Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 12
Is periodontitis a comorbidity of COPD or can associations be explained by shared risk factors/behaviors?
Authors Hobbins S, Chapple ILC, Sapey E ![]() , Stockley RA
, Stockley RA ![]()
Received 16 November 2016
Accepted for publication 7 February 2017
Published 4 May 2017 Volume 2017:12 Pages 1339—1349
DOI https://doi.org/10.2147/COPD.S127802
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Richard Russell
Stephanie Hobbins,1 Iain LC Chapple,2 Elizabeth Sapey,1,3 Robert A Stockley4
1Institute of Inflammation and Aging, 2School of Dentistry, University of Birmingham, Birmingham, 3Centre for Translational Inflammation Research, Institute of Inflammation and Aging, Queen Elizabeth Hospital, 4University Hospital Birmingham NHS Foundation Trust, Edgbaston, Birmingham, UK
Abstract: COPD is recognized as having a series of comorbidities potentially related to common inflammatory processes. Periodontitis is one of the most common human inflammatory diseases and has previously been associated with COPD in numerous observational studies. As periodontitis and COPD are both chronic, progressive conditions characterized by neutrophilic inflammation with subsequent proteolytic destruction of connective tissue, it has been proposed that they share common pathophysiological processes. The mechanisms proposed to link COPD and periodontitis include mechanical aspiration of oral contents into the respiratory tree, overspill of locally produced inflammatory mediators into systemic circulation or oral or lung-derived bacteremia activating an acute-phase response with subsequent reactive oxygen species (ROS) and cytokine release by systemic neutrophils at distant sites. Studies of systemic neutrophils in COPD and chronic periodontitis describe altered cellular functions that would predispose to inflammation and tissue destruction both in the lung and in the mouth, again potentially connecting these conditions. However, COPD and periodontitis also share risk factors such as age, chronic tobacco smoke exposure, and social deprivation that are not always considered in observational and interventional studies. Furthermore, studies reporting associations have often utilized differing definitions of both COPD and periodontitis. This article reviews the current available evidence supporting the hypothesis that COPD and inflammatory periodontal disease (periodontitis) could be pathologically associated, including a review of shared inflammatory mechanisms. It highlights the potential limitations of previous studies, in particular, the lack of uniformly applied case definitions for both COPD and periodontitis and poor recognition of shared risk factors. Understanding associations between these conditions may inform why patients with COPD suffer such a burden of comorbid illness and new therapeutic strategies for both the diseases. However, further research is needed to clarify factors that may be directly causal as opposed to confounding relationships.
Keywords: inflammation, emphysema, dental health, smoking, neutrophil
Introduction
COPD is defined as a chronic, progressive disease characterized by airflow obstruction due to an enhanced chronic inflammatory response within the airways.1,2 COPD is an important cause of morbidity and mortality and is now recognized as having a series of comorbidities hypothesized to be related to a common systemic or organ-specific inflammatory process. This concept is reflected in the updated Global initiative for chronic Obstructive Lung Disease (GOLD) classification systems for COPD. GOLD now recognizes the significance of extra-pulmonary effects of COPD and that comorbidities contribute to the overall severity of disease and as such should be actively sought and treated.3 An observational study found that >97% of a COPD cohort had at least one other comorbidity, with >50% having at least 4.4 This impacts the health care costs of COPD patients with multiple comorbidities that are 4.7 times higher than those with no comorbidity.5
Respiratory comorbidities associated with COPD include bronchiectasis,6 asthma, pulmonary fibrosis,7 and lung cancer,7 but extra thoracic chronic inflammatory disease also co-occurs.8 Cardiovascular diseases are the most common,9 including hypertension,7 venous thromboembolism, ischemic heart disease, arrhythmias,7,10 and heart failure.7,11 This high prevalence may, in part, reflect the presence of common risk factors including age, smoking, and sedentary lifestyle, but even when such factors are accounted for, there remains increased risk, supporting linked pathogenic drivers of disease. Cerebrovascular disease,9,10,12 cognitive impairment,13 diabetes,9,14 skeletal muscle dysfunction,15 low bone mineral density and osteoporosis11,16 also frequently accompany COPD and support the hypothesis that the pathophysiology underlying these conditions relates to systemic inflammation and hypoxia.11,13 Recently, there has been increasing interest in the association between several chronic diseases including COPD and the chronic inflammatory oral disease, periodontitis, although the underlying nature of the association is unclear.
Periodontitis
Teeth are attached to the surrounding and supporting alveolar bone by periodontal ligament fibers that run from the bone into the cementum on the entire root surface of teeth. Periodontitis is a chronic, bacteria-initiated inflammatory disease, affecting ~50% of adults, with 11.2% suffering from severe periodontitis.17 Sites with periodontitis exhibit clinical signs of gingival inflammation and loss of connective tissue attachment. Plaque biofilm accumulation above and below the gingival margin leads to a dysbiosis (a microbial imbalance or maladapted inflammatory response to bacteria), which in turn drives a dysfunctional host-immune-inflammatory response, connective tissue destruction, and the formation of periodontal pockets.18 The active periodontal lesion is neutrophil and B-cell dominant and associated with systemic inflammation, probably arising due to repeated bacteremias of periodontal origin activating an acute-phase response.19 This subsequently triggers priming and activation of peripheral blood neutrophils with an exaggerated release of reactive oxygen species (ROS) and pro-inflammatory cytokines.20 Finally, the local destructive inflammatory processes lead to loss of alveolar bone and subsequent tooth loss.18
Generally, a diagnosis of periodontitis is based on the combination of periodontal and gingival inflammation, determined as the presence of sub-gingival bleeding on probing (BOP), the number and depth of periodontal pockets, the amount of clinical attachment loss (CAL), and presence of radiologically evaluated alveolar bone loss.21–23 Probing pocket depth (PPD) and CAL are measured by using manual or controlled force periodontal probes. The PPD is the distance from the gingival margin to the base of the gingival sulcus (or periodontal pocket). CAL is the distance from the cemento–enamel junction to the base of the sulcus or periodontal pocket.21 Figure 1 summarizes these parameters.
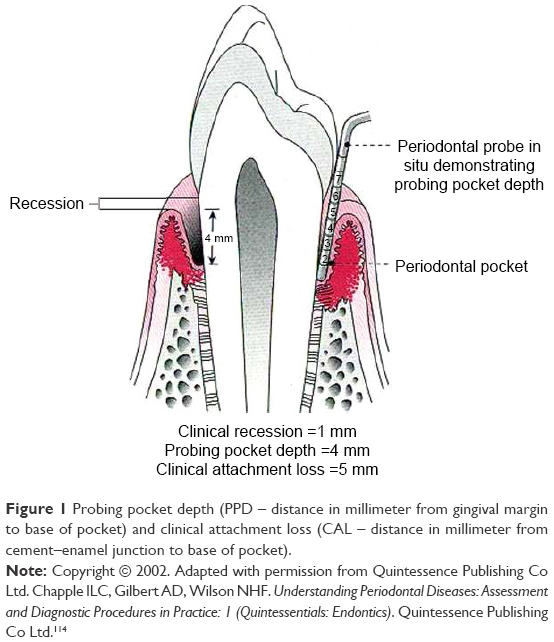 | Figure 1 Probing pocket depth (PPD – distance in millimeter from gingival margin to base of pocket) and clinical attachment loss (CAL – distance in millimeter from cement–enamel junction to base of pocket). |
Periodontitis has also been linked to a number of other chronic systemic diseases as well as to COPD. These include diabetes mellitus,24,25 rheumatoid arthritis,25,26 and cardiovascular diseases. The increased prevalence of cardiovascular disease,27,28 hypertension,25 and stroke in periodontal disease25,29 has been attributed to a common susceptibility hypothesis, that of direct bacterial invasion of blood vessels, and/or a systemic inflammatory response leading to increased circulating cytokines that damage vascular endothelium.30 Importantly, treating periodontitis in patients with common comorbidities has shown to be an effective way of reducing both financial costs and admissions for cardiovascular disease and diabetes mellitus,31 lending credence to this hypothesis. Thus, both periodontitis and COPD occur with a similar spectrum of comorbid disease. This needs recognition and documentation when reporting associations between periodontitis and COPD as it likely reflects a subgroup of patients with a predisposition or the presence of common etiological factors for this spectrum of diseases.
In keeping with this, there are important similarities in both the mechanisms underpinning periodontitis and COPD and the risk factors for developing these conditions that are worthy of exploration. Both periodontitis and COPD are chronic and progressive conditions characterized by neutrophilic inflammation with subsequent proteolytic destruction of connective tissues.32–34 Of note, not all patients with common risk factors (chronic cigarette smoke inhalation for both condition and poor dental hygiene for periodontitis) develop disease, for example, only 20% of smokers go on to develop significant COPD,35 supporting the concept of susceptibility. They also share several other common risk factors including age and poor socioeconomic status.33,36
As both the conditions result in tissue destruction by neutrophilic inflammation, it has been suggested that they share common pathophysiological processes. If true, this would have important implications for the diagnosis and potentially even the treatment of these conditions, as diagnosing and controlling one with an appropriate intervention may reduce severity and/or progression in the other.
Mechanisms
There are several mechanisms that may potentially link respiratory disease with periodontitis.
Aspiration of potentially pathogenic bacteria
The first is mechanical; the aspiration of oral contents including microbial pathogens into the respiratory tract with airway inflammation and remodeling (caused by COPD) supporting adherence, immune evasion, colonization, and subsequent pulmonary infections. Micro-aspiration is common even in healthy patients and frequently occurs during sleep. Studies have shown that typical volumes aspirated are of a magnitude likely to contain bacterial pathogens.37 In healthy patients, there are defense mechanisms to ensure that, despite aspiration, the distal airway and lung parenchyma remain protected.38 This clearance is mediated by both mechanical and immunological mechanisms and includes cough and mucociliary clearance of pathogens and phagocytosis of microorganisms by local immune cells.37 However, in patients with underlying chronic health problems, aspiration of oral secretions containing bacterial pathogens may not always be cleared.39,40 In these cases, the periodontium could act as a repository of bacteria, enhancing respiratory tract colonization and inflammation. For COPD patients, this is important as recurrent infections causing exacerbations are known to amplify the decline in lung function41 but current evidence to support the link between COPD and periodontitis remains, at best, indirect.
Oral colonization by respiratory pathogens, fostered by poor oral hygiene and periodontal diseases, seems to be associated with nosocomial pneumonia in some studies. A recent meta-analysis identified 11 observational studies of intubated patients where periodontal disease was assessed with respiratory infections as an outcome.42 Approximately half of these studies reported an association between dental status and ventilator-associated pneumonia, with one study43 reporting that the pathogen thought to cause pneumonia had first colonized the dental plaque biofilm. However, these studies were conducted in extremely unwell patients who were at significant risk of pneumonia and some did not include a control population. Studies have associated pneumonia with poor dental hygiene and periodontitis in both the community and the outpatient setting but most do not provide information about potentially confounding factors (comorbidities, smoking status, etc) or information about microbial pathogens. There are five intervention trials44–48 where a mixed population of patients received treatments to lower oropharyngeal bacterial load, and a meta-analysis suggested that oral hygiene and antibiotic intervention significantly reduced the risk of developing pneumonia in these populations, supporting a causal link between oral pathogens and lower respiratory tract infections.42 However, once again, the majority of studies were conducted in an intensive care setting, and it is unclear whether the findings are generalizable.
Approximately 10 bacterial species have been identified as putative pathogens in inflammatory periodontal diseases. These are mainly gram-negative rods and include Aggregatibacter actinomycetemcomitans in aggressive periodontitis and Porphyromonas gingivalis, Treponema denticola, and Tanerella forsythia in chronic periodontitis.49 However, these are not commonly associated with lower respiratory infections in the general population or in COPD, and there are no studies in COPD patients that have directly associated oral pathogens with COPD exacerbations.
Periodontitis as an inflammatory reservoir
Alternative hypotheses linking the two conditions suggest bacterial aspiration is not required to initiate shared pathology. The entry of periodontal bacteria into the vasculature during eating and tooth brushing can trigger an acute-phase response in the systemic circulation as evidenced by elevated serum levels of inflammatory mediators.21 In keeping with this hypothesis, periodontitis is now recognized as a significant and independent risk factor for atherogenic cardiovascular disease, and the mechanisms seem to involve periodontal pathogens disseminating throughout the vasculature.27 Moreover, peripheral blood neutrophils from periodontitis patients exhibit hyper-reactivity to bacterial stimuli and hyperactivity (in absence of a stimulus) with respect to ROS and pro-inflammatory cytokine release.50–54 Alternatively, the local inflammatory response to periodontitis could lead to an “overspill” of periodontal cytokines into the systemic circulation, with subsequent distant inflammatory sequelae. Evidence to support this is weak55 although this may just reflect the rapid distribution and binding of pro-inflammatory cytokines to receptors as argued for COPD.56 Nevertheless pro-inflammatory cytokine networks have been found to be activated in periodontitis tissues57 and many of the same cytokines have been found to be raised in blood58–60 and lung secretions from patients with COPD61–65 at least supporting a common inflammatory process.
If present, any shared mechanisms are likely to impact on both the diseases, with periodontal inflammation and infection impacting on lung pathology and inflammatory lung disease amplifying periodontal disease risk. These mechanisms are summarized in Figure 2.
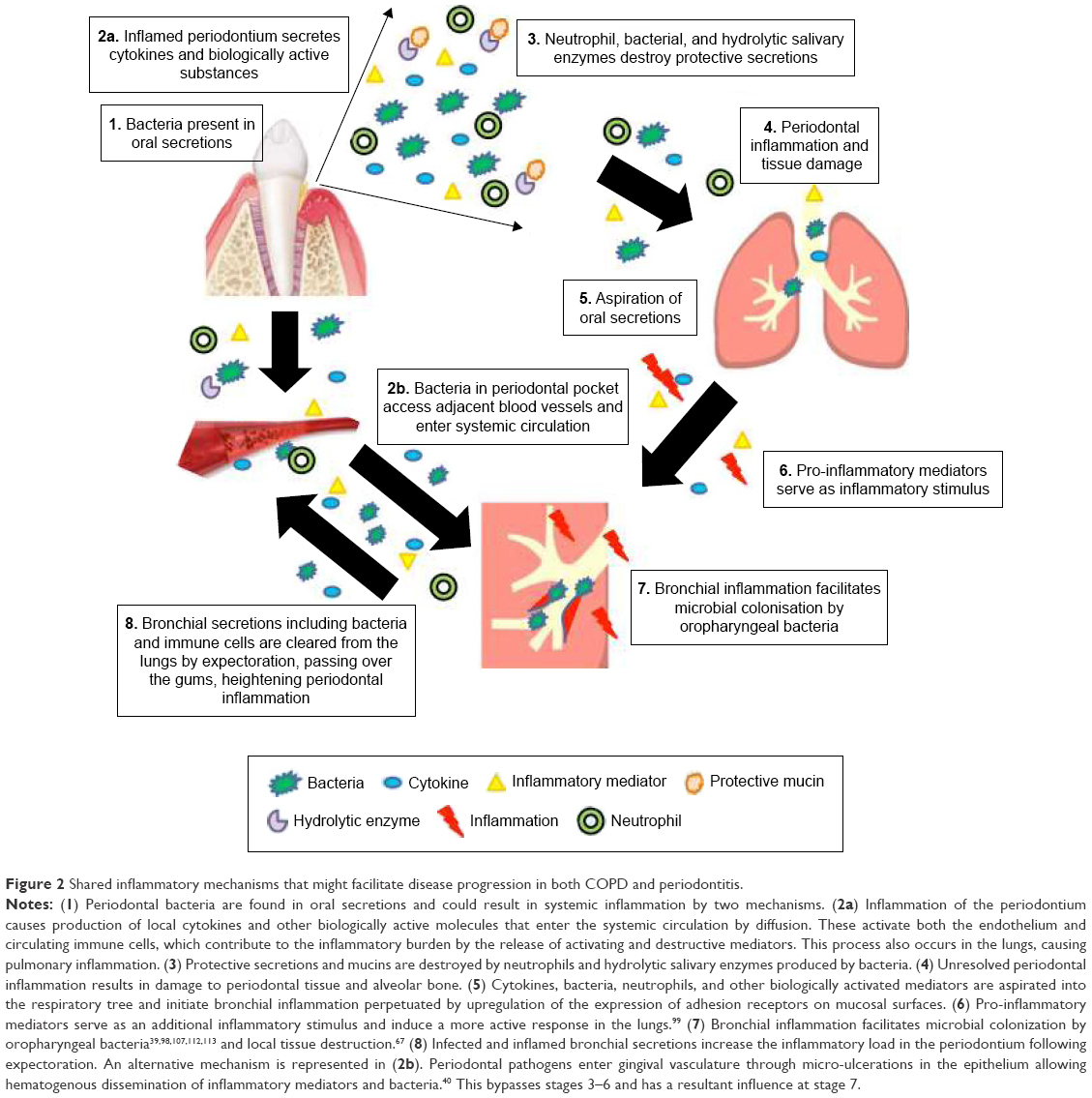 | Figure 2 Shared inflammatory mechanisms that might facilitate disease progression in both COPD and periodontitis. |
The most compelling evidence for a common mechanism causing both periodontitis and COPD is that of the neutrophilic inflammation and neutrophil-mediated tissue damage that characterizes both the diseases.
Neutrophilic inflammation in COPD
Neutrophils have a key role in inflammatory processes, and there is compelling evidence to support their role in the pathogenesis of COPD. Neutrophils have been implicated in the development and progression of all of the pulmonary features of COPD in animal and cell studies, including emphysema through the release of destructive mediators such as neutrophil elastase and matrix metalloproteinases (MMPs)66 and chronic bronchitis through mucous gland hyperplasia.68 Pulmonary neutrophilic inflammation is a feature of cigarette smoking, but importantly, in patients with COPD it is sustained even following smoking cessation,68,69 though not in smokers without airflow obstruction.
As well as being increased in number in bronchial tissue and airway secretions, there is evidence to suggest that neutrophils in COPD patients are also dysfunctional. Circulating neutrophils produce more ROS than those from healthy nonsmokers or smokers with normal lung function.70 The cells also have enhanced, rapid, and tortuous chemotactic responses to inflammation and evidence of increased degranulation and tissue destruction both in vitro and systemically.71,72
The level of neutrophilic inflammation in airways correlates with disease severity and progression in COPD whether measured by lung physiology, radiologically, or health status.63,73–76 These data support the concept that activated neutrophils play an important role in lung injury and emphysema through neutrophilic release of inflammatory cytokines, destructive proteases such as neutrophil elastase, proteinase 3, MMP-8, and MMP-977–79 all of which are capable of degrading components of the extracellular matrix, promoting inflammatory responses in the surrounding tissue80 and increasing the generation of ROS.70 Cigarette smoking further enhances recruitment of these activated neutrophils with more subsequent release of tissue destructive proteinases,81 supporting the faster physiological decline seen in COPD patients who continue to smoke.
Neutrophils in periodontitis
Periodontitis is also characterized by a neutrophilic infiltrate.82 A review by Hajishengallis83 highlighted the increased prevalence of periodontitis in conditions characterized by abnormal neutrophil function. These include defective neutrophil production as seen in autoimmune neutropenia or chemotherapy-induced neutropenia, defective neutrophil release from the bone marrow as seen in WHIM (warts, hypogammaglobulinemia, infections, and myelokathexis), inherited leukocyte adhesion deficiency, excessive neutrophil activation, deficient clearance of neutrophils as in autoimmune inflammatory diseases including systemic lupus erythematosus and the Papillon–Lefevre syndrome (neutrophil cathepsin C deficiency).
There is evidence of both hyper-reactive and hyperactive neutrophil functions in periodontitis. Systemic neutrophil chemotaxis studies in patients with chronic periodontitis showed reduced speed (movement in any direction), velocity (speed of movement toward the chemoattractant), and accuracy (accuracy of movement toward the chemoattractant).84 This behavior improved after treatment of periodontitis,85 suggesting a causal link between periodontitis and the systemic cell function. A similar pattern of neutrophil migration has been described in COPD where cells displayed poor migratory accuracy but with random movement in all directions at increased speed.72 Recent publications have also described compromised neutrophil bacterial phagocytosis in periodontitis86 (studies in COPD are less clear); enhanced neutrophil-derived ROS87,88 and increased NET formation in both COPD and periodontitis,89,90 all of which could contribute to the sustained inflammation that characterizes both the conditions.34,53 The cause for such injurious cell behavior is unclear, and it is unknown whether neutrophil dysfunction precedes or is a consequence of disease. However, these impaired neutrophil functions may mean that the host is less able to deal efficiently with bacteria either in the periodontium or in the lung. Furthermore, as neutrophils release both proteinases91,92 and ROS while migrating through complex tissues,93 it is possible that the tortuous abnormal migratory pathways lead to increased by-stander tissue damage, thereby perpetuating the inflammatory response leading to excess tissue damage seen as alveolar bone loss in the mouth and emphysema and COPD in the lung. This concept has been supported in COPD where there is systemic evidence of increased proteinase activity,94 but remains unconfirmed in periodontitis.
COPD and periodontitis
There is evidence of a clinical association between periodontitis and COPD including a meta-analysis of 14 observational studies.36 The overall sample size represented 3,988 COPD patients and 22,871 controls. The authors reported a pooled odds ratio (OR) for having COPD in populations with periodontitis as 2.08 (95% confidence interval [CI] 1.48–2.91; P<0.001).
Alternatively, data from a large longitudinal COPD cohort study found that patients had a significantly increased cumulative incidence of periodontitis with adjusted hazard ratio (HR) of 1.20 (95% CI 1.15–1.25) rising to 3.17 (95% CI 2.81–3.57) in the subgroup that had required hospitalization for COPD.33 The risk of developing periodontitis increased proportionately with the annual number of emergency room visits and admissions for COPD.33 A further in-patient study supported these findings, noting significantly increased markers of periodontitis in a hospitalized COPD group compared with an in-patient control group (probing depth [PD] 2.57±0.85 vs 1.76±0.7, P<0.0001; and clinical attachment level [CAL] 3.16±0.53 vs 2.12±0.92, P<0.0001).95
A recent, large cross-sectional study found that after adjustment for the confounders of age, smoking, socio-economic status, and dental habits, moderate periodontitis was still associated with COPD in male patients (relative risk =1.38, 95% CI 1.12–2.05), but not female patients, and the relative risk of severe periodontitis in the male group was 1.23 (95% CI 1.06–1.56).96 Prasanna also found significantly higher periodontal indices in COPD patients in comparison with non-COPD controls that persisted after correction for age.40 A Chinese case–control study of 306 dentate COPD patients and 328 dentate healthy controls found the COPD cohort had fewer remaining teeth and a greater percentage of the remaining teeth had CAL ≥4 mm.97 In addition, an Indian observational study of 150 dentate patients also found patients with COPD had significantly higher mean CAL measurements.98
Using a different measure of periodontitis, Leuckfeld et al assessed orthopantomograms in a retrospective cross-sectional study of patients evaluated for lung transplant. They found a significant difference in radiographically determined periodontitis, with 44% of COPD patients affected compared with 7% of non-COPD patients. The results also remained significantly different after adjustment for age, gender, and smoking.99
Several studies have also found associations between indices of periodontitis and lung function measurements. An observational study of 102 dentate COPD patients found significantly worse indices of periodontitis after adjusting for age and smoking.100 The same study confirmed a significant correlation between several adverse periodontal outcomes; CAL, PD, and gingival index (GI) and forced expiratory volume in 1 s (FEV1) (r=−0.63, −0.60, and −0.44, respectively; P<0.0001). A large German prospective cohort study of 1,268 people found that pulmonary function parameters were significantly associated with CAL measurements after adjustment for confounding factors.101
However, other studies have failed to find such associations. Bergstrom et al102 found that smoking rather than COPD was the major risk factor for periodontal pocket depth (OR 24.2; 95% CI 2.0–286.8). One analysis of the NHANES III survey found no relationship between the two diseases, although subgroup analysis found an association in current smokers aged ≥30 years with CAL ≥4 mm.103 Finally, a cross-sectional study of 392 dentate patients with COPD was also unable to find significant differences between periodontal measurements and exacerbation rates.104 However, the lack of consistency in findings also reflects the lack of consistency in case definitions, data collection, and analysis (eg, case definitions vs continuous outcome variables such as PPD, CAL, etc).
Potential limitations of these studies
The data summarized above remains controversial as there are several potential limitations in the studies that have assessed associations between COPD and periodontitis.
First, many studies have not accounted for common risk factors that might contribute to the presence of both periodontitis and COPD. For example, the longitudinal study by Shen et al failed to correct for smoking status, dental habits, or socioeconomic status,33 and no adjustment was made for socioeconomic factors in the radiographic study by Leuckfeld et al.99 Where appropriate allowance had occurred, the OR for having COPD in the populations with periodontitis (OR 1.78; 95% CI 1.23–2.58) was lower than initially reported.36
Second, the manner in which the diseases are diagnosed in the epidemiological studies has a significant impact on the results of prevalence and associations. For epidemiological studies, a case definition needs to be easily and accurately measurable21 and a uniform definition is essential to allow calculations and comparisons of relative risks to be made across studies.105
Most studies investigating associations between COPD and periodontitis base the diagnosis of COPD on post-bronchodilator spirometry and GOLD staging.95,96,100,102,103,106 However, some authors used pre-bronchodilator spirometry,107 which could lead to an overestimation of the prevalence of COPD, others report lung function measurements but not whether pre- or post-bronchodilation,40,98 and one study failed to mention how COPD was confirmed.99 A German prospective study used body plethysmography to determine lung function,101 whereas Shen et al used International Classification of Diseases, Ninth Revision, Clinical Modification (ICD-9-CM) codes that do not provide details of how the diagnosis was established.33
Third, there is also great variation in how authors define periodontitis. Savage et al conducted a systematic review of 3,472 papers analyzing 104 of these in detail and found that only 15 studies gave a definition of periodontitis.23 This is a continuing problem in the literature studying COPD and periodontitis. Published studies have used various periodontal measurement parameters for analysis but do not always give a specific definition for the disease,40,95,100,104,107 which likely reflects the lack of a uniformly accepted definition of periodontitis. When defined, measurements to determine the presence or severity of periodontitis vary and include one or a combination of the PPD, CAL, bleeding index (BI), gingival index (GI), plaque index (PI), or oral hygiene index (OHI), reflecting the lack of an agreed GOLD standard. Wang et al measured PPD, CAL, and BI, but in their statistical analysis report, the percentage of sites with PD of ≥4 and 5 mm and CAL ≥3 and 4 mm,97 since these values are regarded as representing active periodontitis (PPD >4 mm). Deo et al measured PPD, CAL, and OHI but reported only on CAL and OHI.98 Other studies have favored the use of PD and CAL in combination.103,106 However, Hyman et al only found significant associations with CAL alone.103 This may reflect the fact that PPD used in isolation accounts only for current disease status, whereas CAL represents the cumulative periodontal tissue damage with age.102
The studies by Chung et al and Kim et al used the crude screening index called the World Health Organization community periodontal index to screen for periodontitis in large populations, defining the disease as present with a score of 3 (>3.5 mm pocket) or 4 (>5.5 mm pocket).96,108 Mean pocket depth of ≥4 mm was arbitrarily chosen as a cut-off point to define periodontitis in the Bergstrom et al’s study,102 since this value represents clinical pathology, and pocket depths of missing teeth were set as the values of neighboring tooth sites. Interestingly, when analyzed separately, recession was more prevalent in smokers, suggesting measurement of pocket depth alone may underestimate disease severity in smoking groups.102
Two further studies employed very different methods to define periodontitis. Leuckfeld et al used mean marginal bone level ≥4 mm on orthopantomograms,99 and Shen et al used ICD-9-CM codes.33 Although radiographs allow assessment of cumulative effects on alveolar bone loss, they do not allow assessment of current disease activity, they are not always available, and radiation exposure for epidemiological purposes is unlikely to be acceptable.22
In younger populations, PPD and CAL correlate well, but as people age, gingival recession and therefore measures of attachment loss become a normal part of the ageing process. In addition, increased PPD may arise as a false-positive clinical finding. This can occur when gingival tissues become swollen due to inflammation (in the absence of periodontitis), thus moving the gingival margin upwards (false pocketing). Periodontitis can progress without significant pockets if gingival recession is also present, as frequently found in smokers.23 The use of CAL alone may also overestimate periodontitis because attachment loss can occur with non-inflammatory gingival recession, such as that induced by trauma from over tooth brushing.21
During the 5th European Workshop in Periodontology, a two threshold level criterion was proposed to diagnose periodontitis. This was the presence of proximal attachment loss of ≥3 mm in ≥2% of nonadjacent teeth to diagnose incipient cases and the presence of proximal attachment loss of ≥5 mm in ≥30% of teeth to determine severe disease. However, the authors of this proposal also stated that a single-index measurement of periodontitis does not uniformly reflect the disease and that these criteria are designed for use in studies determining risk factors for periodontitis and not the prevalence of periodontitis.105
In an attempt to produce a standard case definition for periodontitis, the Division of Oral Health at the Centers for Disease Control and Prevention (CDC) alongside the American Academy of Periodontology appointed a working group in 200321 to define severe and moderate periodontitis. Severe periodontitis required involvement of two or more interproximal sites with CAL ≥6 mm and one or more interproximal sites with PD ≥5 mm. Moderate periodontitis required involvement of two or more interproximal sites with CAL ≥4 mm or ≥2 interproximal sites with PD ≥5 mm.21 Since the development of these definitions, Eke et al as part of the group at the CDC have added a further definition classifying mild periodontitis as ≥2 interproximal sites with attachment loss ≥3 mm and ≥2 interproximal sites with PD ≥4 mm, or one with PD ≥5 mm.109 These authors applied their criteria to a group of 456 dentate individuals and found that the addition of mild cases increased the total prevalence of periodontitis from 22.3% to 29.4%. They also applied the criteria established by the 5th European Workshop and found the European criteria gave a greater prevalence of 42% for incipient cases, compared with 24.6% for the CDC criteria, although the prevalence was the same for severe periodontitis (4.8%).109
The current American classification system was developed in 1999 at the International Workshop for a Classification of Periodontal Diseases and Conditions. This led to the addition of a subcategory for gingival conditions, recognition of subtypes of periodontitis, and importantly the clarification of periodontitis as the manifestation of systemic disease.110 In 2014, a task force developed a clinical interpretation of this classification, addressing the concept of attachment levels in the diagnosis of periodontitis. This recognized the importance of attachment levels but also difficulties in accurate measurement and interpretation especially in those with non-inflammatory gum recession. Clarification was also made regarding localized versus generalized periodontitis, and these criteria are now widely adopted in the USA.111
Importantly, these variations in definition of periodontal disease will influence prevalence rates and hence ORs of both COPD in periodontitis and vice versa.
Socioeconomic status
Both COPD and periodontitis share the common risk factors of poor socioeconomic status and smoking,33,36 and it is possible that the link seen between the two conditions also reflects poor oral health in combination with other factors such as smoking, poor nutrition, poor self-care behaviors, and exposure to other environmental factors such as pollutants.
Kim et al found COPD patients with and without periodontitis were significantly older, less well educated, took less exercise, had lower income, smoked more, and had a greater alcohol intake.108 Chung et al also noted that the COPD patients were generally less well educated, had a lower income, smoked more, and drank more alcohol.96 The COPD patients in Prasanna’s study were also noted to belong to a lower income group, were more illiterate, and were of a lower socioeconomic status regardless of periodontal status.40 Holtfreter et al found that patients with higher indices of periodontitis were less well educated and more often current smokers and had higher levels of systemic inflammation and worse lung function.101 All these studies indicate significant confounders that likely impact on results published to date even though Bhavsar et al did not find any differences in socioeconomic status or educational qualifications between their COPD cohort and control groups.95
Dental habits
It seems intuitive that poor dental hygiene would also increase the prevalence of periodontal diseases and may reflect education and socioeconomic status. Thus, it is important that future studies assessing specific comorbid associations between COPD and periodontitis also include the potential confounding effect of dental habits.
In support of this, Bhavsar et al found patients in the COPD group, as a whole, had significantly reduced brushing frequency compared with a non-COPD group (independent of periodontal status).95 Similarly, Liu et al found that a higher proportion of patients with frequent COPD exacerbations brushed their teeth less than once per day and had higher PI scores after adjusting for other confounding factors.104 Wang et al also found that their COPD group generally had lower tooth brushing times (P<0.0001) and oral health knowledge scores (P<0.0001) and were also less likely to use dental floss and have regular dental visits than controls.97 All these studies indicate that lifestyle factors are also likely to be a crucial confounder when seeking associations and/or a common pathophysiological process.
Conclusion
The available evidence still provides some support for the hypothesis that COPD and periodontitis could be causally linked; raising the possibility that treatment of one could influence the severity and progression of the other. There are similarities in disease mechanisms (that of dysfunctional neutrophil behaviors, sustained neutrophilic inflammation, and connective tissue loss) that suggest a shared pathophysiology and support epidemiological evidence of association. However, some studies conducted to date have significant limitations that need to be overcome in order to provide a clear picture. First, the presence and severity of periodontitis must be defined using robust criteria, and second, the diagnosis of COPD must be confirmed according to international standards; this will require close collaboration between dentists and physicians. Third, known relevant confounding factors must be recorded and taken into account, and this will require a significant sample size to allow appropriate statistical modeling. Finally, the inflammatory profiles (systemically or locally) in groups of patients with COPD and periodontitis need to be compared with that of matched patients with COPD and no periodontitis. Such studies could then be used to design appropriately powered interventional trials to determine whether treating periodontitis is of benefit to COPD-related lung disease.
Acknowledgments
SH is funded by an FP7 EU grant, and RAS is the principal investigator (PI). ES is funded by the Medical Research Council. Both SH and ES are supported by the National Institute for Health Research (NIHR) Wellcome Birmingham Clinical Research Facility.
Author contributions
All authors contributed toward data analysis, drafting and revising the paper and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
Sapey E, Bayley D, Ahmad A, Newbold P, Snell N, Stockley RA. Inter-relationships between inflammatory markers in stable COPD patients with bronchitis; the intra and inter patient variability. Thorax. 2008;63:493–499. | ||
Stockley RA. Neutrophils and the pathogenesis of COPD. Chest. 2002;121:151S–155S. | ||
Global Strategy for the Diagnosis, Management, and Prevention of Chronic Obstructive Pulmonary Disease; 2015. Available from: http://goldcopd.org/. Accessed January 14, 2017. | ||
Vanfleteren LE, Spruit MA, Groenen M, et al. Clusters of comorbidities based on validated objective measurements and systemic inflammation in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2013;187(7):728–735. | ||
Mapel DW, Hurley JS, Frost FJ, Petersen HV, Picchi MA, Coultas DB. Health care utilization in chronic obstructive pulmonary disease: a case-control study in a health maintenance organization. Arch Intern Med. 2000;160(17):2653–2658. | ||
Du Q, Jin J, Liu X, Sun Y. Bronchiectasis as a comorbidity of chronic obstructive pulmonary disease: a systematic review and meta-analysis. PLoS One. 2016;11(3):e0150532. | ||
Hillas G, Perlikos F, Tsiligianni I, Tzanakis N. Managing comorbidities in COPD. Int J Chron Obstruct Pulmon Dis. 2015;10:95–109. | ||
Li X, Kong L, Li F, et al. Association between psoriasis and chronic obstructive pulmonary disease: a systematic review and meta-analysis. PLoS One. 2015;10(12):e0145221. | ||
Feary JR, Rodrigues LC, Smith CJ, Hubbard RB, Gibson JE. Prevalence of major comorbidities in subjects with COPD and incidence of myocardial infarction and stroke: a comprehensive analysis using data from primary care. Thorax. 2010;65(11):956–962. | ||
Schneider C, Bothner U, Jick SS, Meier CR. Chronic obstructive pulmonary disease and the risk of cardiovascular diseases. Eur J Epidemiol. 2010;25(4):253–260. | ||
Patel AR, Hurst JR. Extrapulmonary comorbidities in chronic obstructive pulmonary disease: state of the art. Expert Rev Respir Med. 2011;5(5):647–662. | ||
Truelsen T, Prescott E, Lange P, Schnohr P, Boysen G. Lung function and risk of fatal and non-fatal stroke. The Copenhagen City Heart Study. Int J Epidemiol. 2001;30(1):145–151. | ||
Dodd JW, Getov SV, Jones PW. Cognitive function in COPD. Eur Respir J. 2010;35(4):913–922. | ||
Mannino DM, Thorn D, Swensen A, Holguin F. Prevalence and outcomes of diabetes, hypertension and cardiovascular disease in COPD. Eur Respir J. 2008;32(4):962–969. | ||
Seymour JM, Spruit MA, Hopkinson NS, et al. The prevalence of quadriceps weakness in COPD and the relationship with disease severity. Eur Respir J. 2010;36(1):81–88. | ||
Chen SJ, Liao WC, Huang KH, et al. Chronic obstructive pulmonary disease and allied conditions is a strong independent risk factor for osteoporosis and pathologic fractures: a population-based cohort study. QJM. 2015;108(8):633–640. | ||
Kassebaum NJ, Bernabe E, Dahiya M, Bhandari B, Murray CJ, Marcenes W. Global burden of severe periodontitis in 1990–2010: a systematic review and meta-regression. J Dent Res. 2014;93(11):1045–1053. | ||
Meyle J, Chapple I. Molecular aspects of the pathogenesis of periodontitis. Periodontol 2000. 2015;69(1):7–17. | ||
Cekici A, Kantarci A, Hasturk H, Van Dyke TE. Inflammatory and immune pathways in the pathogenesis of periodontal disease. Periodontol 2000. 2014;64(1):57–80. | ||
Ling MR, Chapple IL, Matthews JB. Peripheral blood neutrophil cytokine hyper-reactivity in chronic periodontitis. Innate Immun. 2015;21(7):714–725. | ||
Page RC, Eke PI. Case definitions for use in population-based surveillance of periodontitis. J Periodontol. 2007;78(7 Suppl):1387–1399. | ||
Leroy R, Eaton KA, Savage A. Methodological issues in epidemiological studies of periodontitis – how can it be improved? BMC Oral Health. 2010;10:8. | ||
Savage A, Eaton KA, Moles DR, Needleman I. A systematic review of definitions of periodontitis and methods that have been used to identify this disease. J Clin Periodontol. 2009;36(6):458–467. | ||
Wang TF, Jen IA, Chou C, Lei YP. Effects of periodontal therapy on metabolic control in patients with type 2 diabetes mellitus and periodontal disease: a meta-analysis. Medicine (Baltimore). 2014;93(28):e292. | ||
Chrysanthakopoulos NA, Chrysanthakopoulos PA. Association between indices of clinically-defined periodontitis and self-reported history of systemic medical conditions. J Investig Clin Dent. 2016;7(1):27–36. | ||
Scher JU, Bretz WA, Abramson SB. Periodontal disease and subgingival microbiota as contributors for rheumatoid arthritis pathogenesis: modifiable risk factors? Curr Opin Rheumatol. 2014;26(4):424–429. | ||
Dietrich T, Sharma P, Walter C, Weston P, Beck J. The epidemiological evidence behind the association between periodontitis and incident atherosclerotic cardiovascular disease. J Periodontol. 2013;84(4 Suppl):S70–S84. | ||
Widen C, Holmer H, Coleman M, et al. Systemic inflammatory impact of periodontitis on acute coronary syndrome. J Clin Periodontol. 2016;43:713–719. | ||
Sfyroeras GS, Roussas N, Saleptsis VG, Argyriou C, Giannoukas AD. Association between periodontal disease and stroke. J Vasc Surg. 2012;55(4):1178–1184. | ||
Seymour GJ, Ford PJ, Cullinan MP, Leishman S, Yamazaki K. Relationship between periodontal infections and systemic disease. Clin Microbiol Infect. 2007;13 (Suppl 4):3–10. | ||
Jeffcoat MK, Jeffcoat RL, Gladowski PA, Bramson JB, Blum JJ. Impact of periodontal therapy on general health: evidence from insurance data for five systemic conditions. Am J Prev Med. 2014;47(2):166–174. | ||
Fokkema SJ, Timmerman MF, van der Weijden FA, Wolffe GN, Renggli HH. A possible association of alpha1-antitrypsin deficiency with the periodontal condition in adults. J Clin Periodontol. 1998;25(8): 617–623. | ||
Shen TC, Chang PY, Lin CL, et al. Risk of periodontal diseases in patients with chronic obstructive pulmonary disease: a nationwide population-based cohort study. Medicine (Baltimore). 2015;94(46):e2047. | ||
Stockley JA, Walton GM, Sapey E. Aberrant neutrophil functions in stable chronic obstructive pulmonary disease: the neutrophil as an immunotherapeutic target. Int Immunopharmacol. 2013;17(4):1211–1217. | ||
Tashkin DP, Clark VA, Coulson AH, et al. The UCLA population studies of chronic obstructive respiratory disease. VIII. Effects of smoking cessation on lung function; a prospective study of a free-living population. Am Rev Respir Dis. 1984;130:707–715. | ||
Zeng XT, Tu ML, Liu DY, Zheng D, Zhang J, Leng W. Periodontal disease and risk of chronic obstructive pulmonary disease: a meta-analysis of observational studies. PLoS One. 2012;7(10):e46508. | ||
Gleeson K, Eggli DF, Maxwell SL. Quantitative aspiration during sleep in normal subjects. Chest. 1997;111(5):1266–1272. | ||
Bansal M, Khatri M, Taneja V. Potential role of periodontal infection in respiratory diseases – a review. J Med Life. 2013;6(3):244–248. | ||
Azarpazhooh A, Leake JL. Systematic review of the association between respiratory diseases and oral health. J Periodontol. 2006;77(9):1465–1482. | ||
Prasanna SJ. Causal relationship between periodontitis and chronic obstructive pulmonary disease. J Indian Soc Periodontol. 2011;15(4):359–365. | ||
Donaldson GC, Seemungal TA, Bhowmik A, Wedzicha JA. Relationship between exacerbation frequency and lung function decline in chronic obstructive pulmonary disease. Thorax. 2002;57(10):847–852. | ||
Scannapieco FA, Bush RB, Paju S. Associations between periodontal disease and risk for nosocomial bacterial pneumonia and chronic obstructive pulmonary disease. A systematic review. Ann Periodontol. 2003;8(1):54–69. | ||
Fourrier F, Dubois D, Pronnier P, et al. Effect of gingival and dental plaque antiseptic decontamination on nosocomial infections acquired in the intensive care unit: a double-blind placebo-controlled multicenter study. Crit Care Med. 2005;33(8):1728–1735. | ||
Fourrier F, Cau-Pottier E, Boutigny H, Roussel-Delvallez M, Jourdain M, Chopin C. Effects of dental plaque antiseptic decontamination on bacterial colonization and nosocomial infections in critically ill patients. Intensive Care Med. 2000;26(9):1239–1247. | ||
Genuit T, Bochicchio G, Napolitano LM, McCarter RJ, Roghman MC. Prophylactic chlorhexidine oral rinse decreases ventilator-associated pneumonia in surgical ICU patients. Surg Infect (Larchmt). 2001;2(1):5–18. | ||
Bergmans DC, Bonten MJ, Gaillard CA, et al. Prevention of ventilator-associated pneumonia by oral decontamination: a prospective, randomized, double-blind, placebo-controlled study. Am J Respir Crit Care Med. 2001;164(3):382–388. | ||
Yoneyama T, Hashimoto K, Fukuda H, et al. Oral hygiene reduces respiratory infections in elderly bed-bound nursing home patients. Arch Gerontol Geriatr. 1996;22(1):11–19. | ||
Yoneyama T, Yoshida M, Ohrui T, et al. Oral care reduces pneumonia in older patients in nursing homes. J Am Geriatr Soc. 2002;50(3):430–433. | ||
Haffajee AD, Socransky SS. Microbial etiological agents of destructive periodontal diseases. Periodontol 2000. 1994;5:78–111. | ||
Geivelis M, Turner DW, Pederson ED, Lamberts BL. Measurements of interleukin-6 in gingival crevicular fluid from adults with destructive periodontal disease. J Periodontol. 1993;64(10):980–983. | ||
Shimada Y, Komatsu Y, Ikezawa-Suzuki I, Tai H, Sugita N, Yoshie H. The effect of periodontal treatment on serum leptin, interleukin-6, and C-reactive protein. J Periodontol. 2010;81(8):1118–1123. | ||
Matthews JB, Wright HJ, Roberts A, Ling-Mountford N, Cooper PR, Chapple IL. Neutrophil hyper-responsiveness in periodontitis. J Dent Res. 2007;86(8):718–722. | ||
Wright HJ, Matthews JB, Chapple IL, Ling-Mountford N, Cooper PR. Periodontitis associates with a type 1 IFN signature in peripheral blood neutrophils. J Immunol. 2008;181(8):5775–5784. | ||
Dias IH, Matthews JB, Chapple IL, Wright HJ, Dunston CR, Griffiths HR. Activation of the neutrophil respiratory burst by plasma from periodontitis patients is mediated by pro-inflammatory cytokines. J Clin Periodontol. 2011;38(1):1–7. | ||
Li X, Kolltveit KM, Tronstad L, Olsen I. Systemic diseases caused by oral infection. Clin Microbiol Rev. 2000;13(4):547–558. | ||
Sinden NJ, Stockley RA. Systemic inflammation and comorbidity in COPD: a result of ‘overspill’ of inflammatory mediators from the lungs? Review of the evidence. Thorax. 2010;65(10):930–936. | ||
Preshaw PM, Taylor JJ. How has research into cytokine interactions and their role in driving immune responses impacted our understanding of periodontitis? J Clin Periodontol. 2011;38 (Suppl 11):60–84. | ||
Schols AM, Buurman WA, Staal van den Brekel AJ, Dentener MA, Wouters EF. Evidence for a relation between metabolic derangements and increased levels of inflammatory mediators in a subgroup of patients with chronic obstructive pulmonary disease. Thorax. 1996;51(8):819–824. | ||
Eid AA, Ionescu AA, Nixon LS, et al. Inflammatory response and body composition in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2001;164(8 Pt 1):1414–1418. | ||
Karadag F, Karul AB, Cildag O, Yilmaz M, Ozcan H. Biomarkers of systemic inflammation in stable and exacerbation phases of COPD. Lung. 2008;186(6):403–409. | ||
Sapey E, Bayley D, Ahmad A, Newbold P, Snell N, Stockley RA. Inter-relationships between inflammatory markers in patients with stable COPD with bronchitis: intra-patient and inter-patient variability. Thorax. 2008;63(6):493–499. | ||
Hacievliyagil SS, Gunen H, Mutlu LC, Karabulut AB, Temel I. Association between cytokines in induced sputum and severity of chronic obstructive pulmonary disease. Respir Med. 2006;100(5):846–854. | ||
He Z, Chen Y, Chen P, Wu G, Cai S. Local inflammation occurs before systemic inflammation in patients with COPD. Respirology. 2010;15(3):478–484. | ||
Kuschner WG, D’Alessandro A, Wong H, Blanc PD. Dose-dependent cigarette smoking-related inflammatory responses in healthy adults. Eur Respir J. 1996;9(10):1989–1994. | ||
Stone H, McNab G, Wood AM, Stockley RA, Sapey E. Variability of sputum inflammatory mediators in COPD and alpha1-antitrypsin deficiency. Eur Respir J. 2012;40(3):561–569. | ||
Caramori G, Adcock IM, Di Stefano A, Chung KF. Cytokine inhibition in the treatment of COPD. Int J Chron Obstruct Pulmon Dis. 2014;9:397–412. | ||
Kim V, Cornwell WD, Oros M, Durra H, Criner GJ, Rogers TJ. Plasma chemokine signature correlates with lung goblet cell hyperplasia in smokers with and without chronic obstructive pulmonary disease. BMC Pulm Med. 2015;15:111. | ||
Louhelainen N, Rytila P, Haahtela T, Kinnula VL, Djukanovic R. Persistence of oxidant and protease burden in the airways after smoking cessation. BMC Pulm Med. 2009;9:25. | ||
Gamble E, Grootendorst DC, Hattotuwa K, et al. Airway mucosal inflammation in COPD is similar in smokers and ex-smokers: a pooled analysis. Eur Respir J. 2007;30(3):467–471. | ||
Noguera A, Batle S, Miralles C, et al. Enhanced neutrophil response in chronic obstructive pulmonary disease. Thorax. 2001;56(6):432–437. | ||
Burnett D, Chamba A, Hill SL, Stockley RA. Neutrophils from subjects with chronic obstructive lung disease show enhanced chemotaxis and extracellular proteolysis. Lancet. 1987;2(8567):1043–1046. | ||
Sapey E, Stockley JA, Greenwood H, et al. Behavioral and structural differences in migrating peripheral neutrophils from patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2011;183(9):1176–1186. | ||
Stanescu D, Sanna A, Veriter C, et al. Airways obstruction, chronic expectoration, and rapid decline of FEV1 in smokers are associated with increased levels of sputum neutrophils. Thorax. 1996;51(3):267–271. | ||
Di Stefano A, Capelli A, Lusuardi M, et al. Severity of airflow limitation is associated with severity of airway inflammation in smokers. Am J Respir Crit Care Med. 1998;158(4):1277–1285. | ||
Vernooy JH, Kucukaycan M, Jacobs JA, et al. Local and systemic inflammation in patients with chronic obstructive pulmonary disease: soluble tumor necrosis factor receptors are increased in sputum. Am J Respir Crit Care Med. 2002;166(9):1218–1224. | ||
Higashimoto Y, Iwata T, Okada M, Satoh H, Fukuda K, Tohda Y. Serum biomarkers as predictors of lung function decline in chronic obstructive pulmonary disease. Respir Med. 2009;103(8):1231–1238. | ||
Kheradmand F, Shan M, Xu C, Corry DB. Autoimmunity in chronic obstructive pulmonary disease: clinical and experimental evidence. Expert Rev Clin Immunol. 2012;8(3):285–292. | ||
Boschetto P, Quintavalle S, Zeni E, et al. Association between markers of emphysema and more severe chronic obstructive pulmonary disease. Thorax. 2006;61(12):1037–1042. | ||
Barnes PJ, Shapiro SD, Pauwels RA. Chronic obstructive pulmonary disease: molecular and cellular mechanisms. Eur Respir J. 2003;22(4):672–688. | ||
Hogg JC, Senior RM. Chronic obstructive pulmonary disease – part 2: pathology and biochemistry of emphysema. Thorax. 2002;57(9):830–834. | ||
Houghton AM. Endogenous modifiers of cigarette smoke exposure within the lung. Proc Am Thorac Soc. 2012;9(2):66–68. | ||
Scott DA, Krauss J. Neutrophils in periodontal inflammation. Front Oral Biol. 2012;15:56–83. | ||
Hajishengallis E, Hajishengallis G. Neutrophil homeostasis and periodontal health in children and adults. J Dent Res. 2014;93(3):231–237. | ||
Roberts HM, Ling MR, Insall R, et al. Impaired neutrophil directional chemotactic accuracy in chronic periodontitis patients. J Clin Periodontol. 2015;42(1):1–11. | ||
Kumar RS, Prakash S. Impaired neutrophil and monocyte chemotaxis in chronic and aggressive periodontitis and effects of periodontal therapy. Indian J Dent Res. 2012;23(1):69–74. | ||
Carneiro VMA, Bezerra ACB, GuimarÃEs MdCM, Muniz-Junqueira MI. Decreased phagocytic function in neutrophils and monocytes from peripheral blood in periodontal disease. J Appl Oral Sci. 2012;20(5):503–509. | ||
Sammut D, Lewis A, Budd R, et al. Increased reactive oxygen species generation in quiescent and activated peripheral blood neutrophils in chronic obstructive pulmonary disease. B31 Inflamm COPD. 2015:A2710 Abstract. | ||
Chapple ILC. Reactive oxygen species and antioxidants in inflammatory diseases. J Clin Periodontol. 1997;24(5):287–296. | ||
White P, Cooper P, Milward M, Chapple I. P95 – differential activation of neutrophil extracellular traps by specific periodontal bacteria. Free Rad Biol Med. 2014;75 (Suppl 1):S53. | ||
Wright TK, Gibson PG, Simpson JL, McDonald VM, Wood LG, Baines KJ. Neutrophil extracellular traps are associated with inflammation in chronic airway disease. Respirology. 2016;21(3):467–475. | ||
Stockley RA. Neutrophils and protease/antiprotease imbalance. Am J Respir Crit Care Med. 1999;160(5 Pt 2):S49–S52. | ||
Sharafkhaneh A, Hanania NA, Kim V. Pathogenesis of emphysema: from the bench to the bedside. Proc Am Thorac Soc. 2008;5(4):475–477. | ||
Barnes PJ. Mediators of chronic obstructive pulmonary disease. Pharmacol Rev. 2004;56(4):515–548. | ||
Carter RI, Ungurs MJ, Mumford RA, Stockley RA. Aalpha-Val360: a marker of neutrophil elastase and COPD disease activity. Eur Respir J. 2013;41(1):31–38. | ||
Bhavsar NV, Dave BD, Brahmbhatt NA, Parekh R. Periodontal status and oral health behavior in hospitalized patients with chronic obstructive pulmonary disease. J Nat Sci Biol Med. 2015;6 (Suppl 1):S93–S97. | ||
Chung JH, Hwang HJ, Kim SH, Kim TH. Associations between periodontitis and chronic obstructive pulmonary disease; the 2010–2012 Korean National Health and Nutrition Examination Survey (KNHANES). J Periodontol. 2016;87(8):864–871. | ||
Wang Z, Zhou X, Zhang J, et al. Periodontal health, oral health behaviors, and chronic obstructive pulmonary disease. J Clin Periodontol. 2009;36(9):750–755. | ||
Deo V, Bhongade ML, Ansari S, Chavan RS. Periodontitis as a potential risk factor for chronic obstructive pulmonary disease: a retrospective study. Indian J Dent Res. 2009;20(4):466–470. | ||
Leuckfeld I, Obregon-Whittle MV, Lund MB, Geiran O, Bjortuft O, Olsen I. Severe chronic obstructive pulmonary disease: association with marginal bone loss in periodontitis. Respir Med. 2008;102(4):488–494. | ||
Peter KP, Mute BR, Doiphode SS, Bardapurkar SJ, Borkar MS, Raje DV. Association between periodontal disease and chronic obstructive pulmonary disease: a reality or just a dogma? J Periodontol. 2013;84(12):1717–1723. | ||
Holtfreter B, Richter S, Kocher T, et al. Periodontitis is related to lung volumes and airflow limitation: a cross-sectional study. Eur Respir J. 2013;42(6):1524–1535. | ||
Bergstrom J, Cederlund K, Dahlen B, et al. Dental health in smokers with and without COPD. PLoS One. 2013;8(3):e59492. | ||
Hyman JJ, Reid BC. Cigarette smoking, periodontal disease: and chronic obstructive pulmonary disease. J Periodontol. 2004;75(1):9–15. | ||
Liu Z, Zhang W, Zhang J, et al. Oral hygiene, periodontal health and chronic obstructive pulmonary disease exacerbations. J Clin Periodontol. 2012;39(1):45–52. | ||
Tonetti MS, Claffey N. Advances in the progression of periodontitis and proposal of definitions of a periodontitis case and disease progression for use in risk factor research. Group C consensus report of the 5th European Workshop in Periodontology. J Clin Periodontol. 2005;32 (Suppl 6):210–213. | ||
Barros SP, Suruki R, Loewy ZG, Beck JD, Offenbacher S. A cohort study of the impact of tooth loss and periodontal disease on respiratory events among COPD subjects: modulatory role of systemic biomarkers of inflammation. PLoS One. 2013;8(8):e68592. | ||
Zhou X, Han J, Liu Z, Song Y, Wang Z, Sun Z. Effects of periodontal treatment on lung function and exacerbation frequency in patients with chronic obstructive pulmonary disease and chronic periodontitis: a 2-year pilot randomized controlled trial. J Clin Periodontol. 2014;41(6):564–572. | ||
Kim SW, Han K, Kim SY, Park CK, Rhee CK, Yoon HK. The relationship between the number of natural teeth and airflow obstruction: a cross-sectional study using data from the Korean National Health and Nutrition Examination Survey. Int J Chron Obstruct Pulmon Dis. 2016;11:13–21. | ||
Eke PI, Page RC, Wei L, Thornton-Evans G, Genco RJ. Update of the case definitions for population-based surveillance of periodontitis. J Periodontol. 2012;83(12):1449–1454. | ||
Armitage GC. Development of a classification system for periodontal diseases and conditions. Ann Periodontol. 1999;4(1):1–6. | ||
American Academy of Periodontology Task Force Report on the Update to the 1999 Classification of Periodontal Diseases and Conditions. J Periodontol. 2015;86(7):835–838. | ||
Scannapieco FA, Ho AW. Potential associations between chronic respiratory disease and periodontal disease: analysis of National Health and Nutrition Examination Survey III. J Periodontol. 2001;72(1):50–56. | ||
Eriksson S. Pulmonary emphysema and alpha1-antitrypsin deficiency. Acta Med Scand. 1964;175:197–205. | ||
Chapple ILC, Gilbert AD, Wilson NHF. Understanding Periodontal Diseases: Assessment and Diagnostic Procedures in Practice: 1 (Quintessentials: Endontics). Quintessence Publishing Co Ltd. 2002. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.