Back to Journals » International Journal of Nanomedicine » Volume 21
Intratracheal Administration of pH-Responsive Nanomicelles: A More Effective Strategy for Enhanced Intracellular Drug Delivery in Lung Cancer Treatment
Authors Lin L, Wang G ![]() , Zhang J, Luo J
, Zhang J, Luo J ![]() , Fan X, Gao J, Cai X
, Fan X, Gao J, Cai X
Received 16 August 2025
Accepted for publication 6 February 2026
Published 10 March 2026 Volume 2026:21 555824
DOI https://doi.org/10.2147/IJN.S555824
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr RDK Misra
Luting Lin,1– 3 Guowei Wang,4 Jie Zhang,5 Jingming Luo,1,2 Xudong Fan,1 Jianqing Gao,6 Xinjun Cai1,2
1Department of Pharmacy, Hangzhou Red Cross Hospital, Hangzhou, People’s Republic of China; 2School of Pharmaceutical Sciences, Zhejiang Chinese Medical University, Hangzhou, People’s Republic of China; 3Department of Pharmacy, The Fourth Affiliated Hospital of Zhejiang University School of Medicine, Yiwu, People’s Republic of China; 4Department of Ultrasound in Medicine, The Second Affiliated Hospital of Zhejiang University School of Medicine, Hangzhou, People’s Republic of China; 5Department of Pharmaceutics, Jiaxing Key Laboratory for Photonanomedicine and Experimental Therapeutics, College of Medicine, Jiaxing University, Jiaxing, People’s Republic of China; 6College of Pharmaceutical Sciences, Zhejiang University, Hangzhou, People’s Republic of China
Correspondence: Xinjun Cai, Email [email protected] Jianqing Gao, Email [email protected]
Background: The efficacy of chemotherapy for treating lung cancer is hindered by insufficient intracellular drug utilisation. Moreover, non-targeted distribution often leads to severe side effects, resulting in poor prognosis and low patient compliance. Therefore, a more effective strategy is required to achieve effective treatment. In this study, we aimed to develop a pH-responsive nanoplatform for intratracheal administration to enhance drug accumulation in lung cancer tissues and promote the accumulation of drugs within tumour cells.
Results: A self-assembled nanomicelle named SN-38@PEG-PMMSD (PPM) was constructed using a cinnamaldehyde synthetic carrier material loaded with SN-38 and nanoprecipitation. Intratracheal administration enhanced the accumulation of PPM within the lungs and tumors (the fold increase in lung accumulation following intratracheal (i.t.) were 49.63-fold higher than intravenous (i.v.) delivery at the 48-hour timepoint). Owing to its small size, PPM can easily penetrate deep into tumour tissues. The micro-acidic environment characteristic of tumours increases the efficiency of tumour cell uptake of PPM. This triggered a pH-responsive reaction in the acidic lysosomal milieu, leading to dissociation of PPM and the regeneration of cinnamaldehyde while releasing SN-38. Cinnamaldehyde acted as a reactive oxygen species (ROS) amplifier, facilitating ROS generation. Elevated ROS levels, in conjunction with SN-38, resulted in strong antitumor effects.
Conclusion: In summary, Intratracheal administration of pH-responsive PPM is anticipated to enhance drug accumulation in tumour tissues, improve drug uptake by tumour cells, and achieve effective treatment of lung cancer.
Keywords: intratracheal administration, cinnamaldehyde, SN-38, lung cancer
Introduction
Lung cancer is one of the most prevalent malignant tumours globally, ranking second in terms of incidence and first in terms of mortality rates among all cancers.1 In clinical practice, 85% of lung cancer cases are classified as non-small cell lung cancer, which is characterized by poor prognosis and high metastatic potential, resulting in an extremely low 5-year survival rate.2,3 Current treatments for lung cancer, including surgical resection, chemotherapy, radiation therapy, and immunotherapy, have not significantly improved the prognosis of lung cancer. Chemotherapy, the primary treatment method for advanced-stage lung cancer, exhibits limited efficacy owing to its severe side effects and inadequate ability to deliver the therapy to the cancerous tissues.4 This underscores the need for improved local strategies to reduce local recurrence and minimise systemic side effects.
In recent years, pulmonary-targeted therapy has regarded as a promising approach for the treatment of NSCLC, and nanomedicines delivered via the pulmonary route have significant potential for the treatment of lung cancer.5–8 Inhalable nanoscale drug delivery systems offer a non-invasive alternative for direct and effective drug delivery to the lungs, enabling precise control and improvement of the delivery process and overall therapeutic efficacy, while addressing the non-specificity nature and rapid elimination associated with traditional pulmonary drug delivery.9 However, most inhalable therapies still face the formidable physiological barriers. The intricate architecture of the respiratory tract coupled with pulmonary clearance mechanisms poses significant challenges to nanoparticle deposition and retention in the lungs, including: (i) rapid mucociliary clearance in the upper airways, and (ii) phagocytic elimination by alveolar macrophages. Furthermore, drug losses during operation of clinical inhalation devices may compromise the actual delivered dose. Therefore, improving pulmonary retention requires limited mucus and absorptive clearance. Research indicates that nanoparticles coated with polyethylene glycol (PEG) have demonstrated the capability to passively diffuse through the mucus layer, which is attributed to PEG’s hydrophilicity, neutral charge, and non-adhesive properties.10 Therefore, this study will employ PEGylation to engineer mucus-insensitive nanoparticles, reducing mucus adsorption and enhancing clearance evasion. The pulmonary administration in this investigation will be performed via precise intratracheal administration to ensure localized drug deposition. Appropriately sized nanomedicines can effectively enhance the penetration of drugs into tumour sites.11,12 However, targeted delivery of nanomedicine to tumour cells is only one part of the requirements for achieving effective tumour treatment. Suboptimal drug concentrations within tumour cells can significantly affect therapeutic efficacy, underscoring the critical need to enhance intracellular drug accumulation.13,14 Refining the composition, shape, and size of nanoparticles enables enhancement of the uptake efficiency of nanoparticles.15–17
7-Ethyl-10-hydroxycamptothecin (SN-38) is the active metabolite of the first-line chemotherapy drug irinotecan (CPT-11) and exhibits 100–1000 times greater in vitro cytotoxicity than CPT-11, inducing DNA strand breaks in cancer cells.18 However, the clinical application of SN-38 is limited by its low water solubility, poor stability, and potential for inducing toxic side effects.19,20 The effective intracellular utilisation of SN-38 holds great significance for further development of highly efficient antitumour drugs. Elevating the level of reactive oxygen species (ROS) in tumour cells not only enhances antitumour efficacy,21–23 but also facilitates the disruption of the lysosomal membrane, thereby promoting the release of drugs into cells.24
Cinnamaldehyde (CA), which was approved by the Food and Drug Administration (FDA) as a commercial food additive, has been found to have potent antibacterial effects.25–27 Moreover, a significant body of literature has reported that CA and its derivatives demonstrate cytotoxic activity against cancer cells and suppress neoplastic proliferation via induction of apoptosis,28–30 which also function as ROS-generating agent or ROS scavengers under various conditions.31 Some studies have demonstrated that CA inhibits ROS production, reduces oxidative stress, and prevents DNA damage.32–34 While another studies have indicated its ability to stimulate ROS production in mitochondria,35–38 primarily through the depletion of intracellular thiols.35,39 Furthermore, CA demonstrates considerable potential as a carrier design material due to its high susceptibility to coupling and modification.29,36,38,40,41 Capitalizing on its pro-oxidative potential specifically, we engineered pH-responsive cinnamaldehyde-based polymeric nanostructures that leverage the acidic tumor microenvironment for precisely facilitate elevated intracellular ROS levels and enhanced intracellular drug accumulation in malignant cells.
Based on this, as illustrated in Graphical Abstract, we developed and constructed a SN-38@PEG-PMMSD nanomicelle (PPM) acid-responsive nanoplatform for pulmonary delivery to enhance drug accumulation in lung cancer tissues and promote the accumulation of drugs within tumour cells, thereby effectively treating lung cancer. First, PEG-PMMSD was synthesized, and then loaded with SN-38 to prepare PPM. After intratracheal administration, PPM exhibited higher accumulation in the lung and tumour tissues, with preferential uptake by tumour cells. The acidic microenvironment of the tumour further enhanced the uptake efficiency, thereby increasing drug accumulation within cells and facilitating the effective utilisation of PPM. Therefore, pH-responsive delivered via the pulmonary route are anticipated to augment drug accumulation in cancerous lung tissues, enhance the efficacy of drug uptake by tumour cells, and achieve effective treatment of lung cancer.
Materials and Methods
Materials
SN-38 was purchased from Selleck Co., Ltd. (Houston, Texas, USA). The PEG-PMMSD is synthesized by Xi’an Ruixi Biological Co., Ltd (Xi’an, China). FBS and DMEM were obtained from Gibco BRL (California, USA). ICG was obtained from Shanghai Aladdin Co., Ltd. (Shanghai, China). Beyotime Biotechnology Co., Ltd. (Shanghai, China) offers the DCFH-DA probe and lysosome staining kit. A CCK8 Kit was obtained from Shanghai Life iLab Biotech Co., Ltd. (Shanghai, China). TdT In Situ Apoptosis Detection Kit-Fluorescein was obtained from USA R&D Systems, Inc (Minnesota, USA). Ki-67 antibody is from Abcam Biotechnology Ltd. (Cambridge, UK).
Synthesis of MMSD Carrier and Preparation of PPM
The aldehyde group present in the molecular structure of CA was reacted with glycidoxypropane to yield 5-methyl-5-hydroxymethyl-2-styrenyl-[1,3]-dioxane (MHSD). Subsequent reaction of MHSD with methacryloyl chloride was carried out to synthesize end-capped double-bonded 5-methyl-5-methacryloyloxy-methyl-2-styrenyl-[1,3]-dioxane (MMSD). The chemical structure of MMSD was confirmed by 1H nuclear magnetic resonance (NMR) spectroscopy (Figure S1, Supplementary Materials). MMSD was polymerized to form the nanocarrier material, polyethylene glycol-poly(5-methyl-5-methacryloyloxy-methyl-2-styrenyl-[1,3]-dioxane) (PEG-PMMSD). Detailed synthesis procedures are provided in the Supplementary Materials (Supplementary Materials and Methods 1.1–1.3). Hereafter, the abbreviation “MMSD” will be used throughout the text. The chemical structure and purity of the resulting copolymer was characterized (Figures S2 and S3; 1H and 13C NMR spectroscopy), which confirmed the successful synthesis of PEG-PMMSD (MMSD). These analytical data provided valuable evidence for the subsequent preparation of the pH-responsive polymeric micelles (PPM).
PPM were prepared by nanoprecipitation method. A solution was prepared by dissolving 2 mg of SN-38 and 10 mg of PEG-PMMSD in 400 μL of dimethyl sulfoxide (DMSO). The resulting mixture was gradually and uniformly injected into 1.6 mL of ultrapure water using a syringe while being continuously stirred with a magnetic stirrer. The reaction was allowed to proceed in darkness for 1 h after the injection was completed. Unencapsulated SN-38 was removed by dialysis using a pre-treated dialysis membrane (molecular weight cut-off: 8000–14,000 Da). The PPM mother liquor was transferred into the dialysis bag and dialyzed against ultrapure water for 6 h, with the water being replaced hourly. Finally, the purified SN-38-loaded PPM were obtained and stored at 4°C for further use. The same procedure was followed for the preparation of blank, with the omission of SN-38.
Characterization of PPM
The PPM nanocarrier solution, prepared as previously described, was diluted with ultrapure water at a 50-fold ratio and subsequently filtered by a 0.22-μm filter membrane for storage. The size (Size), zeta potential (Zeta), and dispersity index (PDI) of the PPM were determined using a Malvern Zetasizer under conditions of 25°C and an equilibrium time of 120 sec. Each parameter was measured three times for each sample.
The PPM solution was diluted with ultrapure water at a ratio of 1:500 (v/v) and subsequently sonicated for 10 min to ensure complete homogenization prior to further use. A few droplets of the diluted PPM solution were carefully deposited onto a hydrophobic sealing film. A carbon-coated copper mesh (TEM-grade) was then grasped at its edge using fine-tipped tweezers, and its front side was gently brought into contact with the liquid droplet on the film. The sample was allowed to dry under a cool and well-ventilated environment. Subsequently, negative staining was performed using phosphotungstic acid for 30 sec, followed by blotting to remove excess stain. Morphological and size characteristics of the PPM were examined using a transmission electron microscope (TEM).
The concentration of SN-38 was measured by an Agilent high-performance liquid chromatography system equipped with a C18 column., acetonitrile: water (containing 0.25% formic acid) as the mobile phase at a ratio of 32.5: 67.5, a flow rate of 1 mL/min, detection wavelength set to 372 nm and column temperature maintained at 30°C. PPM was diluted tenfold with acetonitrile (containing 10% DMSO), vortexed for ten minutes and centrifuged at 12,000 rpm for 10 minutes to obtain the supernatant. The SN-38 concentration was then determined and used to calculate encapsulation rate while drug loading (DL%) and encapsulation efficiency (EE%) were computed by means of the following formula:

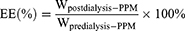
Quantitative HPLC analysis demonstrated that PPM prepared at the SN38:PMMSD ratio of 1:5 exhibited a drug loading capacity (DL) of 19.57±2.17% and encapsulation efficiency (EE) of 88.72±5.11% (Table S1). Comparative evaluation revealed this formulation ratio achieved optimal drug payload and protective encapsulation performance among all tested proportions. Consequently, the SN38: PMMSD = 1: 5 ratio was established as the standard preparation protocol for PPM systems.
Determination of Critical Micelle Concentration
The critical micelle concentration (CMC) of PPM was determined using fluorescence spectroscopy. PPM was diluted and a series of gradient concentrations were prepared. Different concentrations of PPM solution (100 μL each) were mixed with 2 μL of 250 μM Rhodamine 6G (resulting in a final concentration of 5 μM) and then incubated for 48 h. The maximum optical density value wavelength (525 nm ~ 548 nm) was measured to calculate the CMC value.
In vitro Drug Release Study
The dialysis bag method was used to investigate the release of drug in different pH conditions. HPLC-based measurement of SN-38 concentrations (the SN-38 release via PPM cleavage) was utilized to calculate its cumulative release under different pH conditions (pH5.0; pH6.5; pH7.4). A 1 mL aliquot of the PPM solution containing SN-38 at a concentration of 500 μg/mL is to be transferred into the pretreated dialysis bag, which must be secured within an amber glass bottle pre-filled with phosphate-buffered saline (PBS) solutions at pH 7.4, 6.5, and 5.0, respectively. The mixture was shaken at a constant speed of 300 rpm in a water bath incubator maintained at 37 ± 0.5 °C. The external dialysate was collected at predetermined time intervals, with 1 mL aliquots being withdrawn each time and replaced with an equal volume of the corresponding fresh medium to maintain sink conditions. The concentration of SN-38 was quantified via high-performance liquid chromatography (HPLC). Based on the obtained data, the cumulative release rate was calculated, and the drug release profile was subsequently plotted.
Cytotoxicity Test and Hemolysis Test
The cell line Lewis lung carcinoma (LLC); LLC-Luciferase cells and the human bronchial epithelial cells (BEAS-2B) were purchased from Hunan Fenghui Biotechnology Co., Ltd. (Chang Sha, China). The LLC cells/BEAS-2B cells were maintained in DMEM growth medium supplemented with 10% FBS, penicillin and streptomycin (100 U/mL). The in vitro cytotoxicity of PPM and SN-38 on LLC cells/BEAS-2B cells was measured by Cell counting kit-8 (CCK-8) assay. Cells were incubated for 24 h with PBS, PMMSD, SN-38 and PPM (the concentration of SN38 was 0–0.4 μg/mL). Then, 100 μL of DMEM (containing 10% CCK-8 reagent) was added, and incubated for an additional 0.5–1 h. Finally, the absorbance was measured at 450 nm using a multifunctional enzyme marking instrument (Thermo Scientific™ Varioskan™ LUX, USA). Data are presented as mean ± SD. One-way ANOVA was used for intergroup comparisons, followed by appropriate post-hoc tests (Tukey’s HSD).
A 2% (v/v) red blood cell suspension sourced from rabbits was prepared using normal saline. PPM solutions containing SN-38 at concentrations of 0.05, 0.1, 0.2, 0.4 and 0.8 μg/mL were prepared with normal saline. Subsequently, these PPM solutions were mixed with the red blood cell suspension at a 1:1 ratio, followed by incubation at 37°C for 1 h. After that, the mixtures were centrifuged at 3000 rpm for 10 min, and the supernatants were collected. The absorbance at 414 nm was measured by the multifunctional microplate reader. Pure water and saline were utilized as positive and negative controls, respectively. The hemolysis rate of PPM was computed in accordance with the following formula:

Cell Uptake Assay
LLC cells (1.5×105) and BEAS-2B cells (5×104) were individually seeded into laser confocal culture dishes and cultured. The cells were incubated with complete culture medium containing PPM (with an ICG concentration of 5 μg/mL) for 2 hours. After PBS washing and fixation in 4% paraformaldehyde (20 min, dark), the cells were stained with DAPI (15 min, dark), washed again, and observed by CLSM (Leica, Germany) to assess ICG uptake. Furthermore, LLC cells and BEAS-2B cells were co-cultured and subjected to identical drug treatment before capturing images. Data are presented as mean ± SD. The t-test was used to compare differences.
Reactive Oxygen Species Detection Study
The LLC cells were seeded in a confocal dish and incubated overnight with a complete culture medium containing SN-38, PMMSD, and PPM (SN-38: 0.1 μg/mL) for 24 h. Subsequently, the original culture medium was discarded, and the cells were rinsed with PBS. Then, 100 μL of DMEM (containing 0.1% DCFH-DA probe solution) was added and incubated at 37°C in a CO2 incubator for 25 min, washed with DMEM, and observed under the CLSM (excitation wavelength: 488 nm, Leica, Germany).
The LLC cells were seeded in well plates and were incubated for 24 h with PBS, PMMSD, SN-38 and PPM (SN-38: 0.1 μg/mL). Subsequently, cells were collected per the instructions and loaded with the probe (probe loading procedure as described above). The absorbance was measured at 488 nm using a multifunctional enzyme marking instrument (Thermo Scientific™ Varioskan™ LUX, USA). Quantification was performed by: The absorbance measurements were blank-corrected, and cell counts were determined using a hemocytometer. Calculating fluorescence intensity per unit cell quantity. Expressing results as percentage relative to control group mean fluorescence. Data are presented as mean ± SD. One-way ANOVA was used for intergroup comparisons, followed by appropriate post-hoc tests (Tukey’s HSD).
Lysosome Staining Experiment
LLC cells were maintained overnight in confocal-compatible culture dishes. At designated time intervals, incubation was initiated with complete medium supplemented with PPM (containing ICG). Following manufacturer’s protocol for lysosomal staining, the primary medium was aspirated and replaced with 100 μL of basal medium containing LysoTracker Red (1:1000 dilution). Subsequent 30-minute incubation at 37°C was performed, followed by three washes with basal medium. Cellular imaging was ultimately conducted using the CLSM (LysoTracker Red excitation wavelength: 561 nm; ICG excitation wavelength: 638 nm).
In vivo Distribution Study
The male C57 mice of SPF-grade were procured from the Laboratory Animal Research Center of Zhejiang Chinese Medical University. All animal-related experiments were carried out in accordance with the guidance of the Zhejiang Chinese Medical University Animal Care and Use Committee (IACUC-20230227-02). After 2 weeks of acclimation, animals were inoculated with LLC-Luc cells (3×105 cells for each mouse) using direct injection into the lungs method to establish an in situ model of lung cancer.9 Bioluminescence imaging using the IVIS Spectrum system (PerkinElmer, USA) demonstrated successful engraftment of lung cancer xenografts. After intraperitoneal injection of D-luciferin (150 mg/kg) with 5 min biodistribution, tumor progression was monitored via the in vivo imaging system, with subsequent histological confirmation by H&E staining (Figure S4).
At 10 days post-implantation of orthotopic lung cancer cells, the tumour-bearing mice were categorized into three groups: ICG group (intrabronchial administration, i.t)., PPM group (intrabronchial administration, i.t)., and PPM group (intravenous administration, i.v). The drugs containing ICG at 400 μg/kg (calculated by ICG) were administered to the respective groups. Tracheal instillation was performed for intrabronchial administration as follows: After anesthetizing the mice, tracheal intubation was conducted followed by slow injection of the drug using a needle-free syringe. Approximately 500 μL of air was then slowly injected to ensure even distribution of the drug. The mice were sacrificed at 24 h and 48 h post-injection to collect fluorescence intensity signals from major tissues (heart, liver, spleen, tumour-bearing lung tissue, kidney) using IVIS (PerkinElmer, USA). Data are presented as mean ± SD. One-way ANOVA was used for intergroup comparisons, followed by appropriate post-hoc tests (Tukey’s HSD).
Study of Aggregation and Osmosis in vivo
Following 24- and 48-hour drug administration (dosing protocol detailed in Section 2.10) to lung cancer-bearing mice, the tumour-bearing lung tissues were fixed in a solution of 4% paraformaldehyde, dehydrated, embedded, sectioned, and subsequently dewaxed using water. Following these procedures, DAPI staining was conducted to visualize the penetration of PPM (containing ICG) into the tumour under the fluorescence confocal microscope.
In vivo Antitumour Study
By employing the in situ injection method, a murine model of in situ lung cancer was established. One week later, mice were randomly divided into four groups (five mice each) as follows: Control, PMMSD, SN-38 (0.5 mg/kg), PPM group (containing 0.5 mg/kg SN-38). The mice in each group were given the drug by intrabronchial administration every 3 days for a total of 9 days. The general condition of the mice was recorded, and the tumour size was observed by IVIS imaging system. Data are presented as mean ± SD. One-way ANOVA was used for intergroup comparisons, followed by appropriate post-hoc tests (Tukey’s HSD).
Intracellular Nanomicelles Observation Experiment
LLC cells were treated with PPM at different times, fixed with glutaraldehyde and collected, washed, fixed with 1% osmic acid, dehydrated, embedded, stained, and observed by TEM (Hitachi-7650).
Immunohistochemistry Study
Mice lung cancer tissue samples were fixed, dehydrated and embedded. Next, the sections underwent dewaxing, hydration, and were repaired and inactivated, incubated with primary antibodies (Ki-67, 1:200), washed, incubated with secondary antibodies. Diaminobenzidine was used for development. Then, the slides were stained with hematoxylin and sealed. Finally, observing by a light microscope (Leica, Germany). Data are presented as mean ± SD. One-way ANOVA was used for intergroup comparisons, followed by appropriate post-hoc tests (Tukey’s HSD).
TUNEL Assay
The sections of mice lung cancer tissues were processed exactly according to the instructions, and finally sealed with DAPI-containing capping tablets. The samples were observed by the CLSM. Data are presented as mean ± SD. One-way ANOVA was used for intergroup comparisons, followed by appropriate post-hoc tests (Tukey’s HSD).
Statistical Analysis
GraphPad 8 was utilized for statistical analysis. The number of cells for constructing the in vivo models in this study was determined by previous experiments, and the in vitro assays were repeated at least three times. Data are showed as the mean ± SD (x ± s), with the One-Way ANOVA and t-test being used to compare differences between groups. Statistical significance was set at p < 0.05.
Results
Preparation and Construction of PPM
In this study, we developed a novel carrier based on CA (Figure 1A). The SN-38@PEG-PMMSD nanomicelles were prepared using a simple nanoprecipitation method (PPM; see Graphical Abstract). The morphology of the PPM was analysed using transmission electron microscopy (TEM, Figure 1B). The hydrated particle size and surface zeta potential of PPM were analysed by dynamic light scattering (DLS) techniques. The results showed that PPM exhibited a spherical shape with a relatively uniform dispersion, with a mean size of 106.4 ± 10.1 nm and a PDI of 0.179 ± 0.0313 by DLS (Figure 1C). The zeta potentials were −15.27 ± 1.8 mV (Figure 1D). Additionally, the critical micelle concentration (CMC) of PPM was determined using fluorescence spectroscopy, as depicted in Figure 1E, which was 0.064 μg/mL, indicating the excellent dilution capability of PPM. Furthermore, the DL capacity of PPM reached 19.57 ± 2.17%, accompanied by the EE of 88.72 ± 5.11% (Table S1), demonstrating its effective encapsulation and protection of drugs. This indicated that the material can effectively encapsulate and protect drugs. Furthermore, the stability of the PPM was sufficient, which helped to minimise the premature release of SN-38 and mitigate non-specific toxicity to adjacent normal lung parenchymal tissue.
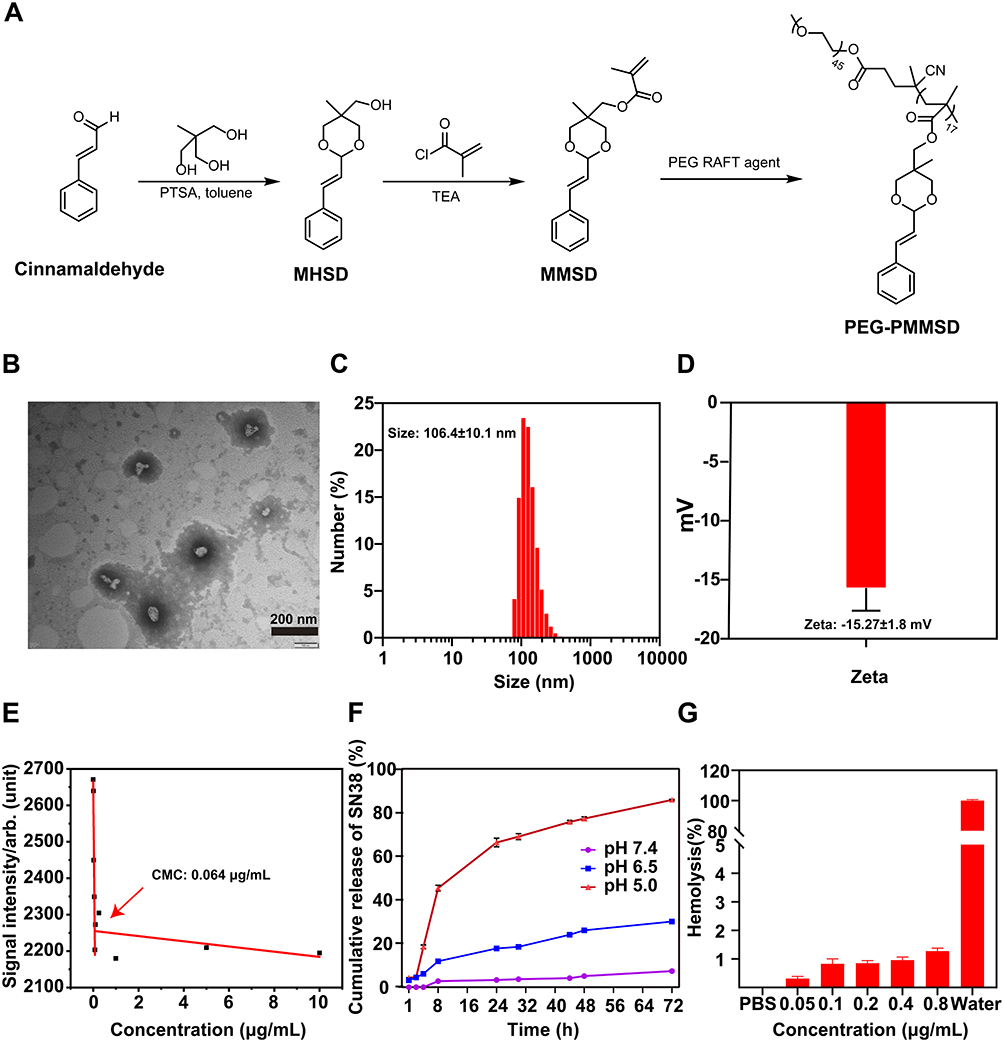 |
Figure 1 Characterization of SN-38@PEG-PMMSD (PPM) nanomicelles was conducted as follows: (A) Synthesis process of PEG-PMMSD. (B) Morphology was evaluated using transmission electron microscopy (TEM). Scale bar = 200 nm. The size (C) and zeta (D) potential of PPM, as determined by dynamic light scattering (DLS). (E) Determination of critical micelle concentration in PPM. (F) In vitro release characteristics of PPM. (G) In vitro haemolysis analysis of PPM. The data are presented as the mean ± SD (n = 3). |
The in vitro release profile was determined by dialysis, as illustrated in Figure 1F, which demonstrated that PPM demonstrates remarkable pH-dependent behavior. In general, the tumour microenvironment exhibits a lower pH level compared to the normal physiological environment due to the rapid proliferation of cells and increased proton excretion during tumourigenesis.42 At pH 6.5, which reflects that of the tumour microenvironment, PPM exhibited a gradual release of SN-38; however, this release rate was markedly lower than that at pH 5.0 (which is an acidic lysosomal environment). This discrepancy can be attributed to the increased propensity for bond cleavage in the styrene moiety of the MMSD dioxane structure under acidic conditions, leading to the regeneration of CA and the subsequent release of SN-38.
Prior to conducting in vivo and in vitro evaluations of antitumour efficacy, the biocompatibility of the PPM was assessed. As illustrated in Figure 1G, the results from the in vitro hamolysis test indicated that the hamolysis rate for 0.8 µg/mL PPM was less than 2% when compared to distilled water, thereby demonstrating its favourable blood compatibility.
In vitro Anti‑tumour Therapy and Intracellular Transport of PPM
The cytotoxic effects of PPM on the BEAS-2B and LLC cell lines were assessed using a CCK-8 assay. Following 24-hour incubation with SN-38/PPM, PPM exhibited significantly stronger cytotoxicity against LLC cells than SN-38 (p < 0.0001; Figure 2A). Dose-response analysis identified 0.1 µg/mL as the optimal concentration for subsequent experiments, where SN-38 alone demonstrated significant inhibitory effects on LLC cells (p < 0.0001). As shown in Figure 2B, PPM demonstrated significantly higher cytotoxicity toward LLC cells than BEAS-2B cells (p < 0.05), whereas SN-38 exhibited comparable toxicity in both cell lines (p > 0.05). This tumor-selective cytotoxicity of PPM contrasted sharply with the nonspecific activity of free SN-38, underscoring the targeting advantage of the PPM.
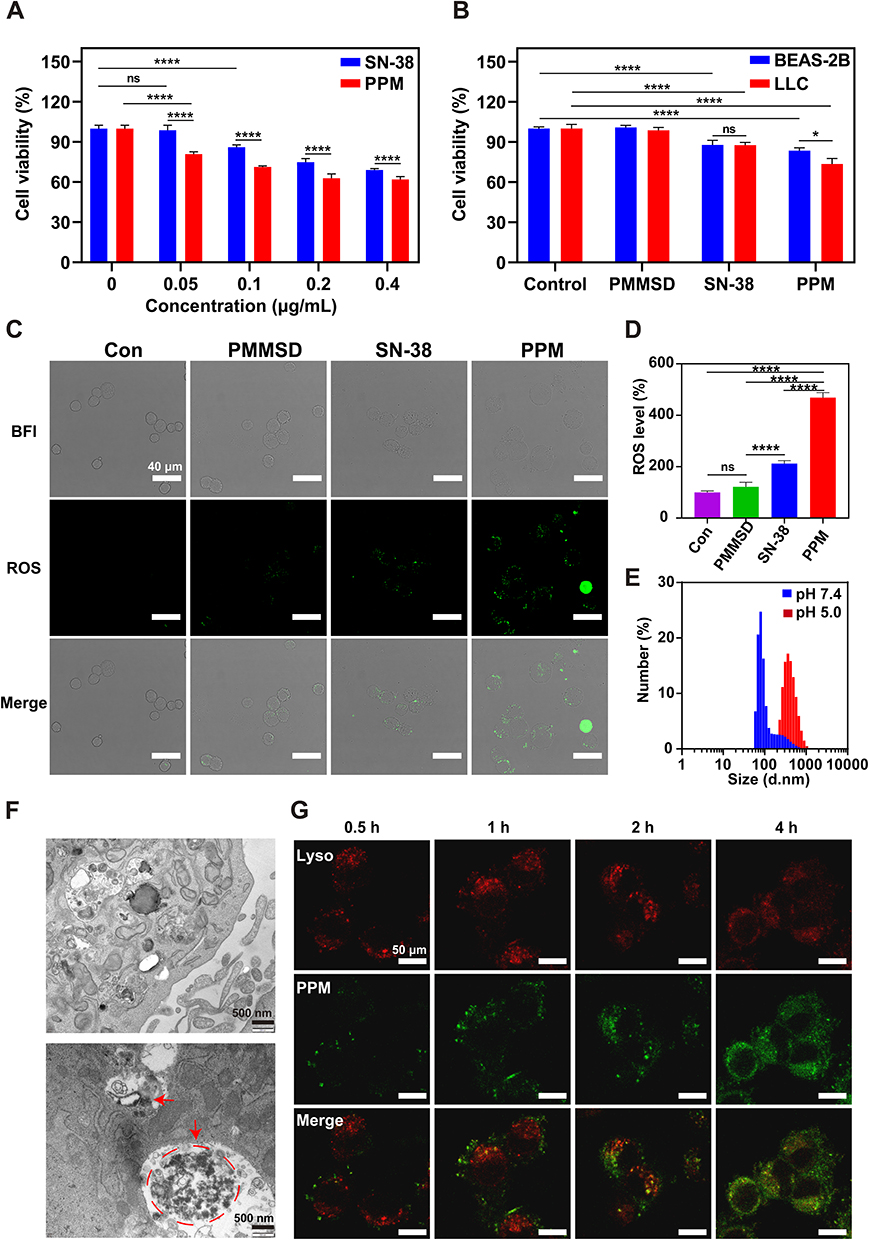 |
Figure 2 In vitro antitumour therapy and intracellular transport of PPM. (A) Cell viability of LLC cells treated with different concentrations of SN-38/PPM for 24 h, n = 6. (B) Cellular activity of BEAS-2B and LLC cells under different dosing regimens (including SN-38 0.1 μg/mL) (n = 5). The values of cell viability represent viability relative to the untreated control group (mean set as 100%). (C) Fluorescence images of ROS in LLC cells after 24 h of drug intervention. Scale bar = 40 μm. (D) Calculating fluorescence intensity of ROS per unit cell quantity (n=3). The values of cell viability represent viability relative to the untreated control group (mean set as 100%). (E) The size of PPM particles after incubation for 2 h in various pH environments. (F) Transmission electron microscopy images of PPM in LLC cells; the red arrow and red colored dotted circle indicate the position of PPM. Scale bar = 500 nm. (G) Subcellular distribution of the PPM in tumour cells. Lysosomes were stained with LysoTracker Green (red), and PPM was labelled with ICG (green). Scale bar = 50 μm. Data are presented as mean ± SD. One-way ANOVA was used for intergroup comparisons, followed by appropriate post-hoc tests (Tukey’s HSD), nsp > 0.05, *p < 0.05, ****p < 0.0001. |
The effect of PPM on ROS generation in LLC cells was evaluated. Compared to SN-38, PPM markedly elevated intracellular ROS levels, whereas PEG-PMMSD had limited effect (Figure 2C and D), likely due to cellular redox homeostasis counteracting CA effect. Although CA alone demonstrated modest ROS-inducing capacity in LLC cells, its combination with SN-38—under conditions of SN-38-induced cellular damage—significantly amplified ROS production (p < 0.0001), correlating with enhanced tumor cell lethality.
The pH-dependent hydrodynamic size variation of PPM was characterized by DLS. Notably, PPM exhibited significantly larger hydrated diameters under acidic conditions (pH 5.0) compared to physiological pH 7.4 (Figure 2E), consistent with the enhanced aggregation propensity observed in Figure S4. Intracellular trafficking analysis via TEM revealed PPM accumulation within lysosomes, forming aggregates up to 2 μm in diameter (Figure 2F). This phenomenon likely stems from acid-triggered hydrolysis of the polystyrene segments in lysosomal compartments, leading to SN-38 release. The pronounced hydrophobicity of liberated SN-38 further promoted aggregation, potentially contributing to the escape of PPM from the organelle. Subsequently, to monitor intracellular trafficking, PPM was fluorescently labeled with indocyanine green (ICG; with an excitation wavelength of 760 nm, denoted as green fluorescence). Confocal imaging revealed time-dependent PPM internalization, with progressive accumulation of green fluorescence (ICG-PPM) in the cytoplasm (Figure 2G). Concomitantly, the signal intensity of lysosomal tracer red (with an excitation wavelength of 560 nm) diminished, suggesting the occurrence of lysosomal escape.
In vitro Cellular Uptake
To evaluate the tumor-selective uptake of PPM, LLC (murine Lewis lung carcinoma) and BEAS-2B (human normal bronchial epithelial) cells were treated with PPM and analyzed via confocal microscopy and quantitative assays (Figure 3A-C, Figure S5 showed the size bar values for Figure 3). Notably, LLC cells exhibited significantly higher PPM internalization than BEAS-2B cells (p < 0.05; Figure 3B and C). Further, acidic conditions (pH 6.5) enhanced PPM uptake in LLC cells by approximately 2.47-fold compared to physiological pH (7.4), confirming its pH-responsive tumor-targeting capability (Figure S6).
 |
Figure 3 The internalization of PPM in LLC/BEAS-2B cells was investigated in both single and co-culture systems under varying pH conditions for 2 h. (A) Uptake of PPM by LLC and BEAS-2B cells was examined using confocal microscopy at pH 7.4 and pH 6.5. Scale bar = 50 μm. The fluorescence intensity of PPM uptake was quantified at pH 7.4 (B) and pH 6.5 (C). The uptake of PPM by LLC/BEAS-2B cells within the co-culture system was assessed via confocal microscopy under both pH 7.4 (D) and pH 6.5 (E). The yellow arrow indicates LLC cells, and the white arrow indicates BEAS-2B cells. Scale bar = 50 μm. The data are presented as the mean ± SD (n = 3). T-test was used to compare differences between groups, *p < 0.05. |
Subsequently, to visually assess the selective uptake of PPM, LLC cells and BEAS-2B cells were co-cultured, and the pH-dependent internalization kinetics were investigated. Figure 3D and E and Figure S7 confirmed that LLC cells preferentially internalised PPM aligning with prior findings. In conclusion, these results demonstrate PPM’s pronounced tumor-selective internalization in vitro, highlighting its potential as a pH-responsive, cancer-targeting candidate.
Penetration of Drugs Delivered by SN-38@PEG-PMMSD into the Lung Cancer Parenchyma
After confirming the uptake of the PPM in vitro, we investigated their fate in an in vivo using a mouse model of in situ lung cancer (Figure S8). Initially, the tissue distribution of PPM after intrapulmonary administration and intravenous injection was compared using ex vivo fluorescence imaging. The fluorescence images in Figure 4A demonstrate that the intratracheally delivered PPM were primarily distributed in the tumours and lung tissues of mice with lung cancer. The fluorescence intensity (FI) was relatively higher in tumour-bearing lung tissue, with high accumulation observed after 48 h. However, intravenously administered PPM was primarily distributed to extrapulmonary organs (such as the liver, spleen, and kidneys), exhibiting relatively lower fluorescence intensity in tumours and lung tissue. Furthermore, we conducted a semi-quantitative analysis of fluorescence intensity in the lungs, tumour tissues, and other peripheral organs following pulmonary administration and intravenous injection at 24 and 48 h. After pulmonary administration of 40 μL of PPM with an ICG concentration of 80 μg/mL for 24 h and 48 h, the area under the receiver operating characteristic curve (AUC) values of lung tissue fluorescence intensity were 14.83-fold and 49.63-fold higher than those following intravenous injection of the same dose of PPM, respectively (Figure 4B and C).
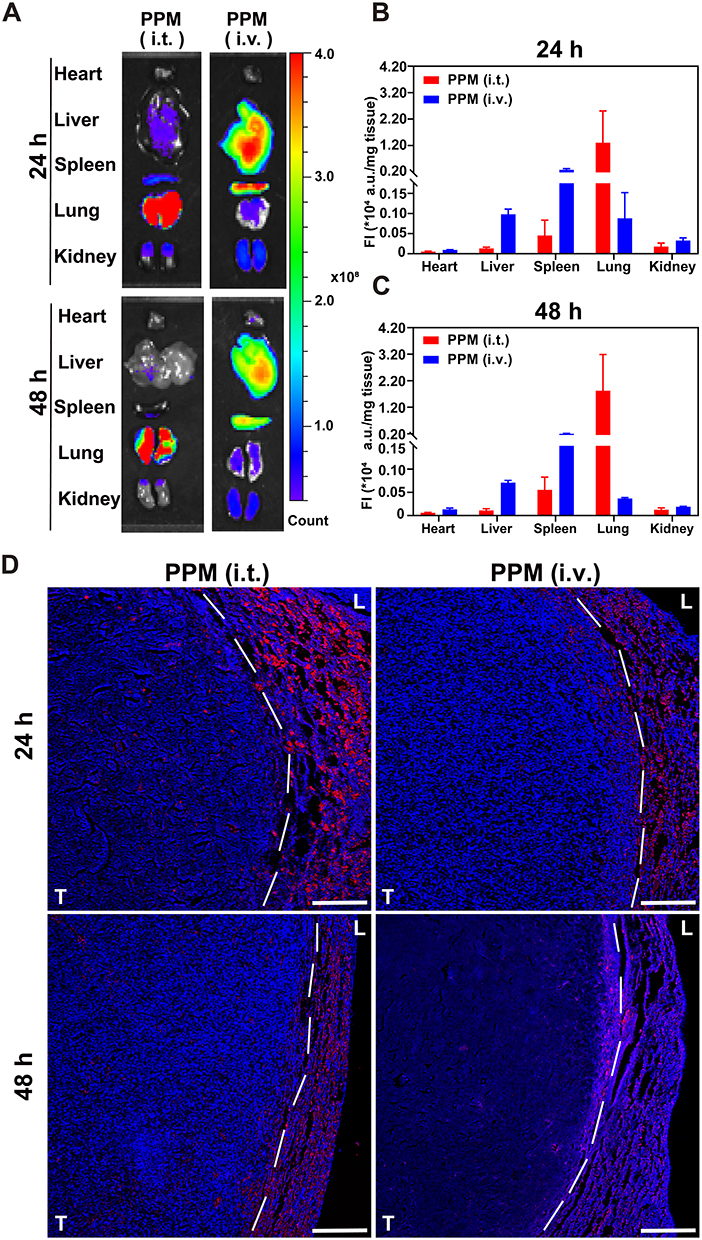 |
Figure 4 Intra-body process of PPM. (A) Ex vivo images of tumorous pulmonary tissues and major organs after 24 h/48 h of administration. Quantitative analysis of fluorescence intensity of tumours and major organs after 24 h (B)/48 h (C) of administration (p/s/cm2/sr/mg), n = 3. (D) Localization of PPM (red) in lung and tumour tissues of orthotopic lung tumour-bearing mice after intratracheal (i.t.) or intravenous (i.v.) administration. Cell nuclei were stained with DAPI (blue); scale bar = 100 μm; L and T represent lung and tumour, respectively. |
Additionally, PPM demonstrated deep penetration into the tumour (Figure 4D). The accumulation of PPM in tumour-bearing lung tissues was consistent with its distribution in ex vivo tissues. After intraluminal administration of PPM to mice with localised lung cancer, the lung and tumour tissues were examined using DAPI-stained staining. In the figure, the dashed line indicates the boundary between the tumour and lung tissues based on the differences in cell nuclear density. The fluorescence intensity within the tumour decreased over time, indicating sustained high distribution and good drug retention of PPM (depicted by red fluorescence) penetrating deep into the lung tumours within 24 h after administration, as shown in Figure 4D. In contrast, PPM exhibited diminished fluorescence intensity at the tumour site and reduced penetration into the tissues following intravenous administration. It accumulated at the interface between the tumour and lung tissue within 48 h, indicating its rapid clearance from the circulation.
Antitumour Effect in vivo
To further explore their in vivo therapeutic effects, PBS, PMMSD, SN-38, and PPM at a dose of 0.5 mg/kg were administered intratracheally to the LLC-Luc in situ lung cancer model. Tumor-bearing mice were randomized into four treatment groups (Figure 5A) one week post tumor inoculation. Tumor volume was monitored via a small-animal live imaging system. Treatment was administered every 3 days for a total of three cycles, and tumor progression was assessed on days 5 and 9 (Figure 5B–D). In the treatment groups, both SN-38 and PPM showed anti-tumor activity against lung cancer, and PPM had superior tumor-suppressive effects. In contrast, PMMSD showed minimal therapeutic impact (Figure 5B–E).
 |
Figure 5 Antitumour effect in vivo. (A) Intrabronchial instillation of nanomicellar PPM for the treatment of LLC-Luc in situ carcinoma mouse models. (B) Bioluminescent images of primary lung cancer mice in vivo on days 5 and 9 post-administration, and quantification of fluorescence signals in tumours on day 5 (C) and day 9 (D), n = 5. (E) Changes in the weight tumour-bearing mice in each group, n = 5. (F) Weights of the lungs and tumours in each group, n = 5. (G) H&E, immunohistochemical (Ki-67) and TUNEL staining of tumour tissues in each group after drug treatment; scale bar = 50 μm. (H) LDH levels in the BALF after treatment with PBS, free SN-38, PMMSD, and PPM, and the liver-related serum levels of (I) ALT and (J) AST, n = 3. Data are presented as mean ± SD. One-way ANOVA was used for intergroup comparisons, followed by appropriate post-hoc tests (Tukey’s HSD), *p < 0.05. |
The control group showed the earliest weight loss during the treatment period (Figure 5F). The antitumour effects of PPM were investigated using a series of histological studies. Tumour tissue sections were stained with hematoxylin and eosin (H&E), as showed in Figure 5G. Tumour cells in the control group exhibited full and densely packed characteristics, whereas those treated with PPM displayed severe nuclear shrinkage and fragmentation, which are typical morphological features of apoptosis. Similar alterations were observed in the SN-38 group but to a significantly lower degree. Additionally, Ki-67 protein expression and TUNEL staining were performed on the tumour tissues to assess the impact of different treatments on cell proliferation inhibition and apoptosis. Compared with the other groups, the PPM group exhibited a significant decrease in the number of Ki-67 positive cells (as indicated by brown granules) (Figures 5G and S9A), suggesting effective inhibition of tumour cell growth. TUNEL staining revealed a significantly higher number of apoptotic cells in the tumour tissues of the PPM group compared with those in the other groups, as indicated by green fluorescence (Figures 5G and S9B). The substantial therapeutic effect of PPM on lung cancer tissues in vivo was strongly consistent with the in vitro cytotoxicity data. This demonstrates that the pH-responsive nanomicelles of PPM, when administered via the lungs, can significantly increase drug accumulation in cancerous lung tissues and exhibit strong selectivity for tumour cells, thus demonstrating their potential for tumour therapy.
As previously mentioned, this strategy for intratracheal administration with acid-responsive release can minimise the nonspecific targeting of the drug to normal tissues. In vivo pulmonary toxicity was assessed by quantifying the levels of lactate dehydrogenase (LDH) in the bronchoalveolar lavage fluid of mice across various treatment groups (Figure 5H). Additionally, serum levels of alanine aminotransferase (ALT) and aspartate aminotransferase (AST) were measured to evaluate the liver function toxicity associated with each treatment regimen (Figure 5I-J). Notably, the LDH, ALT, and AST levels observed in the PPM group did not differ significantly from those in the control group and remained within normal physiological ranges. The results presented in Figure S10 further corroborates this conclusion. These findings indicate that PPM does not exhibit significant liver toxicity and demonstrates favourable biocompatibility and biosecurity. Concurrently, compared to those in the control group, ALT levels in the SN-38 group exhibited a marked increase, whereas AST levels showed an upward trend, suggesting potential chemotherapy-induced liver toxicity (Figure 5I). In conclusion, PPM outperformed SN-38 in terms of biocompatibility and antitumour efficacy.
Discussion
To address the clinical challenges of inadequate drug delivery and suboptimal therapeutic outcomes in lung cancer, we developed an intratracheal self-assembling nanomicellar system (PPM) co-loaded with CA and SN-38. This system enhanced tumor accumulation via intratracheal administration. The pH-sensitive property of PPM triggers a size-switching mechanism, facilitating lysosomal escape and significantly enhancing intracellular drug accumulation in tumor cells. Moreover, PPM further enhanced reactive oxygen species (ROS) generation compared to free SN-38 alone, with CA and SN-38 synergistically or additively achieving significant antitumor efficacy.
Our findings supported the notion that intratracheal administration strategies would minimize systemic exposure while maintaining therapeutic concentrations at the tumor site. Following intratracheal administration, the AUC values of lung tissue fluorescence intensity exhibited 14.83-fold (24 h post-dose) and 49.63-fold (48 h post-dose) increases compared to intravenous injection (Figure 4B and C). In contrast to pulmonary drug delivery targeting the tumour-bearing lung tissues, intravenous drug delivery involves the circulation of PPM in the blood system, which appears to be more susceptible to metabolism.43 The prolonged tumor retention of intratracheally administered PPM establishes an extended therapeutic window for in vivo antitumor therapy, partially attributable to their pH-responsive aggregation propensity in acidic microenvironments (Figures 2E and F and S4).
Larger nanoparticles exhibit limited penetration into the deep tissues of tumours, leading to inadequate delivery within the tumour.44 In this context, the diminutive size of the PPM creates advantageous conditions for improving tumour permeability. It is important to note that the premature release of SN-38 may result in non-specific toxicity to the adjacent normal lung parenchymal tissues. The tumour microenvironment exhibits a lower pH level compared to the normal physiological environment due to the rapid proliferation of cells and increased proton excretion during tumourigenesis.42 Consequently, pH-responsive nanomicelles confer the advantage of tumor-targeted drug delivery. In this study, capitalizing on the acidic tumor microenvironment, we engineered acid-sensitive PPM. This design not only enhances cellular uptake efficiency in malignant cells but also significantly mitigates off-target toxicity to healthy pulmonary tissues.
While tumor-targeted delivery represents the first critical step for nanomedicine efficacy, subsequent intracellular drug trafficking ultimately determines therapeutic success. Three key parameters must be optimized: (1) enhanced cellular internalization, (2) suppressed drug efflux, and (3) sustained high intracellular drug concentrations. Our data demonstrate that PPM exhibit pronounced tumor cell selectivity. Quantitative evaluation revealed a significantly higher cellular uptake in LLC cells (2.29-fold increase, p < 0.05) compared to BEAS-2B cells at physiological pH (7.4). This differential uptake was further amplified by 1.84-fold under mildly acidic conditions (pH 6.5) (Figures 3A–C and S6). This preferential accumulation correlates with significantly stronger cytotoxicity in LLC cells (Figure 2B). The findings from prior in vitro release experiments indicated (Figure 1F) that PPM can gradually release SN-38 in a mildly acidic environment (pH 6.5), which may explain why this environment enhances the uptake efficiency of tumour cells. Tumour cells not only internalise PPM but also absorb free SN-38, thereby facilitating the targeted cytotoxicity of PPM against tumour cells. Although heightened metabolic activity in tumor cells may contribute to this disparity, the dominant role of pH-triggered nanoparticle/nanomicelles behavior is evident.
Furthermore, the pH-sensitive properties of PPM was confirmed by its swelling and aggregation under acidic conditions (Figures 2E and F and S4). The styrene moiety in PPM was liable to be cleaved within the acidic milieu of lysosomes. This cleavage event caused the core layer of the nanomicelles to gradually transform from hydrophobicity to hydrophilicity. Consequently, the polymer micelles gradually swelled and their aggregation was facilitated. Simultaneously, SN-38 was released. Characterized by distinct hydrophobicity, the property of SN-38 promoted its aggregation. Overall, this expansion likely facilitates lysosomal escape via a mechanical disruption mechanism.45 The result of lysosomal escape directly contributes to elevated intracellular drug concentrations. In conclusion, PPM demonstrated favourable pH-responsive properties and displayed increased selectivity for homologous tumour cells in vitro, making it a promising candidate for targeted uptake by tumour cells.
Higher ROS levels likely induced apoptosis,46 as supported by our in vitro and in vivo pharmacodynamic studies (Figures 2 and 5), implying that redox imbalance contributes to the antitumor response. Although SN-38 could elevate ROS levels through DNA damage,47 the increase was insufficient to produce significant effects at low doses or when used alone, resulting in negligible cytotoxicity. In contrast, the PPM (CA-based carrier loaded with SN-38) developed in this study not only retained SN-38’s DNA-damaging capacity but also enabled CA to further stimulate ROS generation. CA could form a thiol derivative with reduced GSH,48 which may explain its promoting effect on ROS generation. In other words, CA and SN-38 could promote ROS generation through distinct pathways, thereby enhancing intracellular ROS to a greater extent and more effectively inducing apoptosis. Recent research has shown that increasing levels of reactive oxygen species (ROS) in tumour cells not only enhances the effectiveness of antitumour treatment,49,50 but also facilitates the disruption of lysosomal membranes.51 These findings indicated that CA amplifies cellular ROS levels in a self-reinforcing manner, which may facilitate lysosomal escape, thereby enhancing intracellular drug release.
While our data demonstrate that tracheal instillation of PPM prolongs pulmonary and tumor retention compared to intravenous administration, we acknowledge that comprehensive pharmacokinetic studies are required to fully validate the advantages of pulmonary delivery. The limited sample size in animal cohorts may affect the statistical robustness of our findings, and a positive control could provide additional validation. Subsequent investigations will optimize model construction parameters, incorporate expanded the sample size, implement longitudinal tumor monitoring via IVIS over a longer period observation period, and elucidating the molecular mechanisms underlying preferential PPM uptake by tumor cells. Moreover, The synergistic effect of CA and SN38 on ROS generation merits deeper mechanistic exploration, particularly in relation to potential crosstalk with cellular uptake machinery. Advanced quantitative methods are needed to precisely evaluate lysosomal escape dynamics, such as calcein release assays, galectin puncta formation assays, or highly sensitive split-luciferase reporter systems. Notably, clinical translation of PPM faces multifaceted challenges: physiological discrepancies between instillation (non-uniform deposition) and true inhalation delivery, impact of nebulization devices on actual delivery efficiency, industrial-scale sterilization, and physiological variances between animal models and human patients.
Conclusions
In this study, we developed a pH-sensitive carrier material, PEG-PMMSD, using the FDA-approved and nontoxic CA as the foundational component. This carrier was been loaded with the potent antitumour agent SN-38 and self-assembled into nanomicelles designated as PPM. Nanomicelles were administered via the pulmonary route to enhance drug accumulation in lung tumours, and their retention was improved by modifying the PEG surface, thereby minimising mucus adsorption and clearance absorption. Small nanomicelles can easily penetrate deep into the tumour tissues and are preferentially internalised by tumour cells, thereby augmenting their cytotoxic effects. The acidic tumour microenvironment further enhances the efficacy of PPM uptake. Additionally, PPM undergoes bond cleavage in response to acidity, regenerating CA and releasing SN-38. CA functions as an ROS amplifier by further boosting ROS generation within tumour cells, leading to highly effective antitumour outcomes when combined with SN-38. This strategy facilitates the precise modulation of drug release, enhances the accumulation of nanomicelles within tumour cells, and ultimately results in a more effective anticancer effect. This study not only highlights the advantages of pulmonary administration but also lays the groundwork for more effective cancer treatment strategies.
Ethical Approval
This study was reviewed and approved by the Zhejiang Chinese Medical University Animal Care and Use Committee (IACUC-20230227-02). The guidance of the Zhejiang Chinese Medical University Animal Care and Use Committee on Humane Care and Use of Laboratory Animals was strictly followed throughout the animal study.
Acknowledgments
This work was done at the Laboratory of Hangzhou Integrative Medicine Hospital Affiliated to Zhejiang Chinese Medical University (Hangzhou Red Cross Hospital), China. The authors are grateful to the staff in the laboratory for their technical assistance.
Funding
This work is financially supported by the National Natural Science Foundation of China (No. 82003669), Zhejiang Province Traditional Chinese Medicine Science and Technology Plan Project (No.2024ZR025).
Disclosure
The authors declare no competing interests.
References
1. Siegel RL, Giaquinto AN, Jemal A. Cancer statistics, 2024. Ca A Cancer J Clin. 2024;74(1):12–18. doi:10.3322/caac.21820
2. Andrew MDW, Richard CW, Ruth BE, et al. American cancer society guideline for the early detection of prostate cancer: update 2010. CA Cancer J Clin. 2010;60(2):70–98. doi:10.3322/caac.20066
3. Thai AA, Solomon BJ, Sequist LV, Gainor JF, Heist RS. Lung cancer. Lancet. 2021;398(10299):535–554. doi:10.1016/s0140-6736(21)00312-3
4. Yadav A, Singh S, Sohi H, Dang S. Advances in delivery of chemotherapeutic agents for cancer treatment. AAPS Pharm Sci Tech. 2021;23(1):25. doi:10.1208/s12249-021-02174-9
5. Liu J, Deng Y, Fu D, et al. Sericin microparticles enveloped with metal-organic networks as a pulmonary targeting delivery system for intra-tracheally treating metastatic lung cancer. Bioact Mater. 2020;6(1):273–284. doi:10.1016/j.bioactmat.2020.08.006
6. Mangal S, Gao W, Li T, Zhou Q. Pulmonary delivery of nanoparticle chemotherapy for the treatment of lung cancers: challenges and opportunities. Acta Pharmacol Sin. 2017;38(6):782–797. doi:10.1038/aps.2017.34
7. Anderson CF, Grimmett ME, Domalewski CJ, Cui H. Inhalable nanotherapeutics to improve treatment efficacy for common lung diseases. WIREs Nanomed Nanobiotech. 2020;12(1):e1586. doi:10.1002/wnan.1586
8. García-Fernández A, Sancenón F, Martínez-Máñez R. Mesoporous silica nanoparticles for pulmonary drug delivery. Adv Drug Delivery Rev. 2021;177:113953. doi:10.1016/j.addr.2021.113953
9. Ma S, Cong Z, Wei J, et al. Pulmonary delivery of size-transformable nanoparticles improves tumor accumulation and penetration for chemo-sonodynamic combination therapy. J Control Release. 2022;350:132–145. doi:10.1016/j.jconrel.2022.08.003
10. Liu C, Liu W, Liu Y, et al. Versatile flexible micelles integrating mucosal penetration and intestinal targeting for effectively oral delivery of paclitaxel. Acta Pharm Sin B. 2023;13(8):3425–3443. doi:10.1016/j.apsb.2023.05.029
11. Sun Q, Zhou Z, Qiu N, Shen Y. Rational design of cancer nanomedicine: nanoproperty integration and synchronization. J Control Release. 2022;350:132–145. doi:10.1002/adma.201606628
12. Wang J, Mao W, Lock LL, et al. The role of micelle size in tumor accumulation, penetration, and treatment. ACS Nano. 2015;9(7):7195–7206. doi:10.1021/acsnano.5b02017
13. Huang X, Mu N, Ding Y, et al. Targeted delivery and enhanced uptake of chemo-photodynamic nanomedicine for melanoma treatment. Acta Biomater. 2022;147:356–365. doi:10.1016/j.actbio.2022.05.015
14. Ho T, Guidolin K, Makky A, et al. Novel strategy to drive the intracellular uptake of lipid nanoparticles for photodynamic therapy. Angew Chem Int Ed. 2023;62(16):e202218218. doi:10.1002/anie.202218218
15. Xie R, Wang Y, Tong F, et al. Hsp70-targeting and size-tunable nanoparticles combine with PD-1 checkpoint blockade to treat glioma. Small. 2023;19(37):2300570. doi:10.1002/smll.202300570
16. Ma J, Ma C, Huang X, et al. Preparation and cellular uptake behaviors of uniform fiber-like micelles with length controllability and high colloidal stability in aqueous media. Fundam Res. 2023;3(1):93–101. doi:10.1016/j.fmre.2022.01.020
17. Chen Y-Q, Xue M-D, Li J-L, Huo D, Ding H-M, Ma Y. Uncovering the importance of ligand mobility on cellular uptake of nanoparticles: insights from experimental, computational, and theoretical investigations. ACS Nano. 2024;18(8):6463–6476. doi:10.1021/acsnano.3c11982
18. Qi Q-R, Tian H, Yue B-S, Zhai B-T, Zhao F. Research progress of SN38 drug delivery system in cancer treatment. Int J Nanomed. 2024;26(19):945–964. doi:10.2147/IJN.S435407
19. Bala V, Rao S, Boyd BJ, Prestidge CA. Prodrug and nanomedicine approaches for the delivery of the camptothecin analogue SN38. J Control Release. 2013;172(1):48–61. doi:10.1016/j.jconrel.2013.07.022
20. Jiang Y, Chen H, Lin T, et al. Ultrasound-activated prodrug-loaded liposome for efficient cancer targeting therapy without chemotherapy-induced side effects. J Nanobiotechnol. 2024;22(1):2. doi:10.1186/s12951-023-02195-5
21. DeNicola GM, Karreth FA, Humpton TJ, et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature. 2011;475(7354):106–109. doi:10.1038/nature10189
22. Xin Q, Ji Q, Zhang Y, et al. Aberrant ROS served as an acquired vulnerability of cisplatin-resistant lung cancer. Oxid Med Cell Longev. 2022;2022(1):1112987. doi:10.1155/2022/1112987
23. Hou Y, Wang H, Wu J, Guo H, Chen X. Dissecting the pleiotropic roles of reactive oxygen species (ROS) in lung cancer: from carcinogenesis toward therapy. Med Res Rev. 2024;44(4):1566–1595. doi:10.1002/med.22018
24. Chang S-H, Lin P-Y, Wu T-K, et al. Imiquimod-induced ROS production causes lysosomal membrane permeabilization and activates caspase-8-mediated apoptosis in skin cancer cells. J Dermatological Sci. 2022;107(3):142–150. doi:10.1016/j.jdermsci.2022.08.006
25. Yin LA-O, Gou Y, Dai Y, et al. Cinnamaldehyde restores ceftriaxone susceptibility against multidrug-resistant salmonella. Int J Mol Sci. 2023;24(11):9288. doi:10.3390/ijms24119288
26. Khadke SK, Lee J-H, Kim Y-G, Raj V, Lee J. Appraisal of cinnamaldehyde analogs as dual-acting antibiofilm and anthelmintic agents. Frontiers in Microbiology. 2022;13:818165. doi:10.3389/fmicb.2022.818165
27. Zhao X, Miao R, Xu T, et al. Changing cinnamaldehyde skeleton achieves antibacterial nanoswitch. ACS Appl Mater Interfaces. 2024;16(14):17838–17845. doi:10.1021/acsami.3c18277
28. Jiahua P, Xin S, Wenbin Y, et al. The role and mechanism of cinnamaldehyde in cancer. J Food Drug Anal. 2024;32(2):140–154. doi:10.38212/2224-6614.3502
29. Gong N, Ma X, Ye X, et al. Carbon-dot-supported atomically dispersed gold as a mitochondrial oxidative stress amplifier for cancer treatment. Nature Nanotechnol. 2019;14(4):379–387. doi:10.1038/s41565-019-0373-6
30. Zhang WY, Lei W, Shen FK, Wang MK, Li LL, Chang JM. Cinnamaldehyde induces apoptosis and enhances anti-colorectal cancer activity via covalent binding to HSPD1. Phytother Res. 2024;38(10):4957–4966. doi:10.1002/ptr.7840
31. Hong S-H, Ismail IA, Kang S-M, Han DC, Kwon B-M. Cinnamaldehydes in cancer chemotherapy. Phytother Res. 2016;30(5):754–767. doi:10.1002/ptr.5592
32. Kumar R, Pandey AK. P-219 - antioxidant and hepatoprotective activity of cinnamaldehyde against isoniazid induced hepatotoxicity. Free Radic Biol Med. 2018;120:S111. doi:10.1016/j.freeradbiomed.2018.04.366
33. Kim NY, Trinh NT, Ahn SG, Kim SA. Cinnamaldehyde protects against oxidative stress and inhibits the TNF‑α‑induced inflammatory response in human umbilical vein endothelial cells. Int J Mol Med. 2020;46(1):449–457. doi:10.3892/ijmm.2020.4582
34. Yan-Guang L, Jiang-Hong L, Hai-Qin W, Junhua L, Xiao-Ya D. Cinnamaldehyde protects cardiomyocytes from oxygen-glucose deprivation/reoxygenation-induced lipid peroxidation and DNA damage via activating the Nrf2 pathway. Chem Biol Drug Des. 2024;103(2):e14489. doi:10.1111/cbdd.14489
35. Ka H, Park H-J, Jung H-J, et al. Cinnamaldehyde induces apoptosis by ROS-mediated mitochondrial permeability transition in human promyelocytic leukemia HL-60 cells. Cancer Lett. 2003;196(2):143–152. doi:10.1016/S0304-3835(03)00238-6
36. Ma S, Song W, Xu Y, et al. Rationally designed polymer conjugate for tumor-specific amplification of oxidative stress and boosting antitumor immunity. Nano Lett. 2020;20(4):2514–2521. doi:10.1021/acs.nanolett.9b05265
37. Chang W-L, Cheng F-C, Wang S-P, Chou S-T, Shih Y. Cinnamomum cassia essential oil and its major constituent cinnamaldehyde induced cell cycle arrest and apoptosis in human oral squamous cell carcinoma HSC-3 cells. Environ Toxicol. 2017;32(2):456–468. doi:10.1002/tox.22250
38. Tu Y, Xiao X, Dong Y, et al. Cinnamaldehyde-based poly(thioacetal): a ROS-awakened self-amplifying degradable polymer for enhanced cancer immunotherapy. Biomaterials. 2022;289:121795. doi:10.1016/j.biomaterials.2022.121795
39. Hong SH, Kim J, Kim J-M, et al. Apoptosis induction of 2′-hydroxycinnamaldehyde as a proteasome inhibitor is associated with ER stress and mitochondrial perturbation in cancer cells. Biochem Pharmacol. 2007;74(4):557–565. doi:10.1016/j.bcp.2007.05.016
40. Zheng S, Li M, Xu W, et al. Dual-targeted nanoparticulate drug delivery systems for enhancing triple-negative breast cancer treatment. J Control Release. 2024;371:371–385. doi:10.1016/j.jconrel.2024.06.012
41. Zhang J, Sun X, Xu M, et al. A self-amplifying ROS-sensitive prodrug-based nanodecoy for circumventing immune resistance in chemotherapy-sensitized immunotherapy. Acta Biomater. 2022;149:307–320. doi:10.1016/j.actbio.2022.06.035
42. Wang L, He S, Liu R, et al. A pH/ROS dual-responsive system for effective chemoimmunotherapy against melanoma via remodeling tumor immune microenvironment. Acta Pharm Sin B. 2024;14(5):2263–2280. doi:10.1016/j.apsb.2023.12.001
43. Wilhelm S, Tavares AJ, Dai Q, et al. Analysis of nanoparticle delivery to tumours. Nature Rev Mater. 2016;1(5):16014. doi:10.1038/natrevmats.2016.14
44. Cabral H, Matsumoto Y, Fau - Mizuno K, et al. Accumulation of sub-100 nm polymeric micelles in poorly permeable tumours depends on size. Nat Nanotechnol. 2011;6(12):815–823. doi:10.1038/nnano.2011.166
45. Zhou LY, Lv FT, Liu LB, Wang S. Polarity conversion of conjugated polymer for lysosome escaping. ACS Appl Mater Interfaces. 2017;9(33):27427–27432. doi:10.1021/acsami.7b10105
46. Yuhui L, Chengjian S, Zheng H, et al. Inhibition of PI3K/AKT signaling via ROS regulation is involved in Rhein-induced apoptosis and enhancement of oxaliplatin sensitivity in pancreatic cancer cells. Int J Biol Sci. 2021;17(2). doi:10.7150/ijbs.49514
47. Srinivas US, Tan BWQ, Vellayappan BA, Jeyasekharan AD. ROS and the DNA damage response in cancer. Redox Biol. 2019;25:101084. doi:10.1016/j.redox.2018.101084
48. Zhanwei Z, Huan L, Ruoxi Y, et al. Glutathione depletion-induced activation of dimersomes for potentiating the ferroptosis and immunotherapy of “cold”. Tumor Angew Chem Int Ed Engl. 2022;61(22). doi:10.1002/anie.202202843
49. Shi J, Nie W, Zhao X, et al. An intracellular self-assembly-driven uninterrupted ROS generator augments 5-aminolevulinic-acid-based tumor therapy. Adv Mater. 2022;34(30):2201049. doi:10.1002/adma.202201049
50. Yuan Y, Chen B, An X, et al. MOFs-based magnetic nanozyme to boost cascade ROS accumulation for augmented tumor ferroptosis. Adv Healthc Mater. 2024;13(20):e2304591. doi:10.1002/adhm.202304591
51. Liu M-X, Zhang X-L, Yang J-B, Lu Z-L, Zhang Q-T. Highly water-dispersible PCN nanosheets as light-controlled lysosome self-promoting escape type non-cationic gene carriers for tumor therapy. J Mat Chem B. 2022;10(28):5430–5438. doi:10.1039/D2TB00440B
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.