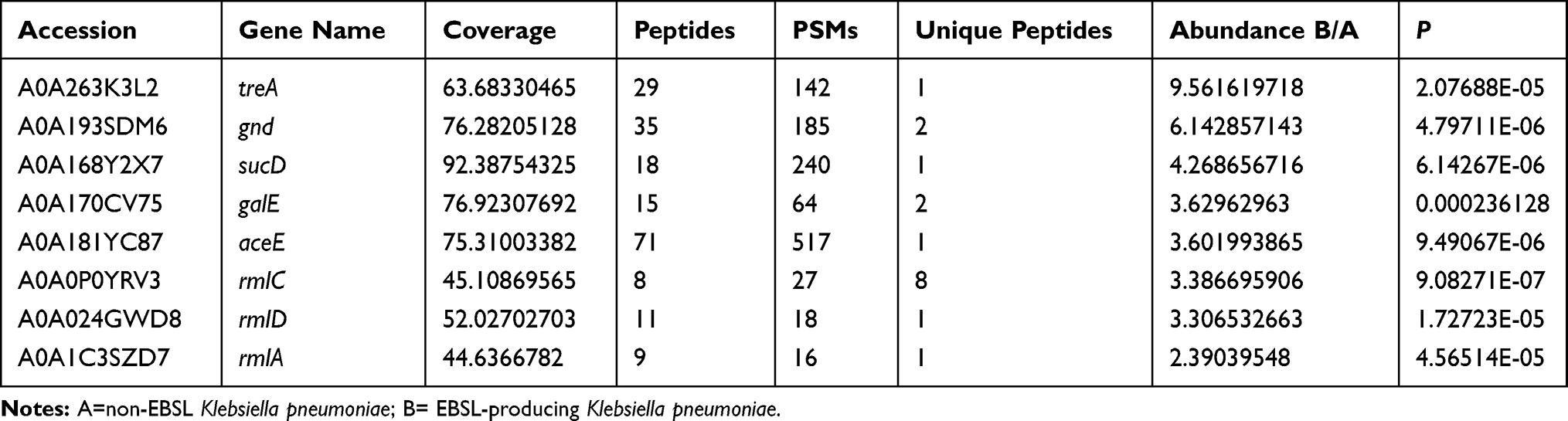 |
Table 1 Top 9 Proteins Potential to Be Anti-Virulence Targets in ESBL-Producing Klebsiella pneumoniae |
Western Blot Analysis to Validate the Representative Differentially Expressed Proteins
Three proteins (treA, gnd, galE) were selected as representative biomarkers of ESBL-producing K. pneumoniae, and validated using Western blot to confirm the proteomic data. Consistently, ESBL-producing K. pneumoniae isolates showed higher expression levels of treA, gnd, and galE (Figure 4A–C).
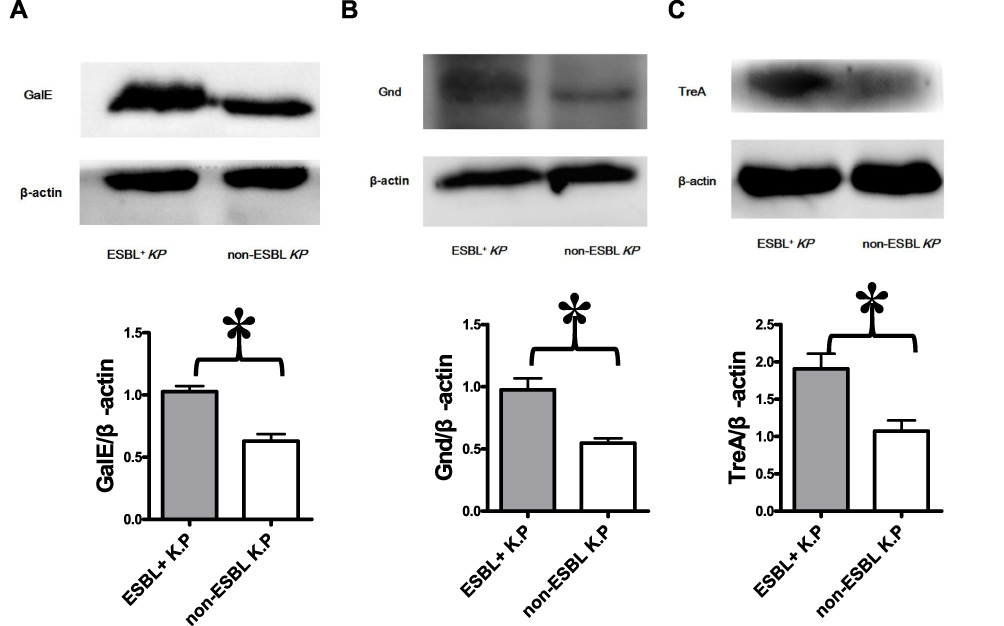 |
Figure 4 Validation of proteomics data by Western blot. The sediments of every 10 isolates of ESBLs+ Klebsiella pneumoniae or non-ESBL Klebsiella pneumoniae were mixed as one group of samples. The protein from each group was analyzed for treA, galE and Gnd levels by Western blot. The blots were reprobed with β-actin antibody to confirm equal loading of the samples. Representative Western blot images from three independent experiments for Gale (A), Gnd (B), and TreA (C) levels with β-actin as a loading control are shown. The expression levels of Gale (A), Gnd (B) and TreA (C) are higher in ESBLs+ Klebsiella pneumoniae group. *P<0.05. |
Discussion
K. pneumoniae causes a wide range of infections in humans, including pneumonia, urinary tract infections, bacteremia, and liver abscesses. The alarming increase in the prevalence of drug-resistant K. pneumoniae, such as ESBL- or carbapenems-producing K. pneumoniae, has raised our awareness of the urgency of non-antibiotic based strategies against antibiotic-resistant infections.
Virulence is an important strategy for K. pneumoniae to grow in the tissues and protect itself from the host immune response. Four major classes of virulence factors, including capsule, lipopolysaccharide, fimbriae, and siderophores, have been well characterized in K. pneumoniae.8 Nevertheless, much is still unknown about additional virulence factors in K. pneumoniae. In this study, by performing comparative proteomics of clinically isolated ESBL-producing K. pneumoniae versus non-ESBL strains, we revealed 9 highly expressed virulence factors of ESBL-producing K. pneumoniae.
Trehalose is a disaccharide formed by two glucose molecules, and is widely distributed in nature in bacteria, fungi, plants, and invertebrates.9 Trehalose not only serves as an energy storage molecule in bacteria, but also plays a role in the virulence of some microorganisms.10 Mutations that abolish trehalose production in various fungi may reduce virulence.11
In the periplasm, trehalose can be degraded into two molecules of glucose by periplasmic trehalase (treA).10 Vanaporn10 showed that treA gene deletion in B. pseudomallei increases the tolerance to thermal stress and results in lower biofilm production than in the wild type. Moreover, mice challenged with treA-deleted B. pseudomallei survived until the end of their study, whereas mice challenged with wild type B. pseudomallei succumbed to the infection. In our study, the ratio of treA between ESBL+ and non-ESBL K. pneumoniae was the highest among all differentially expressed proteins. Therefore, we speculate that the over-expression of treA plays an important role in ESBL+ K. pneumoniae virulence. To date, only a few studies have explored the role of treA in K. pneumoniae, especially in the antibiotic-resistant strains. Further studies are thus needed to reveal its suitability as anti-virulence target.
OmpK35 and OmpK36 are porins closely related to the antibiotic resistance in K. pneumoniae, including ESBL-producing and carbapenem-resistant strains.12,13 The porins can also act as virulence factors during K. pneumoniae infection.14 Most drug-sensitive K. pneumoniae strains express both OmpK35 and OmpK36, whereas most ESBL+ K. pneumoniae strains express only OmpK36.15 Deletion of OmpK36 reduces the virulence of K. pneumoniae by increasing neutrophil-mediated phagocytosis in vivo. However, the deletion also leads to increased antimicrobial resistance, especially associated with decreased susceptibility to carbapenems.12,14,16,17 In this study, OmpK36 ranked second (ratio=8.38) among the differentially expressed proteins, causing the strong virulence of the ESBL+ K. pneumoniae analyzed here. However, due to the increased antibiotic resistance that it induces, OmpK36 should not be selected as an anti-virulence target for treatment of ESBL+ K. pneumoniae infection.
Our data also reveal a significant increase of galE protein in ESBL+ K. pneumoniae strains. The gene galE encodes UDP-glucose 4-epimerase, which epimerizes UDP-galactose and UDP-glucose for galactose metabolism, as well as LPS and EPS formation, thus influencing both bacterial growth and virulence.18 When UDP-galactose accumulates, the concentration of UTP or CTP is reduced in cells and the bacterial growth will be cessative.19 Over-expressed galE has been associated with increased virulence and biofilm formation in numerous bacteria.20–22 Therefore, disrupting or antagonizing galE may be lethal to ESBL+ K. pneumoniae, highlighting this protein as a promising antimicrobial target.
Capsule, the most thoroughly studied virulence factor, is necessary for K. pneumoniae virulence.23 Cps is the conserved gene cluster encoding capsule in K. pneumoniae strains, which harbors a number of genes involved in capsule production, including wza, wzb, wzc, wzi, gnd, ugd, wca, cpsB, cpsG, and galF.24–26 In K. pneumoniae, wza, wzb, and wzc are involved in wzy-dependent K-antigen biosynthesis and export pathway. Our data revealed higher expression of wza, wzb, wzc, gnd, and ugd in ESBL+ K. pneumoniae strains compared to the non-ESBL strains. The gnd-ugd region includes genes involved in the synthesis of GDP-D-mannose (manB and manC) or dTDP-L-rhamnose (rmlA, rmlB, rmlC, and rmlD).24 Expression of rmlA, rmlC, and rmlD is significantly higher in ESBL+ K. pneumoniae than in non-ESBL strains. Considering that rml genes do not exist in human, producing specific dTDP-L-rhamnose biosynthesis inhibitors may directly interfere with ESBL+ K. pneumoniae viability, suggesting the possibility to develop a new class of vaccines targeting nucleotide sugar production against this type of infection. In this study, the expression of sucD in ESBL+ K. pneumoniae was also much higher than in non-ESBL strains. SucD encodes succinyl-CoA synthase α subunit. Succinyl-CoA is part of the tricarboxylic acid (TCA) cycle, whose metabolic products also contribute to the synthesis of capsular polysaccharide in K. pneumoniae. Capsular polysaccharide production contributes to biofilm formation also in K. pneumoniae strains.27 SucD has therefore been identified as an important enzyme in the formation of biofilm in K. pneumoniae.28 Biofilm is a complicated structure comprising bacterial cells embedded in a self-produced EPS matrix,27 which renders the bacteria less susceptible to antibiotics and enables them to counterattack the host defense. Consequently, inhibiting biofilm formation in drug-resistant K. pneumoniae may reduce their resistance to host-mediated defenses29 and weaken their colonizing ability, thereby increasing their susceptibility to antimicrobial agents.30
Nevertheless, there are some limitations of this study. First, the sample size was limited. The number of K. pneumoniae strains used is too small to generalize the findings in the study to a large population. Although we have validated some typical differentially expressed proteins between ESBL-producing and non-ESBL-producing K. pneumoniae by Western blot analysis, the results still need to be further confirmed in a study with a larger sample size. Second, we have not explored the interaction of these differentially expressed proteins with the immune response in vivo. Further studies focusing on this aspect should be undertaken, in order to gain new insights to strengthen the treatment of ESBL-producing K. pneumoniae infections.
Conclusion
In conclusion, our proteomic analysis identified at least 9 plausible virulence-associated proteins that represent promising targets for combating ESBL-producing K. pneumoniae, particularly treA, galE, and gnd. Moreover, our study also demonstrates that proteomics can offer new avenues for exploring new antimicrobial compounds.
Data Sharing Statement
The data used and analyzed during the current study are available from the corresponding author on reasonable request.
Acknowledgments
This work was supported by the Special Funding for Clinical Research of Wu Jieping Medical Foundation [grant numbers 320.6750.18357] and the Science and Technology Development Plan Project of Jilin Province [grant numbers 20190303162SF] to Ke Wang; and the Science and Technology Development Plan Project of Jilin Province [grant numbers: 20180101103JC; 20191102012YY] to Ranwei Li.
Disclosure
The authors declare that they have no conflicts of interest in this work.
References
1. Cantas L, Shah SQ, Cavaco LM, et al. A brief multi-disciplinary review on antimicrobial resistance in medicine and its linkage to the global environmental microbiota. Front Microbiol. 2013;4:96. doi:10.3389/fmicb.2013.00096
2. Rasko DA, Sperandio V. Anti-virulence strategies to combat bacteria-mediated disease. Nat Rev Drug Discov. 2010;9(2):117–128. doi:10.1038/nrd3013
3. Wu HJ, Wang AH, Jennings MP. Discovery of virulence factors of pathogenic bacteria. Curr Opin Chem Biol. 2008;12(1):93–101. doi:10.1016/j.cbpa.2008.01.023
4. Tseng CP, Wu HS, Wu TH, Lin YT, Fung CP. Clinical characteristics and outcome of patients with community-onset Klebsiella pneumoniae bacteremia requiring intensive care. J Microbiol Immunol Infect. 2013;46(3):217–223. doi:10.1016/j.jmii.2012.06.001
5. Lin HA, Huang YL, Yeh KM, Siu LK, Lin JC, Chang FY. Regulator of the mucoid phenotype A gene increases the virulent ability of extended-spectrum beta-lactamase-producing serotype non-K1/K2 Klebsiella pneumonia. J Microbiol Immunol Infect. 2016;49(4):494–501. doi:10.1016/j.jmii.2014.08.023
6. Sahly H, Aucken H, Benedi VJ, et al. Increased serum resistance in Klebsiella pneumoniae strains producing extended-spectrum beta-lactamases. Antimicrob Agents Chemother. 2004;48(9):3477–3482. doi:10.1128/AAC.48.9.3477-3482.2004
7. Wisniewski JR, Zougman A, Nagaraj N, Mann M. Universal sample preparation method for proteome analysis. Nat Methods. 2009;6(5):359–362. doi:10.1038/nmeth.1322
8. Paczosa MK, Mecsas J. Klebsiella pneumoniae: going on the offense with a strong defense. Microbiol Mol Biol Rev. 2016;80(3):629–661.
9. Schiraldi C, Di Lernia I, De Rosa M. Trehalose production: exploiting novel approaches. Trends Biotechnol. 2002;20(10):420–425. doi:10.1016/S0167-7799(02)02041-3
10. Vanaporn M, Sarkar-Tyson M, Kovacs-Simon A, et al. Trehalase plays a role in macrophage colonization and virulence of Burkholderia pseudomallei in insect and mammalian hosts. Virulence. 2017;8(1):30–40. doi:10.1080/21505594.2016.1199316
11. Tournu H, Fiori A, Van Dijck P, Goldman WE. Relevance of trehalose in pathogenicity: some general rules, yet many exceptions. PLoS Pathog. 2013;9(8):e1003447. doi:10.1371/journal.ppat.1003447
12. Chen JH, Siu LK, Fung CP, et al. Contribution of outer membrane protein K36 to antimicrobial resistance and virulence in Klebsiella pneumoniae. J Antimicrob Chemother. 2010;65(5):986–990. doi:10.1093/jac/dkq056
13. Shin SY, Bae IK, Kim J, et al. Resistance to carbapenems in sequence type 11 Klebsiella pneumoniae is related to DHA-1 and loss of OmpK35 and/or OmpK36. J Med Microbiol. 2012;61(Pt 2):239–245. doi:10.1099/jmm.0.037036-0
14. Tsai Y-K, Fung C-P, Lin J-C, et al. Klebsiella pneumoniae outer membrane porins OmpK35 and OmpK36 play roles in both antimicrobial resistance and virulence. Antimicrob Agents Chemother. 2011;55(4):1485–1493. doi:10.1128/AAC.01275-10
15. Hernandez NE, Tereschuk ML, Abdala LR. Antimicrobial activity of flavonoids in medicinal plants from Tafi del Valle (Tucuman, Argentina). J Ethnopharmacol. 2000;73(1–2):317–322. doi:10.1016/S0378-8741(00)00295-6
16. Zhang Y, Jiang X, Wang Y, et al. Contribution of beta-lactamases and porin proteins OmpK35 and OmpK36 to carbapenem resistance in clinical isolates of KPC-2-producing Klebsiella pneumoniae. Antimicrob Agents Chemother. 2014;58(2):1214–1217. doi:10.1128/AAC.02045-12
17. Hamzaoui Z, Ocampo-Sosa A, Fernandez Martinez M, et al. Role of association of OmpK35 and OmpK36 alteration and blaESBL and/or blaAmpC genes in conferring carbapenem resistance among non-carbapenemase-producing Klebsiella pneumoniae. Int J Antimicrob Agents. 2018;52(6):898–905. doi:10.1016/j.ijantimicag.2018.03.020
18. Csiszovszki Z, Krishna S, Orosz L, Adhya S, Semsey S, Hendrix R. Structure and function of the D-galactose network in enterobacteria. mBio. 2011;2(4):e00053–e00011. doi:10.1128/mBio.00053-11
19. Lee SJ, Trostel A, Le P, Harinarayanan R, Fitzgerald PC, Adhya S. Cellular stress created by intermediary metabolite imbalances. Proc Natl Acad Sci U S A. 2009;106(46):19515–19520. doi:10.1073/pnas.0910586106
20. Zou Y, Feng S, Xu C, et al. The role of galU and galE of Haemophilus parasuis SC096 in serum resistance and biofilm formation. Vet Microbiol. 2013;162(1):278–284. doi:10.1016/j.vetmic.2012.08.006
21. Chai Y, Beauregard PB, Vlamakis H, Losick R, Kolter R, Greenberg EP. Galactose metabolism plays a crucial role in biofilm formation by Bacillus subtilis. mBio. 2012;3(4):e00184–e00112. doi:10.1128/mBio.00184-12
22. Niou YK, Wu WL, Lin LC, et al. Role of galE on biofilm formation by Thermus spp. Biochem Biophys Res Commun. 2009;390(2):313–318. doi:10.1016/j.bbrc.2009.09.120
23. Lawlor MS, Handley SA, Miller VL. Comparison of the host responses to wild-type and cpsB mutant Klebsiella pneumoniae infections. Infect Immun. 2006;74(9):5402–5407. doi:10.1128/IAI.00244-06
24. Shu HY, Fung CP, Liu YM, et al. Genetic diversity of capsular polysaccharide biosynthesis in Klebsiella pneumoniae clinical isolates. Microbiology. 2009;155(Pt 12):4170–4183. doi:10.1099/mic.0.029017-0
25. Arakawa Y, Wacharotayankun R, Nagatsuka T, Ito H, Kato N, Ohta M. Genomic organization of the Klebsiella pneumoniae cps region responsible for serotype K2 capsular polysaccharide synthesis in the virulent strain chedid. J Bacteriol. 1995;177(7):1788–1796. doi:10.1128/JB.177.7.1788-1796.1995
26. Pan YJ, Lin TL, Chen CT, et al. Genetic analysis of capsular polysaccharide synthesis gene clusters in 79 capsular types of Klebsiella spp. Sci Rep. 2015;5:15573. doi:10.1038/srep15573
27. Wu MC, Lin TL, Hsieh PF, Yang HC, Wang JT. Isolation of genes involved in biofilm formation of a Klebsiella pneumoniae strain causing pyogenic liver abscess. PLoS One. 2011;6(8):e23500. doi:10.1371/journal.pone.0023500
28. Kong Q, Beanan JM, Olson R, et al. Biofilm formed by a hypervirulent (hypermucoviscous) variant of Klebsiella pneumoniae does not enhance serum resistance or survival in an in vivo abscess model. Virulence. 2012;3(3):309–318. doi:10.4161/viru.20383
29. Roilides E, Simitsopoulou M, Katragkou A, Walsh TJ. How biofilms evade host defenses. Microbiol Spectr. 2015;3(3). doi:10.1128/microbiolspec.MB-0012-2014
30. Verma A, Bhani D, Tomar V, Bachhiwal R, Yadav S. Differences in bacterial colonization and biofilm formation property of uropathogens between the two most commonly used indwelling urinary catheters. J Clin Diagn Re. 2016;10(6):PC01–PC03. doi:10.7860/JCDR/2016/20486.7939
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.