")
Back to Journals » International Journal of Nanomedicine » Volume 12
Iron oxide nanoparticles induce cytokine secretion in a complement-dependent manner in a human whole blood model
Authors Wolf-Grosse S, Rokstad AM, Ali S, Lambris JD, Mollnes TE, Nilsen AM, Stenvik J
Received 8 March 2017
Accepted for publication 11 April 2017
Published 23 May 2017 Volume 2017:12 Pages 3927—3940
DOI https://doi.org/10.2147/IJN.S136453
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Thomas Webster
Susann Wolf-Grosse,1 Anne Mari Rokstad,1–3 Syed Ali,4 John D Lambris,5 Tom E Mollnes,2,6–9 Asbjørn M Nilsen,1,* Jørgen Stenvik1,2,*
1Department of Cancer Research and Molecular Medicine, 2Centre of Molecular Inflammation Research (CEMIR), Faculty of Medicine and Health Sciences, Norwegian University of Science and Technology, Trondheim, 3Central Norway Regional Health Authority, Stjørdal, Norway; 4Division of Neurotoxicology, US FDA/National Center for Toxicological Research, Jefferson, AR, 5Department of Pathology and Laboratory Medicine, University of Pennsylvania, Philadelphia, PA, USA; 6Department of Immunology, Oslo University Hospital, Rikshospitalet, Oslo, 7Research Laboratory, Nordland Hospital, Bodø, 8K.G. Jebsen Inflammatory Research Center, University of Oslo, Oslo, 9Faculty of Health Sciences, K.G. Jebsen Thrombosis Research and Expertise Center, University of Tromsø, Tromsø, Norway
*These authors contributed equally to this work
Abstract: Iron oxide nanoparticles (IONPs) are promising nanomaterials for biomedical applications. However, their inflammatory potential has not been fully established. Here, we used a lepirudin anti-coagulated human whole blood model to evaluate the potential of 10 nm IONPs to activate the complement system and induce cytokine production. Reactive oxygen species and cell death were also assessed. The IONPs activated complement, as measured by C3a, C5a and sC5b-9, and induced the production of pro-inflammatory cytokines in a particle-dose dependent manner, with the strongest response at 10 µg/mL IONPs. Complement inhibitors at C3 (compstatin analog Cp40) and C5 (eculizumab) levels completely inhibited complement activation and secretion of inflammatory mediators induced by the IONPs. Additionally, blockade of complement receptors C3aR and C5aR1 significantly reduced the levels of various cytokines, indicating that the particle-induced secretion of inflammatory mediators is mainly C5a and C3a mediated. The IONPs did not induce cell death or reactive oxygen species, which further suggests that complement activation alone was responsible for most of the particle-induced cytokines. These data suggest that the lepirudin anti-coagulated human whole blood model is a valuable ex vivo system to study the inflammatory potential of IONPs. We conclude that IONPs induce complement-mediated cytokine secretion in human whole blood.
Keywords: iron oxide nanoparticles, human whole blood, complement activation, cytokines, reactive oxygen species, complement inhibitors
Background
Due to their magnetic properties, iron oxide nanoparticles (IONPs) have been explored for their use in biomedical applications, for example, as contrast agent for MRI, in tissue repair,1 in targeted drug delivery systems and for induced hyperthermia cancer treatment.2 Such applications might involve intravenous injection. Thus, it is of importance to assess the interactions of these nanoparticles with cellular and humoral components of the innate immune system in the blood.
On contact with blood, nanoparticles may be recognized by elements of the immune system, in particular, the plasma cascades like the complement, coagulation and contact system. It has long been known that the plasma cascade systems not only interact with each other, but also exhibit extensive crosstalk with the cellular elements in the blood.3
The complement system, which consists of over 40 plasma and membrane-bound proteins (receptors and regulators),4 destroys and removes foreign substances, either directly or by activating leukocytes and initiating pro-inflammatory responses via mediators such as cytokines. In general, complement activation can be initiated by either the classical, lectin or alternative pathway. Activation of either pathway results in the turnover of the C3 protein, which is followed by the production of the anaphylatoxins C3a and C5a, and the formation of the C5b-9 complex (also named terminal complement complex or membrane attack complex). Uncontrolled complement activation and pro-inflammatory responses induced by biomaterials or nanomaterials may initiate adverse infusion-related reactions.3
Complement activation can play a major role in immune recognition of nanoparticles,5 which is dependent on particle characteristics, such as size, shape and surface properties.6 Previous studies demonstrated complement activation by dextran-coated superparamagnetic iron oxide nanoparticles (SPIONs) and nanoworms in human serum7,8 and complement-mediated SPIO nanoworm recognition and uptake by leukocytes.9 Furthermore, IONPs may induce cytokine production in various immune cells. For example, γ-Fe2O3 nanoparticles can increase TNF in RAW264.7 cells10 and carboxydextran-coated Fe3O4/Fe2O3 nanoparticles induced TNF in THP-1 cells.11 Moreover, non-coated and polyacrylic acid-coated IONPs induced IL-1β, IL-8, IL-6, TNF and IL-10 in primary human blood cells12 and amino-polyvinyl alcohol-coated SPION increased the secretion of IL-1β, IL-4, IL-8, IL-6, MIP-1β and RANTES in whole blood samples.13 In contrast, we did not observe pro-inflammatory cytokine production by IONPs in primary human monocytes,14 which might be due to different properties of the IONPs and different cell types used.
In order to characterize the effects of IONPs in a more physiologically complex system, we used an ex vivo human whole blood model in the present study. This model uses the hirudin analog lepirudin as anti-coagulant, which specifically inhibits thrombin, but has no effect on the complement system, induction of pro-inflammatory cytokines, or the interplay between them.15 Previous studies used this model to examine cholesterol crystal-induced inflammasome activation,16 or to study the effects of implantable sensors17 and polycation-containing microspheres.18 It has also been used to study polystyrene nanoparticles,19 but this human whole blood model has not been previously used to evaluate the biological effects of IONPs.
Here, we report the effects of IONPs in human whole blood with a focus on complement activation and induction of cytokines. We also evaluated if the IONPs induced cytotoxicity as measured by the production of reactive oxygen species (ROS) and cell viability.
Methods
Nanoparticle suspensions
The 10 nm sized IONPs with an amphiphilic polymer coating (catalog numbers SHP-10–10 and SXP-10-05) were purchased from Ocean Nanotech, LLC (Springdale, AR, USA). The IONPs are coated with a monolayer of oleic acid and a monolayer of amphiphilic polymer. The reactive group on the surface is carboxylic acid (Figure S1).
Characterization of IONPs
The size, shape, chemical composition, hydrodynamic diameter and zeta potential of the IONPs have been previously characterized.14 In the present study, additional measurements of the particles’ hydrodynamic diameter in phosphate-buffered saline (PBS) without Ca2+/Mg2+ have been performed. Transmission electron microscopy (TEM) was used to assess the size and agglomeration status of the IONPs in human lepirudin-plasma incubated at 37°C for 2 h. Formvar-coated copper grids were placed on a drop of the IONP sample (100 μg/mL) for 5 min. The grids were gently drained with filter paper and the sample was subsequently inspected without further treatment using a JEOL TEM (JEOL, Tokyo, Japan) at 80 kV.
We verified in our previous study that the particles were not contaminated with endotoxin/lipopolysaccharide (LPS) or Toll-like receptor 2 ligand.14
Human whole blood model
An ex vivo lepirudin-based human whole blood model, as first described by Mollnes et al,15 was adapted to assess the inflammatory effects of IONPs in a complex biological environment. The ex vivo whole blood model is presently one of the systems to closest mimic an in vivo situation. This model uses lepirudin as an anti-coagulant as it specifically inhibits thrombin, but has no effect on the activation of the complement system, as opposed to heparin. Whole blood from voluntary, healthy donors was collected in low activating polypropylene vials (Nunc 4.5 mL) containing lepirudin (50 μg/mL). Immediately afterward, blood (250 μL) was incubated with 50 μL PBS and 50 μL PBS containing IONPs (1, 10 and 100 μg/mL) in 1.8 mL polypropylene vials (Nunc). Zymosan (Zym; 10 μg) served as positive control for complement activation. The samples were incubated for 2, 4 or 6 h at 37°C in a heat cabinet under continuous, slow rotation around the vertical axis using a Rock’n Roller (Labinco B.V., Breda, the Netherlands). In the experiments with the complement inhibitors, blood was pre-incubated for 7 min with C3 inhibitor compstatin analog Cp40 (final concentration 20 μM), a 14-amino acid cyclic peptide ((D)Tyr-Ile-[Cys-Val-Trp(Me)-Gln-Asp-Trp-Sar-Ala-His-Arg-Cys]-mIle-NH2; synthesized as previously described20), C5 inhibitor Soliris® (eculizumab; Alexion Pharmaceuticals, Zürich, Switzerland; final concentration 100 μg/mL), C5a receptor antagonist (C5aRA) PMX53 (final concentration 10 μg/mL; as previously described21) or C3a receptor antagonist (C3aRA; final concentration 50 μM; kindly provided by Robert S Ames) or PBS in the ratio blood to inhibitor or PBS equal to 5:1. IONPs and controls were then added to the pre-incubated blood and further incubated for various time periods. At the end of the incubation time, further complement activation was inhibited with ethylenediaminetetraacetic acid (EDTA) at a final concentration of 10 mM and plasma was prepared by centrifugation at 1,880× g for 15 min. Plasma samples were stored at −20°C until analysis. To assess the baseline values (T0) for cytokine production and complement activation, one blood sample was immediately centrifuged at 1,880× g for 15 min after blood withdrawal and addition of EDTA.
Complement activation assays
C3a and C5a were assessed by measuring the amount of C3a/C3a desArg and C5a/C5a desArg using the C3a PlusEIA kit from MicroVue (Quidel, San Diego, CA, USA) and the C5a enzyme-linked immunosorbent assay (ELISA) kit II from BD BioSciences (San Diego, CA, USA), respectively.
The terminal soluble C5b-9 complement complex (sC5b-9) was measured by an ELISA. This assay, modified after,22 is based on a monoclonal capture antibody, which is specific for a neoepitope exposed in C9 after activation. Briefly, 96-well plates (Costar 3690, high-binding polystyrene plates) were coated with the capture monoclonal antibody aE11. After sample incubation and a washing step, a second biotinylated anti-C6 antibody (mAb 9C4) was added. Following another washing step, Streptavidin-horse radish peroxidase and tetramethylbenzidine substrate were added and the absorbance was measured at 450 and 655 nm (for background correction).
Cytokine analysis
Cytokine production in plasma samples was measured using a 17-plex cytokine assay (BioPlex Multiplex human cytokine 17-plex panel; Bio-Rad, Hercules, CA, USA) containing the following analytics: IL-1β, IL-2, IL-4, IL-5, IL-6, IL-7, IL-8 (CXCL8), IL-10, IL-12 (p70), IL-13, IL-17, granulocyte colony stimulating factor, granulocyte macrophage colony stimulating factor, interferon gamma, MCP-1 (also known as CCL2), MIP-1β (also known as CCL4) and TNF-α. The analysis was performed according to the manufacturer’s instructions. A Bio-Plex 200 instrument (Bio-Rad) and its software (Bio-Plex 6.1) were used for analysis.
Cell viability
Cell viability in the whole blood samples was determined using fixable viability dye eFluor780 (eBioscience, Affymetrix, Santa Clara, CA, USA). Briefly, following incubation of whole blood samples with the various stimuli and controls, 100 μL blood were added to new Nunc tubes containing 5 μL anti-CD14-FITC and 1 μL fixable viability dye eFluor780. The samples were incubated for 30 min on ice and then washed 2× with PBS. Following lysis of red blood cells with BD FACS lysing solution, the samples were resuspended in PBS, before they were analyzed using a FACSCalibur flow cytometer (BD BioSciences).
Reactive oxygen species
Oxidative burst in whole blood samples was determined using the Phagoburst kit (Glycotope Biotechnology, Heidelberg, Germany), with small changes to the manufacturer’s protocol. Whole blood samples were incubated with PBS, IONPs and Zymosan for 2 or 4 h, whereas opsonized Escherichia coli (provided in the kit) was incubated for 10 min according to the manufacturer’s instructions. Following lysis of red blood cells and staining with anti-CD14-PE for 15 min, the samples were run on a FACSCalibur flow cytometer (BD BioSciences).
Statistical analysis
Statistical analysis was done with GraphPad Prism 6 (GraphPad Software Inc., La Jolla, CA, USA). The data was analyzed using one-way, repeated measures analysis of variance with Dunnett’s multiple comparison test. In order to eliminate uncertainties concerning normal distribution of the data due to a low sample number between 3 and 14, the data was log transformed before analysis. Results were considered statistically significant when P<0.05.
Ethics
The use of human whole blood for our experiments was approved by the Regional Committee for Medical and Health Research Ethics in Central Norway (REC Central), The Norwegian Ministry of Education and Research, 2009/2245. Experiments were conducted according to their regulations and guidelines. Informed written consent was obtained from each voluntary donor prior to blood sampling.
Results
Physicochemical characterization of the IONPs
An initial characterization of the IONPs as to their hydrodynamic diameter, zeta potential and chemical composition was done in our previous study14 and is shown in Table 1. In the present study, we additionally determined the hydrodynamic diameter of the IONPs in PBS as the IONPs were dispersed in PBS before adding to the whole blood samples. The particles retained almost the same hydrodynamic diameter in PBS as in water (as supplied) (Table 1). Furthermore, visual examination of the particle size and agglomeration status in lepirudin-plasma using TEM confirmed the primary particle size of 10 nm and indicated only few particle agglomerates with sizes up to 100 nm (Figure 1).
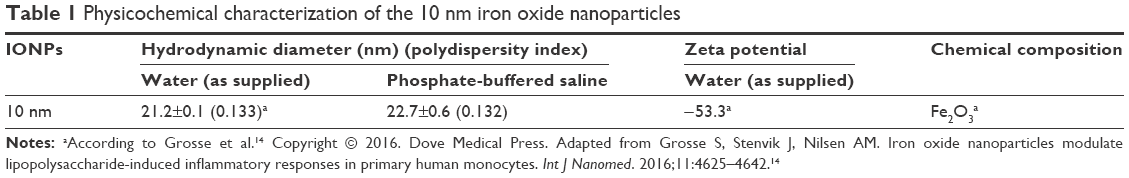 | Table 1 Physicochemical characterization of the 10 nm iron oxide nanoparticles |
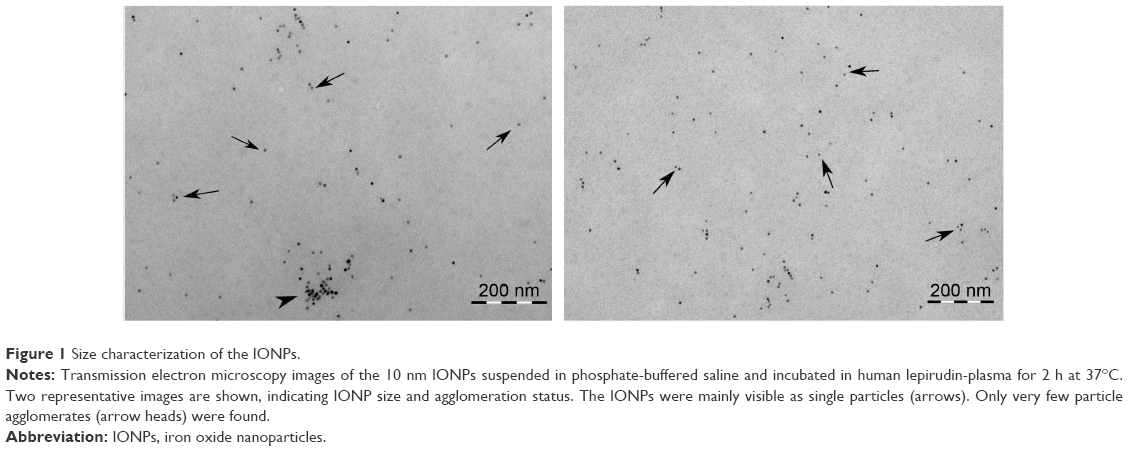 | Figure 1 Size characterization of the IONPs. |
Complement activation in human whole blood
The IONPs were suspended in PBS without Ca2+/Mg2+ and added to lepirudin anti-coagulated peripheral blood from healthy donors. In order to assess if the IONPs activate the complement system, we determined the plasma concentrations of C3a, C5a and sC5b-9 after various time points (Figure 2). Incubation for 2 and 4 h with IONPs at lower concentrations resulted in a significant increase in C5a and sC5b-9 (Figure 2B and C), whereas after 6 h, all three activation products, C3a, C5a and sC5b-9, were significantly increased (Figure 2). The strongest activation of complement was seen at 10 μg/mL IONPs, while no clear activation was seen at 100 μg/mL.
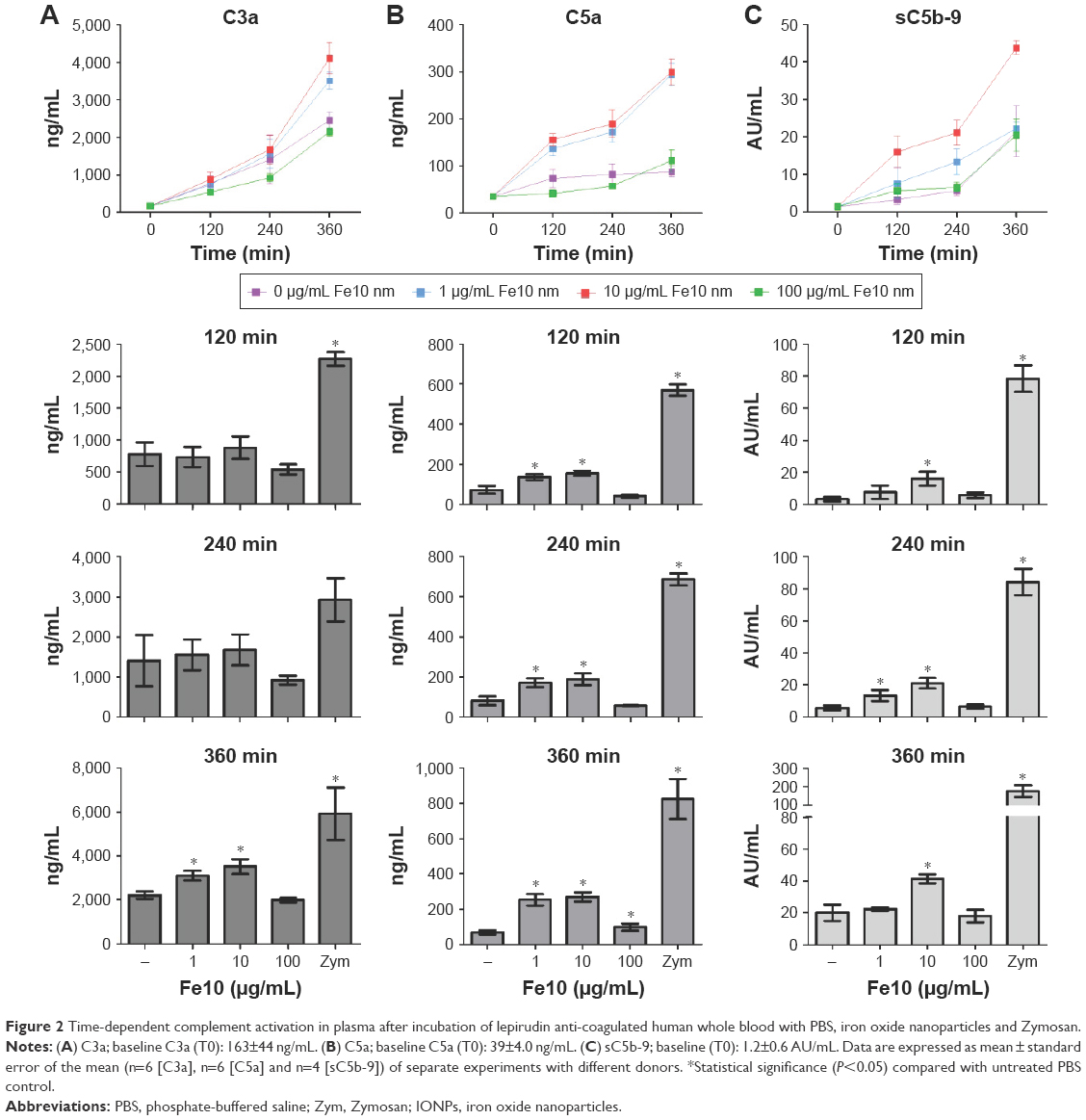 | Figure 2 Time-dependent complement activation in plasma after incubation of lepirudin anti-coagulated human whole blood with PBS, iron oxide nanoparticles and Zymosan. |
Cytokine response in human whole blood
We further analyzed the levels of 17 cytokines in the plasma following IONP exposure. A significant increase of IL-1β, TNF, IL-6, MCP-1, IL-8 and MIP-1β was apparent after 6 h (Figure 3). The other cytokines were either below the detection limit or not significantly induced by IONPs after 6 h, and are thus not shown. The IL-1β, TNF, IL-6 and IL-8 secretion correlated with the particle-induced complement activation, with an induction optimum at 10 μg/mL IONPs (Figure 2). The chemokines MCP-1 and MIP-1β showed a gradual dose-dependent increase, with the highest potency at 100 μg/mL IONPs (Figure 3). The activation optimum for MCP-1 and MIP-1β was thus different from the complement peaking at 10 μg/mL IONPs. The cytokine secretion was initially measured after 2, 4 and 6 h (Figure S2). Following IONP exposure, the earliest cytokine induction above saline control was apparent at 4 h for IL-8 and MIP-1β.
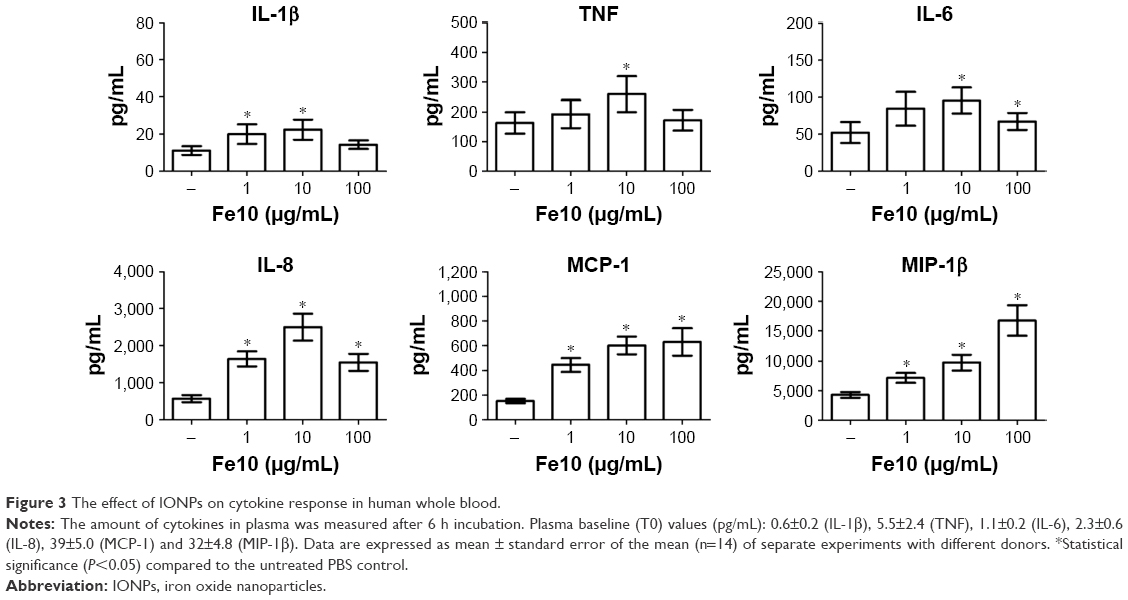 | Figure 3 The effect of IONPs on cytokine response in human whole blood. |
Particle-induced cytokine response with complement inhibitors
In order to test if the particle-induced cytokine production was complement mediated, we used two inhibitors of the complement components C3 and C5, respectively, and further on receptor antagonists for C3aR and C5aR1. The C3 inhibitor compstatin analog Cp40 significantly decreased the particle-induced production of all cytokines examined (Figure 4). In addition, statistically significant inhibition of the IL-1β, TNF, IL-6 and IL-8 response by compstatin analog Cp40 was detected in the PBS control samples, consistent with the background activation of complement induced by the polypropylene vials. The particle-induced cytokine induction exceeded the background activation, thus the results altogether indicate that complement activation was responsible for the particle-induced cytokine production.
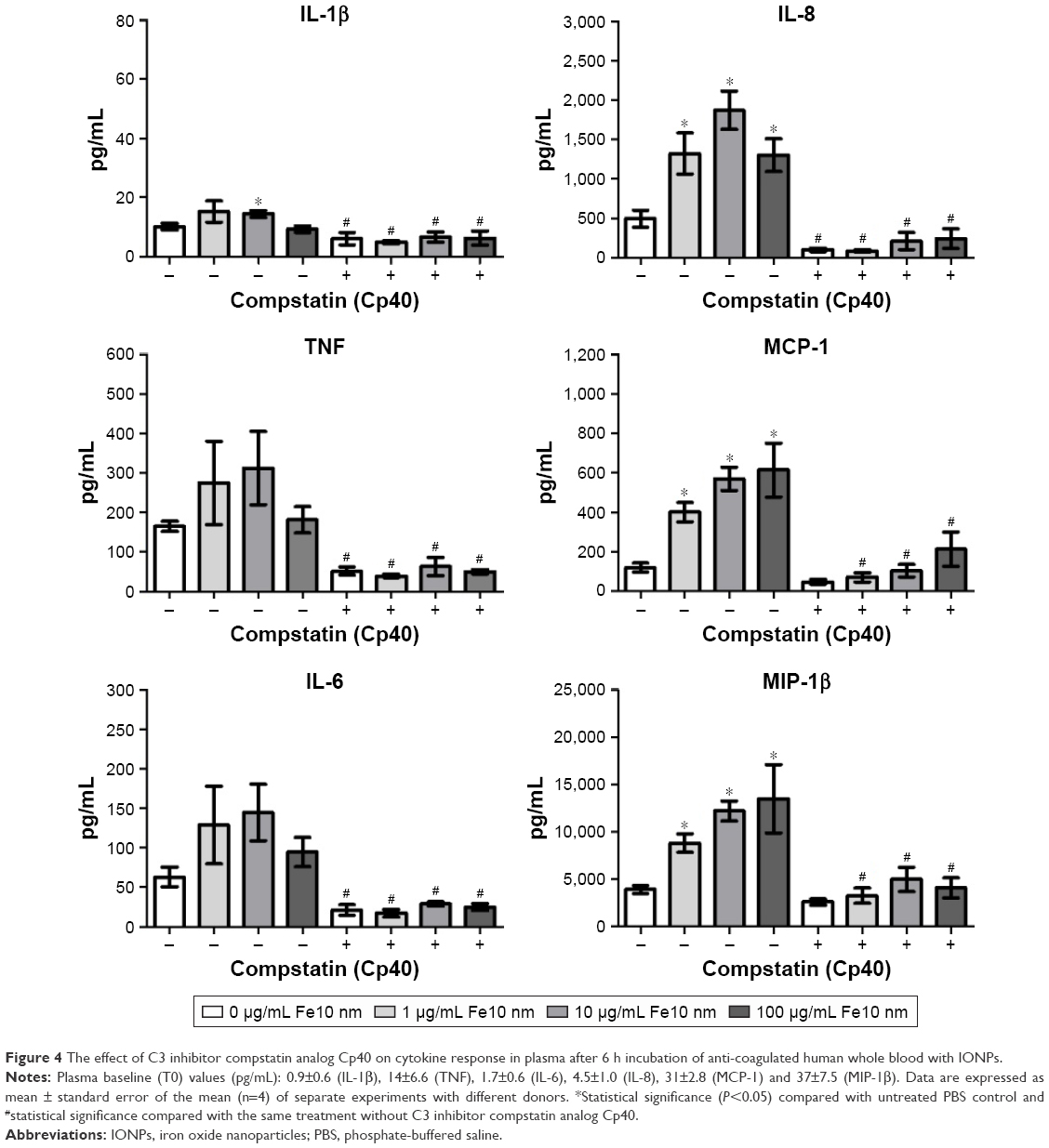 | Figure 4 The effect of C3 inhibitor compstatin analog Cp40 on cytokine response in plasma after 6 h incubation of anti-coagulated human whole blood with IONPs. |
Inhibition of C5 activation with eculizumab significantly decreased the production of MCP-1 and IL-8 (Figure 5). Moreover, TNF, IL-6 and IL-1β were lower, though not significant, after treatment with eculizumab. In contrast, the particle-mediated MIP-1β production appeared to be unaffected by the addition of the C5 inhibitor. It has to be noted that the particle-induced cytokine production in these experiments, utilizing another set of donors, was not as strong as in the experiments with compstatin analog Cp40 (Figure 4). We conclude that the particle-mediated activation of C5 is involved in the cytokine response induced by IONPs. Of note, compstatin analog Cp40 and eculizumab also completely abolished the sC5b-9 formation by Zymosan exposure, included as a control (Figure S3).
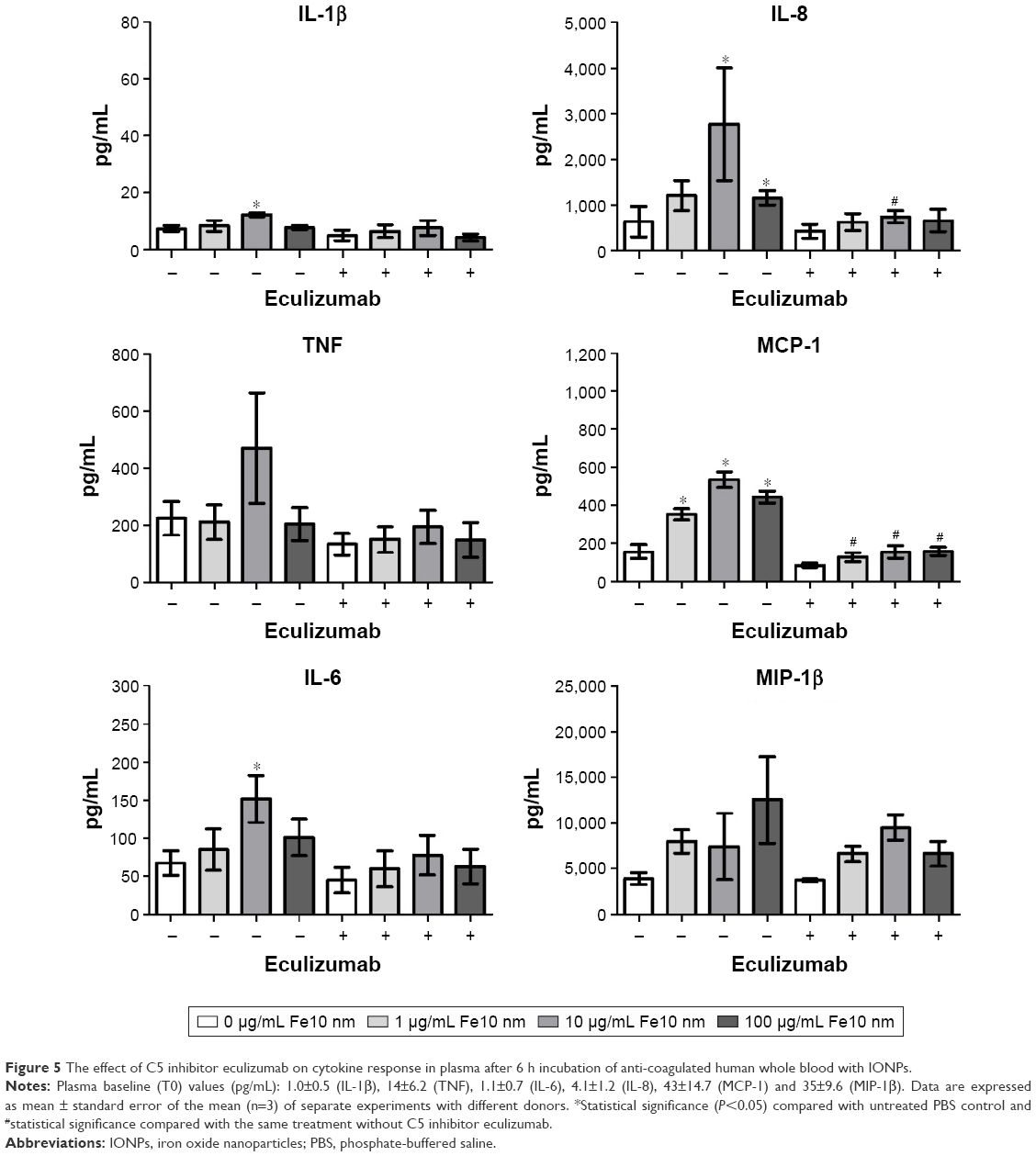 | Figure 5 The effect of C5 inhibitor eculizumab on cytokine response in plasma after 6 h incubation of anti-coagulated human whole blood with IONPs. |
To further validate our results indicating that the particle-induced complement activation triggers the cytokine response, we assessed the cytokine secretion in the presence of two complement receptor antagonists. For these experiments, eculizumab was included as an internal control and we chose 10 μg/mL IONPs, as this particle concentration induced the strongest cytokine response. Blocking of either the C3a receptor (C3aR) or the C5a receptor (C5aR1) significantly reduced the particle-induced IL-6, MCP-1 and IL-8 production (Figure 6). Additionally, the production of TNF and IL-1β was abolished by addition of C5aRA. In contrast, the MIP-1β production was unaffected by both receptor antagonists as well as eculizumab (Figure 6). These data indicate that complement activation, mainly via C5aR activation, but also involving the C3aR, is responsible for the particle-induced cytokine responses.
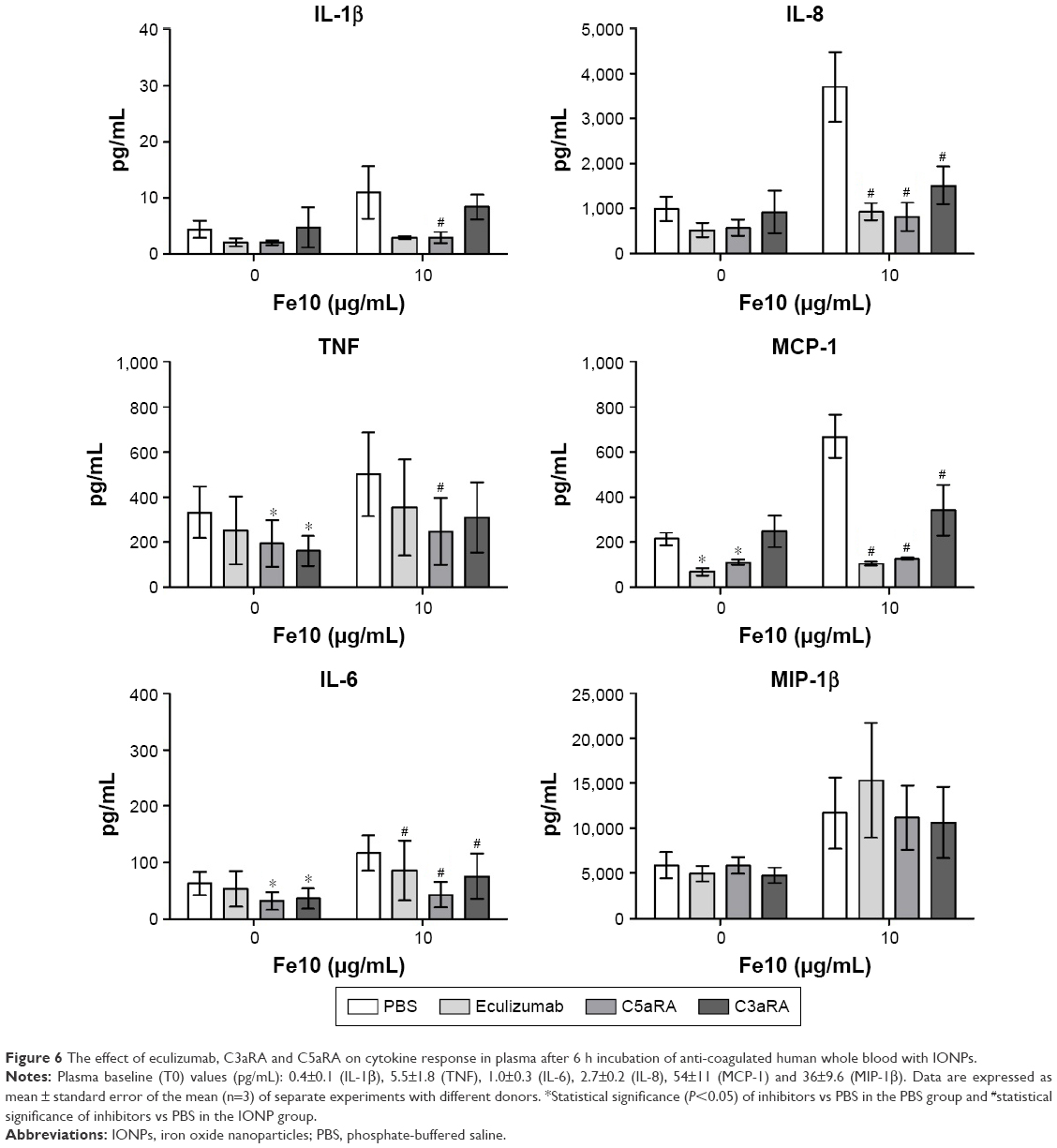 | Figure 6 The effect of eculizumab, C3aRA and C5aRA on cytokine response in plasma after 6 h incubation of anti-coagulated human whole blood with IONPs. |
ROS production and viability in human whole blood
We finally assessed if the production of ROS or loss of viability could play a role in IONP-mediated complement activation and cytokine production. IONPs did not significantly increase ROS in monocytes or granulocytes compared with the PBS control, while addition of opsonized E. coli and Zymosan elicited strong responses within 10 and 120 min of incubation, respectively, in both cell types (Figure 7A).
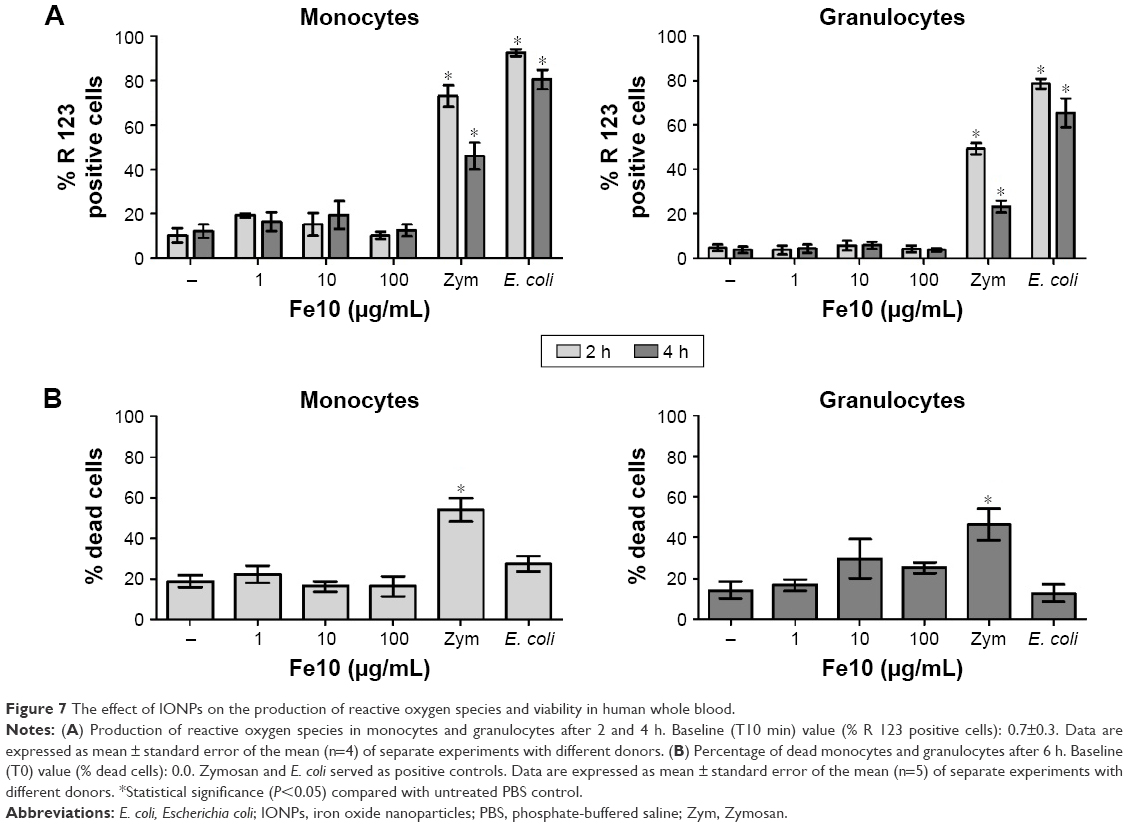 | Figure 7 The effect of IONPs on the production of reactive oxygen species and viability in human whole blood. |
Incubation of human whole blood with PBS for 6 h resulted in ~20% dead or dying monocytes and granulocytes (Figure 7B). Addition of IONPs did not significantly change the percentage of dead monocytes. There was a slightly higher number of dying granulocytes with high concentrations of IONPs, but the difference was not significant. In contrast, stimulation with Zymosan gave ~50% dead or dying cells (P<0.05), while E. coli did not after 6 h incubation in whole blood.
Discussion
We examined the biological effects of 10 nm IONPs on complement activation, cytokine induction, ROS and cell viability in human whole blood. The lepirudin anti-coagulated human blood model is a complex biological model containing various humoral factors and cell types close to an in vivo environment.15 Thus, this ex vivo model is particularly suited for studying complement activation and its consequences in inflammatory crosstalk. To the best of our knowledge, this is the first study that investigates the inflammatory effects of IONPs in human whole blood.
Previous studies associated nanoparticlulate systems, including IONPs, with in vivo complement activation and complement-related side effects.23,24 Here, we showed that IONPs provoked complement activation resulting in the generation of the anaphylatoxins C3a and C5a as well as the soluble terminal complement complex sC5b-9 in an ex vivo human whole blood model. Moreover, the IONPs induced the secretion of pro-inflammatory cytokines in whole blood from healthy donors. Particle contamination with endotoxin/LPS or bacterial lipoproteins could potentially be the reason for the cytokine production. However, as previously demonstrated,14 contamination of the IONPs can be excluded.
By using specific inhibitors of complement proteins and cell surface receptors, we investigated if there is a direct link between complement activation and secretion of cytokines. Our results demonstrated that both C3a and C5a are major contributors to the release of pro-inflammatory cytokines, since both inhibition of C3 and C5 as well as C3aR and C5aR1 blockers significantly reduced IONP-induced IL-1β, TNF, IL-6, IL-8, MCP-1 and MIP-1β production. It has previously been demonstrated that biomaterials, like polyvinyl chloride tubing, induced complement-dependent secretion of chemokines IL-8, MIP-1α and MCP-1.25 Furthermore, a complete dependency on complement has previously also been found for the inflammatory cytokines IL-1β, TNF, IL-6, IL-8, MIP-1β and MCP-1 during exposure to poly-L-lysine containing microcapsules.18 To the best of our knowledge, this is the first study that showed that engineered nanoparticles, like IONPs, induce cytokine secretion in a complement-dependent manner, thus underscoring the potential of inhibiting complement to prevent detrimental coagulation- and inflammation-related effects induced by nanoparticles.
The C3 inhibitor compstatin analog Cp40 completely inhibited the cytokine response, whereas C5 inhibitor eculizumab only partly reduced the production of the inflammatory mediators, which is in agreement with previous findings.26,27 While compstatin analog Cp40 inhibits both the formation of C3a and C5a, eculizumab only inhibited the formation of C5a, thus our data could be explained by a dual involvement of C3a and C5a for optimal cytokine response. In a study by Orning et al,27 the differences between compstatin and eculizumab were also shown to be connected to the C3b/iC3b formation with subsequent cell-adhesion (by complement receptor CR3). Although this study investigated non-phagocytable microcapsules and the effects of cell adhesion, it also illustrates the possibility for the involvement of other complement activating products such as C3b/iC3b.
In the present study, the cytokine response was generally mostly due to C5aR1 activation by C5a. The complement cleavage byproduct and anaphylatoxin C5a is the most potent pro-inflammatory mediator released upon complement activation and far more potent than C3a. It is able to induce monocyte and neutrophil chemotaxis and activation, epithelial and endothelial cell leakage, mast cell degranulation and production of cytokines and chemokines.28 By binding to its main C5a receptor, C5aR1, C5a initiates signaling cascades that result in NFκB activation and ROS production.29 Our results show that low concentrations of IONPs are able to induce the generation of C5a, which was a major driver of the observed cytokine response.
Unexpectedly, high concentrations of IONPs had a weaker effect on C5a generation compared with lower particle concentrations. This “concentration effect” was also observed for C3a and sC5b-9, and corresponded well with the cytokine profile of IL-1β, TNF, IL-6 and IL-8. In contrast, MCP-1 and MIP-1β levels increased with increasing particle concentrations. This suggests that additional, yet unidentified, mechanisms contribute to the induction of these particular cytokines.
The stability of a dispersion decreases with increasing concentration and high particle concentrations could give rise to cytotoxicity. However, our results suggest that neither particle agglomeration nor cytotoxicity were main factors influencing the cytokine response due to treatment of human whole blood with IONPs. It cannot be excluded that complement factors and regulators adsorbed to the particles at the highest IONP concentration examined, thus counteracting the stimulatory effects at lower concentrations. Such a negative regulatory effect might be similar to the adsorption and inactivation of LPS that we previously described using the same IONPs in cell culture.14
In order to examine if the cytokine response in our study was accompanied by ROS production, we measured intracellular ROS in the whole blood samples after 2 and 4 h. The IONPs did not significantly increase ROS compared with the control samples, which is in agreement with previous findings.30,31 However, our results are in contrast to other studies that show IONP-induced ROS production.32–35 Possible explanations for the discrepancies include one or more of the following factors: different types and sizes of IONPs used; different surface properties (eg, surface coatings) of the IONPs that affect the degradation potential and thus the exposure to the naked iron oxide core and the liberation of iron ions that might induce ROS;32 different cell types used; higher IONP concentration; different (mostly longer) incubation times after which ROS production was measured and/or different methods for assessing intracellular ROS.
We conclude that the IONPs examined here induced a pro-inflammatory response via complement-mediated cytokine secretion without signs of cytotoxicity and ROS production in human whole blood. Furthermore, the ex vivo human whole blood model is a highly suitable model for studying the immunomodulatory effects of engineered nanoparticles. Although this model lacks the complexity of a whole organism, it has major advantages over monoculture models: it minimizes nanoparticle sedimentation, which has shown to have great influence in in vitro nanotoxicity studies,36 and, most importantly, it allows for intercellular communication and crosstalk with the protein cascades in the blood that are important for initial inflammatory responses.
Acknowledgments
The authors thank all volunteers for donating blood samples. Furthermore, we thank Nan E Tostrup Skogaker for her technical support with TEM, which was conducted at the Cellular and Molecular Imaging Core Facility (CMIC), Norwegian University of Science and Technology.
SW-G was financed by NanoLab, Norwegian University of Science and Technology. The Research Council of Norway is acknowledged for the support to the Norwegian Micro- and Nano-Fabrication Facility, NorFab, project number 245963/F50. The work was supported by the Research Council of Norway Grant 223255/F50 through its Centres of Excellence funding scheme and by the National Institutes of Health Grants AI068730 and AI030040, and the European Community’s Seventh Framework Programme under Grant 602699 (DIREKT).
Disclosure
JDL is the inventor of patents and/or patent applications that describe the use of complement inhibitors for therapeutic purposes. He is also the founder of Amyndas Pharmaceuticals, which is developing complement inhibitors for clinical applications. The authors report no other conflicts of interest in this work.
References
Pareta RA, Taylor E, Webster TJ. Increased osteoblast density in the presence of novel calcium phosphate coated magnetic nanoparticles. Nanotechnology. 2008;19(26):265101. | ||
Huber DL. Synthesis, properties, and applications of iron nanoparticles. Small. 2005;1(5):482–501. | ||
Nilsson B, Ekdahl KN, Mollnes TE, Lambris JD. The role of complement in biomaterial-induced inflammation. Mol Immunol. 2007;44(1–3):82–94. | ||
Carroll MV, Sim RB. Complement in health and disease. Adv Drug Deliv Rev. 2011;63(12):965–975. | ||
Moghimi SM. Cancer nanomedicine and the complement system activation paradigm: anaphylaxis and tumour growth. J Control Release. 2014;190:556–562. | ||
ShadiFarhangrazi Z, MoeinMoghimi S. Materials etiquette and complement responses. Curr Bionanotechnol. 2016;2(1):6–10. | ||
Banda NK, Mehta G, Chao Y, et al. Mechanisms of complement activation by dextran-coated superparamagnetic iron oxide (SPIO) nanoworms in mouse vs. human serum. Part Fibre Toxicol. 2014;11:64. | ||
Wang G, Chen F, Banda NK, et al. Activation of human complement system by dextran-coated iron oxide nanoparticles is not affected by dextran/Fe ratio, hydroxyl modifications, and crosslinking. Front Immunol. 2016;7:418. | ||
Inturi S, Wang G, Chen F, et al. Modulatory role of surface coating of superparamagneticiron oxide nanoworms in complement opsonization and leukocyte uptake. ACS Nano. 2015;9(11):10758–10768. | ||
Park EJ, Choi DH, Kim Y, et al. Magnetic iron oxide nanoparticles induce autophagy preceding apoptosis through mitochondrial damage and ER stress in RAW264.7 cells. Toxicol In Vitro. 2014;28(8):1402–1412. | ||
Laskar A, Eilertsen J, Li W, Yuan XM. SPION primes THP1 derived M2 macrophages towards M1-like macrophages. Biochem Biophys Res Commun. 2013;441(4):737–742. | ||
Couto D, Freitas M, Porto G, et al. Polyacrylic acid-coated and non-coated iron oxide nanoparticles induce cytokine activation in human blood cells through TAK1, p38 MAPK and JNK pro-inflammatory pathways. Arch Toxicol. 2015;89(10):1759–1769. | ||
Strehl C, Gaber T, Maurizi L, et al. Effects of PVA coated nanoparticles on human immune cells. Int J Nanomed. 2015;10:3429–3445. | ||
Grosse S, Stenvik J, Nilsen AM. Iron oxide nanoparticles modulate lipopolysaccharide-induced inflammatory responses in primary human monocytes. Int J Nanomed. 2016;11:4625–4642. | ||
Mollnes TE, Brekke OL, Fung M, et al. Essential role of the C5a receptor in E coli-induced oxidative burst and phagocytosis revealed by a novel lepirudin-based human whole blood model of inflammation. Blood. 2002;100(5):1869–1877. | ||
Samstad EO, Niyonzima N, Nymo S, et al. Cholesterol crystals induce complement-dependent inflammasome activation and cytokine release. J Immunol. 2014;192(6):2837–2845. | ||
Sokolov A, Hellerud BC, Johannessen EA, Mollnes TE. Inflammatory response induced by candidate biomaterials of an implantable microfabricated sensor. J Biomed Mater Res A. 2012;100(5):1142–1150. | ||
Rokstad AM, Brekke OL, Steinkjer B, et al. The induction of cytokines by polycation containing microspheres by a complement dependent mechanism. Biomaterials. 2013;34(3):621–630. | ||
Wibroe PP, Anselmo AC, Nilsson PH, et al. Bypassing particle-mediated adverse injection reactions through shape modification and erythrocyte “hitch-hiking”. Nat Nanotechnol. 2017. Available from: http://www.readcube.com/articles/10.1038/nnano.2017.47. Accessed May 5, 2017. | ||
Qu H, Magotti P, Ricklin D, et al. Novel analogues of the therapeutic complement inhibitor compstatin with significantly improved affinity and potency. Mol Immunol. 2011;48(4):481–489. | ||
Finch AM, Wong AK, Paczkowski NJ, et al. Low-molecular-weight peptidic and cyclic antagonists of the receptor for the complement factor C5a. J Med Chem. 1999;42(11):1965–1974. | ||
Mollnes TE, Redl H, Hogasen K, et al. Complement activation in septic baboons detected by neoepitope-specific assays for C3b/iC3b/C3c, C5a and the terminal C5b-9 complement complex (TCC). Clin Exp Immunol. 1993;91(2):295–300. | ||
Andersen AJ, Hashemi SH, Andresen TL, Hunter AC, Moghimi SM. Complement: alive and kicking nanomedicines. J Biomed Nanotechnol. 2009;5(4):364–372. | ||
Szebeni J. Complement activation-related pseudoallergy: a new class of drug-induced acute immune toxicity. Toxicology. 2005;216(2–3):106–121. | ||
Lappegard KT, Bergseth G, Riesenfeld J, et al. The artificial surface-induced whole blood inflammatory reaction revealed by increases in a series of chemokines and growth factors is largely complement dependent. J Biomed Mater Res A. 2008;87(1):129–135. | ||
Harder MJ, Kuhn N, Schrezenmeier H, et al. Incomplete inhibition by eculizumab: mechanistic evidence for residual C5 activity during strong complement activation. Blood. 2017;129(8):970–980. | ||
Orning P, Hoem KS, Coron AE, et al. Alginate microsphere compositions dictate different mechanisms of complement activation with consequences for cytokine release and leukocyte activation. J Control Release. 2016;229:58–69. | ||
Ward PA. The harmful role of c5a on innate immunity in sepsis. J Innate Immun. 2010;2(5):439–445. | ||
Ibrahim FB, Pang SJ, Melendez AJ. Anaphylatoxin signaling in human neutrophils. A key role for sphingosine kinase. J Biol Chem. 2004;279(43):44802–44811. | ||
Lindemann A, Ludtke-Buzug K, Fraderich BM, Grafe K, Pries R, Wollenberg B. Biological impact of superparamagnetic iron oxide nanoparticles for magnetic particle imaging of head and neck cancer cells. Int J Nanomed. 2014;9:5025–5040. | ||
Hildebrand H, Kuhnel D, Potthoff A, Mackenzie K, Springer A, Schirmer K. Evaluating the cytotoxicity of palladium/magnetite nano-catalysts intended for wastewater treatment. Environ Pollut. 2010;158(1):65–73. | ||
Petters C, Thiel K, Dringen R. Lysosomal iron liberation is responsible for the vulnerability of brain microglial cells to iron oxide nanoparticles: comparison with neurons and astrocytes. Nanotoxicology. 2016;10(3):332–342. | ||
Konczol M, Weiss A, Stangenberg E, et al. Cell-cycle changes and oxidative stress response to magnetite in A549 human lung cells. Chem Res Toxicol. 2013;26(5):693–702. | ||
Khan MI, Mohammad A, Patil G, Naqvi SA, Chauhan LK, Ahmad I. Induction of ROS, mitochondrial damage and autophagy in lung epithelial cancer cells by iron oxide nanoparticles. Biomaterials. 2012;33(5):1477–1488. | ||
Apopa PL, Qian Y, Shao R, et al. Iron oxide nanoparticles induce human microvascular endothelial cell permeability through reactive oxygen species production and microtubule remodeling. Part Fibre Toxicol. 2009;6:1. | ||
Joris F, Manshian BB, Peynshaert K, De Smedt SC, Braeckmans K, Soenen SJ. Assessing nanoparticle toxicity in cell-based assays: influence of cell culture parameters and optimized models for bridging the in vitro-in vivo gap. Chem Soc Rev. 2013;42(21):8339–8359. |
Supplementary materials
 | Figure S1 Schematic drawing of the IONPs received from Ocean Nanotech, LLC (Springdale, AR, USA). The particles are coated with a monolayer of oleic acid and amphiphilic polymer. The reactive group on the surface is carboxylic acid. |
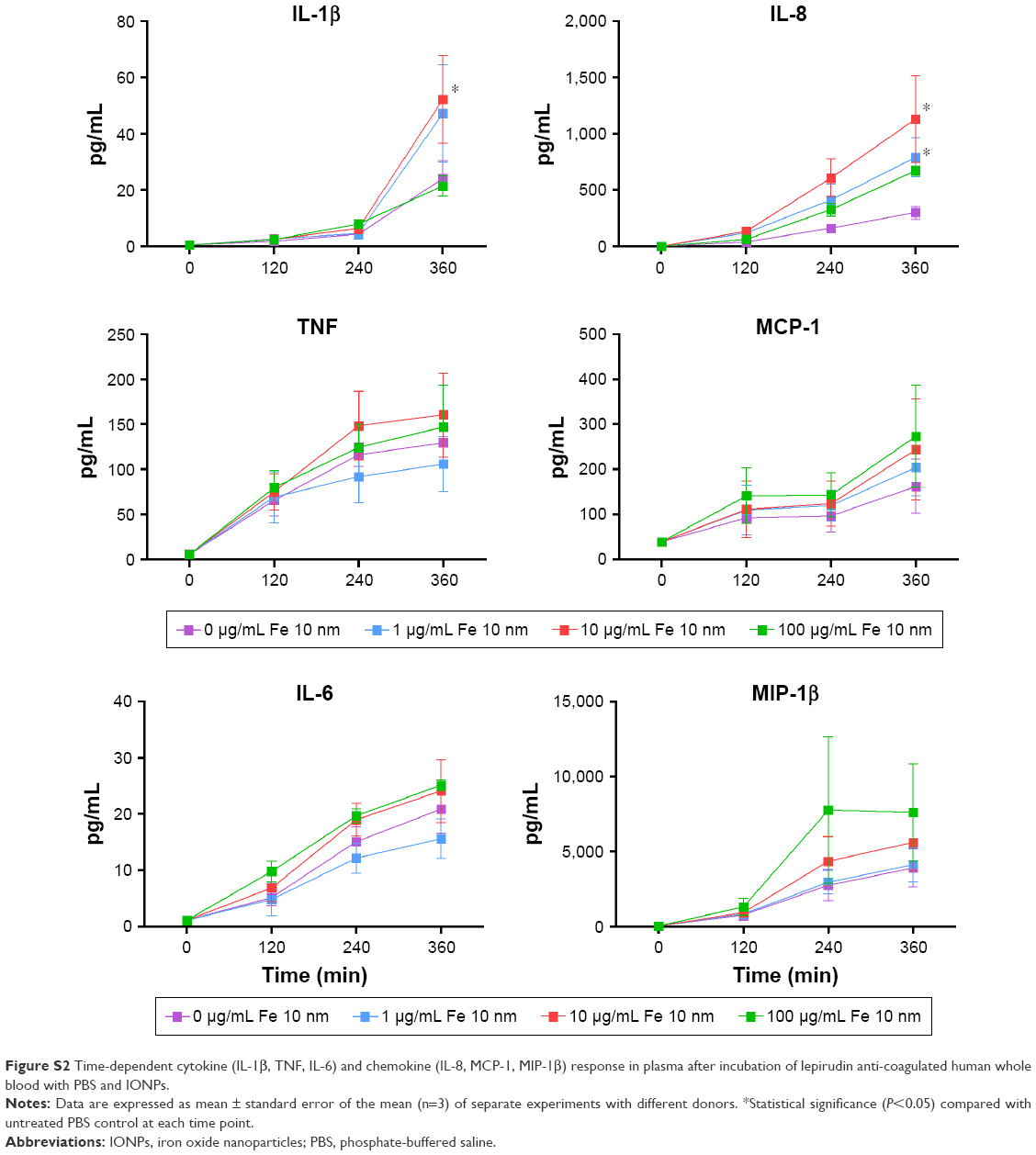 | Figure S2 Time-dependent cytokine (IL-1β, TNF, IL-6) and chemokine (IL-8, MCP-1, MIP-1β) response in plasma after incubation of lepirudin anti-coagulated human whole blood with PBS and IONPs. |
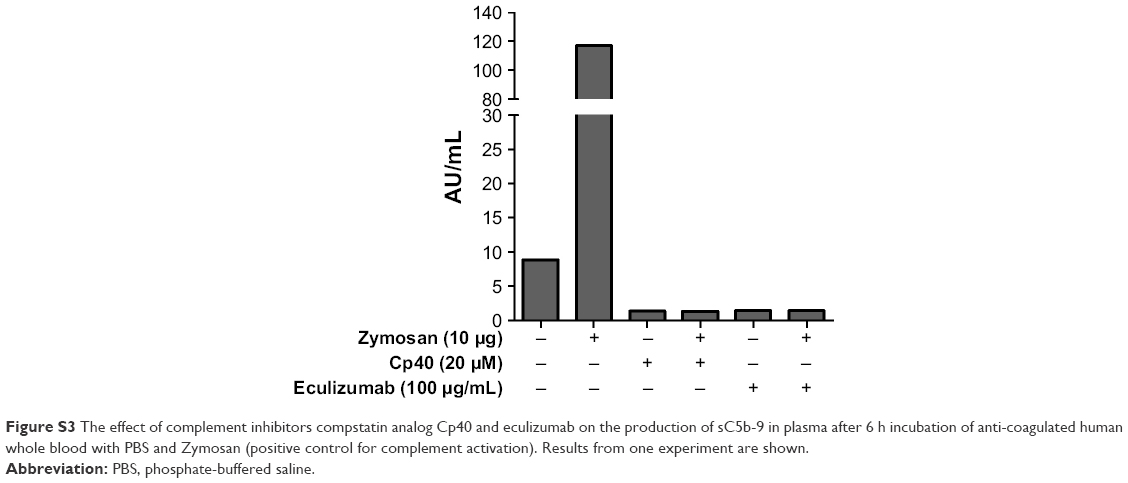 | Figure S3 The effect of complement inhibitors compstatin analog Cp40 and eculizumab on the production of sC5b-9 in plasma after 6 h incubation of anti-coagulated human whole blood with PBS and Zymosan (positive control for complement activation). Results from one experiment are shown. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.