Back to Journals » International Journal of Nanomedicine » Volume 12
Investigating the use of curcumin-loaded electrospun filaments for soft tissue repair applications
Authors Mouthuy PA ![]() , Somogyi Škoc M
, Somogyi Škoc M ![]() , Čipak Gašparović A, Milković L, Carr AJ, Žarković N
, Čipak Gašparović A, Milković L, Carr AJ, Žarković N ![]()
Received 26 January 2017
Accepted for publication 15 March 2017
Published 25 May 2017 Volume 2017:12 Pages 3977—3991
DOI https://doi.org/10.2147/IJN.S133326
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Thomas Webster
Pierre-Alexis Mouthuy,1,2 Maja Somogyi Škoc,3 Ana Čipak Gašparović,1 Lidija Milković,1 Andrew J Carr,2 Neven Žarković1
1Laboratory for Oxidative Stress, Rudjer Boskovic Institute, Zagreb, Croatia; 2Botnar Research Centre, Nuffield Department of Orthopaedics, Rheumatology and Musculoskeletal Sciences, Medical Science Division, University of Oxford, Oxford, UK; 3Department of Materials, Fibres and Textile Testing, University of Zagreb, Zagreb, Croatia
Abstract: Electrospun filaments represent a new generation of medical textiles with promising applications in soft tissue repair. A potential strategy to improve their design is to combine them with bioactive molecules. Curcumin, a natural compound found in turmeric, is particularly attractive for its antioxidant, anti-inflammatory, and antimicrobial properties. However, investigating the range of relevant doses of curcumin in materials designed for tissue regeneration has remained limited. In this paper, a wide range of curcumin concentrations was explored and the potential of the resulting materials for soft tissue repair applications was assessed. Polydioxanone (PDO) filaments were prepared with various amounts of curcumin: 0%, 0.001%, 0.01%, 0.1%, 1%, and 10% (weight to weight ratio). The results from the present study showed that, at low doses (≤0.1%), the addition of curcumin has no influence on the spinning process or on the physicochemical properties of the filaments, whereas higher doses lead to smaller fiber diameters and improved mechanical properties. Moreover, filaments with 0.001% and 0.01% curcumin stimulate the metabolic activity and proliferation of normal human dermal fibroblasts (NHDFs) compared with the no-filament control. However, this stimulation is not significant when compared to the control filaments (0%). Highly dosed filaments induce either the inhibition of proliferation (with 1%) or cell apoptosis (with 10%) as a result of the concentrations of curcumin found in the medium (9 and 32 µM, respectively), which are near or above the known toxicity threshold of curcumin (~10 µM). Moreover, filaments with 10% curcumin increase the catalase activity and glutathione content in NHDFs, indicating an increased production of reactive oxygen species resulting from the large concentration of curcumin. Overall, this study suggested that PDO electrospun filaments loaded with low amounts of curcumin are more promising compared with higher concentrations for stimulating tissue repair. This study also highlighted the need to explore lower concentrations when using polymers as PDO, such as those with polycaprolactone and other degradable polyesters.
Keywords: electrospinning, electrospun filaments, polydioxanone, PDO, curcumin, human fibroblasts
Introduction
Over the last few decades, the use of textile materials for medical purposes has rapidly increased, with applications ranging from simple gauze for wound dressing to sutures and implantable patches for soft tissue repair. The global medical textile market was valued at US $14 billion in 2014 and is expected to reach $20 billion by 2022,1 indicating an increasing growth.
Among the latest fiber technologies used for the manufacture of medical textiles, electrospinning has attracted much interest. Electrospinning is the process by which nanoscale and microscale fibers are drawn from a polymer solution using high-voltage electrical charges. With their high surface-to-volume ratio and porosity and their ability to mimic the extracellular environment, electrospun materials have shown great promises for soft tissue repair and tissue engineering applications. Through their physical cues, they have the ability to influence cell adhesion, spreading, proliferation, and differentiation.2,3
Electrospun materials are mostly produced as nonwoven meshes. However, methods to manufacture electrospun filaments have also been developed, which can be assembled into twisted, braided, or woven structures.4–6 In a previous in vitro work,4 it was shown that electrospun yarns lead to an improved cell response compared with existing monofilaments made by plastic extrusion with the same material. In vivo, they have shown a mild, but quantifiable foreign body reaction.4 Although the overall results were promising, further improvements in the material are required, such as minimizing the foreign body reaction and accelerating the repair response. In particular, incorporating antioxidant molecules might help to decrease oxidative stress, known to be a normal response to implanted materials and linked to both the foreign body reaction and the repair process.7 The appropriate management of oxidative stress during the whole life of the implant through the use of antioxidant molecules might help to minimize the foreign body reaction and maximize the healing of the surrounding tissues.
Among the potential candidates, curcumin (1,7-bis(4-hydroxy-3-methoxyphenyl)-1,6-heptadiene-3,5-dione) has been one of the most investigated antioxidant molecules. Curcumin can be found in turmeric, a plant widely used in food preparation. It exhibits strong antioxidant, anti-inflammatory, and antimicrobial properties that make it a great candidate for wound healing applications.8–11 Besides these properties, it is also attractive for its availability, stability, low cost, and safety, which was demonstrated in numerous clinical trials. The effect of curcumin depends mainly on the dose to which cells are exposed. At concentrations >10 μM, curcumin induces apoptosis in a wide variety of cell lines in in vitro wound models, as it becomes oxidizing and produces reactive oxygen species (ROS).12–14 However, at concentrations between 1 μM and 0.01 μM, curcumin can stimulate the proliferation of various cell types and decrease the markers of inflammation and oxidative stress levels.15–18
Numerous studies have proposed electrospun meshes loaded with curcumin, given the potential for health care applications. A wide range of polymers has been used, including polycaprolactone (PCL),19–21 polylactic acid (PLA),22 polylactic-co-glycolic acid,23 polyvinylpyrrolidone (PVP),24 silk,25 and cellulose acetate.26 Because of the hydrophobic nature of curcumin, the use of hydrophobic polymers such as degradable polyesters is particularly attractive to achieve a slow and prolonged release. Among the polymer carriers that have remained unexplored, polydioxanone (PDO) is promising; it is a poly(ester–ether) that degrades within 6 months and commonly used in medical practice as a suture material. Its use in tissue engineering and drug delivery applications has been of increasing interest in the recent years.27
Despite the number of existing studies investigating the use of curcumin in biomaterials, the range of concentrations in the materials that are relevant for wound healing applications has been poorly explored. Relatively high amounts of curcumin have been incorporated in electrospun materials, as most of the studies carried out propose proportions of curcumin >1% of the polymer weight.19–22,25,28 These materials often lead to concentrations near or above the apoptosis threshold of 10 μM. Although several authors claim benefits from such materials to stimulate repair, it is not clear why these amounts have been selected while data from studies carried out with free curcumin suggest that lower concentrations might be more adequate for wound healing applications.15–17
The aims of this paper were therefore to explore a wide range of curcumin doses incorporated in electrospun fibers and to assess the potential of the resulting materials for wound repair applications. In order to achieve these, electrospun filaments loaded with various amounts of curcumin were created. Then, the main physicochemical properties of the filaments were determined, and the response of human fibroblasts in vitro after exposure to the different filaments was assessed.
Materials and methods
Preparation of the electrospinning solutions
Curcumin (Sigma-Aldrich Co., St Louis, MO, USA; ≥94% curcuminoid content, ≥80% curcumin) was added to 1,1,1,3,3,3-hexafluoroisopropanol (HFIP; Apollo Scientific Ltd, Stockport, UK) at different concentrations by successive dilution (by 10×): 24.4, 2.44, 0.244, 0.0244, and 0.00244 mM. PDO (Sigma-Aldrich Co.) was then added to each solution at a concentration of 9% (weight to volume ratio of HFIP). The concentrations of curcumin were selected to correspond to 0%, 0.001%, 0.01%, 0.1%, 1%, and 10% weight to weight (w/w) ratio of PDO. The final solutions were agitated at room temperature on a roller for at least 24 hours to allow for complete dissolution of the polymer.
Fabrication of continuous electrospun filaments
Electrospinning of continuous filaments was carried out using a thin wire as a collector according to a method described previously.4 Briefly, a single-nozzle electrospinning setup with a high-voltage power supply system (30 kV, SL30P30/230; Spellman, West Sussex, UK) and a syringe pump (World Precision Instruments Limited, Goodfellow, Huntingdon, UK) was used for electrospinning. The wire (100 μm in diameter; Goodfellow, Llangollen, UK) was moved underneath the nozzle at a speed of ~0.6 mm/s. The distance was set at 20 cm, and the average voltage applied was 6 kV. The filaments were collected on a spool on the collection unit.4 Samples were named according to the amount (in %) of curcumin (C) added to the solution: C0, C0.001, C0.01, C0.1, C1, and C10. An overnight vacuum treatment was applied to the samples before use to ensure the removal of solvent traces. Then, the filaments were stretched manually to prevent deformation during further uses, annealed at 65°C for 3 hours, and stored in a freezer at −20°C.
Scanning electron microscopy (SEM)
Samples were mounted on aluminum stubs using carbon adhesive tape and were gold/palladium coated using a sputter coater system (SC7620 Sputter Coater Emitech, Quorum Technologies Ltd, East Sussex, UK). High-resolution images were taken using an environmental scanning electron microscope (Tescan, Brno, Czech Republic). The diameters of fibers and filaments were measured by using ImageJ (W.S. Rasband; US National Institutes of Health, Bethesda, MD, USA).
Tensile properties
Specimens were tested for failure by using tensile machine (Strength tester Tensolab 3000; Mesdan, Puegnago del Garda, Italy) at a rate of 50 mm/min. The distance between grips was set at 5 cm. Dried samples were tested minimum 12 hours after taking the samples out of the freezer. For testing in a wet condition, the samples were soaked in phosphate-buffered saline solution for 2 hours prior to the experiments. The measurements of the force at break (N) and breaking strain (%) were recorded, and 10 replicates were tested per sample type for each experimental repeat (n=2).
Differential scanning calorimetry (DSC)
DSC experiments were carried out by using a PerkinElmer Inc., DSC 8000 instrument (Waltham, MA, US). A flow of nitrogen gas was maintained over the samples to create a reproducible and dry atmosphere and to eliminate air oxidation of the samples at higher temperatures; ~5 mg of material was placed in covered aluminum pans (the pans were not sealed). A covered empty pan was used as the reference. DSC scans were performed from 25°C to 140°C with a heating rate of 10°C/min. The sample was held for 2 minutes at 140°C and then cooled from 140°C to 25°C at a rate of 10°C/min. A reheating scan was then performed with the same settings. The crystallinity was calculated as follows: X(%) = Hf/Hf100%*100, where Hf is the measured heat of fusion (J/g), Hf100% is the theoretical heat of fusion of a 100% crystalline material, which was calculated as being 141.2 J/g for PDO.29
Fourier transform infrared (FTIR) spectroscopy
The surface chemistry of the filaments was analyzed by using an FTIR spectrometer (Spectrum 100 FT-IR; Perkin Elmer) equipped with an attenuated total reflection. Each spectrum was acquired with a resolution of 4 cm−1 by an accumulation of 128 scans, and the signal was measured in a range between 400 and 4,000 cm−1.
Curcumin release in vitro
In order to compare the amount of curcumin released after 24 hours from the different filaments, 1 cm of each sample (12 replicates) was incubated in 100 μL of culture medium (fibroblast growth medium [FGM]; Lonza, Basel, Switzerland) at 37°C. After 24 hours, the medium was removed and analyzed directly by using liquid chromatography–mass spectrometry (LC-MS). A standard curve was established by successive dilution of a curcumin–dimethyl sulfoxide (DMSO) stock solution in medium.
LC-MS analysis for identification and quantification of curcumin
The LC-MS protocol used in this study was modified from an existing protocol.30 Chromatographic separation of curcuminoids and metabolites was performed with an LC 1200 series system (Agilent Technologies, Santa Clara, CA, USA), which consisted of a Binary Pump SL and Hi-P-Autosampler (with injection volume of 2 μL). Elution was achieved with an Agilent Poroshell 120 EC-C8 column (2.1×50 mm, with a particle size of 2.7 μm). The mobile phases consisted of 10 mM ammonium acetate with 0.1% (volume to volume ratio) formic acid in water (A) and of pure acetonitrile (B). A linear gradient was applied as follows with a flow rate of 0.25 mL min−1: 0–3 minutes, 20% B; 3–10 minutes, 85% B; 11.5–15 minutes, 20% B, with a total run time of 15 minutes. The concentrations of curcumin released in the medium (see “Curcumin release in vitro” section) were quantified by using a calibration curve established from known concentrations of curcumin in DMSO. The confirmation of the identity of curcumin was obtained by using a 6,460 triple-quadrupole mass spectrometer (Agilent Technologies) equipped with an Agilent Jet Stream electrospray ionization ion source operating in negative mode. A product ion scan was employed for the confirmation of the compound, which was identified at m/z 149. The acquisition parameters were as follows: collision energy (CE), 12 V; fragmentor voltage, 110 V; MS scan range from 50 m/z (minimum) to 500 m/z (maximum); drying N2 temperature, 300°C; gas flow, 7 L min−1; nebulizer pressure, 15 psi; sheath gas temperature, 350°C; sheath gas flow, 10 L min−1; capillary voltage, 2,000 V; nozzle voltage, 1,000 V.
Cell culture
Normal human dermal fibroblasts (NHDFs) isolated from two donors were purchased from Lonza. The cells were thawed and cultured according to the manufacturer’s instructions. Briefly, the cells were seeded in tissue culture flasks (treated polystyrene; Corning, New York, NY, USA) with FGM (Lonza), which contains 10% fetal bovine serum (Lonza), 0.5% human fibroblast growth factors (Lonza), 0.5% insulin (Lonza), and 0.5% GA-1000 (Gentamicin sulfate and amphotericin-B; Lonza). The cells were cultured at 37°C in a humidifled 5% CO2 incubator. At 80% confluence, they were trypsinized and seeded in well plates for experiments.
Cell experiments
For proliferation and metabolic activity assays, NHDFs were seeded at 1×104 per well in 96-well plates and were allowed to attach for 24 hours prior to the experiments. Filaments, cut in pieces of 1 cm (equivalent to ~0.13 mg in average), were sterilized for 20 minutes under ultraviolet lights before being added to the plates. In order to simulate oxidative stress conditions, the cells were cultured in the presence of 50 μM H2O2 (hydrogen peroxide; Kemika, Zagreb, Croatia) to provide oxidative stress conditions. Preliminary work showed that this concentration was less than the IC50 but significantly reduced the metabolic activity of NHDFs.
Cell proliferation
The proliferation of NHDFs in the presence of the filaments was determined with the 5-bromo-2′-deoxyuridine (BrdU) colorimetric assay, which was performed according to the manufacturer’s instructions (Sigma-Aldrich). Briefly, the BrdU was added to each well for 4 hours prior to the end time point to allow DNA incorporation. Next, the supernatant with the filaments was removed, and the cells were washed twice with Hanks solution. Then, the cells were fixed for 30 minutes with FixDenat solution. Subsequently, the anti-BrdU-peroxidase (1:100) was added to each well and incubated at room temperature for 90 minutes. After removing the unbound antibody conjugate, 100 μL of the substrate solution was added and allowed to stand for 5 minutes. The reaction was quenched by adding 25 μL of 1 M H2SO4 solution. The absorbance was directly measured at 450 nm with a reference wavelength of 690 nm by using an ELISA plate reader (Multiskan EX; Thermo Fisher Scientific, Waltham, MA, USA).
Cell metabolic activity
Cell metabolic activity was measured by using the MTT assay (EZ4U; Biomedica, Vienna, Austria) according to the manufacturer’s instructions.31 Briefly, the medium in each well was replaced by 100 μL of Hanks solution (pH =7.4). Dye solution in volume of 20 μL was added to each well and incubated for 2 hours at 37°C. The absorbance was measured directly at 450 nm with a reference wavelength of 620 nm using an ELISA plate reader (Multiskan EX; Thermo Fisher Scientific).
In vitro wound healing assay (scratch test)
The cells were seeded in 12-well plates at a density of 1×105 cells per well and were cultured for 24 hours to allow for adhesion. A scratch was gently introduced in the center of the cell monolayers using a sterile 1,000-μL pipette tip. The medium was replaced to remove the cell debris, and a filament (10 cm) placed in a cell crown insert was fitted into the scratch area. The cells were incubated in the presence of 50 μM H2O2. Cell spreading in the gap area was analyzed by using ImageJ (W.S. Rasband; US National Institutes of Health) using photographs taken over a period of 3 days.
Catalase activity analysis
The cells were seeded in 12-well plates at a density of 1×105 cells per well and were cultured for 24 hours to allow for adhesion. A length of 10 cm of each filament was then placed in each well (triplicates), and the cells were incubated for another 24 hours in the presence of 50 μM H2O2. The catalase activity was then measured by the modified method of Goth.32 For catalase activity assay, 40 μL of cell lysate was mixed with 65 mM H2O2 for the start of the reaction. Different dilutions of H2O2 (0–75 mM) were used for standards. The reaction was stopped after 5 minutes by the addition of 100 μL of 200 mM ammonium molybdate, and color development was measured in a plate reader at 450 nm. One unit of catalase activity is defined as the amount of enzyme needed for the degradation of 1 μmol of H2O2/min at 25°C. Catalase activity is expressed in graphs as values relative to control (CTRL) (no-filament control).
Determination of glutathione (GSH) levels
GSH levels were measured on the same samples as those used for the catalase test. The intracellular GSH content was measured by the modified protocol described by Tietze.33 Briefly, the samples were diluted to 0.03 mg/mL of protein, and 150 μL of each sample was used for the assay. Reduced GSH in serial dilutions (0–20 mg/mL) was used as a standard. Reaction was started by the addition of freshly prepared reaction mix: 1.8 mM 5,5-dithio-bis-2-nitrobenzoic acid, 0.4 U GSH reductase, and 0.6 mM nicotinamide adenine dinucleotide phosphate in phosphate buffer (100 mM NaH2PO4 and 5 mM EDTA, pH 7.4). The formation of 2-nitro-5-thiobenzoic acid was monitored in a plate reader at 450 nm. GSH concentration in cell lysates is expressed in graphs as values relative to CTRL.
ROS assay
The ROS measurement is based on the intracellular oxidation of 2′,7′-dichlorodihydrofluorescein diacetate (DCFH-DA) to 2′,7′-dichlorofluorescein.34 For ROS measurements, the cells were seeded in white 96-well plates at a density of 1×104 cells per well and were left for 24 hours to allow for adhesion. The medium was then replaced by fresh medium with 50 μM H2O2, and a length of 1 cm of each filament was then placed in each well (triplicates). After incubation for 24 hours, the medium was removed, and the cells were rinsed with Hanks solution. Following this step, the cells were preincubated with 200 μL of Hanks solution containing 10 μM DCFH-DA at 37°C for 60 minutes. Then, the cells were treated with 50 μM H2O2. Fluorescence was measured before treatment and 50 minutes after treatment by using a Cary Eclipse Fluorescence Spectrophotometer (Varian Medical Systems of Palo Alto, CA, USA) with excitation at 500 nm and emission detection at 530 nm.
Statistical analysis
Statistical analysis was performed by using GraphPad Prism software (Version 7; GraphPad Software, Inc., La Jolla, CA, USA). Data were expressed as means with standard deviations. The results were analyzed using one-way analysis of variance with significance set at P<0.05.
Results and discussion
Filament preparation
PDO filaments loaded with various amounts of curcumin were successfully prepared by electrospinning, as shown in Figure 1. Curcumin is a hydrophobic molecule, which consists of two phenol groups connected by two α,β-unsaturated carbonyl groups as shown in Figure 1A. The powder, which has a yellow-orange color (Figure 1B), was easily dissolved in HFIP. Its addition to the PDO solution did not influence visibly the spinning process (sketched in Figure 1C) and did not affect the average voltage applied, which remained at ~6 kV for all experiments. The resulting filaments display similar dimensions (width: 0.3 mm; thickness: 0.1 mm) and show a gradual change in color as the concentration of curcumin increases (Figure 1D).
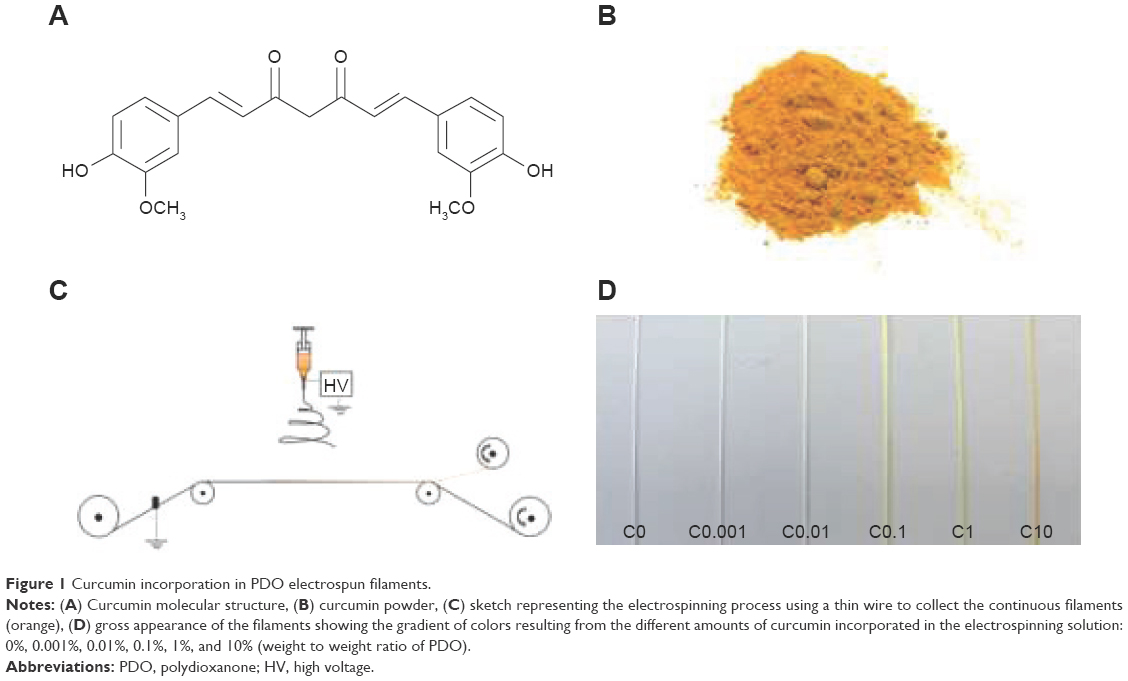 | Figure 1 Curcumin incorporation in PDO electrospun filaments. |
Although the possibility of electrospinning flat meshes loaded with curcumin was demonstrated with various polymers (see the “Introduction” section), this is the first time that pure PDO has been used as a carrier and that curcumin-enriched materials have been produced in the form of long continuous filaments. It is also worth noticing that the detachment of the material from the collecting surface (wire) is performed without any solvents. This is possible because the surface of contact between the material and the wire is small and because the mesh produced on the wire is made of a strong, interconnected fibrous network.4 It can therefore be assumed that no leaching of curcumin occurs during the detachment step.
Morphology of the filaments
Figure 2 shows the microscopic images of the filaments loaded with different amounts of curcumin. The fibrous morphology was maintained for all samples, but more bridges and merged fibers were observed at the concentrations of curcumin of 1% and 10%. As shown in Table 1, the average diameter of the electrospun fibers remains ~2 μm for concentrations <0.1% but significantly decreases at higher concentrations, reaching values of 1.6±0.6 μm (significantly different to 0.1%, P<0.001) and 1.1±0.6 μm (significantly different when compared to all the groups, P<0.001) at 1% and 10% of curcumin, respectively.
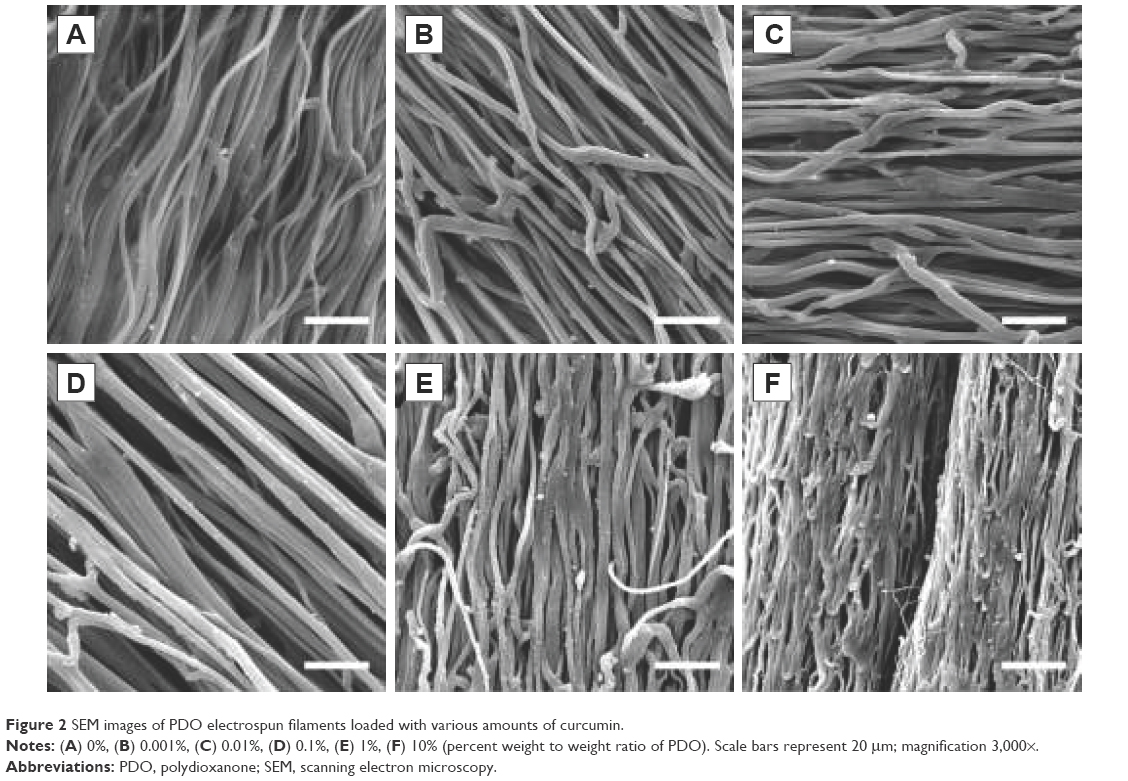 | Figure 2 SEM images of PDO electrospun filaments loaded with various amounts of curcumin. |
 | Table 1 Average diameters of electrospun fibers observed in the different filaments |
Similar observations were found in the literature. For instance, the average diameter of PVP fibers was decreased following the incorporation of curcumin.24 The same was observed for PLA22 and for poly(ε-caprolactone)-poly(ethylene glycol)-poly(ε-caprolactone) fibers.35 It was shown that the incorporation of curcumin actually increases the conductivity of electrospinning solution, thereby enhancing the thinning of the spinning jet and decreasing the fiber diameter.22
Mechanical, chemical, and thermal properties of filaments
Mechanical properties
Figure 3A and B shows the mechanical properties of filaments. Both the maximal force (force at break) and the maximum elongation (elongation at break) increase significantly at the concentrations of curcumin >1%. The strength increases by a third while the elongation is doubled. No significant difference was observed between the dry and wet tests. This increase in both strength and elongation might be explained by the reduction in fiber diameter observed for 1% and 10% and the more interconnected network of fibers (due to a higher number of bridges and crossings).
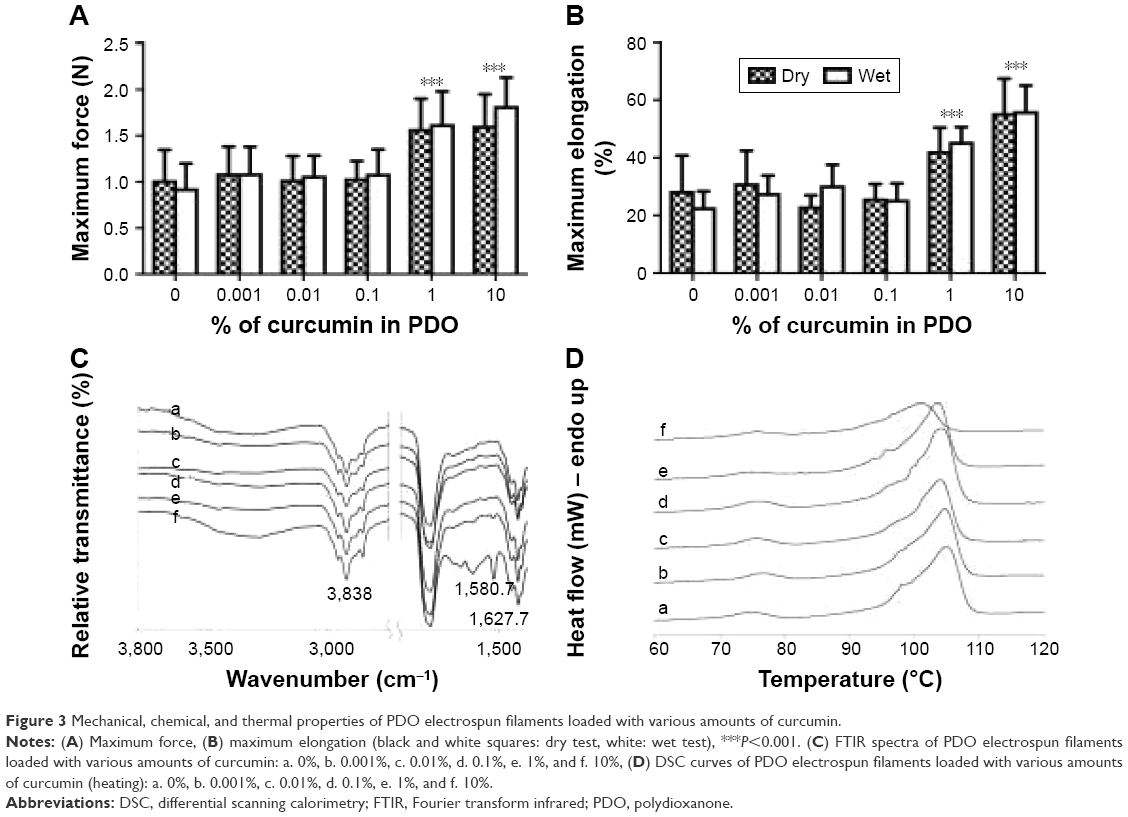 | Figure 3 Mechanical, chemical, and thermal properties of PDO electrospun filaments loaded with various amounts of curcumin. |
Surface chemistry
Figure 3C shows the spectra of the different filaments. Characteristic bands of pure curcumin (see Figure S1) are observed at 3,510 cm−1 (O–H stretching of the phenol group), 1,599 cm−1 (aromatic C=C stretching), and 1,501 cm−1 (C=O stretching, C–C=O bending).36 The main characteristic bands of pure PDO (Figure 3C, Curve a) are observed at 2,927 cm−1 (C–H stretching), 1,734 cm−1 (C=O stretching of ester groups), and 1,429 cm−1 (C–H bending).37
After incorporation of curcumin in PDO, the typical O–H absorption bands of curcumin were not observed in the filament spectra spectrum. This could be attributed to the formation of intermolecular hydrogen bonding between O–H of curcumin and C=O of PDO. The effect of adding curcumin in the PDO matrix is best seen in the region 1,500–1,600 cm−1 although it is difficult to detect <10%. In C10, the bands at 1,588 cm−1 and 1,518 cm−1 found in the FTIR spectra correspond to the bands observed in pure curcumin at 1,599 cm−1 and 1,501 cm−1, respectively. This shift may reveal hydrophobic interactions between curcumin and PDO, such as C=C interactions. These hydrophobic interactions may have contributed to improving the strength of the materials at concentration >1%.
Thermal properties
Figure 3D and Table 2 show the thermal properties of electrospun filaments obtained from the DSC curves. The main differences between samples are observed for C10, as the onset of the melting peak appears earlier and the melt requires less energy than other filaments. This means that the polymer has become more amorphous, as shown by the lower crystallinity value (~26%, whereas all other samples have ~40%). This shows that the presence of curcumin at large concentrations diminishes the ability of the polymer to form crystalline structures, probably due to its ability to bind to PDO chains, as indicated by the FTIR data. It also suggests that curcumin probably interacts mainly with polymer chains in the amorphous phase, as reported previously.38 This change in crystallinity might have further implications such as affecting the degradation properties of the material.39
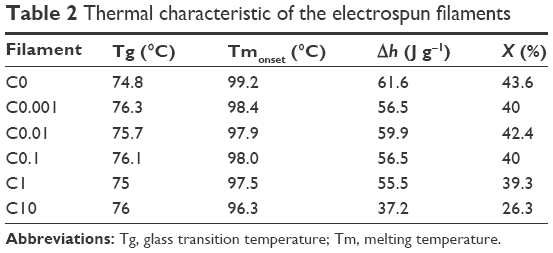 | Table 2 Thermal characteristic of the electrospun filaments |
Release of curcumin in culture medium
Figure 4 shows the results related to the release of curcumin in culture medium. The standard curve, shown in Figure 4A, was validated over a concentration range of 0.005–10 μM. Figure 4B shows the calculated concentrations of curcumin released from the filaments in the medium after 24 hours. While C0.001 led to concentrations below the limit of detection, C0.01 and C0.1 released curcumin at concentrations ~0.01 and 0.1 μM, respectively. Interestingly, C1 releases curcumin at ~9 μM, and C10 leads to concentrations ~32 μM (the sample was diluted 10× for measurements).
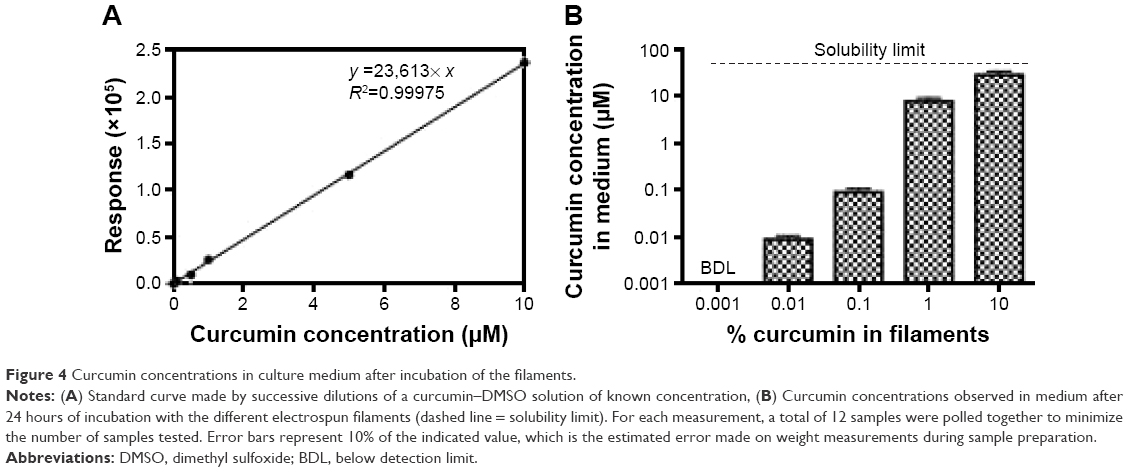 | Figure 4 Curcumin concentrations in culture medium after incubation of the filaments. |
Although C0.001 is below the limit of detection (0.005 μM), it is expected that the filament releases curcumin at ~0.001 μM as it contains 10× less than that of C0.01. Future work with larger amount of material should help to determine this fact. The increase in concentration between C0.1 and C1 is surprising (1 μM was expected, given the value of C0.01 and C0.1), but it might be due to the significantly lower diameter of C1 compared with C0.1, which results in larger surface area and therefore faster release. The fact that C10 is not released 10× the amount of C1 may be because the concentration of curcumin is near the limit of solubility, known to be at ~50 μM in culture medium.12 This may slow down the diffusion of curcumin out of the material.
It is worth noticing that the concentrations provided here do not take into account the degradation of curcumin that has occurred during the incubation period, which is a limitation. Curcumin is known to degrade spontaneously by autoxidation in culture medium.40 Although the 24 hours of incubation was performed in the dark, the degradation of curcumin is still expected.41 This makes it difficult to calculate the percentage of curcumin released from the fibers based on the concentrations calculated. With the values available, the concentrations measured represent ~3% of the total amount incorporated for C0.01 and C0.1, whereas they represent 28% for C1. Although these relatively low percentages (compared with those in the literature) may indicate the potential of PDO as a carrier for the slow delivery of curcumin, future work should investigate how much has actually been degraded. Future work will also examine different time points to look at the release profiles over an extended period of time. Figure S2 presents the preliminary data showing the release profiles for C0.1 and C1.
Cell metabolic activity and proliferation of NHDFs
NHDFs were incubated with the filaments, and Figure 5 shows the effect of released curcumin on their metabolic activity (MTT) and proliferation (BrdU). The MTT results indicate that C0.001 and C0.01 lead to significantly higher values (P≤0.05 and P≤0.01, respectively) compared with the CTRL at Day 1. However, the values were not significant when compared with the filament control (C0, no curcumin). Only C10 was significantly different from all other samples, as it showed very little metabolic activity. At Day 7, the time by which cell populations have normally reached confluence, no difference was observed between samples except for C10, which shows no metabolic activity. Results from the BrdU assay showed that C0.001 leads to significantly higher values (P≤0.05) compared with CTRL at Day 1, but the increase was not significant when compared to C0. BrdU results also indicated that both C1 and C10 were significantly reducing the proliferation of NHDFs at both Day 1 (P≤0.01 and P≤0.001, respectively) and Day 7 (P≤0.001 for both). As shown in Figure 5C and D, the scratch test showed that the cells fill the wound area by Day 3 with filaments C0 to C0.1. With C1, however, despite the fact that BrdU showed inhibition of proliferation, the scratch test results indicated that the average gap area of ~50% observed at Day 3 was not significantly different from lower concentrations (as reflected by the large standard deviation). However, C10 clearly prevented cell proliferation and migration in the vicinity of the filament (placed in the wound area) compared with the controls and other filaments (P≤0.01 at Day 3).
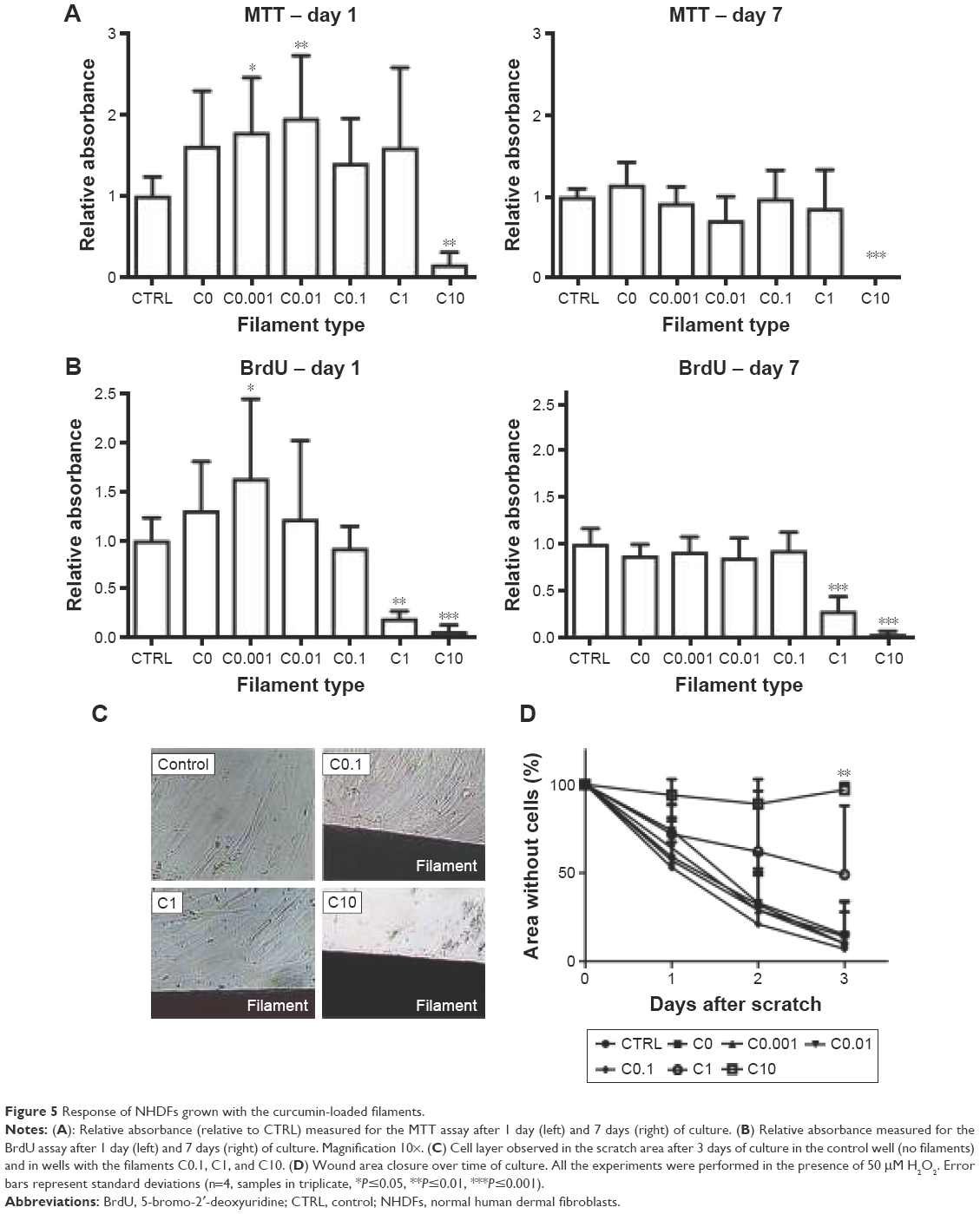 | Figure 5 Response of NHDFs grown with the curcumin-loaded filaments. |
Although cells were not grown directly onto the materials, it seems clear that high concentrations of curcumin in the filaments (1% and 10%) were not favorable to wound healing. The MTT and BrdU assays indicated that cells do not survive in the presence of C10, as values were close to zero at both Days 1 and 7. Moreover, C1 seemed to inhibit proliferation although it did not induce cell death. This was shown by a metabolic activity similar to C0 at both Days 1 and 7 (no significant difference), despite the low BrdU values. This result, together with the mixed results obtained from the scratch test, indicated that C1 might be at the threshold of being toxic for NHDFs. Therefore, although such concentrations might be attractive for anti-cancer therapies35 or antimicrobial applications,24 they are unlikely to be encouraging tissue regeneration. These results are consistent with previously published data, showing that curcumin inhibits cell growth or induces apoptosis in a variety of cell lines, including fibroblasts, at concentrations ranging from 10 to 100 μM.12,13,42 Moreover, similar observations have been made with the experiments carried out with free curcumin (see Figure S3). It was suggested that the toxicity caused by high concentrations of curcumin results from a pro-oxidant effect leading to a large increase in ROS production.13
At low curcumin concentrations (0.001% and 0.01%), the filaments stimulated proliferation of NHDFs compared with the CTRL. However, this effect was not significant when compared to the filament control C0. This means that the polymer itself might contribute to improve the cell metabolic activity and proliferation, such as by reacting with the H2O2 present in the medium (added to simulate oxidative stress conditions). Interestingly, the stimulation effect of free curcumin was observed to peak at ~1 μM (instead of ~0.001 and 0.01 μM for the filaments) in the experiments with free curcumin (see Figure S3B). This suggests that the polymer carrier PDO is efficient at protecting the curcumin from degradation and that slow delivery from the polymer may be more effective than using curcumin in free form. This may be linked to the rapid autoxidation of the molecule in culture medium (see “Release of curcumin in culture medium” section). The role played by the polymer will be explored in more detail in future work. The stimulation effect of curcumin at low concentrations has previously been described in the literature. For instance, it was shown that the application of 0.02 μM of curcumin for 24 hours significantly increased the proliferation of 3T3-L1 preadipocytes.16 In a different study, a concentration of 0.5 μM curcumin was effective to induce the proliferation of embryonic neural progenitor cells.15 Low doses of curcumin (0.1, 0.5, and 1 μM) were also shown to stimulate the proliferation of spinal cord neural progenitor cells.17 In a recent in vivo study, it was shown that electrospun mats made of PCL loaded with 0.5% curcumin significantly increased the rate of wound closure in a mouse compared with a control PCL mat.43 However, the biological response to curcumin concentrations varies largely between animal and human cells.44
It is important to note that, in this study, NHDFs were incubated with the filaments and not actually grown on the material. Further work should also investigate whether culturing cells directly onto the materials may affect the concentration at which curcumin induces apoptosis and stimulates proliferation. In particular, it has been suggested that curcumin might concentrate at the material surface or in the cell membrane in contact with the material rather than being released into the medium.45
Antioxidant activity
As shown in Figure 6A and B, there was no significant change in catalase activity and GSH content between the CTRL and the filaments C0 to C1. However, both tests showed significantly higher values for C10 compared with the control and the other filaments (except for C0 and C0.1 for the catalase assay). Furthermore, as indicated in Figure 6C, a significant reduction in ROS production was observed for C1 and C10 compared with the other samples.
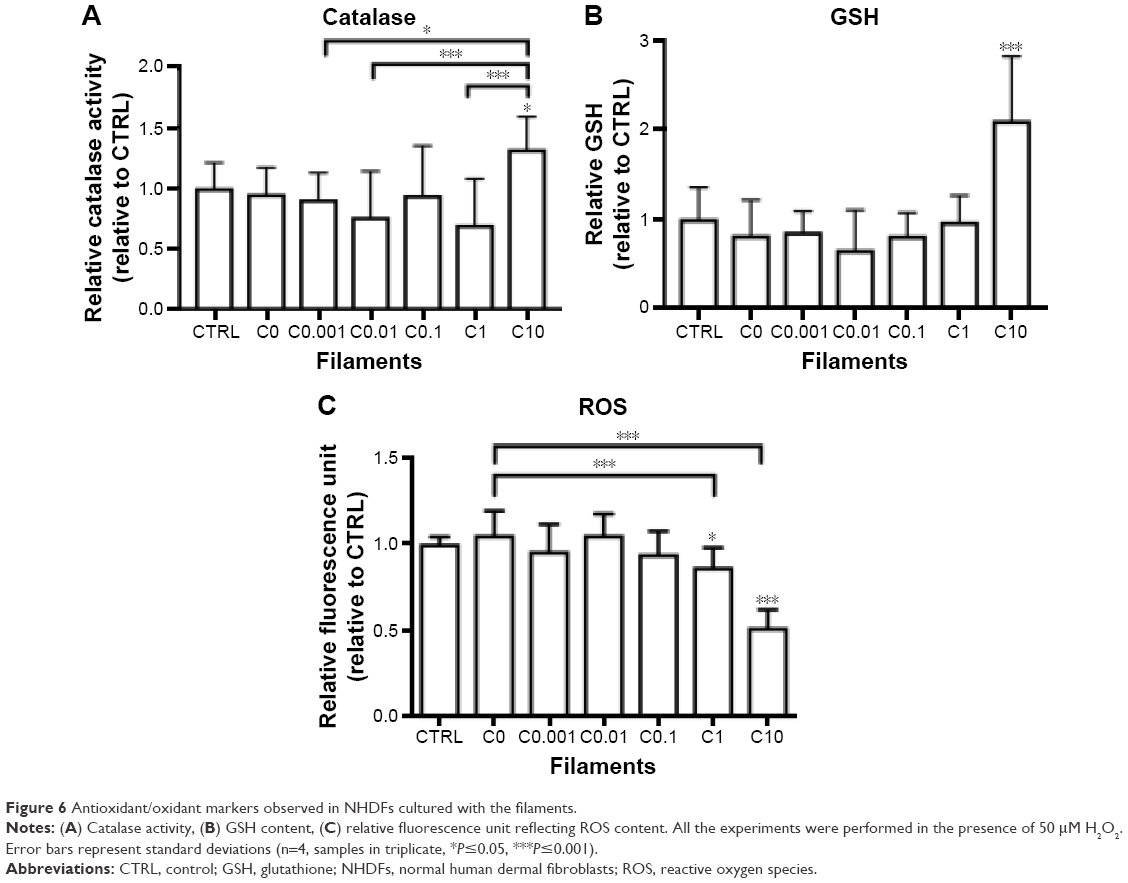 | Figure 6 Antioxidant/oxidant markers observed in NHDFs cultured with the filaments. |
These results suggest that the oxidative levels induced by curcumin released from C10 stimulated the antioxidative defense mechanisms of NHDFs. However, the increases in catalase activity and GSH content were not sufficient to protect the cells, as shown by the low values of MTT and BrdU assays, indicating cell death (see Figure 5A–D). In terms relative to the number of cells, levels of ROS caused by C10 are actually much higher compared with all other filaments (>5× when using MTT values), indicating a high level of oxidative stress that could not be overcome with increased catalase and GSH. Considering this, these results are consistent with the existing data showing that concentration of curcumin >10 μM induces ROS.12–14 Moreover, it is worth noting that ROS are known to react rapidly with other molecules such as lipids, which could lead to more stable signaling molecules further affecting cell growth.31
In the present study, no evidence was showed that curcumin contributes to the antioxidant defense system as a radical scavenger molecule. Future work will investigate the effect of curcumin-loaded filaments on the activities of redox-sensitive transcription factor (eg, nuclear factor kappa-light-chain-enhancer of activated B cells, nuclear factor erythroid 2-related factor 2, and peroxisome proliferator-activated receptor gamma) on which curcumin has been shown to have an effect,46–48 as well as on the content in lipid peroxidation products (such as acrolein and 4-hydroxynonenal).
Conclusion
More care should be taken when determining the concentration of curcumin to incorporate into a biomaterial. Crucial considerations include the intended application, the targeted tissue, and the properties of the implant (eg, size, shape, porosity, and specific surface area). In particular, it is important to ensure that the amount of curcumin released in the surrounding environment (tissues and body fluids) does not reach the apoptosis threshold following the material implantation.
In this paper, we have explored a wide range of curcumin concentrations incorporated into PDO and assessed the potential of the resulting materials for soft tissue repair applications. PDO is a degradable polymer commonly used in medical and dental applications as suture material, and its use as a drug carrier is of increasing interest. PDO filaments were prepared with various amounts of curcumin: 0%, 0.001%, 0.01%, 0.1%, 1%, and 10% (w/w ratio). The results from the present study showed that, although high concentration of curcumin (1% and 10%) might be attractive for their better mechanical properties, they inhibit the proliferation of NHDFs or cause apoptosis. This was confirmed by the concentrations found in the medium after 24 hours of incubation, which are near or above the toxicity threshold of curcumin. Filaments with 10% curcumin were also shown to increase the catalase activity and GSH content, indicating the presence of higher amounts of ROS, a result of curcumin’s toxicity. At lower concentrations (≤0.1%), curcumin had no effect on the physicochemical properties of the filaments. However, the lowest concentrations (0.001% and 0.01%) stimulated both the metabolic activity and the proliferation of NHDFs compared with the CTRL.
Overall, these findings suggested that PDO filaments loaded with low amounts of curcumin, in particular <0.01%, are more promising materials for stimulating tissue repair than those loaded with high concentrations. This paper also indicated the potential of PDO as a drug carrier in the context of tissue repair applications, given the slow release and small amounts of curcumin needed to stimulate cell proliferation. It highlighted the need to explore lower concentrations for other polymers, in particular those similar to PDO, such as those with PCL and other degradable polyesters. Moreover, additional in vitro work studying the behavior of cells grown directly onto the materials and future in vivo studies using relevant animal models should help to confirm the most relevant range of curcumin concentrations for soft tissue repair applications.
Acknowledgments
The authors acknowledge the contribution of the National Institute for Health Research Oxford Biomedical Research Centre and would like to thank Dr Ana Butorac from BIOCentar (Zagreb) for her support with the LC-MS experiment. The authors also gratefully acknowledge the contributions of Dr Koraljka Gall Trošelj, Mrs Tea Vuković, Mrs Tea Kečkeš, and Mrs Antonina Lach at various stages during the project. This project takes part to the Open Research Data Pilot launched by the European Commission. All data sets underpinning the results presented in this paper can be found in the repository of the Rudjer Boskovic Institute, Fulir (www.fulir.irb.hr/), under the project name BIOXYARN.
This work was supported by the individual fellowship grant BIOXYARN funded by the H2020 Marie Skłodowska-Curie Actions of the European Commission (H2020-MSCA-IF-2014-654761).
Disclosure
The authors report no conflicts of interest in this work.
References
Grant View Research. [webpage on the Internet]. Medical textiles market analysis by raw material (non-woven, knitted, woven), by application (implantable goods, non-implantable goods, healthcare & hygiene products) and segment forecasts to 2022;2016:110. Available from: http://www.grandviewresearch.com/industry-analysis/medical-textiles-market. Accessed May 19, 2017. | ||
Rho KS, Jeong L, Lee G, et al. Electrospinning of collagen nanofibers: effects on the behavior of normal human keratinocytes and early-stage wound healing. Biomaterials. 2006;27:1452–1461. | ||
Downing TL, Soto J, Morez C, et al. Biophysical regulation of epigenetic state and cell reprogramming. Nat Mater. 2013;12:1154–1162. | ||
Mouthuy PA, Zargar N, Hakimi O, Lostis E, Carr A. Fabrication of continuous electrospun filaments with potential for use as medical fibres. Biofabrication. 2015;7:025006. | ||
Barber JG, Handorf AM, Allee TJ, Li WJ. Braided nanofibrous scaffold for tendon and ligament tissue engineering. Tissue Eng Part A. 2013;19:1265–1274. | ||
Khil MS, Bhattarai SR, Kim HY, Kim SZ, Lee KH. Novel fabricated matrix via electrospinning for tissue engineering. J Biomed Mater Res B Appl Biomater. 2005;72:117–124. | ||
Mouthuy PA, Snelling SJB, Dakin SG, et al. Biocompatibility of implantable materials: an oxidative stress viewpoint. Biomaterials. 2016;109:55–68. | ||
Aggarwal BB, Harikumar KB. Potential therapeutic effects of curcumin, the anti-inflammatory agent, against neurodegenerative, cardiovascular, pulmonary, metabolic, autoimmune and neoplastic diseases. Int J Biochem Cell Biol. 2009;41:40–59. | ||
Akbik D, Ghadiri M, Chrzanowski W, Rohanizadeh R. Curcumin as a wound healing agent. Life Sci. 2014;116:1–7. | ||
Phan TT, See P, Lee ST, Chan SY. Protective effects of curcumin against oxidative damage on skin cells in vitro: its implication for wound healing. J Trauma. 2001;51:927–931. | ||
Sidhu GS, Singh AK, Thaloor D, et al. Enhancement of wound healing by curcumin in animals. Wound Repair Regen. 1998;6:167–177. | ||
Quitschke WW. Differential solubility of curcuminoids in serum and albumin solutions: implications for analytical and therapeutic applications. BMC Biotechnol. 2008;8:84. | ||
Scharstuhl A, Mutsaers HAM, Pennings SWC, Szarek WA, Russel FGM, Wagener FADTG. Curcumin-induced fibroblast apoptosis and in vitro wound contraction are regulated by antioxidants and heme oxygenase: implications for scar formation. J Cell Mol Med. 2009;13:712–725. | ||
Attari F, Zahmatkesh M, Aligholi H, et al. Curcumin as a double-edged sword for stem cells: dose, time and cell type-specific responses to curcumin. Daru. 2015;23:33. | ||
Kim SJ, Son TG, Park HR, et al. Curcumin stimulates proliferation of embryonic neural progenitor cells and neurogenesis in the adult hippocampus. J Biol Chem. 2008;283:14497–14505. | ||
Kim JH, Park SH, Nam SW, et al. Curcumin stimulates proliferation, stemness acting signals and migration of 3T3-L1 preadipocytes. Int J Mol Med. 2011;28:429–435. | ||
Son S, Kim KT, Cho DC, Kim HJ, Sung JK, Bae JS. Curcumin stimulates proliferation of spinal cord neural progenitor cells via a mitogen-activated protein kinase signaling pathway. J Korean Neurosurg Soc. 2014;56:1–4. | ||
Jain SK, Rains J, Croad J, Larson B, Jones K. Curcumin supplementation lowers TNF-α, IL-6, IL-8, and MCP-1 secretion in high glucose-treated cultured monocytes and blood levels of TNF-α, IL-6, MCP-1, glucose, and glycosylated hemoglobin in diabetic rats. Antioxid Redox Signal. 2009;11:241–249. | ||
Merrell JG, McLaughlin SW, Tie L, Laurencin CT, Chen AF, Nair LS. Curcumin loaded poly(ε-caprolactone) nanofibers: diabetic wound dressing with antioxidant and anti-inflammatory properties. Clin Exp Pharmacol Physiol. 2009;36:1149–1156. | ||
Fu SZ, Meng XH, Fan J, et al. Acceleration of dermal wound healing by using electrospun curcumin-loaded poly(epsilon-caprolactone)-poly(ethylene glycol)-poly(epsilon-caprolactone) fibrous mats. J Biomed Mater Res B Appl Biomater. 2014;102:533–542. | ||
Lian Y, Zhan JC, Zhang KH, Mo XM. Fabrication and characterization of curcumin-loaded silk fibroin/P(LLA-CL) nanofibrous scaffold. Front Mater Sci. 2014;8:354–362. | ||
Chen Y, Lin J, Fei Y, Wang H, Gao W. Preparation and characterization of electrospinning PLA/curcumin composite membranes. Fiber Polym. 2010;11:1128–1131. | ||
Yakub G, Toncheva A, Manolova N, Rashkov I, Kussovski V, Danchev D. Curcumin-loaded poly(l-lactide-co-D,l-lactide) electrospun fibers: Preparation and antioxidant, anticoagulant, and antibacterial properties. J Bioact Compat Polym. 2014;29(6):607–627. | ||
Wang C, Ma C, Wu Z, et al. Enhanced Bioavailability and Anticancer Effect of Curcumin-Loaded Electrospun Nanofiber: In Vitro and In Vivo Study. Nanoscale Res Lett. 2015;10:439. | ||
Elakkiya T, Malarvizhi G, Rajiv S, Natarajan TS. Curcumin loaded electrospun Bombyx mori silk nanofibers for drug delivery. Polym Int. 2014;63:100–105. | ||
Suwantong O, Opanasopit P, Ruktanonchai U, Supaphol P. Electrospun cellulose acetate fiber mats containing curcumin and release characteristic of the herbal substance. Polymer. 2007;48:7546–7557. | ||
Goonoo N, Jeetah R, Bhaw-Luximon A, Jhurry D. Polydioxanone-based bio-materials for tissue engineering and drug/gene delivery applications. Eur J Pharm Biopharm. 2015;97(Pt B):371–391. | ||
Ramalingam N, Natarajan TS, Rajiv S. Preparation and characterization of electrospun curcumin loaded poly(2-hydroxyethyl methacrylate) nanofiber – a biomaterial for multidrug resistant organisms. J Biomed Mater Res A. 2015;103:16–24. | ||
Ishikiriyama K, Pyda M, Zhang G, Forschner T, Grebowicz J, Wunderlich B. Heat capacity of poly-p-dioxanone. J Macromol Sci B. 1997;37:27–44. | ||
Tan S, Rupasinghe TW, Tull DL, et al. Degradation of curcuminoids by in vitro pure culture fermentation. J Agric Food Chem. 2014;62:11005–11015. | ||
Cipak A, Mrakovcic L, Ciz M, et al. Growth suppression of human breast carcinoma stem cells by lipid peroxidation product 4-hydroxy-2-nonenal and hydroxyl radical-modified collagen. Acta Biochim Pol. 2010;57:165–171. | ||
Goth L. A simple method for determination of serum catalase activity and revision of reference range. Clin Chim Acta. 1991;196:143–151. | ||
Tietze F. Enzymic method for quantitative determination of nanogram amounts of total and oxidized glutathione: applications to mammalian blood and other tissues. Anal Biochem. 1969;27:502–522. | ||
Kalinich JF, Ramakrishnan N, McClain DE. The antioxidant Trolox enhances the oxidation of 2′,7′-dichlorofluorescin to 2′,7′-dichlorofluorescein. Free Radic Res. 1997;26:37–47. | ||
Guo G, Fu S, Zhou L, et al. Preparation of curcumin loaded poly(epsilon-caprolactone)-poly(ethylene glycol)-poly(epsilon-caprolactone) nanofibers and their in vitro antitumor activity against Glioma 9L cells. Nanoscale. 2011;3:3825–3832. | ||
Mohan PRK, Sreelakshmi G, Muraleedharan CV, Joseph R. Water soluble complexes of curcumin with cyclodextrins: characterization by FT-Raman spectroscopy. Vib Spectrosc. 2012;62:77–84. | ||
Nishida H, Yamashita M, Hattori N, Endo T, Tokiwa Y. Thermal decomposition of poly(1,4-dioxan-2-one). Polym Degrad Stab. 2000;70:485–496. | ||
Rachmawati H, Yanda YL, Rahma A, Mase N. Curcumin-loaded PLA nanoparticles: formulation and physical evaluation. Sc Pharm. 2016;84:191–202. | ||
Abhari R, Mouthuy PA, Zargar N, Brown C, Carr A. Effect of annealing on the mechanical properties and the degradation of electrospun polydioxanone filaments. J Mech Behav Biomed Mater. 2017;67:127–134. | ||
Schneider C, Gordon ON, Edwards RL, Luis PB. Degradation of curcumin: from mechanism to biological implications. J Agric Food Chem. 2015;63:7606–7614. | ||
Blasius R, Duvoix A, Morceau F, et al. Curcumin stability and its effect on glutathione S-transferase P1-1 mRNA expression in K562 cells. Ann N Y Acad Sci. 2004;1030:442–448. | ||
Atsumi T, Tonosaki K, Fujisawa S. Comparative cytotoxicity and ROS generation by curcumin and tetrahydrocurcumin following visible-light irradiation or treatment with horseradish peroxidase. Anticancer Res. 2007;27:363–371. | ||
Bui HT, Chung OH, Dela Cruz J, Park JS. Fabrication and characterization of electrospun curcumin-loaded polycaprolactone-polyethylene glycol nanofibers for enhanced wound healing. Macromol Res. 2014;22:1288–1296. | ||
Ravindran J, Prasad S, Aggarwal BB. Curcumin and cancer cells: how many ways can curry kill tumor cells selectively? AAPS J. 2009;11:495–510. | ||
Li C, Luo T, Zheng Z, Murphy AR, Wang X, Kaplan DL. Curcumin-functionalized silk materials for enhancing adipogenic differentiation of bone marrow-derived human mesenchymal stem cells. Acta Biomater. 2015;11:222–232. | ||
Gonzalez-Reyes S, Guzman-Beltran S, Medina-Campos ON, Pedraza-Chaverri J. Curcumin pretreatment induces Nrf2 and an antioxidant response and prevents hemin-induced toxicity in primary cultures of cerebellar granule neurons of rats. Oxid Med Cell Longev. 2013;2013:801418. | ||
Biswas SK, McClure D, Jimenez LA, Megson IL, Rahman I. Curcumin induces glutathione biosynthesis and inhibits NF-kappaB activation and interleukin-8 release in alveolar epithelial cells: mechanism of free radical scavenging activity. Antioxid Redox Signal. 2005;7:32–41. | ||
Sandur SK, Ichikawa H, Pandey MK, et al. Role of pro-oxidants and antioxidants in the anti-inflammatory and apoptotic effects of curcumin (diferuloylmethane). Free Radic Biol Med. 2007;43:568–580. |
Supplementary materials
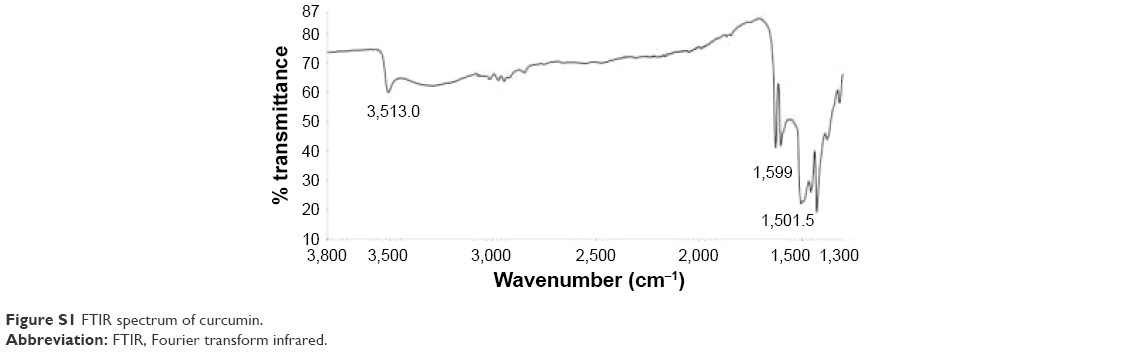 | Figure S1 FTIR spectrum of curcumin. |
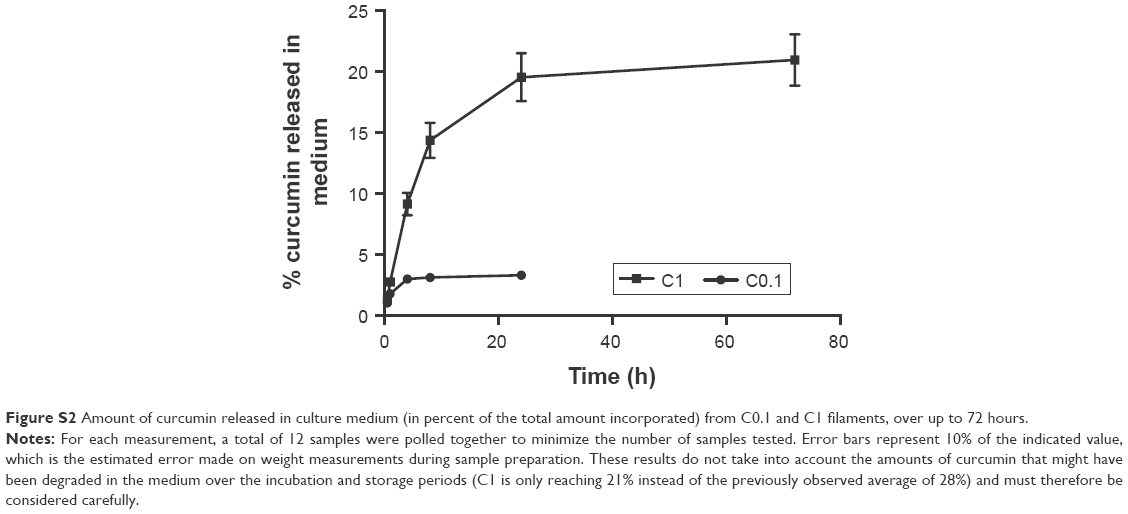 | Figure S2 Amount of curcumin released in culture medium (in percent of the total amount incorporated) from C0.1 and C1 filaments, over up to 72 hours. |
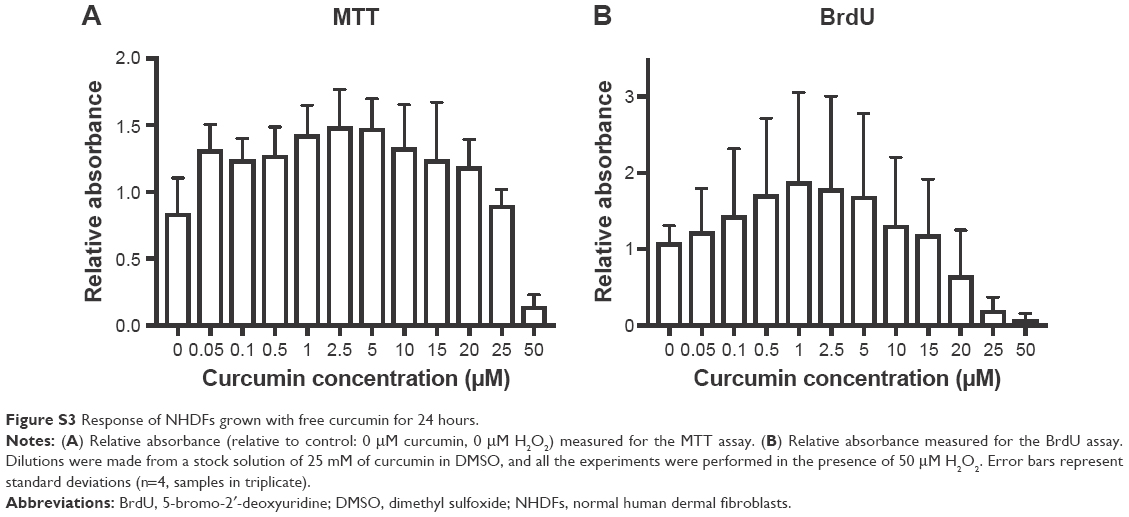 | Figure S3 Response of NHDFs grown with free curcumin for 24 hours. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.