")
Back to Journals » International Journal of Nanomedicine » Volume 10 » Issue 1
Investigation of enzyme-sensitive lipid nanoparticles for delivery of siRNA to blood–brain barrier and glioma cells
Authors Bruun J, Larsen TB, Jølck RI, Eliasen R, Holm R, Gjetting T, Andresen T
Received 25 April 2015
Accepted for publication 20 June 2015
Published 24 September 2015 Volume 2015:10(1) Pages 5995—6008
DOI https://doi.org/10.2147/IJN.S87334
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Thomas Webster
Jonas Bruun,1 Trine B Larsen,1 Rasmus I Jølck,1 Rasmus Eliasen,1 René Holm,2 Torben Gjetting,1 Thomas L Andresen1
1Department of Micro- and Nanotechnology, Center for Nanomedicine and Theranostics, Technical University of Denmark, DTU Nanotech, Lyngby, Denmark; 2H Lundbeck A/S, Biologics and Pharmaceutical Science, Valby, Denmark
Abstract: Clinical applications of siRNA for treating disorders in the central nervous system require development of systemic stable, safe, and effective delivery vehicles that are able to cross the impermeable blood–brain barrier (BBB). Engineering nanocarriers with low cellular interaction during systemic circulation, but with high uptake in targeted cells, is a great challenge and is further complicated by the BBB. As a first step in obtaining such a delivery system, this study aims at designing a lipid nanoparticle (LNP) able to efficiently encapsulate siRNA by a combination of titratable cationic lipids. The targeted delivery is obtained through the design of a two-stage system where the first step is conjugation of angiopep to the surface of the LNP for targeting the low-density lipoprotein receptor-related protein-1 expressed on the BBB. Second, the positively charged LNPs are masked with a negatively charged PEGylated (poly(ethylene glycol)) cleavable lipopeptide, which contains a recognition sequence for matrix metalloproteinases (MMPs), a class of enzymes often expressed in the tumor microenvironment and inflammatory BBB conditions. Proteolytic cleavage induces PEG release, including the release of four glutamic acid residues, providing a charge switch that triggers a shift of the LNP charge from weakly negative to positive, thus favoring cellular endocytosis and release of siRNA for high silencing efficiency. This work describes the development of this two-stage nanocarrier-system and evaluates the performance in brain endothelial and glioblastoma cells with respect to uptake and gene silencing efficiency. The ability of activation by MMP-triggered dePEGylation and charge shift is demonstrated to substantially increase the uptake and the silencing efficiency of the LNPs.
Keywords: matrix metalloproteinase, cleavable PEG-lipid, gene therapy, BBB, angiopep, nanocarrier
Introduction
The blood–brain barrier (BBB) consists of a largely impermeable network of capillary endothelium cells connected by tight junctions. It hinders passive diffusion of all but small, hydrophobic molecules into the brain and thereby provides protection to the underlying cells and preserves brain homeostasis.1 While this physical barrier effectively keeps out toxins and unwanted proteins, it also excludes large-molecule drugs like recombinant proteins and gene-based medicines.2 If the drug manages to pass the BBB, it may be challenged with efflux transporters that clear toxins and maintain the low protein environment in the central nervous system.3
One promising strategy to allow transport of large-molecule drugs across the BBB is to encapsulate them in nanoparticles, thereby protecting them from degradation and immune activation in the bloodstream. Furthermore, the nanoparticles allow for surface engineering and functionalization with targeting ligands to provide cellular uptake through interactions with cell surface receptors expressed on the BBB.4,5 One such ligand is angiopep, a 20 amino acid peptide, which has been reported to interact with the low-density lipoprotein receptor-related protein-1 (LRP-1).6,7 LRP-1 is highly expressed on both brain endothelial cells and the underlying glioblastoma, enabling angiopep-coated nanoparticles to have a dual targeting effect that facilitate both transport across the BBB and uptake by glioblastoma.8–10
Encapsulation of siRNA in nanoparticles is particularly beneficial as the siRNA relies on protection from degradation by ubiquitous nucleases in the bloodstream and has a high molecular weight and negative charge that disfavors passive cellular uptake of naked siRNA.11,12 Through electrostatic interactions with positively charged synthetic materials, such as cationic lipids, siRNA can be complexed in the interior of a nanoparticle thereby obtaining protection from degradation during systemic circulation.13 However, it is necessary to shield the positive charge on the particles with a negatively charged surface coating in order to obtain long systemic circulation and avoid clearance by the mononuclear phagocyte system.14
Long systemic circulation that allows for accumulation in diseased tissue is essential for an efficient delivery system and is commonly achieved by coating the carrier surface with the hydrophilic polymer poly(ethylene glycol) (PEG).15 PEG-coating minimizes binding of serum proteins, activation of the immune system, and unspecific uptake in nontargeted cells and reduces the positive charge on the lipid nanoparticles (LNPs). However, the drawback of PEGylation is limited uptake in targeted cells and hindering of endosomal escape following uptake.16,17 To solve this PEGylation dilemma, different strategies have been reported for triggered dePEGylation at the target site. Examples are conjugation of PEG to the nanoparticle through labile linkers, such as disulfide bonds,18 pH-sensitive groups,19 or esterase-sensitive groups.20
In addition, the unique pathophysiological conditions of tumors can be exploited to trigger removal of PEG.21 One class of tumor-specific enzymes that particularly have been exploited for in situ activation of drug delivery vehicles is matrix metalloproteinases (MMPs),22 which regulate various cell behaviors, including cancer cell growth, differentiation, apoptosis, migration, invasion, and regulation of tumor angiogenesis. While MMP activity needs tight regulation in healthy tissue, the degradation of the surrounding extracellular matrix is a necessity for tumor growth; hence, expression and activation of MMPs are upregulated in most cancer types compared to normal tissue.23 Introducing a MMP-cleavable peptide as a linker between PEG and the nanoparticle provides a trigger mechanism for dePEGylation of the nanoparticle upon arrival at the tumor site, which facilitates increased cellular uptake and endosomal escape.24–28
We have recently reported an alternative approach to introduce cleavable PEG by incorporating four glutamic acid residues in a PEGylated cleavable lipopeptide (PCL) with either cholesterol or fatty acids as lipid anchor.29 The four glutamic acid residues combined with PEG provide efficient charge shielding of the nanoparticle and ensure an overall negative surface charge that is essential for long systemic circulation. Upon MMP cleavage these residues are released together with the PEG coat exposing the underlying positive charges, which results in an increased interaction between the nanoparticle and the cellular membrane.17,30 However, this system relies on passive accumulation at the tumor site, which is not very effective in brain tumors due to the BBB.
Here we present PCL-decorated LNPs as a two-stage vehicle for delivery of siRNA to brain tumors. For receptor-mediated transport across the BBB, the MMP-sensitive LNP vehicle is functionalized with angiopep that provides dual targeting to both brain endothelial and glioma cells (Figure 1, left panel). Once at the tumor site, the receptor-mediated uptake is supported by MMP-triggered proteolytical activation of the LNP, which results in removal of the protective PEG coating and negative charge, thus favoring cellular endocytosis and release of siRNA (Figure 1, right panel). We investigate this double-functionalized siRNA delivery vehicle and evaluate the influence of the lipid anchor for its potential to effectively target and mediate protein knockdown in brain endothelial and glioblastoma cell cultures.
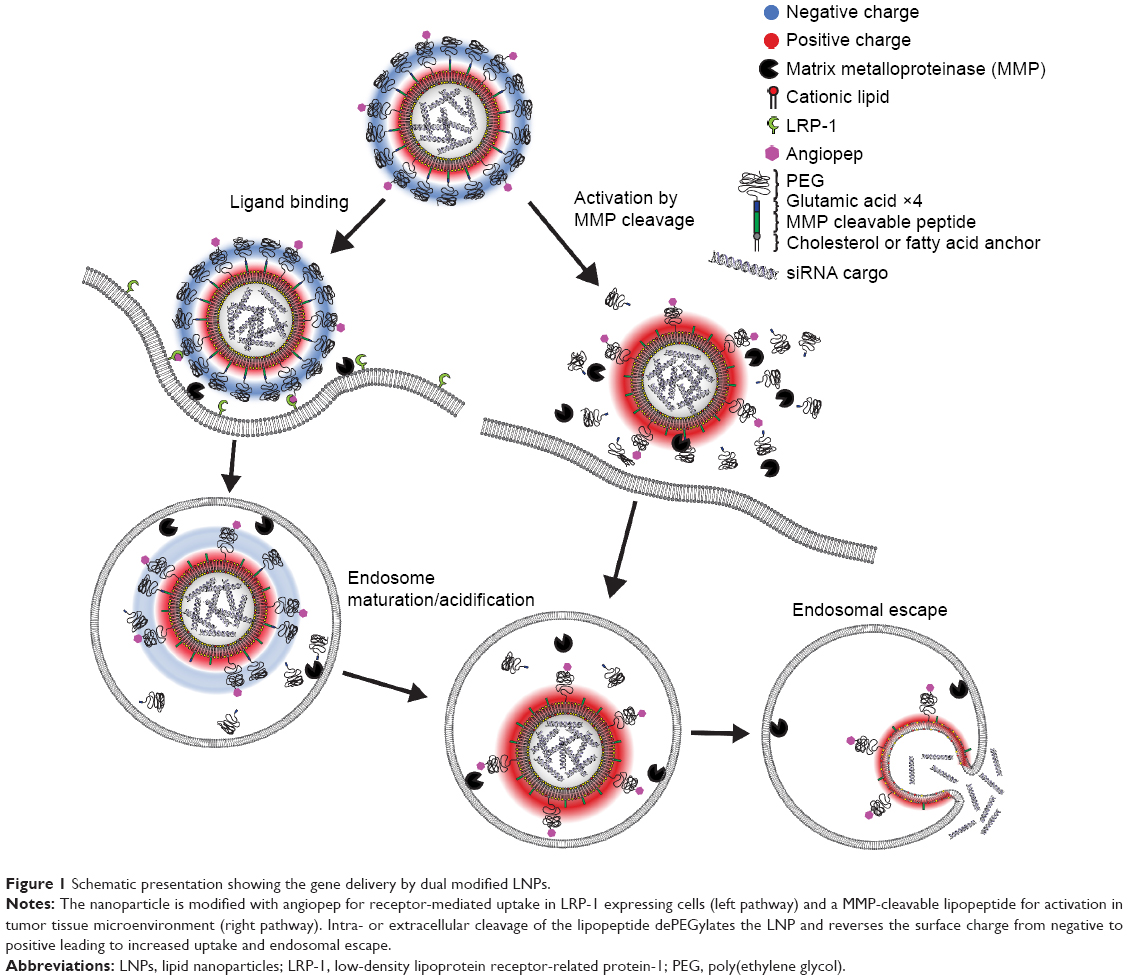 | Figure 1 Schematic presentation showing the gene delivery by dual modified LNPs. |
Materials and methods
Materials
All chemicals were purchased from Sigma-Aldrich Inc. (St Louis, MO, USA) unless otherwise stated. Angiopep (TFFYGGSRGKRNNFKTEEYC) was obtained from Bachem AG (Bubendorf, Switzerland). 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC); 1,2-dioleoyl-3-trimethylammonium-propane (chloride salt) (DOTAP); 1,2-dioleoyl-3-dimethylammonium-propane (DODAP); 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-(lissamine rhodamine B sulfonyl) (ammonium salt) (DOPE-RhB); 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-(polyethylene glycol)2000 (DSPE-PEG2000); 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[maleimide(polyethylene glycol)2000] (DSPE-PEG2000-maleimide); and cholesterol were all purchased from Avanti Polar Lipids Inc. (Alabaster, AL, USA). 9-Fluorenylmethoxycarbonyl (Fmoc) amino acids and O-(7-azabenzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HATU) were purchased from GL Biochem (Shanghai, People’s Republic of China) or Bachem AG. TentaGel PAP2000 resin was custom made by Rapp Polymere GmbH (Tuebingen, Germany). Double-stranded luciferase GL3 siRNA (sense 5′-[CUUACGCUGAGUACUUCGA] RNA [TT] DNA – 3′) and antisense siRNA (siGFP) (5′-GGCUACGUCCAGGAGCGCACC-RNA [TT] DNA – 3′) were purchased from Eurofins (Glostrup, Denmark). 3-(4,5-Dimethylthiazol-2-yl)-5-(3-carboxy-methoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) reagent and reporter lysis buffer were purchased from Promega Inc. (Madison, WI, USA).
Synthesis of lipopeptides
Cholesterol-PEGylated cleavable lipopeptide (Chol-PCL)
The peptide H-Gly-Trp(Boc)-Ile-Pro-Val-Ser(tBu)-Leu-Arg(Pbf)-Ser(tBu)-Gly-Glu(tBu)-Glu(tBu)-Glu(tBu)-Glu(tBu) was synthesized on an Initiator Alstra peptide synthesizer (Biotage, Uppsala, Sweden). TentaGel PEG-attached peptide (PAP) PEG2000-resin (294 mg, 0.1 mmol) was initially swelled in dichloromethane (DCM) for 1 hour. All couplings were conducted for 5 minutes at 75°C using 4 equiv amino acid, 3.9 equiv HATU, and 8 equiv 2,4,6-collidine in N,N-dimethylformamide (DMF). Ile was double coupled. The second coupling was conducted for 30 minutes at room temperature (RT). Fmoc deprotection was done twice using 20% piperidine in DMF for 3 and 10 minutes. The peptide was cleaved for 3 hours using 10 mL trifluoroacetic acid (TFA)/water/triisopropyl silane (TIPS) (95:2.5:2.5), after which the cleavage solvent was removed in vacuo and the peptide precipitated in diethyl ether and centrifuged. The isolated peptide was dissolved in 100 mL dry DCM/THF/DMF (10:10:1) and stirred overnight. The peptide was completely dissolved and 1.1 equiv cholesteryl chloroformate and 10 equiv diisopropylethylamine (DIPEA) was added and stirred. After 30 minutes, THF and DCM were removed in vacuo and the peptide precipitated in diethyl ether. The peptide was dissolved in 100 mL water/acetonitrile (9:1) with 0.1% triethylamine (TEA) and purified using semipreparative high-performance liquid chromatography (HPLC; Waters 600 Pump and Controller and a Waters 2489 UV/Visible Detector, Waters, Milford, MA, USA) employing a Waters XTerra® C8 5 μm (19×150 mm) column (Waters). Eluent: (A) 5% acetonitrile +0.1% TEA in water, (B) 0.1% TEA in acetonitrile. Gradient profile: linear gradient from 0% B to 50% B over 15 minutes. Flow rate: 19 mL/min. Chol-PCL was isolated as a broad peak with retention time of 11.8 minutes. The solvent was removed in vacuo and the product lyophilized from a mixture of water and acetonitrile to give a white fluffy powder (110 mg, 27%). The purity of the product was monitored by analytical HPLC using a Waters XTerra® C8 5 μm (4.6×150 mm) column (Waters). Eluent: (A) 5% acetonitrile +0.1% TFA in water, (B) 0.1% TFA in acetonitrile. Gradient profile: linear gradient from 0% B to 100% B over 15 minutes. Flow rate: 1 mL/min. Purity >98%. MALDI-TOF MS (matrix-assisted laser desorption/ionization time-of-flight mass spectroscopy; m/z): 3,979.3± n×44.0.
Dimyristoyl-PEGylated cleavable lipopeptide (DM-PCL)
The peptide H-Trp(Boc)-Ile-Pro-Val-Ser(tBu)-Leu-Arg(Pbf)-Ser(tBu)-Gly-Glu(tBu)-Glu(tBu)-Glu(tBu)-Glu(tBu) was synthesized manually on TentaGel PAP2000-resin (518 mg, 0.17 mmol) using standard Fmoc chemistry. Each coupling was achieved by 4 equiv Fmoc protected amino acid, 3.95 equiv HATU, and 8 equiv 2,4,6-collidine in DMF. Fmoc deprotection was done using 20% piperidine in DMF for 2×5 minutes. Each acylation and deprotection step was monitored by Kaiser ninhydrin test. The N-terminal end was acylated using Fmoc-dap(Fmoc)-OH in the presence of HATU and 2,4,6-collidine in DMF as already described. Fmoc was deprotected using 20% piperidine in DMF and subsequently acylation with myristic acid with HATU and 2,4,6-collidine in DMF/DCM (1:1). DM-PCL was cleaved for 3 hours using 10 mL TFA/water/TIPS (95:2.5:2.5), after which the cleavage solvent was removed in vacuo. Final purification was achieved by semipreparative HPLC employing a Waters XTerra® C18 5 μm (19×150 mm) column (Waters). Eluent: (A) 5% acetonitrile +0.1% TFA in water, (B) 0.1% TFA in acetonitrile. Gradient profile: linear gradient from 0% B to 100% B over 20 minutes. Flow rate: 19 mL/min. DM-PCL was isolated as a broad peak with retention time of 15.5 minutes. The solvent was removed in vacuo and the product lyophilized from a mixture of water and acetonitrile to give a white fluffy powder (293 mg, 41%). The purity of the product was monitored by analytical HPLC using the same gradient profile and solvent mixtures as described using a Waters XTerra® C18 5 μm (4.6×150 mm) column (Waters), flow rate: 1 mL/min. Purity >98%. MALDI-TOF MS (m/z): 4,061.4± n×44.0.
DSPE-PEG2000-angiopep
Angiopep (10 mg, 4.16 μmol) and DSPE-PEG2000-maleimide (24.5 mg, 8.32 μmol) were dissolved and mixed in 2 mL methanol/water (1:1). TEA (1.6 μL, 12.5 μmol) was added and the reaction was stirred overnight. The solvent was removed in vacuo and the product purified by semi-preparative HPLC employing a Waters XTerra® C8 5 μm (19×150 mm) column (Waters). Eluent: (A) 5% acetonitrile +0.1% TFA in water, (B) 0.1% TFA in acetonitrile. Gradient profile: linear gradient from 0% B to 100% B over 15 minutes. Flow rate: 17 mL/min. DSPE-PEG2000-angiopep was isolated as a broad peak with retention time of 11.5 minutes. The solvent was removed in vacuo and the product lyophilized from a mixture of water and acetonitrile to give a white fluffy powder. The purity of the product was monitored by analytical HPLC using the same gradient profile and solvent mixtures as already described using a Waters XTerra® C8 5 μm (4.6×150 mm) column (Waters), flow rate: 1 mL/min. Purity >98%. MALDI-TOF MS (m/z): 5,441.4± n×44.0.
Preparation of LNPs
The LNPs with different PEG-lipids were dissolved in ethanol and mixed according to the formulation (Table 1) following a procedure outlined by Jeffs et al.31 Briefly, the ethanol concentration was reduced to 90%, by adding water, before mixing one volume of lipids (20 mM) with an equal volume of siRNA solution (0.8 mg/mL) in sodium citrate (0.1 M, pH 5.0) under constant stirring. Immediately after mixing, a double volume of buffered saline (sodium chloride [0.3 M], sodium citrate [20 mM, pH 6.0]) was added. The resulting ethanolic LNP suspension was dialyzed against PBS (1:1,000) in a Slide-A-Lyzer cassette (molecular weight cut-off 10k, Pierce, Rockford, IL, USA) overnight at RT with one buffer exchange after 2 hours.
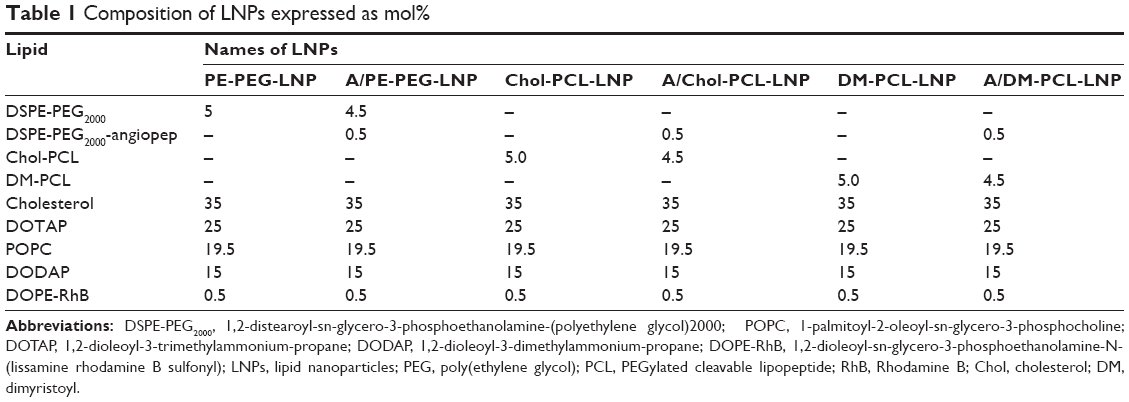 | Table 1 Composition of LNPs expressed as mol% |
Characterization of LNPs
The hydrodynamic diameter and polydispersity index (PDI) of the LNPs were determined using a Zeta PALS, Zeta Potential Analyzer (Brookhaven Instruments, Holtsville, NY, USA) by dissolving 20 μL LNP in 2 mL buffer (10 mM Na-HEPES, 5% w/v glucose, 1 mM CaCl2, pH 7.4). Data were fit using built-in software to estimate size and PDI.
ζ-potential was subsequently measured in the same sample using a conditioned electrode with 10 subruns while observing a fitting model residual <0.04.
For digestion with proteinase, LNPs were mixed with HEPES-buffered saline (1:1) (50 mM HEPES-Na, 100 mM NaCl, 1 mM CaCl2, 2 μM ZnCl2, pH 7.4) supplemented with either the metalloproteinase enzyme thermolysin (20 μg/mL) or buffer as control. Samples were incubated overnight at 37°C before analysis with MALDI-TOF MS and Zeta Potential Analyzer.
The encapsulation efficiency of siRNA was determined using Quant-iT™ PicoGreen® (Invitrogen Inc., Carlsbad, CA, USA) as described previously.29,31 Briefly, a standard curve of free siRNA was constructed in the presence or absence of Triton X-100 (1 vol%). LNPs were diluted 50 times and treated with water, heparin in PBS (1 μg/mL), or Triton X-100 (1 vol%) for 30 minutes, then mixed with Tris-EDTA buffer containing PicoGreen® reagent. Fluorescence from PicoGreen reacting with free siRNA was read (λex =485 nm, λem =535 nm) after 5 minutes using a Victor3 plate reader (Perkin Elmer, Waltham, MA, USA).
Cell lines and plasmid construction
Murine brain endothelial cells (bEnd.3) and human glioblastoma U87MG cells were obtained from ATCC (Boras, Sweden). The cells were cultured in a humidified incubator at 37°C in a 5% CO2 atmosphere, bEnd.3 in DMEM and U87MG in RPMI medium, respectively, supplemented with fetal bovine serum (10%), penicillin (100 units/mL), and streptomycin (100 mg/mL) (Invitrogen Inc.).
Cell lines with constitutive expression of luciferase were established by transfection using the plasmid pLUCneo. This plasmid was prepared from pcDNA3.1+ (Invitrogen Inc.) and pCMVluc as described previously.32 Briefly, the firefly luciferase cDNA was isolated as a HindIII-XbaI fragment and ligated to pcDNA3.1+ and digested with the same two enzymes. Escherichia coli transformation, plasmid preparation, and analysis were carried out using standard molecular biology techniques.33 The neomycin-resistance-cassette present in pLUCneo allows for selection of stable clones using G418.
For establishing stable cell lines of bEnd.3 and U87MG expressing luciferase, transfection was performed using Lipofectamine 2000 (Invitrogen Inc.) according to manufacturer’s instructions. Briefly, cells were plated at 105 cells/well in a six-well plate overnight. The media was replaced by OptiMEM (Invitrogen Inc.) containing 5 μg pLUCneo and 20 μL Lipofectamine 2000. After 6 hours transfection, the media was changed to culture media and the cells were left for 2 days. The cells were trypsinized and transferred to 6 cm petri dishes and the media was changed to selection medium containing 1 or 0.6 mg/mL G418 for bEnd.3 and U87MG cells, respectively. Cloning rings were placed upon single emerging colonies, which were then trypsinized and transferred to 12-well plates and propagated. Each cloned line was analyzed for luciferase activity as described in the “In vitro gene delivery” section below and the highest expressing clone from each cell line was selected and used throughout the study.
Flow cytometry for quantitative uptake study
The bEnd.3 cells were plated in 12-well plates for 24 hours with 8×104 cells/well. LNPs containing 120 pmol siRNA (40 nmol lipid) were added to cells in 1 mL full growth medium and incubated for 4 hours at 37°C. The cells were washed twice with heparin in PBS (0.1 mg/mL) and twice with PBS before being trypsinized and centrifuged at 1,600 rpm for 6 minutes to obtain a cell pellet, which was resuspended in PBS. The uptake was measured by analyzing 12,000 events for each sample with a Gallios flow cytometer (Beckman Coulter, Brea, CA, USA) system using a blue laser (488 nm) for excitation of RhB-labeled lipids in the LNP and a filter of 575±10 nm for emission.
γ-33P labeling of siRNA
Luciferase siRNA (50 pmol) was mixed with adenosine 5′ triphosphate [γ-33P] (Perkin Elmer) (20 pmol, 65 μCi) for labeling of the 5′-OH group of the siRNA strain. Transfer of the γ-phosphate was catalyzed by T4 polynucleotide kinase (T4 PNK) (Pierce) according to the manufacturer’s protocol. After 30 minutes of incubation at 37°C, 1 μL of EDTA (0.5 M, pH 8.0) was added and the temperature was increased to 75°C for additional 10 minutes of incubation. γ-33P labeled siRNA was then separated from unincorporated label with QIAquick nucleotide removal kit according to the manufacturer (Qiagen, Copenhagen, Denmark).
In vitro gene delivery
U87MG and bEnd.3 cells were plated in 24-well plates for 24 hours with 4×104 cells/well. LNPs containing 30, 60, or 120 pmol siRNA (10, 20, or 40 nmol lipid) were added to cells in 0.5 mL full growth medium and incubated for 2 days at 37°C. Lipofectamine® RNAiMAX (Invitrogen Inc.) was included as a positive control and mixed with siRNA according to the manufacturer’s protocol, but at siRNA concentration and incubation time corresponding to the LNP samples. After incubation, 100 μL of media was collected and the cells were washed twice with heparin in PBS (0.1 mg/mL) and twice with PBS before lysed in 100 μL reporter lysis buffer. Lysate and medium samples were analyzed for RhB fluorescence using a Victor3 plate reader (Perkin Elmer) (λex =570 nm, λem =615 nm). Furthermore, 20 μL of the lysate was analyzed for luciferase activity (Luciferase kit, Promega) in a luminometer (Lumat LB9507; Berthold, Bad Wildbad, Germany), total protein concentration was determined for 20 μL lysate using BCA kit (diluted ×10, Pierce). A purified, recombinant firefly luciferase enzyme (Promega) was diluted to construct a standard curve and luciferase activity was expressed as picogram luciferase enzyme per milligram of total protein (pg luc/mg protein). For experiments with γ-33P labeled siRNA, 50 μL of the remaining lysate was transferred to scintillation tubes and 5 mL of Ultima Gold™ (Perkin Elmer) was added before the radioactive concentration was determined by scintillation counting (Beckman Coulter, Fullerton, CA, USA).
Cytotoxicity assay
The cytotoxicity of LNPs was evaluated using MTS assay. Cells (6,000 cells/well) were seeded in 96-well plates and incubated overnight at 37°C in 5% CO2, before addition of LNPs corresponding to siRNA concentrations ranging from 10 to 2,200 nM. After 48 hours incubation, the medium was replaced with MTS reagent (Promega) diluted 5 times in fresh medium, and the plate was incubated for 2 hours before measuring ultraviolet–visible absorbance at 490 nm using a Victor3 plate reader (Perkin Elmer).
Results
Preparation of siRNA-loaded LNPs
LNPs with different PEG-lipids, with or without targeting ligand, (Table 1) were loaded with siRNA by mixing an ethanolic lipid solution with an aqueous siRNA solution. As previously described,29,31 siRNA encapsulation was highly efficient and >95% of the siRNA was encapsulated inside the LNPs, according to PicoGreen measurements (Table 2). Even when challenging the LNPs with physiological concentration of heparin, the siRNA remained unreachable for the PicoGreen stain. This confirmed that siRNA was encapsulated in the interior and not merely loosely associated to the LNP exterior under the PEG layer.
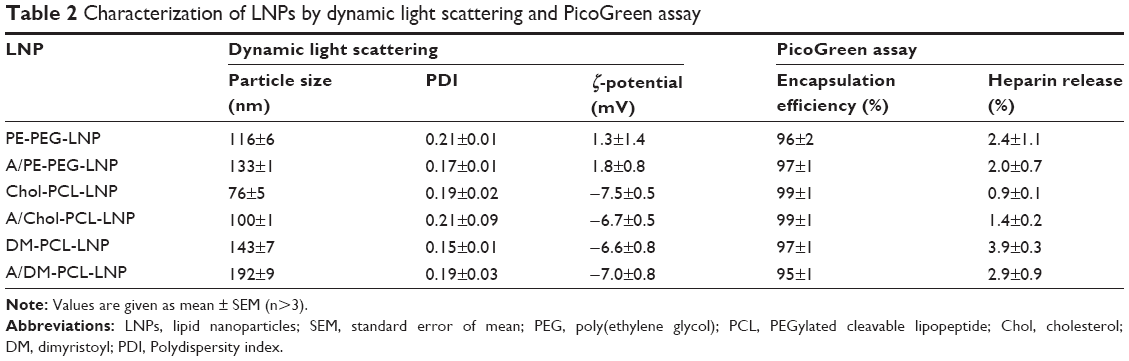 | Table 2 Characterization of LNPs by dynamic light scattering and PicoGreen assay |
Three types of LNPs were prepared with different PEGylated lipids: The noncleavable LNPs designated PE-PEG-LNP and the cleavable LNPs with either Chol-PCL (cholesterol anchored PEGylated cleavable lipopeptide) or DM-PCL (dimyristoyl anchored PEGylated cleavable lipopeptide), designated Chol-PCL-LNP and DM-PCL-LNP, respectively. Each of the three LNP types was prepared in variants with and without angiopep functionalization (Table 1). The hydrodynamic diameter of the LNPs varied between 100 and 200 nm depending on the type of PEGylated lipids included in the formulation (Table 2). LNPs with angiopep conjugated to the surface had an increased diameter of 20–50 nm compared to the nontargeting LNPs. The PDI was low for all LNPs and in the range of 0.1–0.2, which indicated a relatively homogenous particle population.
The charge of the cationic lipids was partly shielded by the PEG layer, ensuring a close to neutral ζ-potential for PE-PEG-LNP and A/PE-PEG-LNP (Table 2). For formulations with PCL, the four incorporated glutamic acid residues caused a reduction of the ζ-potential resulting in a slightly negative surface charge, confirming the charge shielding properties of the designed PCL lipopeptide.
Angiopep-mediated uptake
First step in the designed two-stage delivery system is targeting to the BBB by functionalization of the LNPs with the angiopep ligand known to target LRP-1.6 The functionalization was obtained by coupling angiopep to DSPE-PEG2000-maleimide and incorporating this lipopeptide in the lipid membrane. The angiopep-dependent cellular uptake of the single functionalized LNP (A/PE-PEG-LNP) and the nontargeted PE-PEG-LNP was investigated as an indication of their potential to cross the BBB.
PE-PEG-LNP had an insignificant uptake in bEnd.3 cells over 4 hours, and the fluorescence histogram was inseparable from that of the untreated cells (Figure 2). In contrast, angiopep containing A/PE-PEG-LNP had a 2.4-fold higher mean fluorescence intensity than the PE-PEG-LNP without the angiopep, and this clearly shifted the histogram toward higher fluorescence intensity suggesting enhanced cellular uptake of the targeted LNPs.
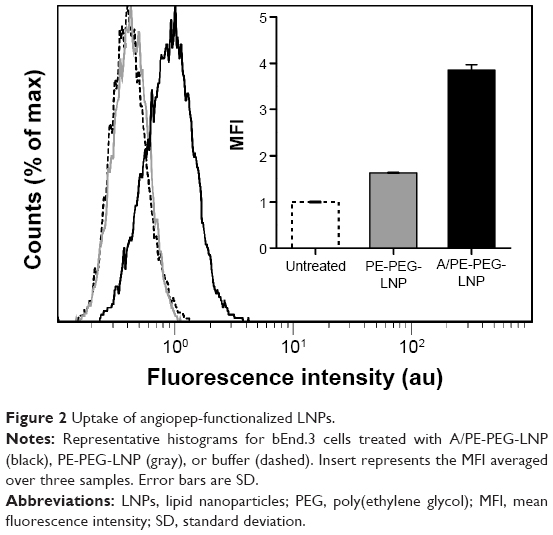 | Figure 2 Uptake of angiopep-functionalized LNPs. |
This confirmed the previously described ability of angiopep to target and mediate uptake of PEGylated nanoparticles in brain endothelial cells and thereby, potentially, transportation over the BBB.34 However, since the uptake is only improved 2.4 times relative to the control LNP, it also shows that the LRP-1 receptor does not have a very high capacity for internalization of angiopep-functionalized LNPs in our in vitro model.
Removal of the PEG coating
In addition to angiopep targeting, proteinase-triggered activation based on MMP hydrolysis was evaluated. The PCL was made in two versions, one with cholesterol (Chol-PCL) and the other with fatty acids (DM-PCL) as their respective lipid membrane anchor. LNPs containing either of these lipopeptides were digested with thermolysin, and analyzed by MALDI-TOF mass spectrometry. Thermolysin cleaves peptides at the same recognition site as MMP-2 and MMP-9, and was therefore used as a model proteinase, as MMP-2 and MMP-9 self-hydrolyze rapidly.35 Their short lifetime complicates in vitro analyses while the constant in situ activity is ensured by continuous secretion.36 As expected, DM-PCL-LNP and Chol-PCL-LNP were cleaved between their leucine and serine residues by thermolysin while the DSPE-PEG2000 coated LNP (PE-PEG-LNP) was unaffected by the presence of the proteinases (Figure 3A). Furthermore, ζ-potential measurements of the samples showed that LNPs with PE-PEG had no change in surface charge following proteinase treatment in contrast to the PCL-modified LNPs that showed a shift in ζ-potential from negative to neutral (Chol-PCL-LNP) or positive (DM-PCL-LNP) values after thermolysin digestion (Figure 3B).
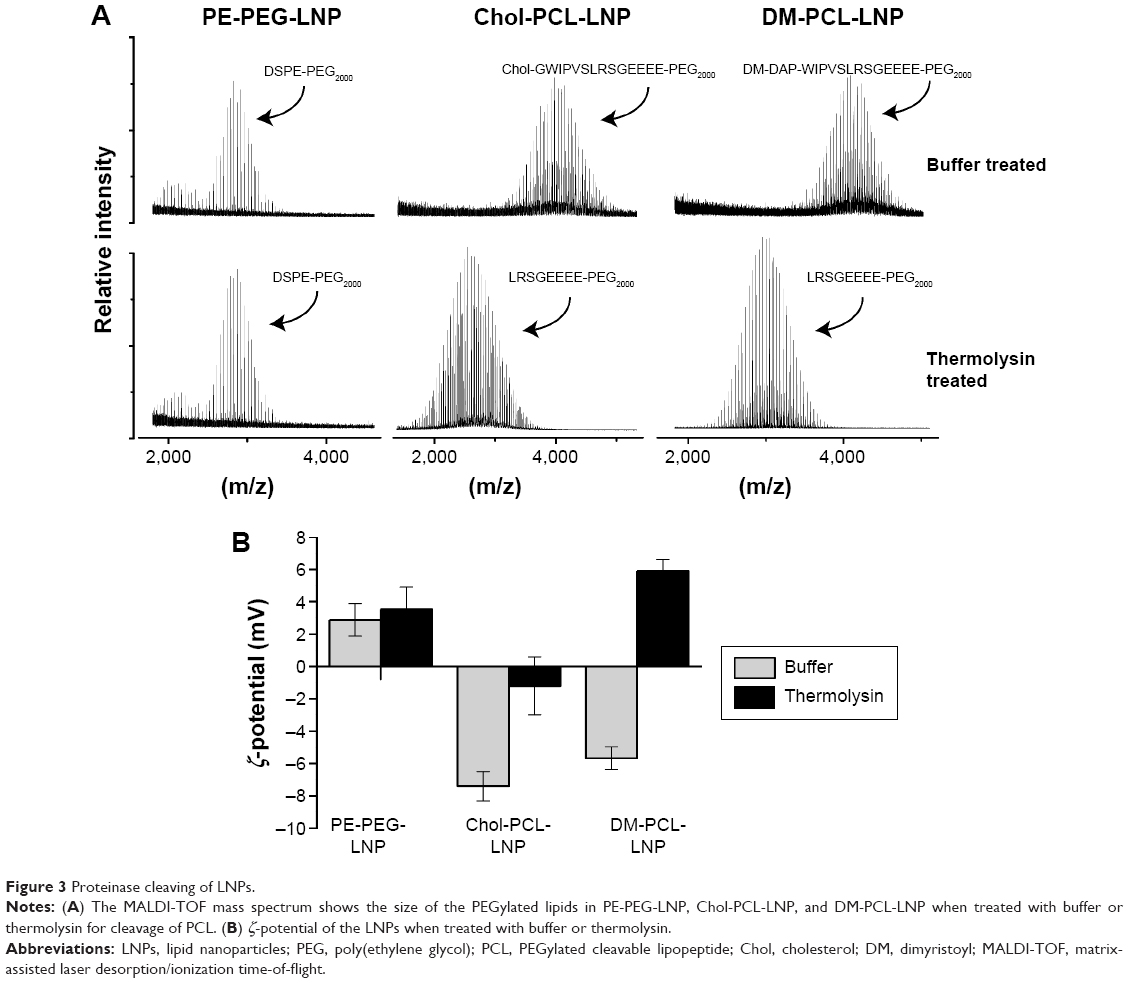 | Figure 3 Proteinase cleaving of LNPs. |
Cholesterol-anchored PCL
The combined effect of angiopep modification and cleavable PEG in the formulation of LNPs was initially evaluated with Chol-PCL for uptake and gene knockdown in U87MG cells expressing LRP-1 and MMP-2. Cellular uptake was evaluated by measuring fluorescence intensity of the cell lysate, and these data confirmed the results from the flow cytometry uptake study (Figure 4A). For both angiopep-functionalized LNPs, A/PE-PEG-LNP and A/Chol-PCL-LNP, a significantly higher uptake was obtained compared to their nontargeted counterpart, PE-PEG-LNP and Chol-PCL-LNP, respectively.
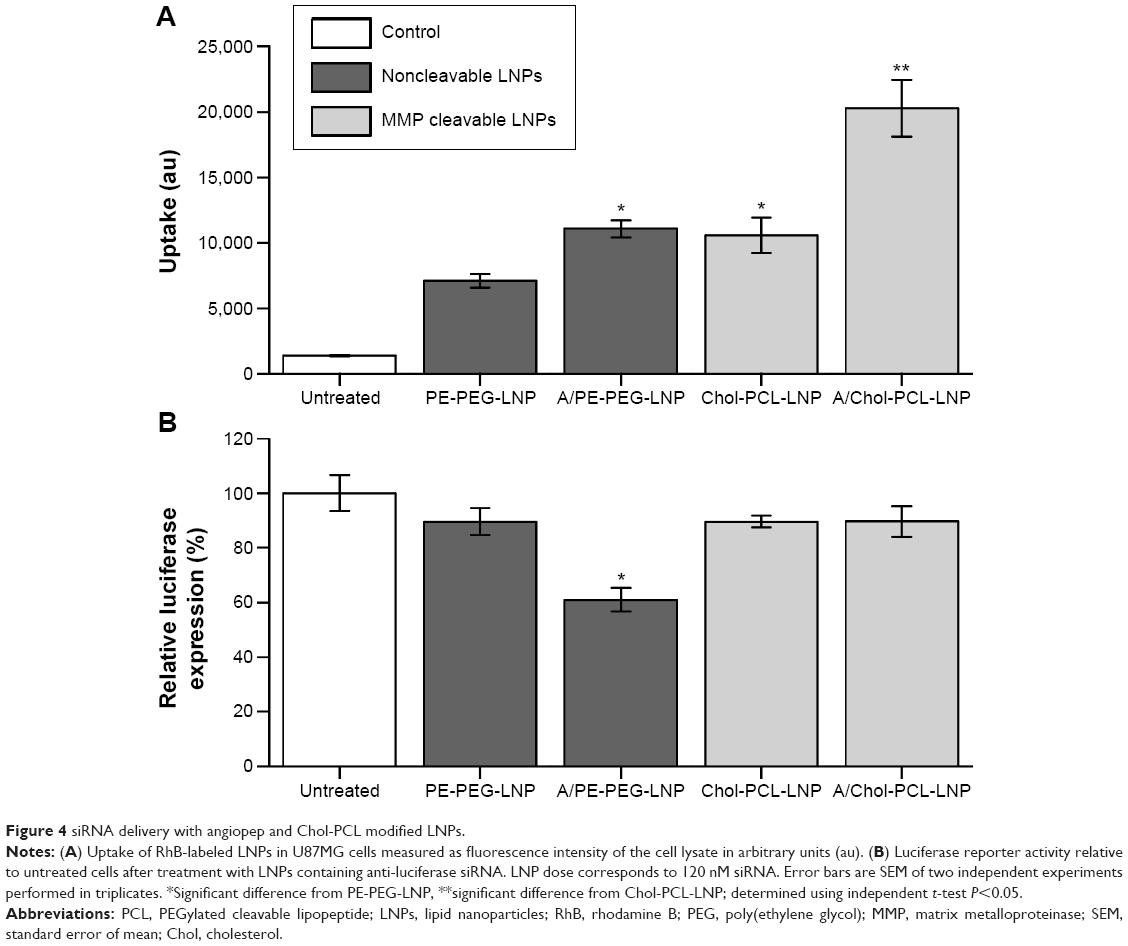 | Figure 4 siRNA delivery with angiopep and Chol-PCL modified LNPs. |
The modification with the MMP-sensitive Chol-PCL had a similar positive effect on the uptake in these MMP-expressing cells (Figure 4A), an effect that has previously been demonstrated to depend on the MMP expression level of the cells.29 When combined with angiopep for a dual-modification (A/Chol-PCL-LNP), the highest uptake was obtained with a fluorescence intensity in the cells that was threefold higher than for PE-PEG-LNP.
Despite both angiopep functionalization and incorporation of Chol-PCL resulted in increased uptake, only A/PE-PEG-LNP facilitated a significant higher luciferase knockdown than PE-PEG-LNP, while the two Chol-PCL containing LNPs resulted in knockdown at the same level as PE-PEG-LNP (Figure 4B). This indicated that even though both Chol-PCL-LNP and A/Chol-PCL-LNP entered the cells to a higher extent than PE-PEG-LNP, the siRNA was not sufficiently released into the cytosol from these LNPs and so they had consequently minimal effect on the luciferase expression. Endosomal escape is an essential factor in successful gene delivery and without this ability, the cholesterol-anchored PCL was categorized as unsuitable for siRNA delivery in the type of formulation used here.
Cellular uptake with DM-anchored PCL
As an alternative lipid anchor to cholesterol, the PCL variant containing two myristic acids (DM) attached to the 2,3-diamino-propionate containing tetradecapeptide was tested. The substitution of the lipid anchor caused a small increase in the size of LNPs (Table 2), but the ability to remove the PEG coat and reverse the surface charge was retained (Figure 2).
The uptake of DM-PCL containing LNPs (DM-PCL-LNP and A/DM-PCL-LNP) was tested in the two MMP-2/9 expressing brain cell lines bEnd.3 and U87MG.37,38 Both LNPs had a tenfold higher uptake than PE-PEG-LNP (Figure 5A), which was three times higher than observed for A/Chol-PCL-LNP (Figure 4A). While it was observed that the LNPs with cholesterol-anchored PCL had an additive effect of dual-modification with both angiopep and PCL, this was not the case for DM-PCL-LNPs. Instead the uptake improvement by the cleavable PEG coating was so much higher than the receptor-mediated uptake that it completely overshadowed the effect of angiopep functionalization. This was evident by an equal uptake of the dual functionalized A/DM-PCL-LNP and the nontargeted DM-PCL-LNP (Figure 5A). It was therefore hypothesized that the difference in uptake between the DM- and Chol-anchored PCLs is caused by a difference in orientation and positioning of the lipopeptides in the LNPs due to their individual size and lipophilicity. This difference in orientation presumably provides better accessibility for MMPs to the cleavage site of DM-PCL than Chol-PCL.
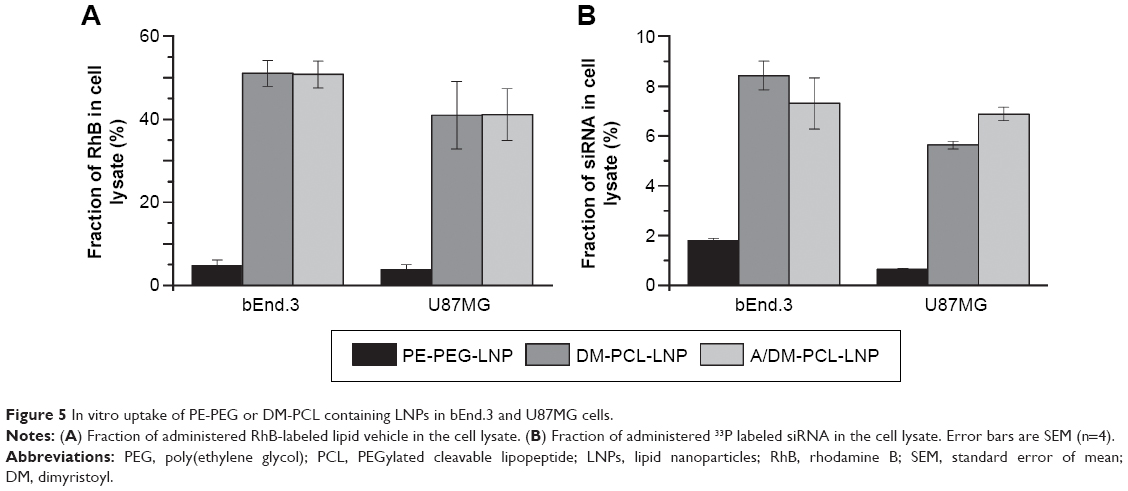 | Figure 5 In vitro uptake of PE-PEG or DM-PCL containing LNPs in bEnd.3 and U87MG cells. |
The fluorescence measurements providing information about the uptake of the lipid vehicle (Figure 5A) were supported by a scintillation measurement of a radioactive label on the siRNA cargo (Figure 5B). Thereby, it was possible to measure the concentration of administered siRNA in the cell lysate compared to that in the media. A notable difference in the fraction of cellular associated lipid vehicle compared to the siRNA cargo was observed (Figure 5). The cells internalized approximately half of the lipids from DM-PCL-LNP and A/DM-PCL-LNP after 48 hours, while more than 90% of the labeled siRNA was found in the cell media. This indicates that the siRNA either escapes the LNP after internalization and is secreted from the cell to a much higher extent than the lipids or is released outside the cells after which the empty LNP is internalized. In the case of an intracellular disassembly of the LNP with following biased secretion of siRNA and lipid vehicle, the difference might be associated with the ability of the lipids to fuse or integrate with the various membranes of the cell. In this case, the measured difference in the cellular content of vehicle and siRNA indicates that at least 85% of internalized siRNA were secreted from the cells. In general for all LNPs, the siRNA uptake resembled that of the lipid vehicle although the difference between PE-PEG-LNP and the two DM-PCL-LNPs was less pronounced.
Knockdown with DM-PCL modified LNPs
The ability to mediate knockdown was examined together with the uptake efficiency, and, as for the uptake, (Figure 5) there was greater knockdown for the DM-PCL containing LNPs in both bEnd.3 cells (Figure 6A) and U87MG cells (Figure 6B). At siRNA concentration of 120 nM, both DM-PCL-LNP and A/DM-PCL-LNP attained a reduction of the luciferase protein expression down to 30% of the expression level of untreated cells (Figure 6), which was a significant improvement from the 90% and 60% previously attained by A/Chol-PCL-LNP and A/PE-PEG-LNP, respectively (Figure 4B). When doubling the concentration (120 nM), it was possible to reach a knockdown of 15% in the bEnd.3 cells that was comparable to the commercial agent RNAiMAX. The increased amount of siRNA did not influence the knockdown in U87MG further as it seemed to have reached a maximum effect at 120 nM (Figure 6B).
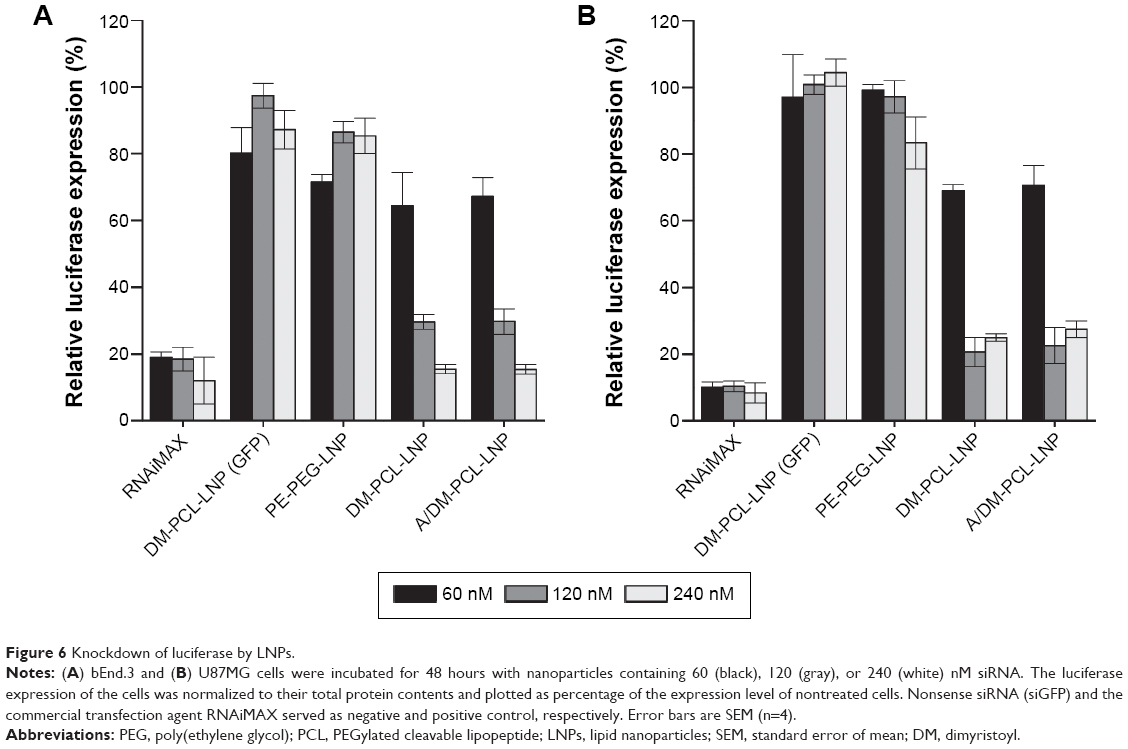 | Figure 6 Knockdown of luciferase by LNPs. |
In good correlation with the uptake profile, there was no additive effect of angiopep functionalization since no significant difference between the knockdown ability of DM-PCL-LNP and A/DM-PCL-LNP was observed. DM-PCL-LNP particles with nonsense siRNA (DM-PCL-LNP [GFP]) were used as a negative control to measure off-target effect of the knockdown. This LNP did not cause any significant changes in the luciferase expression underlined the specificity of the knockdown.
Cytotoxicity of the LNP
Cytotoxicity of the LNPs was determined using MTS assay for the most uptake- and knockdown-efficient DM-PCL-LNP as representative for the LNPs. Proliferation of siRNA-treated cells was normalized to untreated cells after 48 hours incubation with DM-PCL-LNP containing siRNA in concentrations ranging from 10 to 2,200 nM. As shown in Figure 7, proliferation of the LNP-treated cells was matching that of untreated cells up to siRNA concentrations of 2,200 nM where the proliferation was decreased to 88%. This upper concentration was 18 times higher than the working concentration used in the uptake and knockdown experiments. The low effect on proliferation at this concentration underlines the low toxicity of the LNPs. As a comparison, the cationic lipid-based RNAiMAX caused a decrease in proliferation at concentrations above 80 nM. This is also above the concentration recommended by the manufacturer, but underlines the potential of the anionic PCL-based delivery system for in vivo use even at high concentrations.
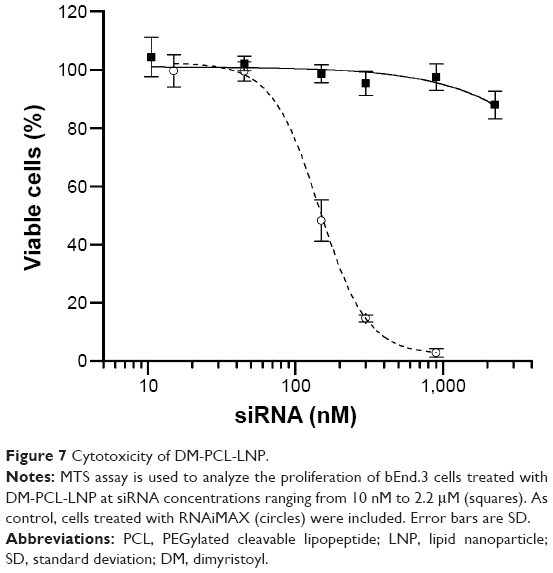 | Figure 7 Cytotoxicity of DM-PCL-LNP. |
Discussion
A number of studies have reported angiopep-mediated uptake of nanoparticles in brain endothelial cells and its ability to facilitate transport across the BBB.5,34,39 Our study supports the reported targeting capability of angiopep by showing a 2.4-fold increased uptake of angiopep-functionalized PEGylated LNPs and furthermore demonstrates that these targeted LNPs have increased ability to mediate knockdown in LRP-1 expressing cells compared to nontargeted LNPs.
Furthermore, the angiopep-mediated uptake combined with DM-PCL for site-specific triggering of dePEGylation and the charge switch proved to be a highly efficient system for in vitro delivery of siRNA. A recent study has shown that angiopep combined with a mechanism for MMP-triggered activation of a cell penetrating peptide results in a twofold increase in nanoparticle uptake.40 In our system, the activation by MMP induces removal of the protective PEG coating concurrently with a shift to a positive surface charge which altogether lead to a 10-fold higher uptake than nontargeted PEGylated LNPs. Furthermore, the protein knockdown of the MMP-sensitive LNPs is comparable to the commercial agent RNAiMAX, which in many cases sets the standard for an optimal in vitro siRNA delivery agent. Where RNAiMAX is only compatible with in vitro settings, the investigated new system has the potential of systemic circulation that allows for in vivo application and accumulation in diseased target tissue. The obtained protein knockdown to 30% was also comparable to other cutting edge studies of similar brain targeting siRNA delivery vehicles.41–43 However, while these systems have a positive charge, which may hamper their in vivo potential, the PCL of the LNP presented here ensures a net negative charge of the vehicle during systemic circulation.
The high uptake and knockdown were at equivalent levels for the nontargeted LNPs and the angiopep-modified DM-PCL-LNP, indicating a redundancy of the angiopep functionalization in this in vitro setting. Our initial studies with noncleavable LNPs and cholesterol-anchored PCL demonstrated nonetheless that angiopep has an impact on siRNA delivery with both a PEGylated and a cleavable LNP. Seen in this light, the lacking additive effect of DM-PCL and angiopep is possibly a result of the high efficiency of the dePEGylation and charge shift on the uptake and siRNA delivery, rather than a disability of the two modifications to work additively. The dePEGylation and positive charge simply have such a pronounced effect that it overshadows the influence of angiopep and diminish it to an undetectable level at the 48 hours cell incubation period studied in this work. In vivo, however, angiopep could possibly have an important effect since cleavage of PCL depends on accumulation of the LNP in areas with high MMP activity, which is not the case at the luminal site of the BBB during the early stage of brain tumor development. At this point, the LNP depends on an active transporter for passage across the BBB, and here, angiopep or other targeting ligands will be essential for the successful transfection of glioblastoma.
The large influence of DM-PCL was seen for both cell types even though bEnd.3 cells only have low extracellular MMP activity. This indicates that other factors like activity of other intra- or extracellular peptidases or protonation of the glutamic acid residues in PCL could influence the transfection in addition to the MMP activation. Increased positive charge due to protonation of the amino acids in the acidic tumor microenvironment increases cell interaction and thereby also the uptake,44,45 even with a noncleaved PCL. Once inside the endosome, the more acidic environment will lead to a more pronounced protonation of the amino acids working together with the titratable DODAP lipids to obtain a high positive charge. This will increase the interaction with the endosome membrane and promote endosomal escape and thereby increase the transfection potency. In the endosomal compartment, high proteinase activity will furthermore increase the probability of PCL cleavage resulting in high efficiency of gene silencing independent of extracellular MMP. Thus, the beneficial effect of PCL could be a combination of factors that act, once the LNP has entered the endosomes and lysosomes, to facilitate the release of siRNA into the cytosol. This hypothesis underlines the potential two-stage delivery system with angiopep for cellular uptake and PCL for endosomal release.
The work presented here focuses on delivery of siRNA across the BBB for treatment of glioblastoma, which therapeutically could be used for the delivery of anti-c-MET or anti-survivin siRNA that have been proved to cause in vitro and in vivo cytotoxicity in glioma cells.46,47 However, the application perspective includes several other brain disorders like ischemic stroke, or Alzheimer’s or Parkinson’s disease. These disorders are often associated with an adverse inflammatory response and impaired BBB, partly caused by a substantial upregulation of MMP expression.48–50 The high transfection ability of A/DM-PCL-LNP in bEnd.3 cells demonstrates that the investigated delivery system is well suited for such purposes and could be utilized for delivery of, eg, MMP-regulatory siRNA into brain endothelial cells.51,52 The combined effect of angiopep for targeting and MMP for activation could potentially direct the LNPs to areas with high MMP activity and function as a regulator for reducing the adverse side effects caused by MMP overexpression.
Conclusion
This study demonstrated that angiopep can facilitate uptake of LNPs in LRP-1 expressing cells, but in order to obtain effective gene delivery it can advantageously be combined with a mechanism for removing the protective PEG coating. This was achieved by incorporating the MMP-cleavable lipopeptide PCL in the LNP formulation, whereby we obtained an efficient siRNA delivery system with high uptake and gene knockdown. PCL was capable of masking the intrinsic positive charge of the LNP and ensuring a low cytotoxicity, while at the same time, it proved so effective for cell uptake and gene knockdown in vitro that it overshadowed the effect of targeting with angiopep. However, for in vivo studies angiopep functionalization could play a more important role as mediator of transport across the BBB and targeting to glioma.
Disclosure
The authors report no conflicts of interest in this work.
References
Abbott NJ, Patabendige AA, Dolman DE, Yusof SR, Begley DJ. Structure and function of the blood–brain barrier. Neurobiol Dis. 2010;37(1):13–25. | ||
Tobias A, Ahmed A, Moon KS, Lesniak MS. The art of gene therapy for glioma: a review of the challenging road to the bedside. J Neurol Neurosurg Psychiatry. 2013;84(2):213–222. | ||
Abbott NJ. Blood-brain barrier structure and function and the challenges for CNS drug delivery. J Inherit Metab Dis. 2013;36(3):437–449. | ||
Pérez-Martínez FC, Guerra J, Posadas I, Ceña V. Barriers to non-viral vector-mediated gene delivery in the nervous system. Pharm Res. 2011;28(8):1843–1858. | ||
Huang R, Ma H, Guo Y, et al. Angiopep-conjugated nanoparticles for targeted long-term gene therapy of Parkinson’s disease. Pharm Res. 2013;30(10):2549–2559. | ||
Demeule M, Currie JC, Bertrand Y, et al. Involvement of the low-density lipoprotein receptor-related protein in the transcytosis of the brain delivery vector angiopep-2. J Neurochem. 2008;106(4):1534–1544. | ||
Demeule M, Régina A, Ché C, et al. Identification and design of peptides as a new drug delivery system for the brain. J Pharmacol Exp Ther. 2008;324(3):1064–1072. | ||
Huang S, Li J, Han L, et al. Dual targeting effect of angiopep-2-modified, DNA-loaded nanoparticles for glioma. Biomaterials. 2011;32(28):6832–6838. | ||
Ren J, Shen S, Wang D, et al. The targeted delivery of anticancer drugs to brain glioma by PEGylated oxidized multi-walled carbon nanotubes modified with angiopep-2. Biomaterials. 2012;33(11):3324–3333. | ||
Xin H, Sha X, Jiang X, Zhang W, Chen L, Fang X. Anti-glioblastoma efficacy and safety of paclitaxel-loading angiopep-conjugated dual targeting PEG-PCL nanoparticles. Biomaterials. 2012;33(32):8167–8176. | ||
Wang J, Lu Z, Wientjes MG, Au JL. Delivery of siRNA therapeutics: barriers and carriers. AAPS J. 2010;12(4):492–503. | ||
Gomes-da-Silva LC, Fonseca NA, Moura V, Pedroso de Lima MC, Simões S, Moreira JN. Lipid-based nanoparticles for siRNA delivery in cancer therapy: paradigms and challenges. Acc Chem Res. 2012;45(7):1163–1171. | ||
Sørensen DR, Leirdal M, Sioud M. Gene silencing by systemic delivery of synthetic siRNAs in adult mice. J Mol Biol. 2003;327(4):761–766. | ||
Li W, Szoka FC. Lipid-based nanoparticles for nucleic acid delivery. Pharm Res. 2007;24(3):438–449. | ||
Senior J, Delgado C, Fisher D, Tilcock C, Gregoriadis G. Influence of surface hydrophilicity of liposomes on their interaction with plasma protein and clearance from the circulation: studies with poly(ethylene glycol)-coated vesicles. Biochim Biophys Acta. 1991;1062(1):77–82. | ||
Torchilin VP, Omelyanenko VG, Papisov MI, et al. Poly(ethylene glycol) on the liposome surface: on the mechanism of polymer-coated liposome longevity. Biochim Biophys Acta. 1994;1195:11–20. | ||
Hatakeyama H, Akita H, Harashima H. The polyethyleneglycol dilemma: advantage and disadvantage of PEGylation of liposomes for systemic genes and nucleic acids delivery to tumors. Biol Pharm Bull. 2013;36(6):892–899. | ||
Ishida T, Kirchmeier MJ, Moase EH, Zalipsky S, Allen TM. Targeted delivery and triggered release of liposomal doxorubicin enhances cytotoxicity against human B lymphoma cells. Biochim Biophys Acta. 2001;1515(2):144–158. | ||
Kale AA, Torchilin VP. Enhanced transfection of tumor cells in vivo using “Smart” pH-sensitive TAT-modified pegylated liposomes. J Drug Target. 2007;15(7–8):538–545. | ||
Xu H, Deng Y, Chen D, Hong W, Lu Y, Dong X. Esterase-catalyzed dePEGylation of pH-sensitive vesicles modified with cleavable PEG-lipid derivatives. J Control Release. 2008;130(3):238–245. | ||
Zhu L, Kate P, Torchilin VP. Matrix metalloprotease 2-responsive multifunctional liposomal nanocarrier for enhanced tumor targeting. ACS Nano. 2012;6(4):3491–3498. | ||
Andresen TL, Thompson DH, Kaasgaard T. Enzyme-triggered nanomedicine: drug release strategies in cancer therapy. Mol Membr Biol. 2010;27(7):353–363. | ||
Egeblad M, Werb Z. New functions for the matrix metalloproteinases in cancer progression. Nat Rev Cancer. 2002;2(3):161–174. | ||
Fujiwara S, Nakagawa K, Harada H, Nagato S, Iwata S, Ohnishi T. Silencing hypoxia-inducible factor-1a inhibits cell migration and invasion under hypoxic environment in malignant gliomas. Int J Oncol. 2007;30:793–802. | ||
Gu G, Xia H, Hu Q, et al. PEG-co-PCL nanoparticles modified with MMP-2/9 activatable low molecular weight protamine for enhanced targeted glioblastoma therapy. Biomaterials. 2013;34(1):196–208. | ||
Hatakeyama H, Akita H, Kogure K, et al. Development of a novel systemic gene delivery system for cancer therapy with a tumor-specific cleavable PEG-lipid. Gene Ther. 2007;14(1):68–77. | ||
Terada T, Iwai M, Kawakami S, Yamashita F, Hashida M. Novel PEG-matrix metalloproteinase-2 cleavable peptide-lipid containing galactosylated liposomes for hepatocellular carcinoma-selective targeting. J Control Release. 2006;111(3):333–342. | ||
Remaut K, Lucas B, Braeckmans K, Demeester J, De Smedt SC. Pegylation of liposomes favours the endosomal degradation of the delivered phosphodiester oligonucleotides. J Control Release. 2007;117(2):256–266. | ||
Gjetting T, Jølck R, Andresen T. Effective nanoparticle based gene delivery by a protease triggered charge switch. Adv Healthc Mater. 2014;3(7):1107–1118. | ||
Miller CR, Bondurant B, McLean SD, McGovern KA, O’Brien DF. Liposome-cell interactions in vitro: effect of liposome surface charge on the binding and endocytosis of conventional and sterically stabilized liposomes. Biochemistry. 1998;37(37):12875–12883. | ||
Jeffs LB, Palmer LR, Ambegia EG, Giesbrecht C, Ewanick S, MacLachlan I. A scalable, extrusion-free method for efficient liposomal encapsulation of plasmid DNA. Pharm Res. 2005;22(3):362–372. | ||
Gjetting T, Arildsen NS, Christensen CL, et al. In vitro and in vivo effects of polyethylene glycol (PEG)-modified lipid in DOTAP/cholesterol-mediated gene transfection. Int J Nanomedicine. 2010;5:371–383. | ||
Sambrook J, Russel DW. Molecular Cloning: A Laboratory Manual. New York, NY: Cold Spring Harbor Laboratory Press; 2001. | ||
Ke W, Shao K, Huang R, et al. Gene delivery targeted to the brain using an angiopep-conjugated polyethyleneglycol-modified polyamidoamine dendrimer. Biomaterials. 2009;30(36):6976–6985. | ||
Turk BE, Huang LL, Piro ET, Cantley LC. Determination of protease cleavage site motifs using mixture-based oriented peptide libraries. Nat Biotechnol. 2001;19(7):661–667. | ||
Okada Y, Morodomi T, Enghild JJ, et al. Matrix metalloproteinase 2 from human rheumatoid synovial fibroblasts. Purification and activation of the precursor and enzymic properties. Eur J Biochem. 1990;194(3):721–730. | ||
Liu J, Jin X, Liu KJ, Liu W. Matrix metalloproteinase-2-mediated occludin degradation and caveolin-1-mediated claudin-5 redistribution contribute to blood-brain barrier damage in early ischemic stroke stage. J Neurosci. 2012;32(9):3044–3057. | ||
Puli S, Lai JC, Bhushan A. Inhibition of matrix degrading enzymes and invasion in human glioblastoma (U87MG) cells by isoflavones. J Neurooncol. 2006;79(2):135–142. | ||
Shao K, Huang R, Li J, et al. Angiopep-2 modified PE-PEG based polymeric micelles for amphotericin B delivery targeted to the brain. J Control Release. 2010;147(1):118–126. | ||
Gao H, Zhang S, Cao S, Yang Z, Pang Z, Jiang X. Angiopep-2 and activatable cell-penetrating peptide dual-functionalized nanoparticles for systemic glioma-targeting delivery. Mol Pharm. 2014;11(8):2755–2763. | ||
Pulford B, Reim N, Bell A, et al. Liposome-siRNA-peptide complexes cross the blood-brain barrier and significantly decrease PrP on neuronal cells and PrP in infected cell cultures. PLoS One. 2010;5(6):e11085. | ||
Resnier P, David S, Lautram N, et al. EGFR siRNA lipid nanocapsules efficiently transfect glioma cells in vitro. Int J Pharm. 2013;454(2):748–755. | ||
Malmo J, Sandvig A, Vårum KM, Strand SP. Nanoparticle mediated P-glycoprotein silencing for improved drug delivery across the blood-brain barrier: a siRNA-chitosan approach. PLoS One. 2013;8(1):e54182. | ||
Engin K, Leeper DB, Cater JR, Thistlethwaite AJ, Tupchong L, McFarlane JD. Extracellular pH distribution in human tumours. Int J Hyperthermia. 1995;11(2):211–216. | ||
Lee ES, Na K, Bae YH. Polymeric micelle for tumor pH and folate-mediated targeting. J Control Release. 2003;91(1–2):103–113. | ||
Jin J, Bae KH, Yang H, et al. In vivo specific delivery of c-Met siRNA to glioblastoma using cationic solid lipid nanoparticles. Bioconjug Chem. 2011;22(12):2568–2572. | ||
Cartellieri M, Hendruschk S, Wiedemuth R, et al. RNA interference targeting survivin exerts antitumoral effects in vitro and in established glioma xenografts in vivo. Neuro Oncol. 2011;13(10):1074–1089. | ||
Rosenberg GA. Matrix metalloproteinases and their multiple roles in neurodegenerative diseases. Lancet Neurol. 2009;8(2):205–216. | ||
Jin R, Yang G, Li G. Molecular insights and therapeutic targets for blood-brain barrier disruption in ischemic stroke: critical role of matrix metalloproteinases and tissue-type plasminogen activator. Neurobiol Dis. 2010;38(3):376–385. | ||
Mahajan SD, Aalinkeel R, Reynolds JL, et al. Suppression of MMP-9 expression in brain microvascular endothelial cells (BMVEC) using a gold nanorod (GNR)-siRNA nanoplex. Immunol Invest. 2012;41(4):337–355. | ||
Zhang G, Fahmy RG, DiGirolamo N, Khachigian LM. JUN siRNA regulates matrix metalloproteinase-2 expression, microvascular endothelial growth and retinal neovascularisation. J Cell Sci. 2006;119(15):3219–3226. | ||
Bonoiu A, Mahajan SD, Ye L, et al. MMP-9 gene silencing by a quantum dot-siRNA nanoplex delivery to maintain the integrity of the blood brain barrier. Brain Res. 2009;1282:142–155. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.