Back to Journals » OncoTargets and Therapy » Volume 7
Involvement of c-KIT mutation in the development of gastrointestinal stromal tumors through proliferation promotion and apoptosis inhibition
Authors Ma Y, Yu S, He X ![]() , Xu Y
, Xu Y ![]() , Wu F, Xia Y, Guo K, Wang H
, Wu F, Xia Y, Guo K, Wang H ![]() , Ye Z, Zhang W, Tao H
, Ye Z, Zhang W, Tao H
Received 10 January 2014
Accepted for publication 26 February 2014
Published 2 May 2014 Volume 2014:7 Pages 637—643
DOI https://doi.org/10.2147/OTT.S60458
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Ying-Yu Ma,1 Sheng Yu,2 Xu-Jun He,1 Yuan Xu,2 Fang Wu,2 Ying-Jie Xia,1 Kun Guo,2 Hui-Ju Wang,1 Zai-Yuan Ye,1,3 Wei Zhang,3 Hou-Quan Tao1,3
1Key Laboratory of Gastroenterology of Zhejiang Province, Zhejiang Provincial People’s Hospital, Hangzhou, 2Wenzhou Medical College, Wenzhou, Zhejiang, 3Department of Gastrointestinal Surgery, Zhejiang Provincial People’s Hospital, Hangzhou, People’s Republic of China
Abstract: The aim of this study was to discuss the role of c-KIT mutation in the pathogenesis of gastrointestinal stromal tumors (GISTs) and analyze its correlation with proliferation and apoptosis. c-KIT and PDGFRA genotypes were examined by deoxyribonucleic acid sequencing. Immunohistochemistry was performed to determine the expression levels of Kit, Ki-67 (proliferation marker), and apoptotic protease-activating factor (APAF)-1 (apoptosis marker) and the relationship between their three genes. In the 68 cases examined, 44 cases (64.7%) showed mutations in one of the four exons of c-KIT. The mutations were most frequently found in exon 11 (30 cases [44.1%]), followed by exon 9 (ten cases [14.7%]) and exon 13 (four cases [5.9%]). c-KIT mutation showed no association with prognostic factors using the classification of risk of aggressive behavior in GIST proposed by Fletcher et al. No cases had mutated exon 17 of c-KIT, and neither did exon 12, 14, or 18 of PDGFRA in our present study. There was a positive correlation between the expression level of Kit and Ki-67 (R=0.282, P=0.020). Conversely, a negative correlation was found between the expression levels of Kit and APAF1 (R=−0.243, P=0.046). In conclusion, most GISTs with Kit expression showed c-KIT mutation. Kit expression has a positive correlation with Ki-67 and a negative correlation with APAF1, showing that c-KIT is involved in GIST occurrence and development through proliferation promotion and apoptosis inhibition.
Keywords: gastrointestinal stromal tumors, c-KIT, mutation, proliferation, apoptosis
Introduction
Gastrointestinal stromal tumors (GISTs) are the most common tumors developed in mesenchymal tissues of the gastrointestinal tract. Two-thirds of cases occur in the stomach, and 25% of cases occur in the small intestine.1 It has been noted that approximately 90% of GIST cases have gain-of-function mutations of either Kit or platelet-derived growth factor receptor, alpha polypeptide (PDGFRα) tyrosine kinases, and such mutations may play a critical role in GIST pathogenesis.2–4 Kit, encoded by the oncogene c-KIT,5 is characterized structurally by five immunoglobulin-like domains extracellularly and contains an adenosine triphosphate-binding domain and a phosphotransferase domain separated by an interkinase sequence in the cytoplasm.6–9 Similar to Kit, PDGFRα is also a member of the type III tyrosine-kinase family, and PDGFRA gene mutations found in GISTs result in constitutive activation of this tyrosine kinase, which contributes to tumorigenesis.10
Corless and Heinrich showed that approximately 60%–70% of GIST cases carry mutations in c-KIT. The four exons involved, ie, 9, 11, 13, and 17, correspond to the extracellular, juxtamembranous, tyrosine kinase 1, and tyrosine kinase 2 domains of the Kit protein, respectively.11 These mutations result in constitutively activated tyrosine-kinase activity of Kit, which is believed to be the primary oncogenic event in GIST.12–15 c-KIT exon 11 deletions were described to be independent adverse prognostic factors in patients with GIST.16–18 Furthermore, in another study, it was found that deletions in the last part of exon 11 (codons 562–579) were more frequently associated with GIST malignancy than deletions in the first part (codons 550–561).1,19 However, the prognostic value of some types of c-KIT mutation is still unclear.
About 15% of GIST cases do not express c-KIT mutations, 5%–7% of which have mutations affecting the genes encoding PDGFRA.10 Therefore, PDGFRA may also play an important role in GIST. In addition to the importance of c-KIT and PDGFRA in development of GIST, the correlation between their mutational status and their responses to tyrosine-kinase inhibitors, which showed their roles in primary and secondary resistance, has also been widely investigated.20,21 Detection and analysis of gene mutations of c-KIT and PDGFRA have become a standard of care for patients diagnosed with GIST. However, the prognostic values of some types of c-KIT and PDGFRA mutations are still unclear. The aim of this paper was to study the clinical significance of the mutational status of c-KIT and PDGFRA and its value as a predictive and prognostic factor in GIST. The correlation between Kit and apoptosis and proliferation was also explored.
Materials and methods
Patients and tumor samples
A total of 68 GIST samples were collected at Zhejiang Provincial People’s Hospital from January 2002 to December 2008. The medical records of all patients were reviewed by our pathologists, who used the US National Institutes of Health consensus criteria for risk stratification in GIST to confirm GIST diagnosis. The use of all human tissues was approved by the ethics committee of Zhejiang Provincial People’s Hospital, and informed consent was obtained from all of the participants.
DNA extraction and polymerase chain reaction conditions
All tumor samples were assessed for mutations in exons 9, 11, 13, and 17 of the c-KIT gene and in exons 12, 14, and 18 of the PDGFRA gene using formalin-fixed and paraffin-embedded tissue sections. Specifically, at least 15 unstained tissue sections 5 μm thick from each sample were deparaffinized and air-dried. Histologically confirmed tumorous areas were microdissected using a sterile scalpel blade and placed in 180 μL of ATL buffer (Qiagen, Venlo, the Netherlands). Deoxyribonucleic acid (DNA) was then extracted using a QIAamp DNA mini kit (Qiagen) following the manufacturer’s protocol.
For DNA sequencing, amplification of the DNA was performed by polymerase chain reaction (PCR) in a final volume of 25 μL in the following conditions: 2.5 μL of 10× PCR buffer, 1 μL each of 1 μmol/μL forward and reverse PCR primers, 1.6 μL of deoxynucleoside triphosphates, 0.5 μL of Taq DNA polymerase, and 200 ng of sample DNA. The PCR conditions were preincubation at 94°C for 5 minutes, followed by 45 cycles of the following reactions: denaturation at 94°C for 30 seconds, annealing for 1 minute (specific annealing temperatures are shown in Table 1), extension at 72°C for 1 minute, and a final extension at 72°C for 10 minutes. All PCR amplicons were stored at 4°C. The primer sequences for c-KIT and PDGFRA are listed in Table 1.
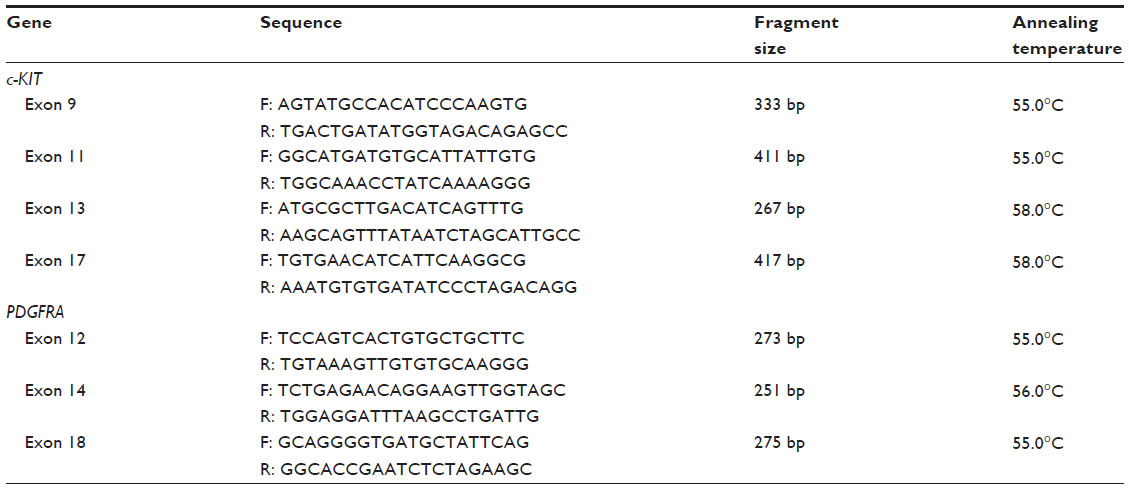 | Table 1 Polymerase chain reaction primers |
All PCR amplicons were sent to the Beijing Genomics Institute, where uncloned PCR products were analyzed by direct sequencing using an ABI 3730XL DNA sequencer (Life Technologies, Carlsbad, CA, USA). Then, mutations of query sequences were detected using the Basic Local Alignment Search Tool (BLAST) when compared to US National Center for Biotechnology Information GenBank sequences. Samples that appeared to contain mutations were further examined by sequencing their reverse primers.
Grading standards of prognostic features
Tumor size was recorded in terms of maximum diameter, and was classified as ≤2 cm, 2–5 cm, 5–10 cm, or >10 cm using the classification of risk of aggressive behavior in GIST proposed by Fletcher et al.22 Tumors that were small (≤2 cm) and showed mitotic activity <5/50 high-power fields (HPFs) were defined as very low risk. Tumors of 2–5 cm in diameter with mitotic activity <5/50 HPFs were defined as low risk. Tumors of 5–10 cm in diameter with mitotic activity <5/50 HPFs or tumors smaller than 5 cm in diameter with mitotic activity of 6–10/50 HPFs were defined as intermediate risk. Tumors larger than 5 cm in diameter with mitotic activity >5/50 HPFs or tumor diameter >10 cm or mitotic activity >10/50 HPFs were defined as high risk.
Selection of markers for proliferation and apoptosis
Apoptotic protease-activating factor (APAF)-1 is one of the most studied proapoptotic proteins. It is the core of the apoptosome, which is formed with the activation of the mitochondrial apoptotic pathway.23 In this study, APAF1 was used as an apoptosis marker to detect the relationship between Kit and apoptosis. As an antigen that corresponds to a nuclear nonhistone protein expressed by cells during tumor proliferation,24 Ki-67 was selected as the marker to detect cell-proliferative fraction.
Immunohistochemical staining
The tumor sections were deparaffinized and rehydrated using graded ethanol. Antigen retrieval was carried out by autoclaving in 0.01 M citrate buffer (pH 6.0) for 3 minutes. The sections were blocked with 3% (volume/volume) H2O2 for 10 minutes, followed by 10% (volume/volume) normal goat serum for 15 minutes at room temperature. They were incubated with primary Kit antibodies (1:100; Santa Cruz Biotechnology, Dallas, TX, USA), Ki-67 (1:50; Bioss Biotechnology, Beijing, People’s Republic of China) and APAF1 (1:50; Bioss Biotechnology) at 4°C overnight. Then, the sections were incubated with biotin-labeled secondary antibody (Life Technologies, Carlsbad, CA, USA) at room temperature for 20 minutes, rinsed with phosphate-buffered saline, and incubated with horseradish peroxidase-conjugated antibody (Life Technologies) for another 20 minutes at room temperature. Finally, the sections were stained with 3,3′-diaminobenzidine tetrahydrochloride and counterstained with hematoxylin.
Immunohistochemical evaluation
Immunohistochemical evaluation was performed independently by two pathologists without knowledge of the clinical data. The immunoreactivity level of each case was estimated by the average signal intensity and the proportion of cells showing positive cytoplasmic staining within the tumor region under light microscopy. The staining intensity was graded on a scale of 0–3+ (0 for no staining, 1+ for weak immunoreactivity, 2+ for moderate immunoreactivity, and 3+ for strong immunoreactivity). The percentage of cells that showed positive staining within the tumor region was scored as follows: 1 for 0%–25% positive, 2 for 26%–50% positive, 3 for 51%–75% positive and 4 for 76%–100% positive. The intensity and proportion scores were then multiplied to obtain a composite score. A composite score of 0–3 was defined as negative, and 4–12 positive.
Statistical analysis
Data analysis was performed using SPSS 13.0 (SPSS Inc., Chicago, IL, USA) software. Categorical variables were expressed as a percentage of the total population and analyzed by the χ2 test. If more than 20% of cells were scored less than 5, Fisher’s exact test was performed. The relationship between Kit, Ki-67, and APAF1 expressions was assessed by Spearman correlation coefficients. The Kaplan–Meier method was used for construction of survival curves, which were compared by the log-rank test. All P-values were obtained from two-tailed statistical tests. Differences were considered significant when P<0.05.
Results
Demographic data
Mutational analysis of c-KIT and PDGFRA was performed on tumor sections of 68 consecutively diagnosed GIST patients. The patients comprised 37 men and 31 women with a mean age of 56.3 years (range 30–84 years) at the time of diagnosis. Of these 68 samples, the primary tumor site was the stomach in 34 cases (50.0%), the small intestine in 20 cases (29.4%), the large intestine in six cases (8.8%), and outside the gastrointestinal tract in eight cases (11.8%).
Tumor genotype
All 68 specimens were screened for mutations in the c-KIT and PDGFRA genes. Overall, 44 cases (64.7%) had mutations in c-KIT in one of the four exons examined. Mutations were most frequently found in exon 11 (30 cases [44.1%]), followed by exon 9 (ten cases [14.7%]) and exon 13 (four cases [5.9%]) (Figure 1). No cases with mutated c-KIT exon 17 were identified in this study. In the 30 cases with exon 11 mutations, there were deletions of amino acid in 15 tumors (50.0%), substitutions in eight (26.7%), both deletions and substitutions in five (16.7%), and insertions in two (6.8%). In the ten cases with exon 9 c-KIT mutations, mutations were restricted to insertions (six tumors [60.0%]) and deletions (four tumors [40.0%]). In the four cases with exon 13 c-KIT mutations, all mutations were substitutions (100.0%). GISTs without c-KIT mutations were further examined for PDGFRA mutations. No cases had mutations in exons 12, 14, or 18 of PDGFRA in this study.
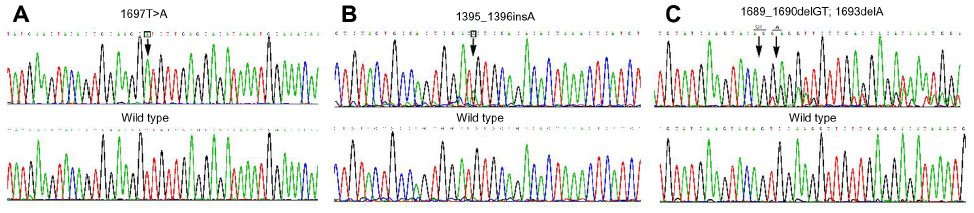 | Figure 1 (A–C) c-KIT mutations in primary gastrointestinal stromal tumors. The mutated nucleotide sequence is indicated in the upper diagram, and the wild-type nucleotide sequence is indicated in the lower diagram. Nucleotide changes are shown in the figure by downward arrows. (A) Substitution; (B) insertion; (C) deletion. |
Correlation between c-KIT mutation and clinicopathological features
Based on the risk of aggressive behavior in GIST, tumors were classified as very low risk (five of 68, 7.4%), low risk (35 of 68, 51.5%), intermediate risk (16 of 68, 23.5%), and high risk (12 of 68, 17.6%) (Table 2). The c-KIT mutation rate was 40.0% in the very low-risk group, 60.0% in the low-risk group, 68.8% in the intermediate-risk group, and 83.3% in the high-risk group. Overall, no significant difference in proportion of mutations was observed between these groups (χ2=3.545, P=0.325). Based on primary tumor sites, the proportion of c-KIT mutations was 61.8% for tumors in the stomach, 60.0% in the small intestine, 83.3% in the large intestine, and 75.0% outside the gastrointestinal tract. No significant difference in proportion of mutations was shown between these groups (χ2=1.419, P=0.731).
 | Table 2 The relationship between clinical parameters of gastrointestinal stromal tumors and c-kit mutation rates |
Survival analysis showed that the mean survival time in groups positive for c-KIT mutation was 71.6±9.3 months, which had no significant difference from that in groups negative for c-KIT mutation (91.3±12.2 months, P=0.234, Figure 2).
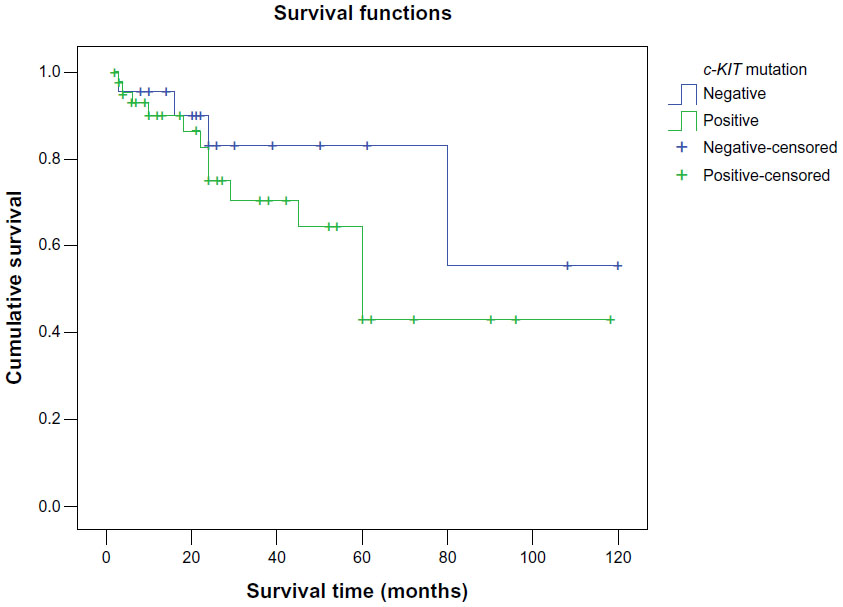 | Figure 2 Kaplan–Meier survival-curve analysis in patients with positive and negative c-KIT mutation. |
Correlation between Kit, Ki-67, and APAF1 in GIST
Positive staining of Kit was identified when specimens showed yellow-brown cytoplasmic granules (Figure 3A). The Kit-positive detection rate was 73.5% (50/68) in the GIST specimens (Figure 3A), which indicated a significant correlation with c-KIT mutations (R=0.324, P=0.007). Ki-67-positive staining was detected in the nucleus in 61.8% (42 of 68) of cases (Figure 3B), while APAF1-positive staining was detected in cytoplasm in 45.2% (28 of 68) of cases (Figure 3C). There was a positive correlation between the expression levels of Kit and Ki-67 (R=0.282, P=0.020, Spearman’s ρ-test). Conversely, a negative correlation was found between the expression levels of Kit and APAF1 (R=−0.243, P=0.046, Spearman’s ρ-test) (Table 3).
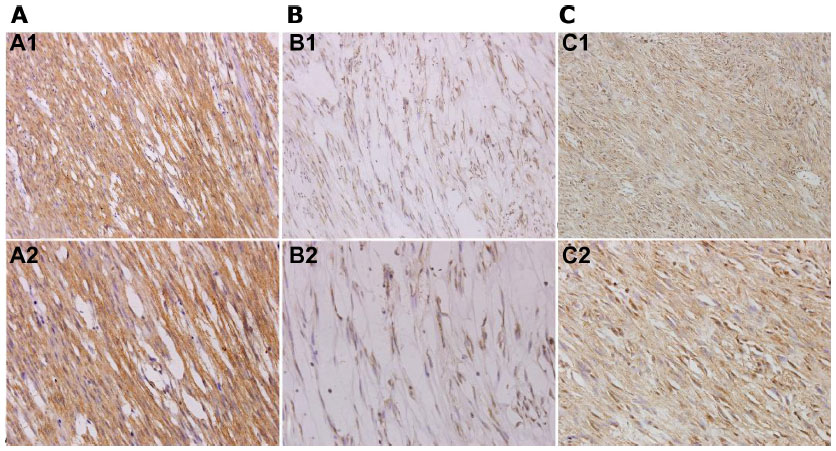 | Figure 3 (A–C) Immunohistochemical analysis of Kit, Ki-67, and apoptotic protease-activating factor (APAF)-1 in gastrointestinal stromal tumors. (A) Positive immunostaining of Kit was mainly in the cytoplasm. Original magnification 100× (A1); original magnification 400× (A2). (B) Positive immunostaining of Ki-67 was mainly in the nucleus. Original magnification 100× (B1); original magnification 400× (B2). (C) Positive immunostaining of APAF-1 was mainly in the cytoplasm. Original magnification 100× (C1); original magnification 400× (C2). |
 | Table 3 Correlations between Kit expression and Ki-67 and apoptotic protease-activating factor (APAF)-1 expression |
Discussion
GIST is a common visceral sarcoma that occurs predominantly in the gastrointestinal tract, and strong expression of the receptor tyrosine kinase Kit is the most specific and widely accepted immunohistochemical marker for this tumor.25,26 Past studies have described many types of gain-of-function mutations of c-KIT and PDGFRA in GISTs, and found that 60% occurred within exon 11 of c-KIT.1,27 Battochio et al found that the sites of PDGFRA mutation correlated with specific anatomical sites.28 Another study reported that tumors with mutated PDGFRA had a prevalent gastric location, epithelioid morphology (pure or prevalent), and low mitotic count.29 In this study, we found that c-KIT gene mutations occurred in 32 (62.7%) of the 68 GIST clinical samples, which is consistent with past results.2–4 However, no cases were found to have mutations of PDGFRA in our study. As this result may be due to our small sample size, it still needs to be validated by well-designed studies using larger number of samples as well as meta-analysis in the future.
The biology of GIST has been widely investigated. Hirota et al demonstrated mutations of the Kit receptor as a pathogenic mechanism of GIST.14 In this study, the Kit-positive detection rate was 73.5%, which showed a positive correlation with c-KIT mutations. We propose that c-KIT mutations elevate Kit expression, thus playing an important role in GIST occurrence and development. To better understand the prognostic significance of the c-KIT mutations, we analyzed the correlation between proportions of c-KIT mutations and prognostic factors using the risk of aggressive behavior classification,22 and no significant association was found. Previous studies described that GIST location was associated with the type of mutation. GISTs with c-KIT exon 9 mutation arise predominantly in the small intestine and colon, while those with PDGFRA mutations most often originate from the stomach.1,30,31 However, our results showed that there was no difference in proportions of mutations among different tumor sites. Bachet et al found that GISTs with mutation (delTyr) arise in small intestine, colon or rectum in about 70% of cases, whereas those with mutation (delWK557–558) occur in stomach in about 75% of cases.32 The results suggest that c-KIT mutations in different parts of the digestive tract may be driven by different types of oncogenic events. Meanwhile, sample size, race, and environment may also affect proportions of mutations. Therefore, further studies with a larger number of samples and quantitative summaries across different races, including specific mutation genotype, are required to evaluate the relationship between c-KIT mutation and risk of tumor site. The correlation between c-KIT mutation and prognosis was reported by many studies, but was still unclear.33,34 In this study, although we found that the mean survival time in the group positive for c-KIT mutation was lower than that in the negative group, no significant difference was found. We believe that our study will serve as a complement for all the previous studies. We hope that all of the findings will allow us to make some definitive conclusions after a systematic review.
Tumor-growth rate depends on rates of apoptosis and proliferation. It has been reported that c-KIT plays a critical role in cell proliferation and differentiation.35,36 Yasuda et al demonstrated that the stem cell factor–Kit pathway enhanced the proliferation and invasiveness of Kit-positive pancreatic cancer cell lines.37 Our results showed that the Ki-67-positive detection rate was 61.8% in GIST specimens, and had a positive correlation with the expression level of Kit. Conversely, the APAF1-positive detection rate was 45.2% in the GIST specimens, and had a negative correlation with the expression level of Kit. These results reveal that Kit is involved in GIST occurrence and development through proliferation promotion and apoptosis inhibition. However, the detailed signal pathway needs to be further investigated.
In conclusion, most GISTs with Kit expression showed c-KIT mutations, and the mutations were most frequently found in exon 11, followed by exon 9 and exon 13. Kit expression had a positive correlation with Ki-67 and a negative correlation with APAF1, showing that c-KIT is involved in GIST development through proliferation promotion and apoptosis inhibition. Identification and validation of more signal pathways will further improve our understanding of the basic mechanism of c-KIT-induced GIST development and facilitate better diagnosis and treatment, and as a result improve the prognosis of patients with GIST.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (81071991, 81372598), the Zhejiang Provincial Program for the Cultivation of High-Level Innovative Health Talents, and the Medicine and Health Research Foundation of Zhejiang Province (2009A026).
Disclosure
The authors report no conflicts of interest in this work.
References
Emile JF, Théou N, Tabone S, et al. Clinicopathologic, phenotypic, and genotypic characteristics of gastrointestinal mesenchymal tumors. Clin Gastroenterol Hepatol. 2004;2(7):597–605. | |
Antonescu CR, Sommer G, Sarran L, et al. Association of KIT exon 9 mutations with nongastric primary site and aggressive behavior: KIT mutation analysis and clinical correlates of 120 gastrointestinal stromal tumors. Clin Cancer Res. 2003;9(9):3329–3337. | |
Hirota S, Ohashi A, Nishida T, et al. Gain-of-function mutations of platelet-derived growth factor receptor alpha gene in gastrointestinal stromal tumors. Gastroenterology. 2003;125(3):660–667. | |
Rubin BP, Heinrich MC, Corless CL. Gastrointestinal stromal tumour. Lancet. 2007;369(9574):1731–1741. | |
Yarden Y, Kuang WJ, Yang-Feng T, et al. Human proto-oncogene c-kit: a new cell surface receptor tyrosine kinase for an unidentified ligand. EMBO J. 1987;6(11):3341–3351. | |
Anderson DM, Lyman SD, Baird A, et al. Molecular cloning of mast cell growth factor, a hematopoietin that is active in both membrane bound and soluble forms. Cell. 1990;63(1):235–243. | |
Flanagan JG, Leder P. The kit ligand: a cell surface molecule altered in steel mutant fibroblasts. Cell. 1990;63(1):185–194. | |
Martin FH, Suggs SV, Langley KE, et al. Primary structure and functional expression of rat and human stem cell factor DNAs. Cell. 1990;63(1):203–211. | |
Zsebo KM, Williams DA, Geissler EN, et al. Stem cell factor is encoded at the Sl locus of the mouse and is the ligand for the c-kit tyrosine kinase receptor. Cell. 1990;63(1):213–224. | |
Heinrich MC, Corless CL, Duensing A, et al. PDGFRA activating mutations in gastrointestinal stromal tumors. Science. 2003;299(5607):708–710. | |
Corless CL, Heinrich MC. Molecular pathobiology of gastrointestinal stromal sarcomas. Annu Rev Pathol. 2008;3:557–586. | |
Besmer P, Murphy JE, George PC, et al. A new acute transforming feline retrovirus and relationship of its oncogene v-kit with the protein kinase gene family. Nature. 1986;320(6061):415–421. | |
Fletcher JA. Role of KIT and platelet-derived growth factor receptors as oncoproteins. Semin Oncol. 2004;31(2 Suppl 6):4–11. | |
Hirota S, Isozaki K, Moriyama Y, et al. Gain-of-function mutations of c-kit in human gastrointestinal stromal tumors. Science. 1998; 279(5350):577–580. | |
Pawson T. Regulation and targets of receptor tyrosine kinases. Eur J Cancer. 2002;38 Suppl 5:S3–S10. | |
Dematteo RP, Gold JS, Saran L, et al. Tumor mitotic rate, size, and location independently predict recurrence after resection of primary gastrointestinal stromal tumor (GIST). Cancer. 2008;112(3):608–615. | |
Martin J, Poveda A, Llombart-Bosch A, et al. Deletions affecting codons 557–558 of the c-KIT gene indicate a poor prognosis in patients with completely resected gastrointestinal stromal tumors: a study by the Spanish Group for Sarcoma Research (GEIS). J Clin Oncol. 2005;23(25):6190–6198. | |
Wardelmann E, Losen I, Hans V, et al. Deletion of Trp-557 and Lys-558 in the juxtamembrane domain of the c-kit protooncogene is associated with metastatic behavior of gastrointestinal stromal tumors. Int J Cancer. 2003;106(6):887–895. | |
Emile JF, Tabone-Eglinger S, Théou-Anton N, Lemoine A. Prognostic value of KIT exon 11 deletions in GISTs. Gastroenterology. 2006;131(3):976–977; author reply 977–978. | |
Heinrich MC, Corless CL, Demetri GD, et al. Kinase mutations and imatinib response in patients with metastatic gastrointestinal stromal tumor. J Clin Oncol. 2003;21(23):4342–4349. | |
Heinrich MC, Maki RG, Corless CL, et al. Primary and secondary kinase genotypes correlate with the biological and clinical activity of sunitinib in imatinib-resistant gastrointestinal stromal tumor. J Clin Oncol. 2008;26(33):5352–5359. | |
Fletcher CD, Berman JJ, Corless C, et al. Diagnosis of gastrointestinal stromal tumors: a consensus approach. Hum Pathol. 2002;33(5):459–465. | |
Soengas MS, Capodieci P, Polsky D, et al. Inactivation of the apoptosis effector Apaf-1 in malignant melanoma. Nature. 2001;409(6817):207–211. | |
Sawhney N, Hall PA. Ki67 – structure, function, and new antibodies. J Pathol. 1992;168(2):161–162. | |
Miettinen M, Lasota J. Gastrointestinal stromal tumors – definition, clinical, histological, immunohistochemical, and molecular genetic features and differential diagnosis. Virchows Arch. 2001;438(1):1–12. | |
Sarlomo-Rikala M, Kovatich AJ, Barusevicius A, Miettinen M. CD117: a sensitive marker for gastrointestinal stromal tumors that is more specific than CD34. Mod Pathol. 1998;11(8):728–734. | |
Corless CL, Fletcher JA, Heinrich MC. Biology of gastrointestinal stromal tumors. J Clin Oncol. 2004;22(18):3813–3825. | |
Battochio A, Mohammed S, Winthrop D, et al. Detection of c-KIT and PDGFRA gene mutations in gastrointestinal stromal tumors: comparison of DHPLC and DNA sequencing methods using a single population-based cohort. Am J Clin Pathol. 2010;133(1):149–155. | |
Lasota J, Dansonka-Mieszkowska A, Sobin LH, Miettinen M. A great majority of GISTs with PDGFRA mutations represent gastric tumors of low or no malignant potential. Lab Invest. 2004;84(7):874–883. | |
Penzel R, Aulmann S, Moock M, Schwarzbach M, Rieker RJ, Mechtersheimer G. The location of KIT and PDGFRA gene mutations in gastrointestinal stromal tumours is site and phenotype associated. J Clin Pathol. 2005;58(6):634–639. | |
Wardelmann E, Hrychyk A, Merkelbach-Bruse S, et al. Association of platelet-derived growth factor receptor alpha mutations with gastric primary site and epithelioid or mixed cell morphology in gastrointestinal stromal tumors. J Mol Diagn. 2004;6(3):197–204. | |
Bachet JB, Hostein I, Le Cesne A, et al. Prognosis and predictive value of KIT exon 11 deletion in GISTs. Br J Cancer. 2009;101(1):7–11. | |
Andersson J, Bumming P, Meis-Kindblom JM, et al. Gastrointestinal stromal tumors with KIT exon 11 deletions are associated with poor prognosis. Gastroenterology. 2006;130(6):1573–1581. | |
Liu XH, Bai CG, Xie Q, Feng F, Xu ZY, Ma DL. Prognostic value of KIT mutation in gastrointestinal stromal tumors. World J Gastroenterol. 2005;11(25):3948–3952. | |
Gospodarowicz D. Growth factors and their action in vivo and in vitro. J Pathol. 1983;141(3):201–233. | |
Karin M. Signal transduction from cell surface to nucleus in development and disease. FASEB J. 1992;6(8):2581–2590. | |
Yasuda A, Sawai H, Takahashi H, et al. The stem cell factor/c-kit receptor pathway enhances proliferation and invasion of pancreatic cancer cells. Mol Cancer. 2006;5:46. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.