Back to Journals » Journal of Pain Research » Volume 13
Intravenous Administration of Triptonide Attenuates CFA-Induced Pain Hypersensitivity by Inhibiting DRG AKT Signaling Pathway in Mice
Authors Ling YJ ![]() , Ding TY, Dong FL, Gao YJ, Jiang BC
, Ding TY, Dong FL, Gao YJ, Jiang BC
Received 3 August 2020
Accepted for publication 19 November 2020
Published 1 December 2020 Volume 2020:13 Pages 3195—3206
DOI https://doi.org/10.2147/JPR.S275320
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Robert B. Raffa
Yue-Juan Ling,1,2 Ting-Yu Ding,1,2 Fu-Lu Dong,3 Yong-Jing Gao,1,2 Bao-Chun Jiang1,2
1Institute of Pain Medicine, Nantong University, Nantong, Jiangsu 226019, People’s Republic of China; 2Institute of Special Environmental Medicine, Nantong University, Nantong, Jiangsu 226019, People’s Republic of China; 3School of Medicine, Nantong University, Nantong, Jiangsu, People’s Republic of China
Correspondence: Bao-Chun Jiang
Institute of Pain Medicine, Nantong University, 9 Seyuan Road, Nantong, Jiangsu 226019, People’s Republic of China
Tel +86-513-55003373
Email [email protected]
Background: Currently, medical treatment of inflammatory pain is limited by a lack of safe and effective therapies. Triptonide (TPN), a major component of Tripterygium wilfordii Hook.f. with low toxicity, has been shown to have good anti-inflammatory and neuroprotective effects. The present study aims to investigate the effects of TPN on chronic inflammatory pain.
Materials and Methods: Inflammatory pain was induced by intraplantar injection of complete Freund’s adjuvant (CFA). TPN’s three different doses were intravenously administered to compare the analgesic efficacy: 0.1 mg/kg, 0.5 mg/kg, and 2.0 mg/kg. The foot swelling was quantitated by measuring paw volume. Mechanical allodynia and thermal hyperalgesia were assessed with von Frey filament testing and Hargreaves’ test, respectively. Western blots, qRT–PCR and immunofluorescence tests were used to analyze the expression of pAKT, tumor necrosis factor-α (TNF-α), interleukin 1 beta (IL-1β), and interleukin 6 (IL-6). Two AKT inhibitors, AKT inhibitor Ⅳ and MK-2206, were used to examine AKT’s effects on pain behavior and cytokines expression.
Results: Intravenous treatment with TPN attenuated CFA-induced paw edema, mechanical allodynia, and thermal hyperalgesia. Western blotting and immunofluorescence results showed that CFA induced AKT activation in the dorsal root ganglion (DRG) neurons. However, these effects were suppressed by treatment with TPN. Furthermore, TPN treatment inhibited CFA-induced increase of pro-inflammatory cytokines, including TNF-α, IL-1β, and IL-6. Consistent with the in vivo data, TPN inhibited LPS-induced Akt phosphorylation and inflammatory mediator production in ND7/23 cells. Finally, intrathecal treatment with AKT inhibitor Ⅳ or MK-2206, attenuated CFA-induced mechanical allodynia and thermal hyperalgesia, and simultaneously decreased the mRNA expression of TNF-α, IL-1β, and IL-6 in DRG.
Conclusion: These data indicate that TPN attenuates CFA-induced pain potentially via inhibiting AKT-mediated pro-inflammatory cytokines production in DRG. TPN may be used for the treatment of chronic inflammatory pain.
Keywords: triptonide, AKT, DRG, inflammatory pain, pro-inflammatory factors
Introduction
Tripterygium wilfordii Hook. f., a member of the Celastraceae family of plants, is one of the fundamental herbs in traditional Chinese herbal medicine. The diterpenoid triepoxide chemicals are the main biologically active ingredients in the root of Tripterygium wilfordii. They have been used as a part of systematic medication to treat a wide variety of diseases, including cancer, lupus, rheumatoid arthritis, Alzheimer’s disease, Parkinson’s disease, and rheumatoid arthritis.1–3
Triptolide (TPL) was the first monomer isolated from Tripterygium wilfordii and structurally characterized in 1972.4 TPL was shown to be able to relieve many different types of pain. It can effectively relieve neuropathic pain by inhibiting microglial activation and astrocytes in the spinal dorsal horn.5,6 TPL also attenuates cancer pain via suppressing the up-regulation of Chemokine (C-C motif) ligand 5 and histone deacetylases in the spinal glial cells.7,8 In addition, TPL inhibits the activation of extracellular signal–regulated kinase (ERK) pathway and the production of inflammatory cytokines in the spinal cord dorsal horn induced by inflammation.9 Recently, TPL was reported to have a potent anti-depressive function by its influences on hippocampal neuroinflammation in a rat model of depression comorbidity of chronic pain.2 However, clinical reports showed that TPL exposure resulted in the injury of some organs, including liver, kidney, heart, testes, and ovary in humans.10–12 Severe hepatotoxicity was also shown after TPL exposure in animals.10,13,14
Triptonide (TPN) is another bioactive component of Tripterygium wilfordii. It was reported that TPN does not induce severe liver toxicity in animals.1,3,15 TPN acts as a novel potent anticancer drugs with low toxicity.16–19 The differences between the chemical structures of TPL and TPN are the substituent groups at C-14 position in which TPL is with C-14-hydroxyl and TPN is with C-14-carbonyl (Figure 1A and B). The metabolomics study shows that the hydroxyl group at C-14 in the molecular structure of TPL plays an important role in its hepatotoxicity.15,20
 |
Figure 1 Chemical structures of triptolide (A) and triptonide (B). |
Sensitization of peripheral nociceptors by pro-inflammatory factors is a major factor of neuropathic pain and inflammation pain.21,22 The tumor necrosis factor-alpha (TNF-α), interleukin-1 beta (IL-1β) and interleukin-6 (IL-6) in the dorsal root ganglia (DRG) contributes to the pathogenesis of chronic pain.23–25 Phosphoinositide 3-kinase/protein kinase B (PI3K/AKT) pathway regulate the TNF-α, IL-1β and IL-6 expression in different cells.26,27 PI3K/AKT pathway could be activated in DRG neurons and spinal glial cells in different pain models.28–30 Recent studies showed that TPN can diminish AKT signaling pathways in cervical cancer cells or lymphoma cells.1,31 Although the analgesic function of TPL has been studied, whether TPN has an analgesic effect and the underlying mechanisms remain unknown.
The intraplantar injection of complete Freund’s adjuvant (CFA) is frequently used to establish a chronic inflammatory pain model since it can trigger inflammatory responses, including the formation of an inflammatory infiltrate, swelling and cytokine release. In this study, we investigated whether systemic treatment with TPN can attenuate nocifensive behaviors in the CFA-induced inflammatory pain model. We also explored the possible analgesic mechanisms of TPN by assaying the activation of AKT pathway and the production of inflammatory cytokines.
Materials and Methods
Animals
Adult ICR male mice (6 to 8 weeks) were obtained from the Animal Care and Use Committee of Nantong University. Animals were kept under a 12/12 h light/dark cycle at a temperature of 23 ± 2°C, humidity (50–60%) with free-feeding. All behavioral experiments were performed between 9 am and 6 pm. All in vivo studies were performed in accordance with the UK Animals Scientific Procedures Act (1986) and were approved by the Animal Care and Use Committee of Nantong University.
CFA Pain Model and Drug Administration
Inflammatory pain was induced by intraplantar injection of 20 μL CFA (100%; Sigma–Aldrich) in the left hind paws under brief anesthesia with isofluorane (2%; RWD Life Science, Shenzhen, China). TPN was purchased from Chengdu Mansite Biotechnology (Sichuan, China). To evaluate if TPN has analgesic effects, We used four experimental treatments: control group (saline injection), light-dose TPN treatment group (0.1 mg/kg), medium-dose TPN treatment group (0.5 mg/kg), and high-dose TPN treatment group (2.0 mg/kg). Each treatment started 1 hour before CFA injection by intravenous injection of a single daily bolus (0.25 mL) for 5 consecutive days (Figure 2A). To analyze CFA’s effect on AKT’s phosphorylation and cytokines’ expression in vivo, we had set a naive group that only received an intraplantar injection of saline (0.9% NaCl, 20 μL/mouse) in the left hind paw. To investigate the involvement of AKT activation in CFA-induced inflammatory pain, intrathecal injection (i.t.) of the AKT inhibitors: AKT inhibitor IV (Sigma-Aldrich, Saint Louis, MO) or MK-2206 (MCE, NJ, USA) was performed in mice.
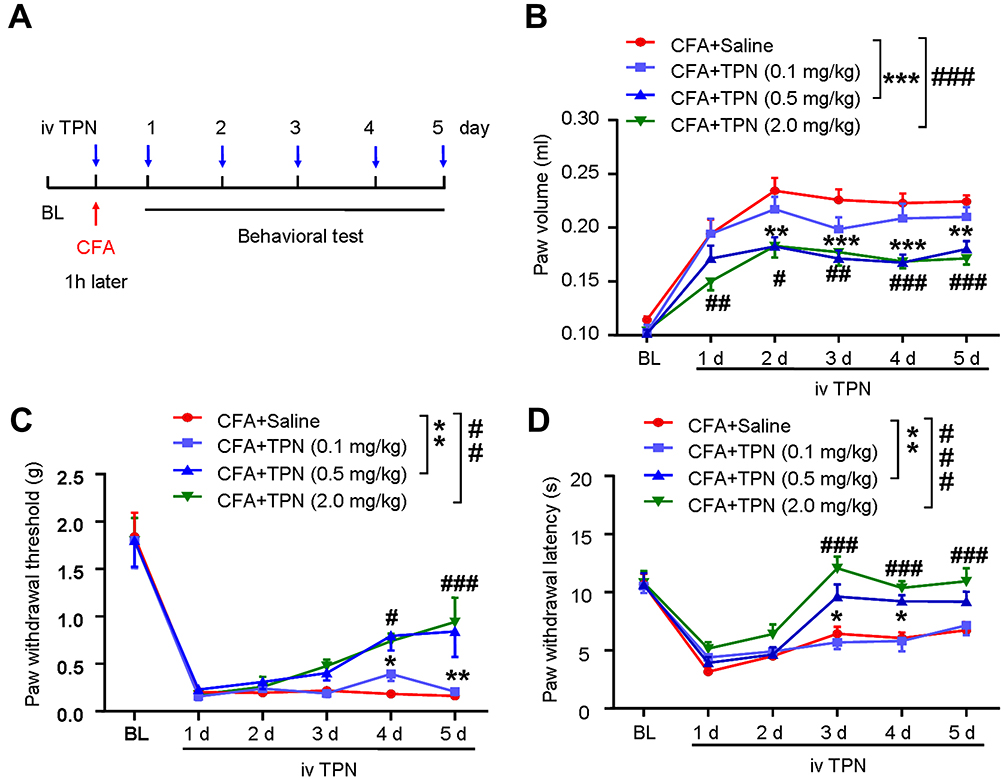 |
Figure 2 TPN treatment attenuates CFA-induced paw swelling, mechanical allodynia, and thermal hyperalgesia. (A) Schematic diagram of the timeline for CFA injection, drug treatment, and behavioral testing. (B) Change in paw volume, (C) Mechanical allodynia and (D) Thermal hyperalgesia of CFA-injected mice treated with various doses of TPN. Two-way ANOVA followed by Bonferroni’s test was applied for statistical comparison between groups, n= 7~8 mice/group, *P < 0.05, **P < 0.01, ***P < 0.001, CFA vs CFA + TPN (0.5mg/kg), #P < 0.05, ##P < 0.01, ###P < 0.001, CFA vs CFA + TPN (2 mg/kg). |
Paw Edema Measurement
Paw volumes were measured by plethysmometry (Ugo Basile Plethysmometer, Comerio, Italy) before CFA injections and 1, 2, 3, 4, and 5 days after TPN administration. Edema was expressed by paw volume (mL).
Behavioral Analysis
For the von Frey test, the animals were put in boxes on an elevated metal mesh floor daily for at least 2 d before baseline testing and allowed 30 min for habituation before the examination. The plantar surface of the left hind paw was stimulated with a series of von Frey hairs with logarithmically incrementing stiffness (0.02–2.56 g, Stoelting). The 50% paw withdrawal threshold was determined using Dixon’s up-down method. For the Hargreaves test, the animals were put in a plastic box placed on a glass plate, and the plantar surface was exposed to a beam of radiant heat through a transparent glass surface (Life Science). The baseline latencies were adjusted to 10–14 s with a maximum of 20 s as a cutoff to prevent potential injury. All the behavioral experimenters were done by individuals who were blinded to the treatment of the mice.
Cell Cultures
Mouse neuroblastoma/rat DRG neuron hybrid ND7/23 (RRID: CVCL_4259) cells were grown under standard cell culture conditions (5% CO2, 37°C) in High Glucose ’Dulbecco’s Modified Eagle Medium (DMEM, Gibco) supplemented with 10% fetal bovine serum and 100 U/mL Penicillin-Streptomycin. ND7/23 cells were incubated with 1 μg/mL Lipopolysaccharides (LPS, Sigma–Aldrich, St. Louis, MO, USA) for 6 h. The treatment of the 1 μg/mL TPN or the treatment of 1 μg/mL AKT inhibitor IV was started 1 h prior to LPS treatment.
Western Blotting
Western blotting analysis of pAKT levels were performed using samples collected from four experimental groups (Naive n = 3, CFA n = 3, CFA + TPN (0.5 mg/kg), n = 3, CFA + TPN (2 mg/kg), n = 3). The tissues or cells were homogenized in a lysis buffer containing protease and phosphatase inhibitors (Sigma-Aldrich) and 30 µg of proteins were loaded for each lane and separated on SDS-PAGE gel (10%). After the transfer, the blots were incubated overnight at 4°C with the pAKT primary antibody (RRID: AB_2716452; rabbit, 1:1000; Cell Signaling Technology, USA). For loading control, the blots were probed with AKT primary antibody (RRID: AB_2225340; rabbit, 1:1000; Cell Signaling Technology, USA). Then the membranes were incubated with secondary antibodies (Dylight 800, Donkey Anti-Rabbit IgG, 1:10,000, Millipore). Protein bands were detected using the Odyssey System (Li-COR, NE, USA), and the intensity of each band was quantified using Image J (NIH, MD, USA).
Immunohistochemistry
Animals were deeply anesthetized with isoflurane and perfused through the ascending aorta with saline followed by 4% paraformaldehyde with 0.1 M phosphate buffer. After the perfusion, the L4-L5 DRG were removed and postfixed in the same fixative overnight. DRG (8–15 µm) were cut in a cryostat and processed for immunofluorescence. DRG was first blocked with 1% bovine serum albumin for 2 h at room temperature, then incubated overnight at 4°C with the pAKT primary antibody (RRID: AB_2716452; rabbit, 1:200; Cell Signaling Technology, USA). The sections were then incubated for 2 h at room temperature with Cy3-conjugated secondary antibodies (1:1000; Jackson ImmunoResearch). The stained sections were examined with Leica SP8 Gated STED confocal microscope (Leica Microsystems, Wetzlar, Germany). The pAKT fluorescence intensity were measured by ImageJ.
Real-Time qPCR
Total RNA was extracted from L4-6 DRG with the Trizol reagent (Invitrogen, Carlsbad, CA, USA). One microgram of total RNA was converted into cDNA using PrimeScript RT reagent kit (Takara, Shiga, Japan). qPCR analysis was performed with the Real-Time Detection System by SYBR green I dye detection (Takara). The cDNA was amplified using the following primers: GAPDH forward, 5′-AAA TGG TGA AGG TCG GTG TGA AC-3′; GAPDH reverse, 5′-CAA CAA TCT CCA CTT TGC CAC TG-3′; TNF-α forward, 5′-GTT CTA TGG CCC AGA CCC TCA C-3′; reverse, 5′-GGC ACC ACT AGT TGG TTG TCT TTG-3′; IL-1β forward, 5′-TCC AGG ATG AGG ACA TGA GCA C-3′; reverse, 5′-GAA CGT CAC ACA CCA GCA GGT TA-3′; IL-6 forward, 5′-TAG TGG ATG CTA CCA AAC TGG A-3′; reverse, 5′-TGT GAC TCC AGC TTA TCT CTT G G-3′.
Quantification and Statistics
All data were analyzed by researchers blinded to the reagents used. All data were analyzed by GraphPad Prism (version 5.01) and presented as mean ± SEM. P < 0.05 was considered statistically significant. Behavioral data were analyzed using two-way ANOVA. Western blotting, immunofluorescence density and qPCR data were compared using one-way ANOVA. Student’s t-test (2-tailed) was used to analyze qPCR data if only 2 groups were applied.
Results
Triptonide Attenuates CFA-Induced Inflammatory Pain
To test the anti-nociceptive effect of TPN in CFA-induced inflammatory pain, different doses of TPN or vehicle were intravenously injected daily for 5 consecutive days, with the first injection of TPN given at 1 h before CFA (Figure 2A). On Day 1, the left hind paw volume of the CFA group was increased by approximately 2-fold compared to the baseline (Figure 2B). TPN at 0.5 mg/kg or 2.0 mg/kg caused a statistically significant decrease of paw swelling from day 1 to day 5 after CFA injection compared with untreated CFA mice [Figure 2B, (Treatment F (3, 150) = 30.08, P < 0.0001; Time F (5, 150) = 61.09, P < 0.0001; treatment * time interaction F (15, 150) = 1.201, P = 0.2770; two-way ANOVA followed by Bonferroni’s multiple comparisons test (compare treatment means), *** P < 0.001, CFA vs CFA+TPN (0.5mg/kg)), ### P < 0.001, CFA vs CFA+TPN (2.0mg/kg)]. Bonferroni’s multiple comparisons test (within each time point, compare treatments) was further used to assess statistical significance between controls and the treatments at the same time point. At almost each time point, both 0.5 mg/kg or 2.0 mg/kg TPN treatments can significantly attenuate CFA-induced footpad edema [Figure 2B, ** P < 0.01, *** P < 0.001, CFA vs CFA+TPN (0.5mg/kg), # P < 0.05, ## P < 0.01, ### P < 0.001, CFA vs CFA+TPN (2.0mg/kg)]. This revealed that TPN alleviated CFA-induced paw edema and had a strong anti-inflammatory effect.
In order to evaluate the effect of TPN on inflammatory pain, behavioral tests were performed in all groups of animals. CFA induced significant mechanical allodynia by decreasing the mechanical threshold induced by mechanical stimuli from day 1 to day 5 after CFA (Figure 2C). The intravenous administration of TPN significantly attenuated mechanical allodynia at 0.5 mg/kg or 2.0 mg/kg, but not at 0.1 mg/kg [(Treatment F (3, 150) = 7.196, P = 0.0002; Time F (5, 150) = 72.60, P < 0.0001; treatment * time interaction F (15, 150) = 1.334, P = 0.1885; two-way ANOVA followed by Bonferroni’s multiple comparisons test (compare treatment means)), ** P < 0.01, CFA vs CFA+TPN (0.5mg/kg), ## P < 0.01, CFA vs CFA+TPN (2.0mg/kg)]. The Bonferroni’s multiple comparisons test (within each time point, compare treatments) showed that TPN at 2 mg/kg or at 0.5 mg/kg had no effect on CFA-induced mechanical allodynia during the first 3 days following CFA injection, but started to show a reversal effect at 4 days and maintained till 5 days [Figure 2C, * P < 0.05, ** P < 0.01, CFA vs CFA+TPN (0.5mg/kg); # P < 0.05, ### P < 0.001, CFA vs CFA+TPN (2.0mg/kg)].
Meanwhile, TPN substantially attenuated CFA-induced thermal hyperalgesia at 2 mg/kg or at 0.5 mg/kg [Figure 2D, (Treatment F (3, 150) = 21.73, P < 0.0001; Time F (5, 150) = 42.25, P < 0.0001; treatment * time interaction F (15, 150) = 2.563, P = 0.002; Two-way ANOVA followed by Bonferroni’s multiple comparisons test (compare treatment means), ** P < 0.01, CFA vs CFA+TPN (0.5mg/kg)), ### P < 0.001, CFA vs CFA+TPN (2.0mg/kg)]. TPN attenuated thermal hyperalgesia at days 3, 4 and 5 after CFA injection [Figure 2D, Bonferroni’s multiple comparisons test (within each time point, compare columns), * P < 0.05, CFA vs CFA+TPN (0.5mg/kg), ### P < 0.001, CFA vs CFA+TPN (2.0mg/kg)]. These data suggest that repeated TPN administration attenuates CFA-induced pain hypersensitivity, both mechanical allodynia and thermal hyperalgesia.
TPN Inhibits CFA-Induced AKT Activation in DRG
To determine whether the analgesic effects of TPN were associated with inhibition of the AKT signaling pathways, we evaluated the expression level of phosphorylation of AKT (pAKT) in the DRG. Western blotting showed that pAKT was significantly increased at 5 days after CFA injection. Treatment with TPN (2 mg/kg) significantly decreased CFA-induced pAKT upregulation (Figure 3A and B, F (3, 8) = 9.506, P = 0.0051, one-way ANOVA followed by ’Bonferroni’s multiple comparisons test, # P < 0.05, Naive vs CFA, ** P < 0.01, CFA vs CFA+TPN). Immunofluorescence analysis also showed upregulation of pAKT expression in DRG from animals injected with CFA. Treatment with TPN (2 mg/kg) significantly attenuated the CFA-induced upregulation of pAKT (Figure 3C and D, Area F (2, 10) = 56.24, P < 0.001, Intensity F (2, 10) = 41.81, P < 0.001, one-way ANOVA followed by ’Bonferroni’s multiple comparisons test, ### P <0.001, Naive vs CFA, *** P < 0.001, CFA vs CFA+TPN), which was consistent with the Western blotting results. Double immunostaining further showed that pAKT was expressed in the DRG neurons (Figure 3E).
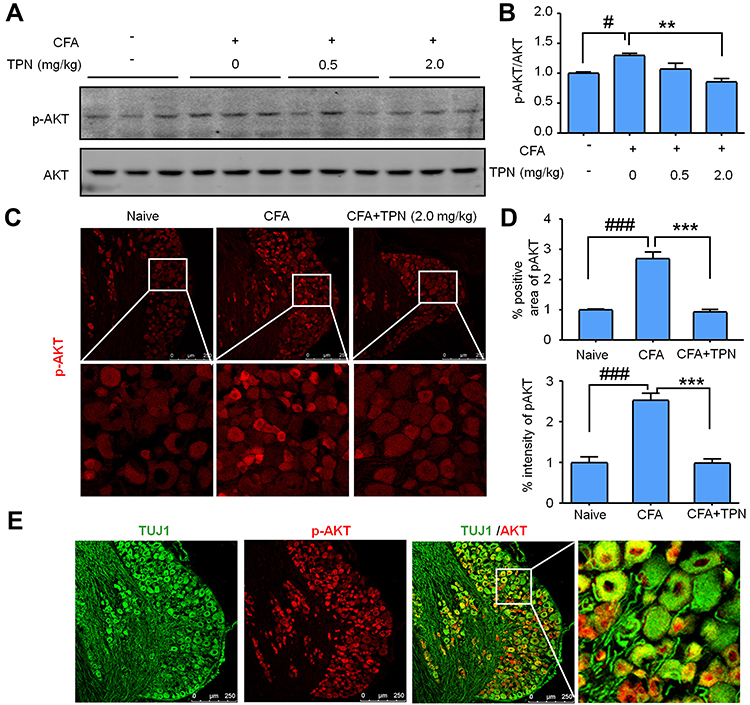 |
Figure 3 TPN treatment decreases pAKT expression in the DRG neurons. (A and B) pAKT expression of CFA-injected mice treated or untreated with TPN. pAKT expression was increased in DRG at 5 days after CFA injection, and recovered in CFA + TPN (2 mg/kg) animals. n = 3, one-way ANOVA followed by Bonferroni’s multiple comparisons test, #P <0.05, Naive vs CFA, **P < 0.01, CFA vs CFA+TPN; (C) Immunostaining of pAKT in the DRG; (D) Statistical data show the number of pAKT positive neurons and pAKT staining intensity in the DRG. n = 5 mice/group, one-way ANOVA followed by Bonferroni’s multiple comparisons test, ###P <0.001, Naive vs CFA, ***P < 0.001, CFA vs CFA+TPN. (E) Double staining of pAKT with a neuronal marker, β-tubulin (TUJ1) in the DRG at 5 days after CFA. Scale bars: 250 μm. |
TPN Inhibits CFA-Induced Increase of TNF-α, IL-1β, and IL-6 in the DRG
Because TPL is involved in the regulation of GFAP and IBA1 expression, which are markers for astrocytes and microglia, respectively, we examined their mRNA expression in the DRG after repeated TPN administration. CFA did not induce GFAP and IBA1 expression, and TPN treatments also did not affect expression levels of them in DRG (Figure 4A and B, GFAP, F (3, 20) = 2.086, P = 0.1343; IBA1, F (3, 20) = 0.5977, P = 0.6239, one-way ANOVA). TNF-α, IL-1β and IL-6 are important pro-inflammatory cytokines in mediating peripheral sensitization and neuropathic pain.32–34 To check whether the anti-nociceptive effect of TPN is associated with the downregulation of pro-inflammatory cytokines, we checked TNF-α, IL-1β, and IL-6 expression. qPCR results showed that, compared with control animals, TNF-α, IL-1β, and IL-6 mRNAs were significantly increased in animals after 5 days of CFA injection. Both the dose of 0.5 mg/kg and 2.0 mg/kg TPN treatments significantly reduced the CFA-induced mRNA increases of TNF-α, IL-1β, and IL-6 (Figure 4C–E, TNF-α, F (3, 15) = 5.568 P = 0.009; IL-1β, F (3, 18) = 9.987, P = 0.0004; IL-6, F (3, 15) = 5.944, P = 0.07; one-way ANOVA followed by Bonferroni’s multiple comparisons test, # P < 0.05, ## P < 0.01, Naive vs CFA, * P < 0.05, ** P < 0.01, *** P < 0.001, CFA vs CFA+TPN). These data indicate TPN’s inhibitory effect on CFA-induced TNF-α, IL-1β, and IL-6 expression in the DRG.
 |
Figure 4 TPN decreases CFA-induced upregulation of TNF-α, IL-1β, and IL-6 increases. (A and B) Effect of TPN treatment on expression of GFAP (A) and IBA1 (B) in DRG from CFA-treated mice. (C–E) Effects of TPN on the mRNA expression levels of TNF-α (C), IL-1β (D), and IL-6 (E). TPN treatment with both medium dose (0.5 mg/kg) and high dose (2 mg/kg) decreased TNF-α, IL-1β, and IL-6 upregulation after CFA injection. n = 5 mice/group, one-way ANOVA followed by Bonferroni’s multiple comparisons test, #P < 0.05, ##P < 0.01, Naive vs CFA, *P < 0.05, **P < 0.01, ***P < 0.001, CFA vs CFA+TPN. |
TPN Attenuates LPS-Induced pAKT Activation, and TNF-α, IL-1β, and IL-6 Expression in the ND7/23 Cells
To further confirm that TPN can attenuate pAKT activation in DRG neurons under inflammatory conditions, we used the classical exogenous toll-like receptor 4 ligand LPS to stimulate ND7/23 cells, a DRG neuron cell line, to mimic inflammatory conditions in vitro. We first checked pAKT expression after incubation with LPS (1 μg/mL) for 6 hours. Western blotting showed that LPS induced a rapid and dramatic increase of pAKT expression. Pre-incubation with TPN, 1 hour before LPS application, markedly inhibited LPS-induced AKT phosphorylation by 43.2% (Figure 5A and B, F (2, 6) = 22.85, P = 0.0016, one-way ANOVA followed by Bonferroni’s multiple comparisons test, ## P < 0.01, Vehicle vs LPS, ** P < 0.01, LPS vs LPS+TPN), which was consistent with the in vivo results (Figure 3A and B).
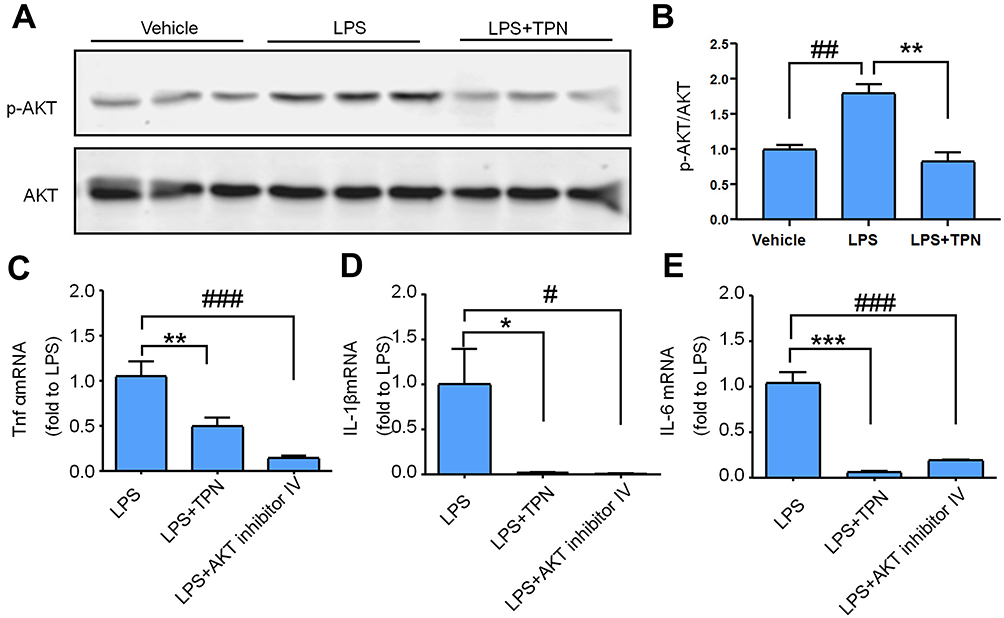 |
Figure 5 TPN decreases LPS-induced pAKT expression and the production of inflammatory cytokines in ND7/23 cells. (A and B) pAKT expression of LPS-induced ND7/23 Cells treated or untreated with TPN (1 μg/mL). pAKT expression was increased after LPS application and recovered in the LPS + TPN group. n = 3, one-way ANOVA followed by Bonferroni’s multiple comparisons test, ##P < 0.01, Vehicle vs LPS, **P < 0.01, LPS vs LPS+TPN. (C–E) Effects of TPN, and AKT inhibitor Ⅳ on the mRNA expression levels of TNF-α (C), IL-1β (D), and IL-6 (E). Both TPN treatment (1 μg/mL) and AKT inhibitor Ⅳ treatment (1 μg/mL) decreased TNF-α, IL-1β, and IL-6 upregulation after LPS incubation. n = 5, one-way ANOVA followed by Bonferroni’s multiple comparisons test, *P < 0.05, **P < 0.01, ***P < 0.001, LPS vs LPS+TPN, #P < 0.05 and ###P < 0.001, LPS vs LPS+ AKT inhibitor Ⅳ . |
To examine whether TPN inhibit TNF-α, IL-1β, and IL-6 expression via pAKT, we pretreated ND7/23 cells with AKT inhibitor, AKT inhibitor Ⅳ, 1 h before LPS application. As shown in Figure 5C–E, the qRT-PCR results showed that TPN (1 µg/mL) treatment blocked CFA-induced TNF-α, IL-1β, and IL-6 increase (TNF-α, F (2, 13) = 20.70, P < 0.0001; IL-1β, F (2, 11) = 8.133, P = 0.0068, IL-6, F (2, 15) = 61.09, P < 0.0001; one-way ANOVA followed by Bonferroni’s multiple comparisons test, * P < 0.05, ** P < 0.01, *** P < 0.001, LPS vs LPS+TPN). In consistent with TPN’s effect, pretreatment with AKT inhibitor Ⅳ (1 µg/mL) also significantly decreased LPS-induced TNF-α, IL-1β, and IL-6 upregulation (Figure 5C–E, ## P < 0.01 and ### P < 0.001, one-way ANOVA). These data suggest that TPN is involved in pAKT-mediated upregulation of TNF-α, IL-1β, and IL-6 in activated ND7/23 cells by LPS.
AKT Inhibitor Attenuates CFA-Induced Pain Hypersensitivity and Upregulation of TNF-α, IL-1β, and IL-6 in DRG
To define whether AKT is associated with CFA-induced pain hypersensitivity and the upregulation of inflammatory cytokines, mice were injected intravenously with the AKT inhibitors AKT inhibitor Ⅳ (1 μg/10 μL) or MK-2206 (1 μg/10 μL) 3 days after CFA injection. The behavioral results showed that the administration of AKT inhibitor Ⅳ slightly reduced mechanical allodynia without significant difference (Figure 6A). But, it can attenuate thermal hyperalgesia at 6 h (Figure 6B, Treatment, F (1, 55) = 7.773 and P = 0.0073, Time, F (4, 55) = 8.480 and P < 0.0001; treatment * time interaction, F (4, 55) = 4.208 and P = 0.0048, two-way ANOVA followed by Bonferroni’s multiple comparisons test, *** P < 0.001). Intravenous administration of MK-2206 attenuated both mechanical allodynia (Figure 6C, Treatment, F (1, 60) = 5.872, P = 0.0184, Time, F (4, 60) = 202.1, P < 0.0001; treatment * time interaction, F(4, 60) = 5.713, P = 0.0006, Two-way ANOVA followed by Bonferroni’s multiple comparisons test, *** P < 0.001) and thermal hyperalgesia (Figure 6D, Treatment, F (1, 60) = 26.76, P < 0.0001, Time, F (4, 60) = 20.52, P < 0.0001; treatment * time interaction, F (4, 60) = 4.355, P = 0.0037, two-way ANOVA followed by Bonferroni’s multiple comparisons test, ** P < 0.01, *** P < 0.001). These results revealed that inhibition of AKT could relieve CFA-induced pain behavior.
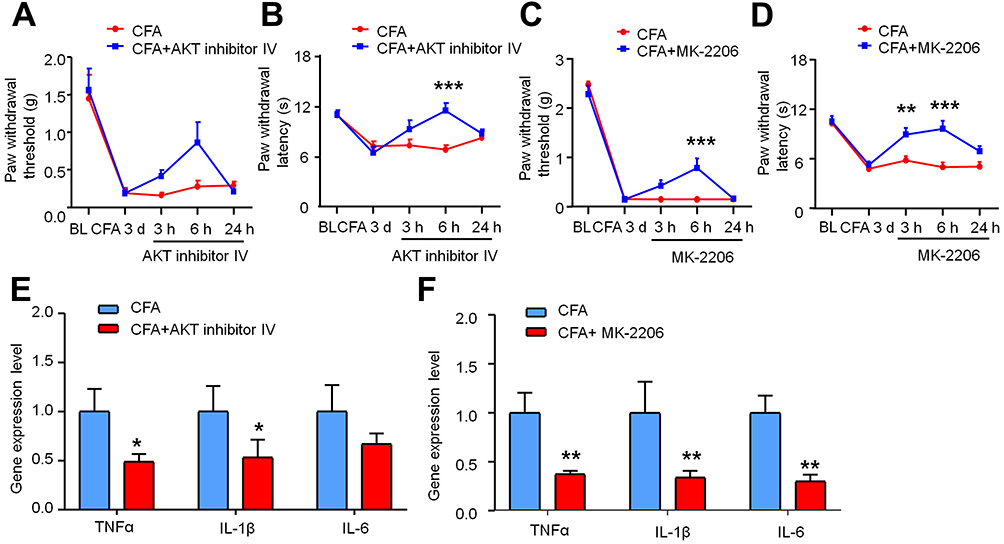 |
Figure 6 AKT inhibitor attenuates CFA-induced pain hypersensitivity and production of TNF-α, IL-1β, and IL-6 in the DRG. (A–D) Effects of AKT inhibitors administration, AKT inhibitor Ⅳ or MK-2206, on mechanical allodynia and thermal hyperalgesia in CFA-induced inflammatory pain mice. Two-way ANOVA followed by Bonferroni’s multiple comparisons test, **P < 0.01, ***P < 0.001. (E and F) Intrathecal injection of AKT inhibitor Ⅳ or MK-2206 decreased expression of TNF-α, IL-1β, and IL-6 in the DRG. n = 6. *P < 0.05, **P < 0.01, AKT inhibitor Ⅳ vs CFA group, or MK-2206 vs CFA, Student’s t-test. |
As shown in Figure 6E and F, AKT inhibitor Ⅳ or MK-2206 treatment also significantly decreased the mRNA expression of TNF-α, IL-1β and IL-6 (* P < 0.05, ** P < 0.01, Student’s t-test). Taken together, these data suggest that the AKT signaling pathway is involved in the development of CFA-induced inflammatory pain, and inhibition of AKT phosphorylation reduces the upregulation of inflammatory cytokines in the DRG.
Discussion
In this study, we investigated the anti-nociceptive effect of systemic injection of TPN on CFA-induced pain hypersensitivity and explored possible mechanisms. Our results demonstrated that TPN administration attenuated CFA-induced edema and pain hypersensitivity. Meanwhile, TPN treatment inhibited the AKT signaling pathway and the expression of TNF-α, IL-1β, and IL-6 in the DRG after CFA injection. AKT pathway acts as an upstream signaling pathway in CFA-induced upregulation of TNF-α, IL-1β, and IL-6. TPN may exert an analgesic effect via inhibition of the CFA-induced DRG AKT-TNF-α/IL-1β/IL-6 signaling pathway (Figure 7).
 |
Figure 7 Schematic representation of the mechanism of action of TPN in analgesia. Black arrows depict promotion. Red T-bar represents inhibition. Intraplantar injection of CFA can activate the AKT signaling pathway in DRG neurons, promoting the expression of pro-inflammatory genes, including TNF-α, IL-1β, and IL-6. Intravenous TPN could inhibit Akt phosphorylation and downstream gene expression, thereby relieving pain. The illustration was created with BioRender (BioRender.com). |
Previous studies showed that TPL could prevent and attenuate neuropathic pain, cancer pain, and inflammatory pain in mice or rats via inhibiting central immune response,35 inhibiting the upregulation of the expression of histone deacetylases in spinal glial cells,8 or inhibiting spinal glial activation.9 However, the role of TPN in chronic pain has not yet been studied. We first demonstration that repeated intravenous injection of TPN before CFA injection persistently attenuated CFA-induced thermal hyperalgesia and mechanical allodynia. TPN dose-dependently inhibited CFA-induced thermal hyperalgesia. However, there is no significant difference in mechanical allodynia between the 0.5 mg/kg and 2.0 mg/kg TPN groups. This may result from the target molecules or signaling pathways of TPN play more important roles in thermal pain sensitivity processing. Besides, the injection of TPN could also effectively reduce paw edema caused by CFA. Overall, these findings indicate that TPN has analgesic properties.
Although the pAKT signaling pathway typically regulates cell growth and survival, increasing evidence indicates the involvement of this pathway in the development and maintenance of chronic pain.36,37 Our data further showed that pAKT was predominantly expressed in DRG neurons after CFA injection. In agreement with our results, pAKT expression in neurons was found in the DRG following paclitaxel treatment.38 Here, intraplantar injection of CFA induced pain hypersensitivity and increased pAKT expression in the DRG, which was inhibited by TPN administration, suggesting that the anti-nociceptive effect of TPN may be mediated by pAKT-mediated peripheral sensitization in the DRG. In accordance with our results, emerging evidence indicates that TPN suppressed the expression of pAKT on in vitro or in vivo models of different kinds of cancers.1,3,31 One report has indicated that TPN inhibits pAKT expression by suppressing a proto-oncogene Lyn in lymphoma.1 Specifically, how the TPN inhibited pAKT signaling pathways in DRG neurons requires further exploration.
TNF-α, IL-1β and IL-6 are well-known pro-inflammatory cytokines that have been implicated in inflammatory pain.39 Our data showed that the expression of TNF-α, IL-1β, and IL-6 was increased in DRG neurons after CFA injection and was inhibited by TPN treatment. TNF-α was expressed in the majority of the voltage-gated sodium channel (Nav) 1.3-positive or Nav1.8-positive neurons and up-regulated the expression of Nav1.3 and Nav1.8 in DRG neurons following peripheral nerve injury.32 Cleaved IL-1β expression was significantly increased in small-sized DRG neurons after CFA injection into the hind paw.40 IL-6 was up-regulated in the ipsilateral L4 and L5 DRG neurons and in the bilateral lumbar spinal cord following L5-ventral root transaction and contributed to the development of neuropathic pain.34 Therefore, TPN may attenuate pain hypersensitivity via the inhibition of TNF-α, IL-1β, and IL-6-mediated neuroinflammation.
Pharmacological inhibition of pAKT inhibited TNF-α, IL-1β, and IL-6 production in vitro. Previous studies have demonstrated that inhibition of pAKT pathway prevented the LPS-induced expression of TNF-α in human bronchial epithelial cells.41 Diesel exhaust particles exposure can activate the AKT signaling pathway and further up-regulate IL-1β protein expression in primary human bronchial epithelial cells.41 Studies from Caco-2 cells and fibroblast-like synoviocytes showed that IL-17-mediated induction of IL-6 was transduced via activation of AKT and NF-κB,42 while mitogen-activated protein kinase (MAPK) are not likely to participate in the process.38 Therefore, it is highly likely that the p-AKT pathway acts as an upstream signaling pathway, up-regulated the expression of cytokines TNF-α, IL-1β, and IL-6 in the different cells. Both AKT Inhibitor IV and MK-2206 can relieve CFA-induced chronic inflammatory pain and the mRNA expression of TNF-α, IL-1β, and IL-6 in vivo. However, AKT Inhibitor IV showed a slightly weaker inhibition effect. Possible reasons are as follows; 1. The two drugs may have different absorption rates and degradation rates in DRG; 2. AKT inhibitor IV targets the ATP-binding site of a kinase upstream of AKT downstream of PI3K. In contrast, MK-2206 inhibits the autophosphorylation of AKT at threonine 308 and serine 473 directly; 3. The different action mechanisms of them may lead to a different degree of compensatory effects. Collectively, these findings suggest that the TPN may exert its analgesic effect through the inhibition of DRG AKT- TNF-α/IL-1β/IL-6 signaling.
Conclusion
Our present study demonstrated that intravenous treatment with TPN significantly attenuated CFA-induced pain hypersensitivity, associated with decreased TNF-α, IL-1β, and IL-6 expression, and decreased pAKT activation in DRG neurons. These data suggest that TPN could attenuate inflammatory pain via the inhibition of pAKT/TNF-α-IL-6-IL-1β signaling pathway axis in DRG neurons. Our work provides valuable information for the preclinical and clinical study of TPN to treat chronic inflammatory pain.
Abbreviations
TPN, triptonide; PI3K, phosphoinositide 3-kinase; AKT, protein kinase B; mTOR, the mammalian target of rapamycin; CFA, complete Freund’s adjuvant; DRG, dorsal root ganglion; TNF-α, tumor necrosis factor alpha; IL-1β, interleukin 1 beta; IL-6, interleukin 6; TPL, triptolide; ERK, extracellular signal-regulated kinase; LPS, lipopolysaccharides; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; Nav, voltage-gated sodium channel.
Data Sharing Statement
The datasets used and/or analyzed during the current study available from the corresponding author on reasonable request.
Ethics Approval
All in vivo studies were performed following the UK Animals Scientific Procedures Act (1986) and were approved by the Animal Care and Use Committee of Nantong University.
Funding
This work was funded by grants from the National Natural Science Foundation of China (NSFC 81771197, 81971054, 81300954 and 81400915), the Natural Science Foundation of Jiangsu Province (BK20171255), the Jiangsu University Qing Lan Project, and the Six talent peaks project in Jiangsu Province (SWYY-070).
Disclosure
The authors report no conflicts of interest for this work.
References
1. Yang P, Dong FL, Zhou QS. Triptonide acts as a novel potent anti-lymphoma agent with low toxicity mainly through inhibition of proto-oncogene Lyn transcription and suppression of Lyn signal pathway. Toxicol Lett. 2017;278:9–17. doi:10.1016/j.toxlet.2017.06.010
2. Hu X, Dong Y, Jin X, et al. The novel and potent anti-depressive action of triptolide and its influences on hippocampal neuroinflammation in a rat model of depression comorbidity of chronic pain. Brain Behav Immun. 2017;64:180–194. doi:10.1016/j.bbi.2017.03.005
3. Dong F, Yang P, Wang R, et al. Triptonide acts as a novel antiprostate cancer agent mainly through inhibition of mTOR signaling pathway. Prostate. 2019;79(11):1284–1293. doi:10.1002/pros.23834
4. Kupchan SM, Court WA, Dailey RG, Gilmore CJ, Bryan RF. Triptolide and tripdiolide, novel antileukemic diterpenoid triepoxides from Tripterygium wilfordii. J Am Chem Soc. 1972;94(20):7194–7195. doi:10.1021/ja00775a078
5. Tang J, Li Z-H, Ge S-N, et al. The inhibition of spinal astrocytic JAK2-STAT3 pathway activation correlates with the analgesic effects of triptolide in the rat neuropathic pain model. Evid Based Complement Alternat Med. 2012;2012:1–13. doi:10.1155/2012/185167
6. Huang Y, Zhu N, Chen T, et al. Triptolide Suppressed the Microglia Activation to Improve Spinal Cord Injury Through miR-96/IKK beta/NF-kappa B Pathway. Spine. 2019;44(12):E707–E714. doi:10.1097/BRS.0000000000002989
7. Hang LH, Li SN, Shao DH, Chen Z, Chen YF, Shu WW. Evidence for involvement of spinal RANTES in the anti-nociceptive effects of triptolide, a diterpene triepoxide, in a rat model of bone cancer pain. Basic Clin Pharmacol Toxicol. 2014;115(6):477–480. doi:10.1111/bcpt.12265
8. Hu X-F, He X-T, Zhou K-X, et al. The analgesic effects of triptolide in the bone cancer pain rats via inhibiting the upregulation of HDACs in spinal glial cells. J Neuroinflamm. 2017;14(1):213. doi:10.1186/s12974-017-0988-1
9. Xu F, Li Y, Li S, et al. Complete Freund’s adjuvant–induced acute inflammatory pain could be attenuated by triptolide via inhibiting spinal glia activation in rats. J Surg Res. 2014;188(1):174–182. doi:10.1016/j.jss.2013.11.1087
10. Mei Z, Wu Q, Hu S, Lib X, Yang X. Triptolide loaded solid lipid nanoparticle hydrogel for topical application. Drug Dev Ind Pharm. 2005;31(2):161–168. doi:10.1081/DDC-200047791
11. Tang X, Wang C, Hsieh Y, et al. Triptolide induces toxicity in inner ear stem cells via promoting DNA damage. Toxicol in Vitro. 2019;61:104597. doi:10.1016/j.tiv.2019.104597
12. Xi C, Peng S, Wu Z, Zhou Q, Zhou J. Toxicity of triptolide and the molecular mechanisms involved. Biomed Pharmacother. 2017;90:531–541. doi:10.1016/j.biopha.2017.04.003
13. Li J, Shen F, Guan C, et al. Activation of Nrf2 protects against triptolide-induced hepatotoxicity. PLoS One. 2014;9(7):e100685. doi:10.1371/journal.pone.0100685
14. Vliegenthart ADB, Wei C, Buckley C, et al. Characterization of triptolide-induced hepatotoxicity by imaging and transcriptomics in a novel zebrafish model. Toxicol Sci. 2017;159(2):380–391. doi:10.1093/toxsci/kfx144
15. Hu DD, Chen XL, Xiao XR, et al. Comparative metabolism of tripolide and triptonide using metabolomics. Food Chem Toxicol. 2018;115:98–108. doi:10.1016/j.fct.2018.03.009
16. Xiang S, Zhao Z, Zhang T, et al. Triptonide effectively suppresses gastric tumor growth and metastasis through inhibition of the oncogenic Notch1 and NF-kappaB signaling pathways. Toxicol Appl Pharmacol. 2019;114870.
17. Wang SS, Lv Y, Xu XC, et al. Triptonide inhibits human nasopharyngeal carcinoma cell growth via disrupting Lnc-RNA THOR-IGF2BP1 signaling. Cancer Lett. 2019;443:13–24. doi:10.1016/j.canlet.2018.11.028
18. Zhang B, Meng M, Xiang S, et al. Selective activation of tumor-suppressive MAPKP signaling pathway by triptonide effectively inhibits pancreatic cancer cell tumorigenicity and tumor growth. Biochem Pharmacol. 2019;166:70–81. doi:10.1016/j.bcp.2019.05.010
19. Zhang M, Tan S, Yu D, et al. Triptonide inhibits lung cancer cell tumorigenicity by selectively attenuating the Shh-Gli1 signaling pathway. Toxicol Appl Pharmacol. 2019;365:1–8. doi:10.1016/j.taap.2019.01.002
20. Zhao Q, Zhang J-L, Li F. Application of metabolomics in the study of natural products. Nat Prod Bioprospect. 2018;8(4):321–334. doi:10.1007/s13659-018-0175-9
21. Jiang BC, Liu T, Gao YJ. Chemokines in chronic pain: cellular and molecular mechanisms and therapeutic potential. Pharmacol Ther. 2020;212:107581. doi:10.1016/j.pharmthera.2020.107581
22. Ji RR, Chamessian A, Zhang YQ. Pain regulation by non-neuronal cells and inflammation. Science (New York, N Y). 2016;354(6312):572–577. doi:10.1126/science.aaf8924
23. Gao Y, Bai L, Zhou W, et al. PARP-1-regulated TNF-α expression in the dorsal root ganglia and spinal dorsal horn contributes to the pathogenesis of neuropathic pain in rats. Brain Behav Immun. 2020;88:482–496. doi:10.1016/j.bbi.2020.04.019
24. Nadeau S, Filali M, Zhang J, et al. Functional recovery after peripheral nerve injury is dependent on the pro-inflammatory cytokines IL-1β and TNF: implications for neuropathic pain. J Neurosci. 2011;31(35):12533–12542. doi:10.1523/JNEUROSCI.2840-11.2011
25. Fang D, Kong LY, Cai J, et al. Interleukin-6-mediated functional upregulation of TRPV1 receptors in dorsal root ganglion neurons through the activation of JAK/PI3K signaling pathway: roles in the development of bone cancer pain in a rat model. Pain. 2015;156(6):1124–1144.
26. Liu Y, Tong C, Tang Y, et al. Tanshinone IIA alleviates blast-induced inflammation, oxidative stress and apoptosis in mice partly by inhibiting the PI3K/Akt/FoxO1 signaling pathway. Free Radic Biol Med. 2020;152:52–60. doi:10.1016/j.freeradbiomed.2020.02.032
27. Han X, Li B, Ye X, et al. Dopamine D2 receptor signalling controls inflammation in acute pancreatitis via a PP2A-dependent Akt/NF-kappaB signalling pathway. Br J Pharmacol. 2017;174(24):4751–4770. doi:10.1111/bph.14057
28. Jiang S-P, Zhang Z-D, Kang L-M, Wang Q-H, Zhang L, Chen H-P. Celecoxib reverts oxaliplatin-induced neuropathic pain through inhibiting PI3K/Akt2 pathway in the mouse dorsal root ganglion. Exp Neurol. 2016;275:11–16. doi:10.1016/j.expneurol.2015.11.001
29. Guo J-R, Wang H, Jin X-J, Jia D-L, Zhou X, Tao Q. Effect and mechanism of inhibition of PI3K/Akt/mTOR signal pathway on chronic neuropathic pain and spinal microglia in a rat model of chronic constriction injury. Oncotarget. 2017;8(32):52923. doi:10.18632/oncotarget.17629
30. Xu JT, Zhao JY, Zhao XL, et al. Opioid receptor-triggered spinal mTORC1 activation contributes to morphine tolerance and hyperalgesia. J Clin Invest. 2014;124(2):592–603. doi:10.1172/JCI70236
31. Kim MJ, Lee TH, Kim SH, Choi YJ, Heo J, Kim YH. Triptolide inactivates Akt and induces caspase-dependent death in cervical cancer cells via the mitochondrial pathway. Int J Oncol. 2010;37(5):1177–1185. doi:10.3892/ijo_00000769
32. He X-H, Zang Y, Chen X, et al. TNF-α contributes to up-regulation of Nav1. 3 and Nav1. 8 in DRG neurons following motor fiber injury. PAIN®. 2010;151(2):266–279. doi:10.1016/j.pain.2010.06.005
33. Copray J, Mantingh I, Brouwer N, et al. Expression of interleukin-1 beta in rat dorsal root ganglia. J Neuroimmunol. 2001;118(2):203–211. doi:10.1016/S0165-5728(01)00324-1
34. Wei X-H, Na X-D, Liao G-J, et al. The up-regulation of IL-6 in DRG and spinal dorsal horn contributes to neuropathic pain following L5 ventral root transection. Exp Neurol. 2013;241:159–168. doi:10.1016/j.expneurol.2012.12.007
35. Wang W, Mei XP, Chen L, et al. Triptolide prevents and attenuates neuropathic pain via inhibiting central immune response. Pain Physician. 2012;15(6):E995–1006.
36. Zhuang Z-Y, Xu H, Clapham DE, Ji -R-R. Phosphatidylinositol 3-kinase activates ERK in primary sensory neurons and mediates inflammatory heat hyperalgesia through TRPV1 sensitization. J Neurosci. 2004;24(38):8300–8309. doi:10.1523/JNEUROSCI.2893-04.2004
37. Chen S-P, Zhou Y-Q, Liu D-Q, et al. PI3K/Akt pathway: a potential therapeutic target for chronic pain. Curr Pharm Des. 2017;23(12):1860–1868. doi:10.2174/1381612823666170210150147
38. Li D, Chen H, Luo X-H, Sun Y, Xia W, Xiong Y-C. CX3CR1-mediated Akt1 activation contributes to the paclitaxel-induced painful peripheral neuropathy in rats. Neurochem Res. 2016;41(6):1305–1314. doi:10.1007/s11064-016-1827-y
39. Cook AD, Christensen AD, Tewari D, McMahon SB, Hamilton JA. Immune cytokines and their receptors in inflammatory pain. Trends Immunol. 2018;39(3):240–255. doi:10.1016/j.it.2017.12.003
40. Matsuoka Y, Yamashita A, Matsuda M, Kawai K, Sawa T, Amaya F. The NLRP2 inflammasome in dorsal root ganglion as a novel molecular platform that produces inflammatory pain hypersensitivity. Pain. 2019;160(9):2149–2160. doi:10.1097/j.pain.0000000000001611
41. Wu W, Peden DB, McConnell R, Fruin S, Diaz-Sanchez D. Glutathione-S-transferase M1 regulation of diesel exhaust particle-induced pro-inflammatory mediator expression in normal human bronchial epithelial cells. Part Fibre Toxicol. 2012;9(1):31. doi:10.1186/1743-8977-9-31
42. Hwang S-Y, Kim J-Y, Kim K-W, et al. IL-17 induces production of IL-6 and IL-8 in rheumatoid arthritis synovial fibroblasts via NF-κB-and PI3-kinase/Akt-dependent pathways. Arthritis Res Ther. 2004;6(2):R120. doi:10.1186/ar1038
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.