Back to Journals » Journal of Pain Research » Volume 14
Involvement of 5-Hydroxytryptamine Receptor 2A in the Pathophysiology of Medication-Overuse Headache
Authors Zheng Z ![]() , Shi X, Xiang Y, Zhang A, Fang Y
, Shi X, Xiang Y, Zhang A, Fang Y ![]()
Received 10 October 2020
Accepted for publication 19 December 2020
Published 16 February 2021 Volume 2021:14 Pages 453—461
DOI https://doi.org/10.2147/JPR.S283734
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Robert B. Raffa
Zhenyang Zheng,1 Xiaolei Shi,2 Yue Xiang,3 Aiwu Zhang,2 Yannan Fang2
1Department of Neurology, Fujian Medical University Union Hospital, Fuzhou, 350001, People’s Republic of China; 2Department of Neurology, The First Affiliated Hospital, Sun Yat-Sen University, Guangzhou, 510080, People’s Republic of China; 3Department of Nursing, Fujian Health College, Fuzhou, 350101, People’s Republic of China
Correspondence: Yannan Fang
Department of Neurology, The First Affiliated Hospital, Sun Yat-Sen University, Guangzhou, Guangdong, People’s Republic of China
Tel +86-20-87755766
Fax +86-20-87335935
Email [email protected]
Background: Recent studies indicated that analgesic overuse upregulated 5-hydroxytryptamine receptor 2A (5-HT2AR) and subsequently activated nitric oxide synthase (NOS) and thus induced latent sensitization, which provided a mechanistic basis for medication-overuse headache (MOH). Moreover, glycogen synthase kinase-3β (GSK-3β) was regulated by serotonin receptors and the phosphorylation of GSK-3β affected NOS activity, indicating that GSK-3β could be involved in the regulation of NOS activity by 5-HT2AR in MOH pathophysiology. Herein, we performed this study to investigate the role of 5-HT2AR in MOH pathophysiology and the role of GSK-3β in the regulation of NOS activity by 5-HT2AR.
Materials and Methods: Wistar rats were daily administered with paracetamol (200 mg/kg) for 30 days to set animal models for pre-clinical MOH research. After the rat MOH models were successfully established, the expression of 5-HT2AR and NOS, GSK-3β activity in trigeminal nucleus caudalis (TNC) were assayed. Then, 5-HT2AR antagonist ketanserin and agonist DOI were applied to investigate the effect of 5-HT2AR on NOS activity in TNC of MOH rats, and GSK-3β antagonist LiCl and agonist perifosine were applied to explore the role of GSK-3β in the activation of NOS by 5-HT2AR.
Results: We found that the expression of 5-HT2AR and NOS, GSK-3β activity were enhanced in TNC of MOH rats. 5-HT2AR modulator regulated the activity of NOS and GSK-3β in TNC of MOH rats, and drugs acting on GSK-3β affected NOS activity.
Conclusion: These data suggest that GSK-3β may mediate the activation of NOS by 5-HT2AR and underline the role of 5-HT2AR in MOH pathophysiology.
Keywords: medication-overuse headache, 5-hydroxytryptamine receptor 2A, nitric oxide synthase, glycogen synthase kinase-3β
Introduction
Medication-overuse headache (MOH) is one of the most common chronic headache disorders occurring from the excessive use of headache abortive medication. MOH is the 20th cause of disability worldwide according to Global Burden of Disease Study 2015,1 which causes the poor general quality of life in sufferers and the mechanism of it remains not clear, as well as the potential therapeutic target remains unfound.2
Previous studies indicated that serotonergic dysfunction was implicated in the pathogenesis of MOH. A pilot study showed that the whole blood 5-hydroxytryptamine (5-HT) levels were reduced in MOH sufferers.3 Depletion of 5-HT from storage, increase of serotonin transporter activity in platelets and suppression of 5-HT uptake following analgesic abuse could result in the hyposerotonergic state.4–6 Moreover, analgesic-induced 5-HT depletion consequently upregulates the pro-nociceptive 5-hydroxytryptamine receptor 2A (5-HT2AR).7 While the upregulation of central and peripheral 5-HT2AR is important to cortical hyper-excitation and nociceptive facilitation in MOH.8 Presumably, the upregulation of 5-HT2AR may facilitate the chronicization of headache and the development of MOH.
Reducing 5-HT level brings an immediate increase in neuronal nitric oxide synthase (nNOS) activity,9 and serotonin depletion induces cortical hyperexcitability and trigeminal nociceptive facilitation via the nitric oxide (NO) pathway.10 The activation of 5-HT2AR leads to an enhancement of nitric oxide synthase (NOS) expression and NO production in the trigeminovascular pathway and consequently may trigger migraine attacks by inducing cerebral vasodilation and nociceptive sensitization.11 NO/NOS signaling may participate in the serotonergic dysfunction in MOH pathogenesis.
Triptan administration enhances the expression of nNOS in trigeminal ganglion dural afferents, which is critical for increased responsiveness to potential migraine triggers and induces latent sensitization, providing a mechanistic basis for MOH.12,13 Additionally, NO stimulates calcitonin gene related peptide (CGRP) synthesis and secretion from trigeminal neurons through activation of T-type calcium channels and mitogen-activated protein kinase signal transduction cascades.14 The enhancement of CGRP may form the basis for decreases in the threshold at which headache attacks can be triggered and could result in more frequent headaches.13
The brain serotonin deficiency is accompanied by reduced phosphorylation of glycogen synthase kinase-3β (GSK-3β) and consequently an increase in kinase activity.15 Further investigation into the effect of serotonin on GSK-3β revealed that serotonergic regulation of the phosphorylation of GSK-3β was achieved by a balance between the opposing actions of the 5-HT receptor subtypes, namely, 5-HT1A receptors mediated increases, while 5-HT2 receptors mediated decreases in phospho-Ser9-GSK-3β levels, thus regulating the activity of the kinase.16 Moreover, GSK-3β regulated the expression of inducible nitric oxide synthase (iNOS) and NO production,17 and GSK-3β phosphorylation modulated the expression of endothelial nitric oxide synthase (eNOS).18
Accordingly, it was hypothesized that analgesic overuse caused upregulation of 5-HT2AR, and the increased 5-HT2AR dephosphorylated GSK-3β and subsequently activated NOS, and induced latent sensitization and finally lead to MOH in the current study. An experimental animal model was employed to investigate the role of 5-HT2AR in MOH pathophysiology and the role of GSK-3β in the regulation of NOS activity by 5-HT2AR and finally to verify the hypothesis in this study.
Materials and Methods
Animals and Drug Treatment
Adult male Wistar rats (aged 8 weeks and weighing 180–200 g) used in the study were maintained under controlled environmental conditions. All experimental procedures were conformed to the specifications of National Institute of Health Guide for the Care and Use of Laboratory Animals and approved by the Institutional Animal Care and Use Committee of Sun Yat-sen University.
The study had three components. The first part was to evaluate the rat MOH model. MOH model had been established in the previous study.19 In brief, rats were administered intraperitoneally paracetamol (200 mg/kg body weight) once daily for 30 days to set the model for pre-clinical MOH research. In this experiment, rats were divided into paracetamol-treated and control groups (10 rats each). Paracetamol (200 mg/kg body weight, intraperitoneally) was administered once daily for 30 days to the paracetamol-treated group, whereas the vehicle (12.5% of 1,2-propanediol in 0.9% sterile saline) of the same volume was given to the control. The tactile sensitivity and the expression of CGRP, C-Fos, 5-HT2AR and NOS, GSK-3β phosphorylation and NO production in TNC were assayed to evaluate the rat MOH model. The second part was to determine the effect of 5-HT2AR on tactile sensitivity, GSK-3β activity and NO/NOS pathway in MOH. MOH rats were established through chronic paracetamol exposure, and then they were divided into 5-HT2AR antagonist ketanserin-treated, agonist DOI-treated and control groups (10 rats each). After 30-day daily paracetamol infusion, ketanserin (10mg/kg body weight, intraperitoneally), DOI (0.1mg/kg body weight, intraperitoneally) or vehicle was administrated once daily for 7 days. The tactile sensitivity of rats, the expression of CGRP, C-Fos and NOS, GSK-3β activity and NO production in TNC were assayed. The third experiment aimed at investigating the effect of GSK-3β on tactile sensitivity, 5-HT2AR and NO/NOS pathway in MOH. MOH rats were divided into GSK-3β antagonist LiCl-treated, agonist perifosine-treated and control groups (10 rats each). After 30-day daily paracetamol infusion, LiCl (100mg/kg body weight, intraperitoneally), perifosine (20mg/kg body weight, intraperitoneally) or vehicle was administrated once daily for 7 days. The tactile sensitivity of rats, the expression of CGRP, C-Fos, 5-HT2AR and NOS, GSK-3β activity and NO production in TNC were assayed. Eighty rats were used in the study in all, 10 rats each group, and there were 8 groups, ie, the first part including 2 groups, the second part including 3 groups, and the third part including 3 groups. These rats were randomized to these groups, and the researchers who performed the detection were blind to the grouping information.
Von Frey Hair Testing
After drug treatment, the tactile sensitivity was determined with calibrated von Frey hairs (Stoelting Co., Wood Dale, Illinois, USA) by the up-down method as described previously.13,20 Briefly, after acclimating the rats in cages for 30 min, a series of von Frey hairs with logarithmically incremental stiffness were applied to the plantar surface of the hind paw for 6–8 s at intervals of 30 s between consecutive stimuli. Quick withdrawal of the hind paw in response to the stimulus was considered as a positive response. The maximum stimulus strength was 15 g.
Immunohistochemical Study
Half of 80 anesthetized rats were transcardially perfused and left medulla oblongata and first cervical cord (C1 level of cervical spinal cord) were isolated for immunohistochemical study as described previously.20 Sections were successively incubated with primary antibody against c-Fos (rabbit polyclonal antibody, dilution 1:500; Abcam, Cambridge, UK) overnight and peroxidase-conjugated goat anti-rabbit immunoglobulin G (IgG, Dako REAL EnVision Detection System, Glostrup, Denmark) for 1 h. c-Fos immunoreactive cells were identified in light of the cell nucleus with an intense brown stain, and the number of c-Fos immunoreactive cells was determined using Image J software (version 1.4.3.67, NIH).
Immunofluorescence Study
Sections were incubated with primary antibody against CGRP (rabbit monoclonal antibody, dilution 1:100; Abcam), 5-HT2AR (rabbit polyclonal antibody, dilution 1:100; Abcam) and nNOS (rabbit monoclonal antibody, dilution 1:200; Cell signaling technology, Danvers, MA, USA) overnight. The secondary antibody was Alexa Fluor® 488 or Alexa Fluor® 555-conjugated goat anti-rabbit IgG (dilution 1:1000; Cell signaling technology) for 1 h. DAPI was used to stain the cell nucleus (1:1000 dilution of 1mg/mL stock solution). Sections were observed under a fluorescence microscope (BX51, Olympus). Immunoreactive cells of CGRP, 5-HT2AR, and nNOS were identified according to the green or red fluorescence labeled in the cells, and the total cells were counted through the blue fluorescence labeled in the cell nucleus. The number of immunoreactive cells was expressed as a percentage of the total cells. The number of immunoreactive cells and the total cells was determined using Image J software. Data from 10 regions sampled from each section (10 sections per rat) were averaged and presented as percentages.
Western Blot Analysis
The other half of 80 anesthetized rats were transcardially perfused and left medulla oblongata and first cervical cord (TNC) were isolated for Western blot analysis as described previously.20 The membranes were successively incubated with primary antibody against CGRP (rabbit monoclonal antibody, dilution 1:500; Abcam), c-Fos (rabbit polyclonal antibody, dilution 1:500; Abcam), 5-HT2AR (rabbit polyclonal antibody, dilution 1:500; Abcam), GSK-3β (rabbit polyclonal antibody, dilution 1:500; Abcam), GSK-3β (phospho S9, rabbit polyclonal antibody, dilution 1:500; Abcam) and nNOS (rabbit monoclonal antibody, dilution 1:500; Cell signaling technology) overnight and horseradish peroxidase-conjugated goat anti-rabbit IgG (dilution 1:5000; Jackson, PA, USA) for 1 h. GAPDH (rabbit monoclonal antibody, dilution 1:3000; Cell signaling technology) was served as a control. Densitometry was analyzed using Image J software.
NO Analysis
Tissue homogenates of the left medulla oblongata and first cervical cord (TNC) were collected for NO analysis using Griess reagent. Briefly, 50μL of the tissue supernatant was reacted with 50μL Griess reagent Ⅰ and 50μL Griess reagent Ⅱ at 37°C incubator for 10 min, respectively. The concentration of nitrite was measured at a wavelength of 540 nm using a microplate reader (Bio-Tek FL600 microplate fluorescence reader).
Statistical Analysis
All statistics were calculated with the IBM SPSS Statistics 23.0 (SPSS Inc., Chicago, IL, USA). Quantitative data were presented as the means ± standard deviation, and the normality of the distributions of data was evaluated with the Kolmogorov–Smirnov test. The quantitative data were analyzed with a one-way ANOVA followed by LSD-t post hoc test when they were normally distributed, otherwise the data were analyzed with the Kruskal–Wallis test. Enumeration data were presented as the frequency or percentage, and they were analyzed with the chi-square test. A two-tailed P value of less than 0.05 was considered as statistically significant.
Results
Establishment and Evaluation of the Rat MOH Models
Chronic paracetamol exposure for 30 days led to a decrease in the hind paw withdrawal thresholds of rats (t-test, t = −3.012, P = 0.006; Figure 1A), that was an increase in the tactile sensitivity of rats, and CGRP and C-Fos immunoreactivity in TNC of rats were significantly increased (Figure 1B and C), and the protein expression of CGRP and C-Fos in TNC of rats were also significantly increased (t-test, t = 22.346, P = 0.000; t = 12.598, P = 0.000; Figure 1D), indicating that the rat MOH models were successfully established. 5-HT2AR and nNOS immunoreactivity in TNC of MOH rats were significantly increased (Figure 1E and F), and the protein expression of 5-HT2AR and nNOS in TNC were also significantly increased (t-test, t = 28.762, P = 0.000; t = 19.982, P = 0.000; Figure 1G). Phosphorylation of GSK-3β in TNC of MOH rats was significantly decreased (t-test, t = −36.820, P = 0.000; Figure 1G), thus leading to an increase in activity of the kinase. Endogenous NO production in TNC was also significantly increased (t-test, t = 5.721, P = 0.000; Figure 1H).
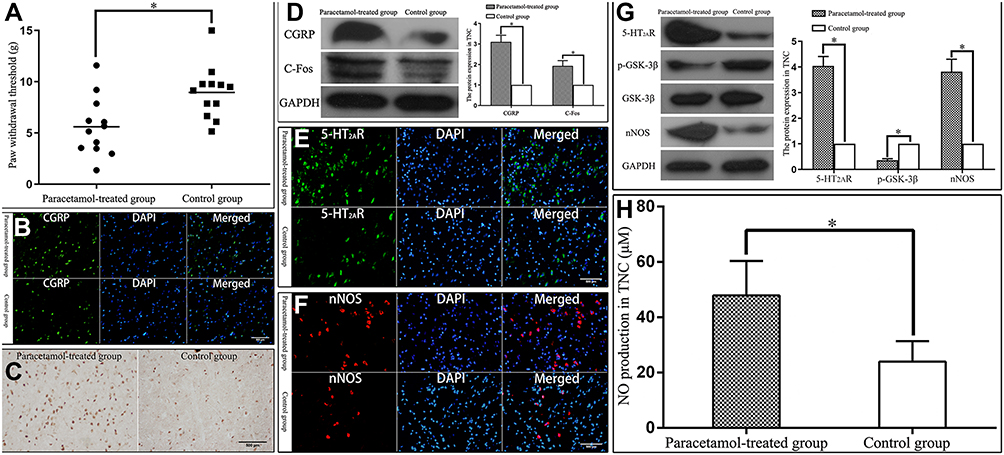 |
Figure 1 Rat MOH model establishment and evaluation. Abbreviations: MOH, medication-overuse headache; CGRP, calcitonin gene related peptide; TNC, trigeminal nucleus caudalis; 5-HT2AR, 5-hydroxytryptamine receptor 2A; nNOS, neuronal nitric oxide synthase; GSK-3β, glycogen synthase kinase-3β. Notes: (A) Chronic paracetamol exposure for 30 days in the paracetamol-treated group significantly reduced (*P = 0.006) the hind paw withdrawal thresholds of rats. (B) Chronic paracetamol exposure significantly increased (P < 0.05) the number of CGRP immunoreactive neurons in TNC of rats. (C) Chronic paracetamol exposure significantly increased (P < 0.05) the number of C-Fos immunoreactive neurons in TNC. (D) Chronic paracetamol exposure significantly increased (*P = 0.000) the protein expression of CGRP and C-Fos in TNC. (E) Chronic paracetamol exposure significantly increased (P < 0.05) the number of 5-HT2AR immunoreactive neurons in TNC. (F) Chronic paracetamol exposure significantly increased (P < 0.05) the number of nNOS immunoreactive neurons in TNC. (G) Chronic paracetamol exposure significantly increased (*P = 0.000) the protein expression of 5-HT2AR and nNOS, while significantly reduced (*P = 0.000) phosphorylation of GSK-3β in TNC. (H) Chronic paracetamol exposure significantly increased (*P = 0.000) endogenous NO production in TNC. |
Activation of nNOS by 5-HT2AR in MOH
MOH rats were established through chronic paracetamol exposure. 5-HT2AR antagonist ketanserin treatment caused decreases in the tactile sensitivity of MOH rats (one-way ANOVA followed by LSD-t post hoc test, F = 15.738, P = 0.021), CGRP, C-Fos and nNOS immunoreactivity in TNC of MOH rats, protein expression of CGRP, C-Fos and nNOS in TNC (Kruskal–Wallis test, χ2 = 36, P = 0.000; one-way ANOVA followed by LSD-t post hoc test, F = 262.059, P = 0.000; one-way ANOVA followed by LSD-t post hoc test, F = 299.013, P = 0.000), and NO production in TNC (one-way ANOVA followed by LSD-t post hoc test, F = 47.691, P = 0.000), and increases in phosphorylation of GSK-3β in TNC (Kruskal–Wallis test, χ2 = 36, P = 0.000); whereas agonist DOI led to the opposite effect, namely, it caused increases in the tactile sensitivity of MOH rats (one-way ANOVA followed by LSD-t post hoc test, F = 15.738, P = 0.003), CGRP, C-Fos and nNOS immunoreactivity in TNC, protein expression of CGRP, C-Fos and nNOS in TNC (Kruskal–Wallis test, χ2 = 36, P = 0.000; one-way ANOVA followed by LSD-t post hoc test, F = 262.059, P = 0.000; one-way ANOVA followed by LSD-t post hoc test, F = 299.013, P = 0.000), and NO production in TNC (one-way ANOVA followed by LSD-t post hoc test, F = 47.691, P = 0.000), and decreases in phosphorylation of GSK-3β in TNC (Kruskal–Wallis test, χ2 = 36, P = 0.000; Figure 2). These data indicated that analgesic overuse could cause upregulation of 5-HT2AR, and consequently activated nNOS and dephosphorylated GSK-3β in MOH.
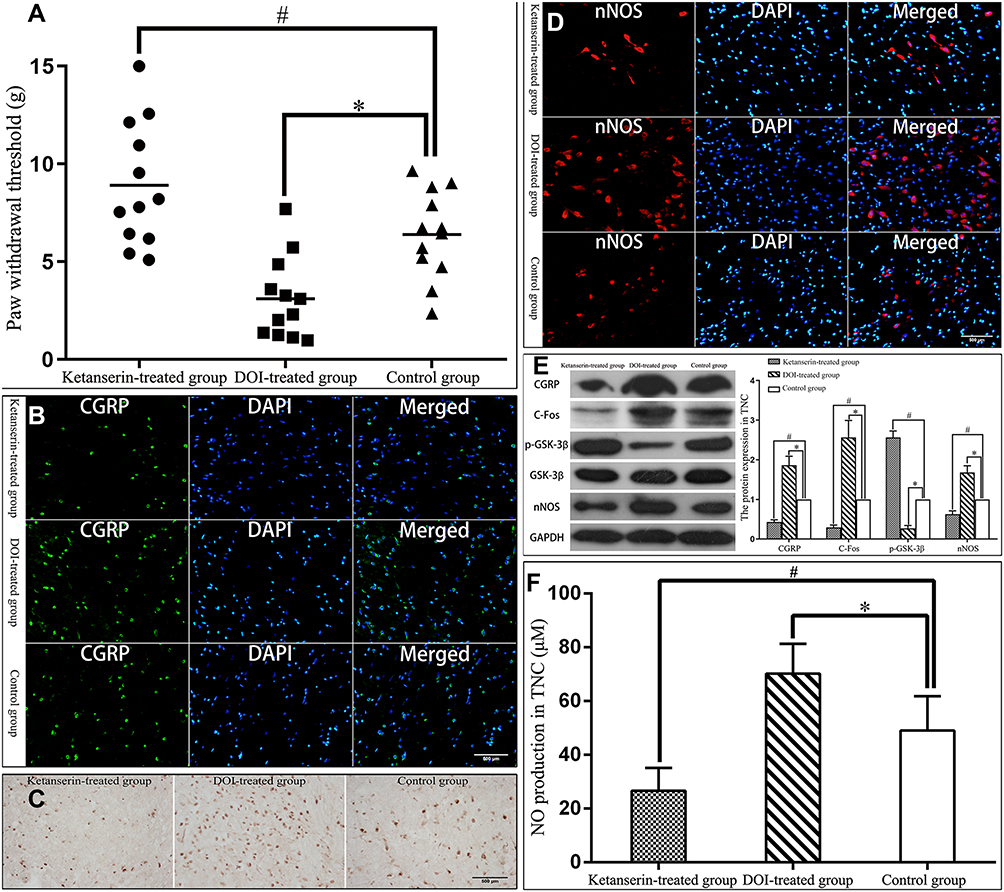 |
Figure 2 Activation of nNOS by 5-HT2AR in MOH. Abbreviations: MOH, medication-overuse headache; 5-HT2AR, 5-hydroxytryptamine receptor 2A; CGRP, calcitonin gene related peptide; TNC, trigeminal nucleus caudalis; nNOS, neuronal nitric oxide synthase; GSK-3β, glycogen synthase kinase-3β. Notes: (A) 5-HT2AR antagonist ketanserin treatment significantly increased (#P = 0.021) the hind paw withdrawal thresholds of rats, while 5-HT2AR agonist DOI treatment significantly reduced (*P = 0.003) the hind paw withdrawal thresholds of rats. (B) Ketanserin treatment significantly reduced (P < 0.05) the number of CGRP immunoreactive neurons in TNC of rats, while DOI treatment significantly increased (P < 0.05) the number of CGRP immunoreactive neurons in TNC of rats. (C) Ketanserin treatment significantly reduced (P < 0.05) the number of C-Fos immunoreactive neurons in TNC, while DOI treatment significantly increased (P < 0.05) the number of C-Fos immunoreactive neurons in TNC. (D) Ketanserin treatment significantly reduced (P < 0.05) the number of nNOS immunoreactive neurons in TNC, while DOI treatment significantly increased (P < 0.05) the number of nNOS immunoreactive neurons in TNC. (E) Ketanserin treatment significantly reduced (#P = 0.000) the protein expression of CGRP, C-Fos, and nNOS, and significantly increased (#P = 0.000) phosphorylation of GSK-3β in TNC. Whereas DOI treatment significantly increased (*P = 0.000) the protein expression of CGRP, C-Fos, and nNOS, and significantly reduced (*P = 0.000) phosphorylation of GSK-3β in TNC. (F) Ketanserin treatment significantly reduced (#P = 0.000) endogenous NO production in TNC, while DOI treatment significantly increased (*P = 0.000) endogenous NO production in TNC. |
Involvement of GSK-3β in the Activation of nNOS by 5-HT2AR
GSK-3β antagonist LiCl treatment caused decreases in the tactile sensitivity of MOH rats (Kruskal–Wallis test, χ2 = 16.667, P = 0.043), CGRP, C-Fos and nNOS immunoreactivity in TNC of MOH rats, protein expression of CGRP, C-Fos and nNOS in TNC (one-way ANOVA followed by LSD-t post hoc test, F = 204.473, P = 0.000; one-way ANOVA followed by LSD-t post hoc test, F = 149.728, P = 0.000; one-way ANOVA followed by LSD-t post hoc test, F = 237.166, P = 0.000), and NO production in TNC (Kruskal–Wallis test, χ2 = 20.667, P = 0.000), and increases in phosphorylation of GSK-3β in TNC (one-way ANOVA followed by LSD-t post hoc test, F = 286.353, P = 0.000); whereas agonist perifosine led to the opposite effect, namely, it caused increases in the tactile sensitivity of MOH rats (Kruskal–Wallis test, χ2 = 16.667, P = 0.003), CGRP, C-Fos and nNOS immunoreactivity in TNC, protein expression of CGRP, C-Fos and nNOS in TNC (one-way ANOVA followed by LSD-t post hoc test, F = 204.473, P = 0.000; one-way ANOVA followed by LSD-t post hoc test, F = 149.728, P = 0.000; one-way ANOVA followed by LSD-t post hoc test, F = 237.166, P = 0.000), and NO production in TNC (Kruskal–Wallis test, χ2 = 20.667, P = 0.043), and decreases in phosphorylation of GSK-3β in TNC (one-way ANOVA followed by LSD-t post hoc test, F = 286.353, P = 0.000). However, the immunoreactivity and protein expression of 5-HT2AR in TNC was not affected by GSK-3β modulator in MOH (one-way ANOVA followed by LSD-t post hoc test, F = 1.620, P = 0.089, P = 0.614; Figure 3). These data indicated that GSK-3β would be involved in the activation of nNOS by 5-HT2AR in MOH.
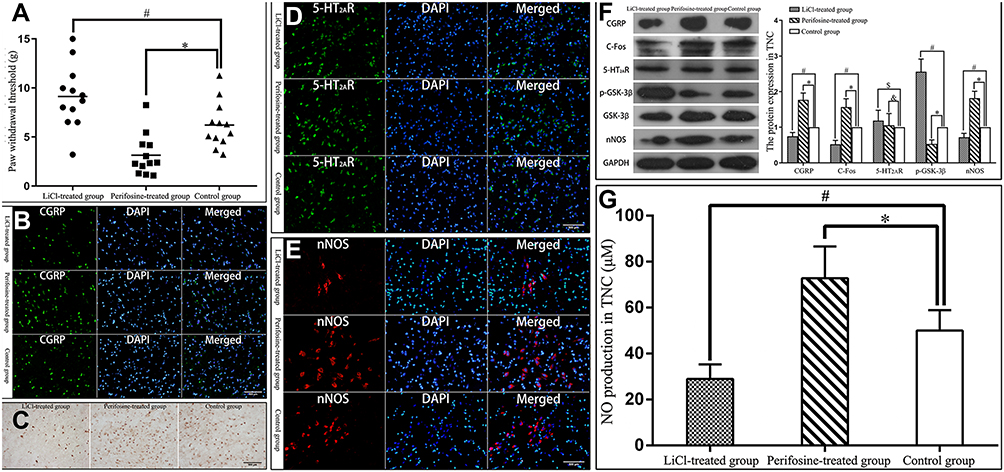 |
Figure 3 Involvement of GSK-3β in the activation of nNOS by 5-HT2AR. Abbreviations: GSK-3β, glycogen synthase kinase-3β; CGRP, calcitonin gene related peptide; TNC, trigeminal nucleus caudalis; nNOS, neuronal nitric oxide synthase; 5-HT2AR, 5-hydroxytryptamine receptor 2A. Notes: (A) GSK-3β antagonist LiCl treatment significantly increased (#P = 0.043) the hind paw withdrawal thresholds of rats, while GSK-3β agonist perifosine treatment significantly reduced (*P = 0.003) the hind paw withdrawal thresholds of rats. (B) LiCl treatment significantly reduced (P < 0.05) the number of CGRP immunoreactive neurons in TNC of rats, while perifosine treatment significantly increased (P < 0.05) the number of CGRP immunoreactive neurons in TNC of rats. (C) LiCl treatment significantly reduced (P < 0.05) the number of C-Fos immunoreactive neurons in TNC, while perifosine treatment significantly increased (P < 0.05) the number of C-Fos immunoreactive neurons in TNC. (D) LiCl or perifosine treatment did not affect the number of 5-HT2AR immunoreactive neurons in TNC (P > 0.05). (E) LiCl treatment significantly reduced (P < 0.05) the number of nNOS immunoreactive neurons in TNC, while perifosine treatment significantly increased (P < 0.05) the number of nNOS immunoreactive neurons in TNC. (F) LiCl treatment significantly reduced (#P = 0.000) the protein expression of CGRP, C-Fos, and nNOS, and significantly increased (#P = 0.000) phosphorylation of GSK-3β in TNC. Whereas perifosine treatment significantly increased (*P = 0.000) the protein expression of CGRP, C-Fos, and nNOS, and significantly reduced (*P = 0.000) phosphorylation of GSK-3β in TNC. LiCl or perifosine treatment did not affect the protein expression of 5-HT2AR in TNC ($P = 0.089, &P = 0.614). (G) LiCl treatment significantly reduced (#P = 0.000) endogenous NO production in TNC, while perifosine treatment significantly increased (*P = 0.043) endogenous NO production in TNC. |
Discussion
Our study showed that chronic paracetamol exposure upregulated the expression of 5-HT2AR and thus activated nNOS in MOH, and GSK-3β participated in the activation of nNOS by 5-HT2AR.
There were two methods to set an animal model for pre-clinical MOH research. One was continuous sumatriptan infusion with Alzet osmotic mini-pumps (0.6mg/kg/day, subcutaneously) for 7 days,12 and the other was daily infusion with paracetamol (200 mg/kg, intraperitoneally) for 30 days.19 The latter animal model treated with chronic paracetamol infusion was easy with a high success rate, thus we chose this method to set an animal model in the current study.
In this study, we found that the 5-HT2AR modulator regulated the activity of nNOS and GSK-3β in TNC of MOH rats, and drugs acting on GSK-3β affected the activity of nNOS, indicating that GSK-3β was implicated in MOH pathophysiology and could mediate the activation of nNOS by 5-HT2AR. GSK-3β is a multifunctional serine/threonine kinase ubiquitously expressed in eukaryotes which is regulated by phosphorylation on tyrosine/serine residues. GSK-3β is highly enriched in the brain and functions in the processes of neural development, neuron polarization, and neurodegeneration.21 Previous studies showed that GSK-3β participated in nociceptive processing. GSK-3β activation contributed to remifentanil-induced postoperative hyperalgesia,22 while GSK-3β inhibition ameliorated remifentanil-induced postoperative hyperalgesia.23 Moreover, GSK-3β served as an important substrate of phosphoinositide 3-kinase (PI3K)/Akt signaling, which was also involved in the nociceptive processing. Akt was a key signaling molecule in sensory neurons and spinal cord after peripheral nerve injury,24 and the Akt signaling pathway was activated in neuropathic pain.25 The activation of Akt signaling pathway in the periphery contributed to pain behavior evoked by noxious stimulation.26 PI3K and PI3K/Akt signaling pathway activation might be engaged in the development of neuropathic pain.27 Moreover, PI3K/Akt/GSK-3β signaling pathway may be activated in heat hyperalgesia and migraine.28,29
5-HT1 receptors and 5-HT2 receptors are involved and play opposing roles in regulating GSK-3β activity. 5-HT could inhibit GSK-3β by acting through 5-HT1 receptors, whereas activation of 5-HT2 receptors would result in GSK-3β activation.16 Moreover, Akt was involved in the regulation of GSK-3 by 5-HT receptors,30 and PI3K/Akt signaling mediated the effect of 5-HT1A receptors on GSK-3β phosphorylation.31 Phosphorylation of Akt/GSK-3β mediated the effect of NOS on myocardial cells,32 and drugs enhancing phosphorylation of Akt/GSK-3β/eNOS induced an increase in NO bioavailability.33 Predictably, PI3K/Akt may mediate the dephosphorylation of GSK-3β by 5-HT2AR and subsequently could activate NOS, inducing latent sensitization and the development of MOH.
Triptans and analgesics are the most common drugs that lead to MOH.2 Previous studies showed that prolonged exposure to sumatriptan produced enhanced excitability of neuronal NOS and subsequent activity of CGRP.12 And in our study, we found that chronic paracetamol exposure led to the activation of neuronal NOS. Neuronal NOS may be the common signaling in the development of MOH caused by different medications. Enhancement of nitric oxide signaling could promote central sensitization, which could provide a mechanistic basis for the transformation of a primary headache to medication overuse headache.13
Being an animal study, our research suffered from several limitations. Firstly, the MOH animal model used in the study was established through chronic paracetamol administration, while MOH patients in the clinical entity overused a variety of analgesics, including non-steroidal anti-inflammatory drugs, triptans, ergotamine, caffeine, and opium, etc. MOH attributed to different drugs may have diverse pathogenesis. MOH animal models established with various kinds of drugs should be evaluated at the same time in the future study. Secondly, paw threshold and periorbital threshold were reliable parameters for evaluation of tactile sensitivity, and periorbital threshold seemed like a more suitable parameter in the study of headache. We only tested the paw threshold in this study because we found that the paw threshold was more reliable and easier to detect in our preliminary study. Thirdly, the present study evaluated the role of GSK-3β in the activation of nNOS by 5-HT2AR solely via drugs acting on the enzymatic activity of GSK-3β. It would be more reasonable if the role of GSK-3β could be investigated in the gene-knockout animals at the same time. The role of GSK-3β signaling in MOH pathophysiology warrants further investigation.
Conclusion
The present findings suggest that GSK-3β may mediate the activation of nNOS by 5-HT2AR and underline the role of 5-HT2AR in MOH pathophysiology. The drug with a high affinity for 5-HT2AR and selective antagonism of 5-HT2AR will be a perfect prophylactic treatment for migraine without the risk of developing MOH. The study will provide insight into the mechanisms of MOH and provide a new target for therapy.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (grant numbers 81171101, 81500965); Training Project for Young and middle-aged core talents in Health system of Fujian province (grant number 2017-ZQN-38); Natural Science Foundation of Fujian province (grant number 2017J05126); and Sailing Fund Project of Fujian Medical University (grant number 2016QH022).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Vos T, Allen C, Arora M, et al. Global, regional, and national incidence, prevalence, and years lived with disability for 310 diseases and injuries, 1990–2015: a systematic analysis for the global burden of disease study 2015. Lancet. 2016;388(10053):1545–1602.
2. Diener HC, Holle D, Solbach K, Gaul C. Medication-overuse headache: risk factors, pathophysiology and management. Nat Rev Neurol. 2016;12(10):575–583. doi:10.1038/nrneurol.2016.124
3. Hering R, Glover V, Pattichis K, Catarci T, Steiner TJ. 5HT in migraine patients with medication-induced headache. Cephalalgia. 1993;13(6):410–412. doi:10.1046/j.1468-2982.1993.1306410.x
4. Srikiatkhachorn A, Maneesri S, Govitrapong P, Kasantikul V. Derangement of serotonin system in migrainous patients with analgesic abuse headache: clues from platelets. Headache. 1998;38(1):43–49. doi:10.1046/j.1526-4610.1998.3801043.x
5. Ayzenberg I, Obermann M, Leineweber K, et al. Increased activity of serotonin uptake in platelets in medication overuse headache following regular intake of analgesics and triptans. J Headache Pain. 2008;9(2):109–112. doi:10.1007/s10194-008-0019-9
6. Srikiatkhachorn A, Anthony M. Platelet serotonin in patients with analgesic-induced headache. Cephalalgia. 1996;16(6):423–426. doi:10.1046/j.1468-2982.1996.1606423.x
7. Srikiatkhachorn A, Puangniyom S, Govitrapong P. Plasticity of 5-HT serotonin receptor in patients with analgesic-induced transformed migraine. Headache. 1998;38(7):534–539. doi:10.1046/j.1526-4610.1998.3807534.x
8. Supornsilpchai W, le Grand SM, Srikiatkhachorn A. Involvement of pro-nociceptive 5-HT2A receptor in the pathogenesis of medication-overuse headache. Headache. 2010;50(2):185–197. doi:10.1111/j.1526-4610.2009.01591.x
9. Ramos AJ, Tagliaferro P, Lopez-Costa JJ, Lopez EM, Pecci SJ, Brusco A. Neuronal and inducible nitric oxide synthase immunoreactivity following serotonin depletion. Brain Res. 2002;958(1):112–121. doi:10.1016/S0006-8993(02)03489-3
10. le Grand SM, Supornsilpchai W, Saengjaroentham C, Srikiatkhachorn A. Serotonin depletion leads to cortical hyperexcitability and trigeminal nociceptive facilitation via the nitric oxide pathway. Headache. 2011;51(7):1152–1160. doi:10.1111/j.1526-4610.2011.01931.x
11. Srikiatkhachorn A, Suwattanasophon C, Ruangpattanatawee U, Phansuwan-Pujito P. 2002 Wolff award. 5 -HT2A receptor activation and nitric oxide synthesis: a possible mechanism determining migraine attacks. Headache. 2002;42(7):566–574. doi:10.1046/j.1526-4610.2002.02142.x
12. De Felice M, Ossipov MH, Wang R, et al. Triptan-induced enhancement of neuronal nitric oxide synthase in trigeminal ganglion dural afferents underlies increased responsiveness to potential migraine triggers. Brain. 2010;133(Pt 8):2475–2488. doi:10.1093/brain/awq159
13. De Felice M, Ossipov MH, Wang R, et al. Triptan-induced latent sensitization: a possible basis for medication overuse headache. Ann Neurol. 2010;67(3):325–337. doi:10.1002/ana.21897
14. Bellamy J, Bowen EJ, Russo AF, Durham PL. Nitric oxide regulation of calcitonin gene-related peptide gene expression in rat trigeminal ganglia neurons. Eur J Neurosci. 2006;23(8):2057–2066. doi:10.1111/j.1460-9568.2006.04742.x
15. Belmaker RH, Agam G, Bersudsky Y. Role of GSK3beta in behavioral abnormalities induced by serotonin deficiency. Proc Natl Acad Sci U S A. 2008;105(20):E23. (). doi:10.1073/pnas.0801168105
16. Li X, Zhu W, Roh MS, Friedman AB, Rosborough K, Jope RS. In vivo regulation of glycogen synthase kinase-3beta (GSK3beta) by serotonergic activity in mouse brain. Neuropsychopharmacol. 2004;29(8):1426–1431. doi:10.1038/sj.npp.1300439
17. Chang Y-T, Chen C-L, Lin C-F, et al. Regulatory role of GSK-3β on NF-κ B, nitric oxide, and TNF-α in group a streptococcal infection. Mediators Inflamm. 2013;2013:720689. doi:10.1155/2013/720689
18. Nakatani K, Horinouchi J, Yabu Y, Wada H, Nobori T. Expression of endothelial nitric oxide synthase is induced by estrogen with glycogen synthase 3beta phosphorylation in MCF-7 cells. Oncol Rep. 2004;12(4):833–836.
19. Supornsilpchai W, le Grand SM, Srikiatkhachorn A. Cortical hyperexcitability and mechanism of medication-overuse headache. Cephalalgia. 2010;30(9):1101–1109. doi:10.1177/0333102409355600
20. Di W, Zheng Z-Y, Xiao Z-J, et al. Pregabalin alleviates the nitroglycerin-induced hyperalgesia in rats. Neuroscience. 2015;284:11–17. doi:10.1016/j.neuroscience.2014.08.056
21. Seira O, Del RJ. Glycogen synthase kinase 3 beta (GSK3beta) at the tip of neuronal development and regeneration. Mol Neurobiol. 2014;49(2):931–944. doi:10.1007/s12035-013-8571-y
22. Yuan Y, Wang JY, Yuan F, Xie KL, Yu YH, Wang GL. Glycogen synthase kinase-3beta contributes to remifentanil-induced postoperative hyperalgesia via regulating N-methyl-D-aspartate receptor trafficking. Anesth Analg. 2013;116(2):473–481. doi:10.1213/ANE.0b013e318274e3f1
23. Li YZ, Tang XH, Wang CY, et al. Glycogen synthase kinase-3beta inhibition prevents remifentanil-induced postoperative hyperalgesia via regulating the expression and function of AMPA receptors. Anesth Analg. 2014;119(4):978–987. doi:10.1213/ANE.0000000000000365
24. Shi TJ, Huang P, Mulder J, Ceccatelli S, Hokfelt T. Expression of p-Akt in sensory neurons and spinal cord after peripheral nerve injury. Neurosignals. 2009;17(3):203–212. doi:10.1159/000210400
25. Guedes RP, Araujo AS, Janner D, Bello-Klein A, Ribeiro MF, Partata WA. Increase in reactive oxygen species and activation of Akt signaling pathway in neuropathic pain. Cell Mol Neurobiol. 2008;28(8):1049–1056. doi:10.1007/s10571-008-9279-9
26. Sun R, Yan J, Willis WD. Activation of protein kinase B/Akt in the periphery contributes to pain behavior induced by capsaicin in rats. Neuroscience. 2007;144(1):286–294. doi:10.1016/j.neuroscience.2006.08.084
27. Xu JT, Tu HY, Xin WJ, Liu XG, Zhang GH, Zhai CH. Activation of phosphatidylinositol 3-kinase and protein kinase B/Akt in dorsal root ganglia and spinal cord contributes to the neuropathic pain induced by spinal nerve ligation in rats. Exp Neurol. 2007;206(2):269–279. doi:10.1016/j.expneurol.2007.05.029
28. Ma W, Chabot JG, Quirion R. A role for adrenomedullin as a pain-related peptide in the rat. Proc Natl Acad Sci U S A. 2006;103(43):16027–16032. doi:10.1073/pnas.0602488103
29. Liu YY, Jiao ZY, Li W, Tian Q. PI3K/AKT signaling pathway activation in a rat model of migraine. Mol Med Rep. 2017;16(4):4849–4854. doi:10.3892/mmr.2017.7191
30. Beaulieu JM. A role for Akt and glycogen synthase kinase-3 as integrators of dopamine and serotonin neurotransmission in mental health. J Psychiatry Neurosci. 2012;37(1):7–16. doi:10.1503/jpn.110011
31. Polter AM, Yang S, Jope RS, Li X. Functional significance of glycogen synthase kinase-3 regulation by serotonin. Cell Signal. 2012;24(1):265–271. doi:10.1016/j.cellsig.2011.09.009
32. Bharti S, Singh R, Chauhan SS, Hussain T, Al-Attas OS, Arya DS. Phosphorylation of Akt/GSK-3beta/eNOS amplifies 5-HT2B receptor blockade mediated anti-hypertrophic effect in rats. FEBS Lett. 2012;586(2):180–185. doi:10.1016/j.febslet.2011.12.015
33. Bharti S, Golechha M, Kumari S, Siddiqui KM, Arya DS. Akt/GSK-3beta/eNOS phosphorylation arbitrates safranal-induced myocardial protection against ischemia-reperfusion injury in rats. Eur J Nutr. 2012;51(6):719–727. doi:10.1007/s00394-011-0251-y
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.