Back to Journals » Drug Design, Development and Therapy » Volume 11
Intranasal agomelatine solid lipid nanoparticles to enhance brain delivery: formulation, optimization and in vivo pharmacokinetics
Authors Fatouh AM, Elshafeey AH, Abdelbary A
Received 15 December 2015
Accepted for publication 31 March 2016
Published 19 June 2017 Volume 2017:11 Pages 1815—1825
DOI https://doi.org/10.2147/DDDT.S102500
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Wei Duan
Ahmed M Fatouh,1 Ahmed H Elshafeey,1,2 Ahmed Abdelbary1
1Department of Pharmaceutics and Industrial Pharmacy, Faculty of Pharmacy, Cairo University, Cairo, Egypt; 2School of Pharmacy, University of Waterloo, Waterloo, ON, Canada
Purpose: Agomelatine is a novel antidepressant drug suffering from an extensive first-pass metabolism leading to a diminished absolute bioavailability. The aim of the study is: first to enhance its absolute bioavailability, and second to increase its brain delivery.
Methods: To achieve these aims, the nasal route was adopted to exploit first its avoidance of the hepatic first-pass metabolism to increase the absolute bioavailability, and second the direct nose-to-brain pathway to enhance the brain drug delivery. Solid lipid nanoparticles were selected as a drug delivery system to enhance agomelatine permeability across the blood–brain barrier and therefore its brain delivery.
Results: The optimum solid lipid nanoparticles have a particle size of 167.70 nm ±0.42, zeta potential of -17.90 mV ±2.70, polydispersity index of 0.12±0.10, entrapment efficiency % of 91.25%±1.70%, the percentage released after 1 h of 35.40%±1.13% and the percentage released after 8 h of 80.87%±5.16%. The pharmacokinetic study of the optimized solid lipid nanoparticles revealed a significant increase in each of the plasma peak concentration, the AUC(0–360 min) and the absolute bioavailability compared to that of the oral suspension of Valdoxan® with the values of 759.00 ng/mL, 7,805.69 ng·min/mL and 44.44%, respectively. The optimized solid lipid nanoparticles gave a drug-targeting efficiency of 190.02, which revealed more successful brain targeting by the intranasal route compared with the intravenous route. The optimized solid lipid nanoparticles had a direct transport percentage of 47.37, which indicates a significant contribution of the direct nose-to-brain pathway in the brain drug delivery.
Conclusion: The intranasal administration of agomelatine solid lipid nanoparticles has effectively enhanced both the absolute bioavailability and the brain delivery of agomelatine.
Keywords: nasal route, direct nose-to-brain pathway, agomelatine
Introduction
Agomelatine (AGM), a melatonin analog, is a potent agonist of melatonin MT1 and MT2 receptors.1 It also shows serotonin 5-HT2C receptor antagonist activity.1–3 AGM is rapidly absorbed (>75%) after oral administration. But it is subjected to extensive first-pass metabolism, leading to a low absolute bioavailability (<5%).4
The two main objectives of the study were: first to decrease the first-pass metabolism of AGM and enhance its absolute bioavailability, and second to enhance its brain delivery. Nasal administration was selected to achieve these objectives because absorption from the nasal cavity is carried out into the systemic circulation by avoiding the hepatic first-pass metabolism, which is expected to enhance AGM absolute bioavailability. On the other hand, AGM can be transported directly from the nasal cavity into the brain avoiding the blood–brain barrier (BBB) through the olfactory region of the nasal epithelium and the trigeminal neural region, which is expected to enhance its brain bioavailability and achieve brain targeting.5,6 The olfactory epithelium is a pseudostratified epithelium, situated between the nasal septum and the lateral walls of each of the two nasal cavities and constitutes about 5% of the total area of the nasal cavity in man.7 Drugs can reach the central nervous system (CNS) via extracellular or intracellular transport along olfactory nerves.8 The trigeminal nerve has branches innervating the respiratory and olfactory epithelium of the nasal cavity. Three branches of the trigeminal nerve (ophthalmic, maxillary and mandibular divisions) merge at the trigeminal ganglion and enter the CNS.9 Due to the large surface area and the high vasculature of the nasal mucosa, the nasal route has a potential to achieve a rapid therapeutic action.10 Based on these advantages, the nasal route was adopted in the study.
Solid lipid nanoparticles (SLNs) are colloidal particles having size ranging from 1 to 1,000 nm. They are prepared using lipids with melting points higher than the room temperature, that is, solid at room temperature. Within the SLN, the lipid usually represents a matrix in which drug is embedded, which can be considered as lipoid nanosphere.11–13 There are three pathways by which AGM can be transported from the nasal cavity into the brain. The first pathway is an indirect one in which AGM has to be absorbed systemically, and then cross the BBB to reach the brain. The second and third pathways are direct pathways in which AGM can reach the brain through the olfactory and trigeminal nerves. To enhance the ability of AGM to cross the BBB and reach the brain indirectly, AGM-loaded SLNs have been prepared. The enhanced SLN permeation through the BBB can be attributed to their endocytosis by the BBB endothelial cells, their inhibitory action on the transmembrane efflux systems or the solubilizing action of the surfactant associated with their preparation.14
Materials and methods
Materials
Agomelatine was kindly provided by Hikma Pharma (Cairo, Egypt), Polyvinylalcohol (molecular weight: 22,000 Da), glyceryl tripalmitate, sodium deoxycholate and glyceryl tristearate were purchased from Sigma-Aldrich Co. (St Louis, MO, USA). Gelucire 43/01 Geleol and Precirol were kindly provided by Gattefosse Co. (Saint-Priest, France). Dichloromethane and stearic acid were purchased from El-Nasr Pharmaceutical Chemicals Co. (Cairo, Egypt). All other reagents were of analytical grade.
Preparation of solid lipid nanoparticles
SLNs were prepared using the emulsification solvent evaporation technique.15 Briefly, AGM and lipid were dissolved in dichloromethane. This organic phase was added drop wise to an aqueous solution containing surface active agent(s). The obtained pre-emulsion was subsequently subjected to ultrasonication using probe sonicator (Ultrasonic processor modelVCX 750) to decrease the globules size to the required nanometer range. The formed emulsion was stirred at the room temperature using a magnetic stirrer at 400 rpm to allow the organic solvent to evaporate and SLNs to be formed. Each formula was replicated twice, and the results are presented as mean ± SD.
Preliminary study for the optimum conditions
During the preliminary trials, different types and concentrations of lipid were screened for preparing AGM-loaded SLNs. The types of lipids used were Gelucire 43/01, glyceryl tripamitate, glyceryl tristearate, Geleol, stearic acid, and Precirol, whereas the concentrations of lipids screened were 1%, 1.5%, 2% and 3% w/v, respectively. When the effect of lipid type was investigated, lipids were used at the concentration of 1% w/v, while Gelucire 43/01 was the lipid used when the effect of lipid concentration was studied. Polyvinyl alcohol (PVA) was used as an emulsifier in all the preliminary trials. One-way analysis of variance (ANOVA) with subsequent least significant difference test was performed to compare the results of these formulations using SPSS 19® software.
A full factorial design
Based on the results of the preliminary trials, a full factorial design was built for further optimization. A 24 full factorial model was developed to study the main effects and interactions of four factors: the type of lipid (X1), the concentration of lipid (X2), the drug:lipid ratio (X3) and the concentration of cosurfactant (X4) as shown in Table 1. The use of a full factorial model avoids possible aliasing of the main effects and interactions, hence 16 formulae (two of each run) of AGM-SLN.
 | Table 1 The independent variables of the full factorial design for optimization of AGM-loaded SLNs |
In vitro characterization of solid lipid nanoparticles
Determination of particle size (PS), polydispersity index (PDI) and zeta potential (ZP)
The mean PS, PDI and ZP were determined by dynamic light scattering (DLS, Zetasizer Nano ZS; Malvern Instruments, Malvern, UK) at 25°C. Before measurement, the formulation was properly diluted with distilled water to have a suitable scattering intensity.
Determination of the entrapment efficiency (EE%)
The EE% of AGM-loaded SLNs was determined by measuring the concentration of the free drug in the aqueous phase of the SLN dispersion. A definite volume of the prepared SLN dispersion was diluted with distilled water and centrifuged using cooling centrifuge (Beckman, Fullerton, CA, USA) at 15,000 rpm for 1 h at 4°C. The unentrapped drug concentration was estimated spectrophotometrically at 276.4 nm using UV/VIS spectrophotometer (UV-1800; Shimadzu, Kyoto, Japan). The EE% was calculated using the following equation:
|
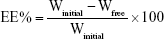
In vitro release study
The in vitro release study was carried out by the dialysis bag diffusion technique. A definite volume of the prepared SLN dispersion was filled into a dialysis bag (molecular weight cutoff: 12,000–14,000 Da). The filled dialysis bag was immersed in a bottle containing 100 mL of phosphate buffer, pH 6.8. The release was carried out using a thermostatic horizontal shaker (GFL; Gesellschatt Laboratories, Berlin, Germany) established at 100 rpm and 37°C±0.5°C. Aliquots were withdrawn at 15 and 30 min, and 1, 2, 4, 6 and 8 h time intervals. The drug concentration was measured spectrophotometrically at 276.4 nm using UV/VIS spectrophotometer (UV-1800). All experiments were repeated three times, and the results were expressed as the mean values ± SD.
Elucidation of optimized formula
Numerical optimization was performed using the statistical program according to the constraints listed in Table 2. The simultaneous optimization technique described by Derringer and Suich16 in 1980 was chosen for optimization of the responses. This method is based on the utilization of desirability functions. Each response is converted into an individual desirability function di that can vary over the range 0< di <1. The design variables are then selected to maximize the overall desirability adopting the following equation:
 | Table 2 The dependent variables of the full factorial design for optimization of AGM-loaded SLNs |
|
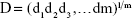
where D is the overall desirability, di is the individual desirability and m is the number of responses to be optimized.
Further investigations of the optimized SLNs formulation
Transmission electron microscopy
The morphology of the selected AGM-loaded SLNs was examined by the transmission electron microscope (JEM-1010; JEOL Ltd., Tokyo, Japan). One drop of diluted SLNs suspension was deposited on a carbon-coated copper grid and negatively stained by 2% (w/v) aqueous phosphotungestic acid solution and then examined at 80 kV.
Differential scanning calorimetry (DSC)
The thermotropic properties and phase transition behavior of the pure components (AGM, Gelucire 43/01, PVA and sodium deoxycholate [SDC]), selected SLN formula as well as the physical mixtures of their components were investigated using DSC (DSC 60; Shimadzu). Samples of 2 mg were sealed in aluminum pans and heated at a rate of 10°C/min to a temperature of 500°C throughout the analysis using indium as the standard reference.
Pharmacokinetic study
Administration of AGM to rats
Seventy-two male rats (weight: 70–250 g) were used in the study. The protocol of the study was reviewed and approved (PI 1197) by the institutional review board (Research Ethics Committee-Faculty of Pharmacy, Cairo University (REC-FOPCU). The rats were divided into 3 groups. Group 1 received intranasal SLN dispersion (SLN-14), Group 2 received intravenous AGM solution in normal saline containing 10% ethanol to enhance drug solubility17 and Group 3 received oral suspension of Valdoxan®. All animals received AGM in a dose of 2.14 μg/g, equivalent to 0.535 μL per gram of the rat weight. The dose was calculated based on the FDA guidelines in the conversion of animal doses to human equivalent doses.18 The calculated dose was given as nasal drops using a micropipette, divided equally into two nostrils. At different time intervals of 2, 5, 10, 15, 30, 60, 120, 180 and 360 min following administration of AGM formulations, 3 rats of each group were sacrificed. Blood was collected from the trunk and put into heparinized tubes, centrifuged at 4,000 rpm for 15 min at 25°C and plasma separated. Brain tissue samples were taken after cutting the skulls, followed by homogenization using Ultra Turrax homogenizer with threefold volumes of saline at 24,000 rpm for 1 min. Homogenized brain and separated plasma tubes were stored at −80°C until being assayed.
Assay of AGM in plasma and brain
AGM was analyzed in plasma and homogenized brain samples using an Applied Biosystems/MDS Sciex liquid chromatography tandem mass spectrometry (LC-MS/MS). Plasma or homogenized brain samples (0.5 mL) were placed in 7 mL glass tubes, and then 50 μL of internal standard solution (100 ng/mL clonazepam) was added. Samples were then vortexed for 1 min. The extraction solvent (4 mL tertiary butylmethyl ether) was added; the tubes were then mixed for 10 min on a rocker-mixer Reax II (Heidolph, Schwabach, Germany). Samples were centrifuged at 1,790 rpm for 10 min at 4°C, using Eppendorf centrifuges 5804 R, and the upper organic layer was transferred into new Wassermann tubes and evaporated to dryness using vacuum concentrator (Eppendorf 5301; Eppendorf AG, Hamburg, Germany). Dry residues were reconstituted by the addition of 0.25 mL of mobile phase, and then tubes were vortex mixed for 1 min and finally placed into the autosampler for LC-MS/MS analysis. An aliquot of 10 μL of the samples was injected into a Shimadzu Prominence (Shimadzu) series LC system equipped with degasser (DGU-20A3) using Agilent C18 column (Agilent, Santa Clara, CA, USA) (50×4.6 mm) with 3.5 mm PS. The isocratic mobile phase (80% acetonitrile +20% water containing 0.1% formic acid) was delivered at a flow rate of 1.0 mL/min into the mass spectrometer’s electrospray ionization chamber. Quantitation was achieved by MS/MS detection in positive ion mode for both AGM and clonazepam IS using an MDS Sciex (Foster City, CA, USA) API-3200 mass spectrometer, equipped with a Turbo Ionspray interface at 500°C. The ion spray voltage was set at 5,500 V. The common parameters, namely, nebulizer gas, curtain gas, auxiliary gas and collision gas, were set at 25, 20, 40 and 6 psi, respectively. The compound parameters, namely, declustering potential, collision energy, entrance potential and collision exit potential, were 41, 6.5, 25 and 4 V for AGM and 51, 9.5, 31 and 6 V for clonazepam (IS), respectively. Detection of the ions was performed in the multiple reaction monitoring mode, monitoring the transition of the m/z 244.03 precursor ion to the m/z 185.30 for AGM and the m/z 315.96 precursor ion to the m/z 270.00 for IS. The Q1 and Q3 quadrupoles were set on unit resolution. The analytical data were processed by Analyst software (version 1.4.2).
Pharmacokinetic analysis
The mean concentrations of AGM in plasma and brain samples were plotted against the time, and the peak plasma and brain concentrations (Cmax) as well as the time to reach these peaks (tmax) were read directly. The area under AGM concentration-time curve (AUC0–360 min) was calculated by the trapezoidal method without extrapolation to infinity. The time to reach half plasma concentration and the mean residence time (MRT) were calculated using Kinetica software program (version 4.4.1). The absolute bioavailability of the intranasal and oral formulations compared to IV solution was calculated.
Transport study using the in vivo rat model
To evaluate AGM brain targeting after nasal dosing, two indices were calculated:
1) | Drug targeting efficiency (%DTE), which compares the delivery of drug to the brain following intranasal administration vs systemic administration and is calculated as follows: |
|

The value of %DTE can range from −8 to 8, and the values >100% indicate a superior drug delivery to the brain following intranasal administration compared to the systemic administration.
2) | Nose-to-brain direct transport percentage (DTP%), which measures the relative contribution of the direct nose-to-brain routes in the overall delivery to the brain (ie, via all the direct routes and via the BBB) is calculated as follows: |
|
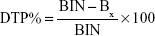
where BIN is AUC0–t (brain) following the intranasal administration and BX is the brain AUC fraction contributed by systemic circulation through the BBB following intranasal administration and equals: where BIV is AUC0–t of AGM in the brain following IV administration, PIV is AUC0–t of AGM in the plasma following IV administration and PIN is AUC0–t of AGM in the plasma following intranasal instillation.
The value of %DTP can range from −8 to 100%; the negative values indicate more efficient drug delivery to the brain through the BBB permeation than the direct nose-to-brain routes, whereas the positive values of the %DTE indicate a significant contribution of the direct routes to the overall brain delivery.
Results and discussion
Preliminary study
Different types of lipids were investigated during the preliminary trials while keeping the lipid concentration at 1% w/v for all the lipid types. Table 3 shows the results of the PS and EE% of the prepared formulations. The statistical analysis using the ANOVA test revealed that Gelucire 43/01, glyceryl tripalmitate and glyceryl tristearate have significantly smaller PS (P<0.0001), whereas Gelucire 43/01, glyceryl tripalmitate and Geleol have significantly larger EE% (P<0.0001). The EE% results may be related to the lipid structure, which had a great influence on the capacity for drug incorporation. Being mixtures of mono-, di- and triglycerides, Gelucire 43/01 and Geleol are expected to form less perfect crystals with many imperfections, offering space to accommodate AGM.19 The reduced PS of Gelucire 43/01 can be related to its content of mono glycerides, diglycerides and polyethylene glycol esters of fatty acids, which may give it surface active properties. Gelucire 43/01 and glyceryl tripalmitate were selected for further trials due to their favored PS and EE%.
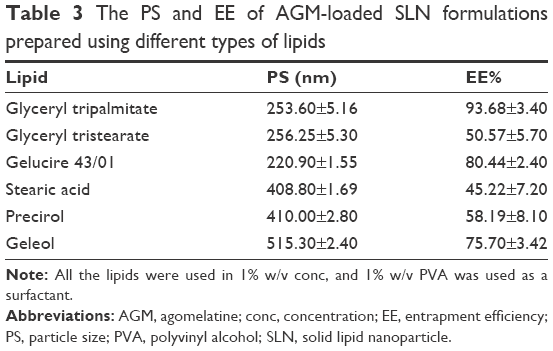 | Table 3 The PS and EE of AGM-loaded SLN formulations prepared using different types of lipids |
When the formulations prepared using Gelucire 43/01 and glyceryl tripalmitate were tested for the in vitro release, rapid release with high percentage of AGM released after 0.25 h (Q0.25h) was obtained (43.73%±1.02% for Gelucire 43/01 and 41.35%±1.62% for glyceryl tripalmitate) (Figure 1). A slower release profile with a lower Q0.25h was desired to ensure that AGM is absorbed systemically and permeates through the BBB, while it is contained within the SLNs to exploit the advantages of the SLNs in the BBB permeation. Therefore, the concentration of lipid was increased from 1% w/v to 1.5% and 3% w/v. As shown in Figure 1, a significant reduction in the Q0.25h was obtained (P<0.0001) (4.39%±0.87% and 4.58%±1.32% for 1.5% and 3% w/v Gel, respectively, and 27.75%±10.26% and 17.90%±2.30% for 1.5% and 3% w/v GTP, respectively). Therefore, these two lipid concentrations were selected for the further trials.
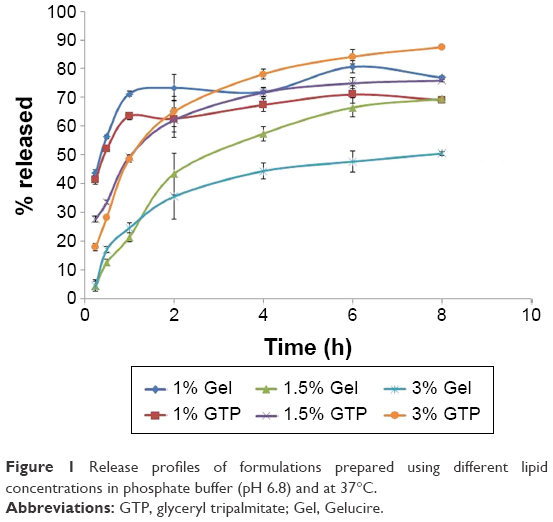 | Figure 1 Release profiles of formulations prepared using different lipid concentrations in phosphate buffer (pH 6.8) and at 37°C. |
The full factorial design results analysis
The composition and results of the dependent variables of the full factorial design AGM-loaded SLN formulations are shown in Table 4.
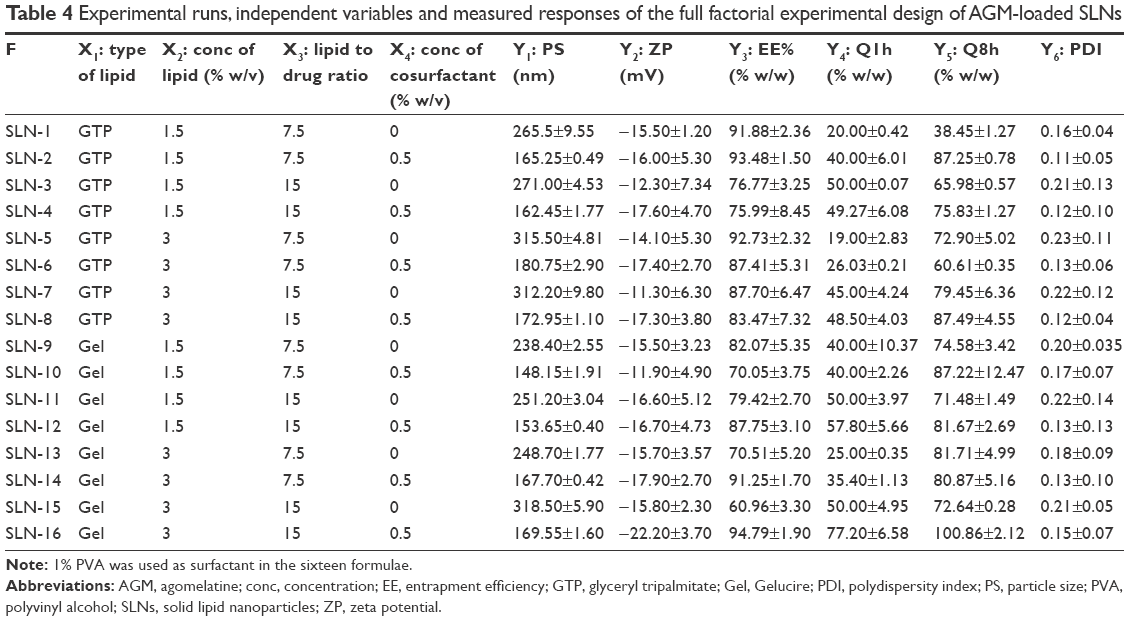 | Table 4 Experimental runs, independent variables and measured responses of the full factorial experimental design of AGM-loaded SLNs |
Particle size
The PS of the design formulae ranged from 148.15 to 318.50 nm. According to the ANOVA test results, the factors, having a significant effect on the PS, are the type of lipid (X1), the concentration of lipid (X2) and the concentration of cosurfactant (X4). It was observed that the formulae prepared using Gelucire 43/01 have significantly smaller PS than the formulae prepared using glyceryl tripalmitate (P=0.0471), which can be attributed to the chemical composition of Gelucire 43/01 that contains mono- and diglycerides in addition to polyethylene glycol esters of fatty acids. These components give Gelucire 43/01 certain surface active properties that can be the reason for the reduced PS. A direct relation was also observed between the PS and the lipid concentration, that is, the formulae containing 3% w/v of the lipid have significantly higher PS (P=0.0100), which is in agreement with previous reports.20 This observation can be attributed to the higher viscosity of the more lipid concentrated SLN dispersion, which decreases the sonication efficiency and increases the rate of particle agglomeration.21 It was also found that the presence of SDC as a cosurfactant leads to a significant decrease in the PS (P<0.0001). This observation can be related to the surface active properties of SDC, which reduced the interfacial tension between the aqueous and organic phases, forming smaller sized emulsion droplets during the pre-emulsion formation stage and causing the formation of smaller sized SLNs upon the organic solvent evaporation. Also, this effect may be due to the cosurfactant steric stabilization of the particles, thus decreasing their tendency to aggregate.22
Zeta potential and polydispersity index
The ZP of the formulae ranged from −11.30 to −22.20 mV, whereas the PDI ranged from 0.11 to 0.23. According to the ANOVA test results, the formulae containing 0.5% w/v SDC have significantly higher negative ZP and lower PDI than the formulae containing no cosurfactant. The higher negative ZP is attributed to the negative charge of SDC, which is an anionic molecule; its hydrophobic group resides in the lipid nanoparticles, whereas its hydrophilic anionic group is adsorbed on the outer surface of the lipid nanoparticles, leading to a higher negative charge. On the other hand, the incorporation of cosurfactant is expected to enhance the stability of the formed pre-emulsion and decrease the tendency of its globules to aggregate. The small sized and uniform pre-emulsion globules are expected, on the organic phase evaporation, to form small and uniform solid particles with low PDI.22
Entrapment efficiency
The EE% of the design formulae ranged from 60.96% to 94.79%. Based on the statistical analysis, there is no factor having a significant effect on the EE (P>0.05). The high EE% obtained in all the formulae in spite of varying the levels of the design factors can be related to the high lipophilicity of AGM (log P=2.8) and the low water solubility (0.358 mg/mL), which increase its localization within the organic phase during the pre-emulsion stage and decrease its release into the surrounding aqueous medium after the organic solvent evaporation and the lipid solidification. The high EE of the highly lipid soluble drugs agrees with the observations of Muller et al.19
In vitro release study
Figure 2 shows the in vitro release profile of the 16 formulae prepared according to the full factorial design. Compared with the release profile of the free drug solution, a significant reduction in the release rate was achieved in the SLN formulae. The percentage released of AGM after 1 h (Q1h) was 89.35% for the free drug solution, whereas it ranged from 19.00% to 77.20% for the SLN formulations. On the other hand, the percentage released of AGM after 8 h (Q8h) was 102.18% for the free drug solution, whereas it ranged from 38.45% to 100.48% for the SLN formulations. The slow release profile of the SLNs can be related to the high EE of the formulations (60.96% to 94.79%), where most of AGM was in the entrapped form while only a small percent of AGM was in the free drug form, which may be the reason for the slow release profile. In the comparison between the release profiles of the full factorial design formulae, the ANOVA test results revealed that the formulae containing 15:1 lipid to drug ratio had significantly higher Q1h than that of the formulae containing 7.5:1 lipid to drug ratio (P=0.0027). This result is contrary to the expectations that it is the lower lipid to drug ratio, which would have faster release as it is believed to have higher drug load per each SLN and therefore higher driving force for release. On the other hand, there was no significant difference between the different formulae regarding Q8h (P>0.05).
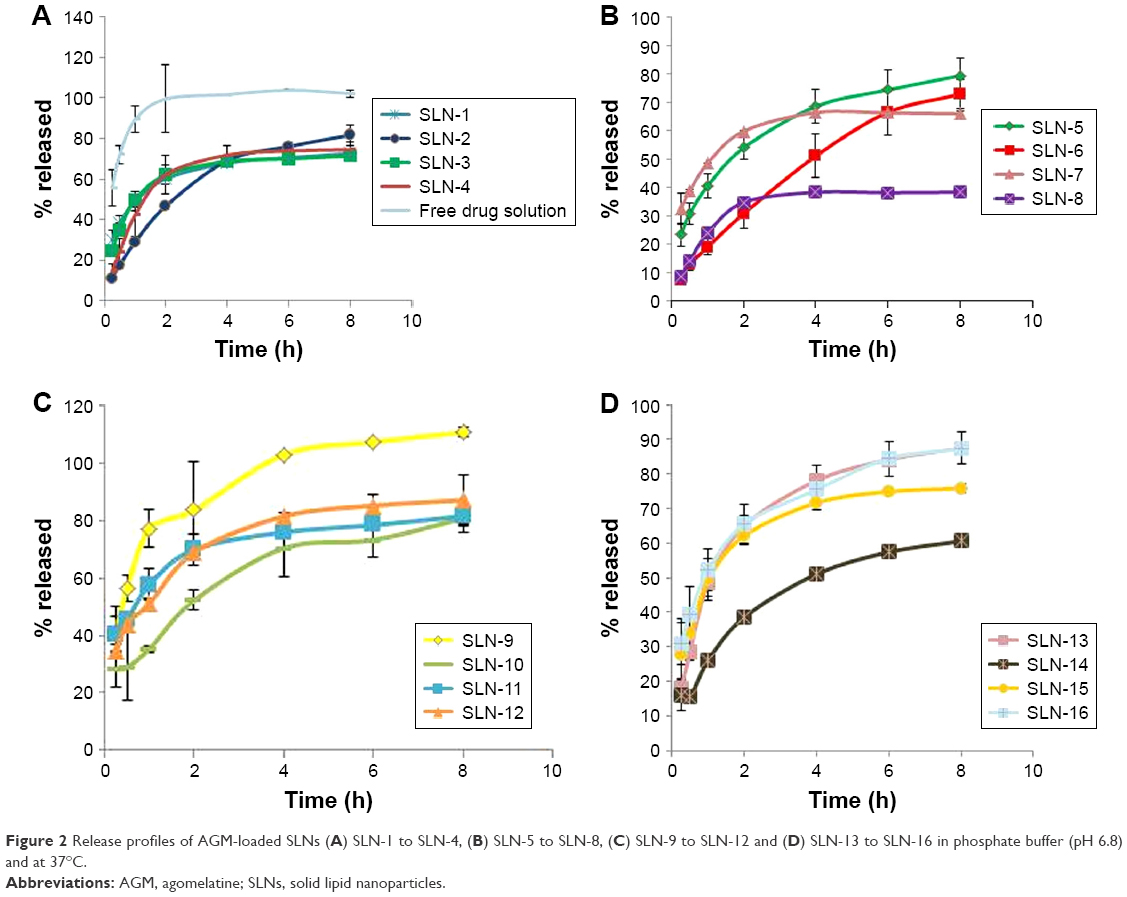 | Figure 2 Release profiles of AGM-loaded SLNs (A) SLN-1 to SLN-4, (B) SLN-5 to SLN-8, (C) SLN-9 to SLN-12 and (D) SLN-13 to SLN-16 in phosphate buffer (pH 6.8) and at 37°C. |
Optimization of the solid lipid nanoparticles formulations
For selection of the optimum formula, the optimization criteria were to minimize PS, PDI and Q1h and maximize EE%, ZP and Q8h as shown in Table 2. As illustrated in Table 5, the formula giving the highest numerical desirability was SLN-14 with a value of 0.77. As shown in Table 6, the predicted responses for this formulation were PS of 158.9 nm, ZP of −17.43 mV, EE% of 85.42%, Q1h of 39.03 %, Q8h of 85.43% and PDI of 0.14, whereas the actual responses were PS of 167.70 nm, ZP of −17.90 mV, EE% of 91.25%, Q1h of 35.40%, Q8h of 80.87% and PDI of 0.13. Results revealed the high similarity between the observed and predicted values of the optimal formulation with bias percentage not exceeding 9.72. Hence, SLN-14 can be considered as a promising SLN formulation; therefore, it was selected for further investigations.
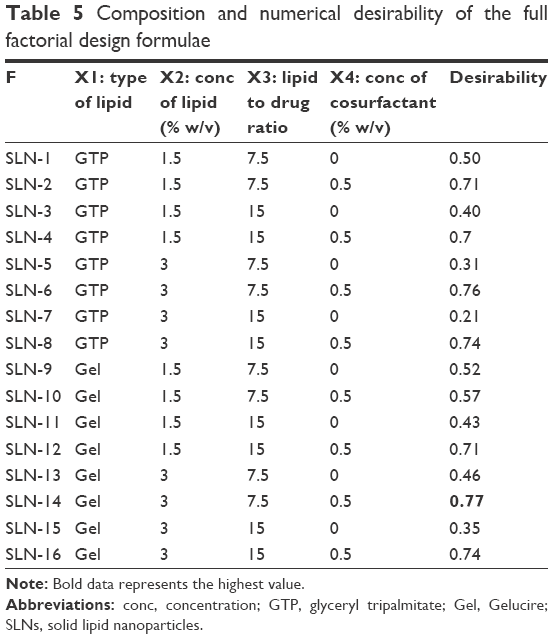 | Table 5 Composition and numerical desirability of the full factorial design formulae |
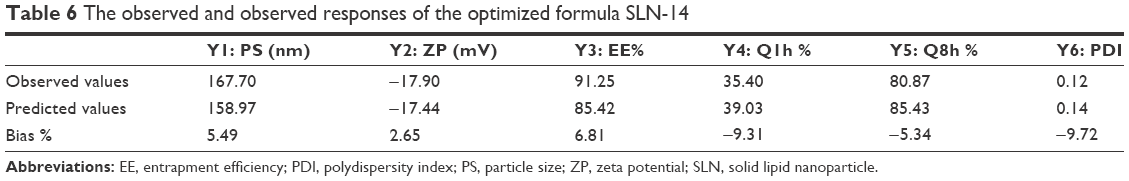 | Table 6 The observed and observed responses of the optimized formula SLN-14 |
Further investigations of the optimized SLNs formulation
Transmission electron microscopy (TEM)
The TEM micrographs of SLN-14 demonstrated that the nanoparticles are well identified and present in a nearly perfect spherical shape (Figure 3). The PS obtained was close to that determined by the dynamic light scattering.
 | Figure 3 Transmission electron micrograph of the optimized formula. |
Differential scanning calorimetry
As illustrated in Figures 4 and 5; the DSC thermogram of AGM shows a sharp endothermic peak at 101.50°C corresponding to the melting point of the drug. Basically, the endothermic peak of AGM is evident in all the thermograms of its physical mixtures with the components of the optimized formula, which might indicate compatibility. On the other hand, the endothermic peak of AGM extensively broadened in the thermogram of the optimized formula SLN-14, which might confirm the presence of the drug either in amorphous state or molecularly dispersed in the lipid matrix.
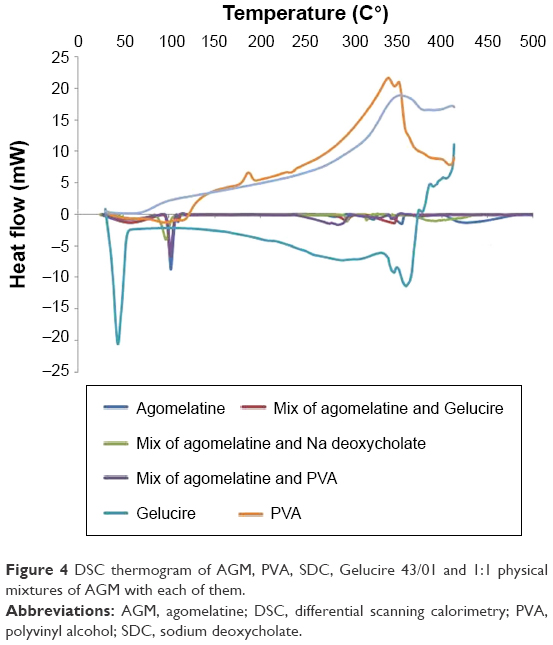 | Figure 4 DSC thermogram of AGM, PVA, SDC, Gelucire 43/01 and 1:1 physical mixtures of AGM with each of them. |
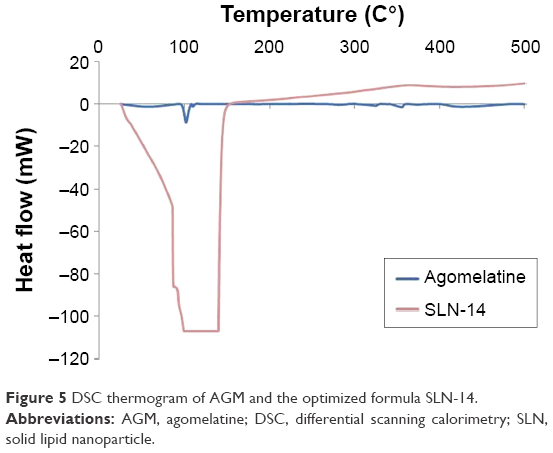 | Figure 5 DSC thermogram of AGM and the optimized formula SLN-14. |
Pharmacokinetic study
The liquid chromatography–mass spectrometry assay has a good linearity from 0.01 to 100 ng/mL with acceptable interday accuracy that ranged from 93.2% to 105.1%, whereas the interday precision ranged from 3.1% to 9.7%. The accuracy of freeze and thaw stability ranged from 87.6% to 95.7%, whereas its precision ranged from 4.8% to 8.9%.
Figure 6 shows the mean AGM concentrations in plasma of rats after administration of intranasal SLN-14, IV AGM solution and oral Valdoxan suspension, whereas Figure 7 shows the mean brain concentrations of these formulations. The pharmacokinetic parameters determined are summarized in Table 7. When AGM pharmacokinetics was analyzed in plasma, SLN-14 was found to have significantly higher Cmax, AUC(0–360 min) and absolute bioavailability (759.00 ng/mL, 7,805.69 ng·min/mL and 44.44%, respectively) than the oral suspension of Valdoxan® (20.73 ng/mL, 2,828.08 ng·min/mL and 16.12%, respectively; P<0.0001). The observed increase in the Cmax, AUC0–360 min and absolute bioavailability of the intranasal SLNs can be attributed to several factors explained as follows: first, the systemic absorption of AGM from the intranasal SLNs is carried out directly into the systemic circulation avoiding the hepatic first-pass metabolism and causing increased amount of AGM to reach the systemic circulation. Second, the lipophilic nature of SLNs being composed of lipids is expected to enhance the ability of the particles to partition into the lipid bilayer of the nasal epithelial cell membrane and pass directly through the cells, thus showing higher systemic absorption. Third, the used cosurfactant SDC is known to enhance nasal absorption via several possible mechanisms, including enhancing permeability of the membrane structure through opening of tight junctions between epithelial cells; extraction of membrane components such as proteins, lactate dehydrogenase enzyme and cholesterol by co-micellization; inhibition of proteolytic enzymes and formation of aqueous pore-type transport pathways, reducing the mucus viscosity and elasticity and thus the barrier function of the layer.23 SLN-14 had also a significantly longer half-life (189.09 min) than the oral suspension of Valdoxan® (91.98 min) (P<0.0001), which can be explained based on most of AGM in the formula SLN-14 is entrapped (EE% =91.25%); thus, it is expected to be released and subsequently eliminated at a slower rate. Also as having PS <200 nm, SLNs are expected to bypass the filtration at the interendothelial slits in the walls of venous sinuses and subsequently have a reduced reticuloendothelial clearance and prolonged circulation time.14,24 In addition, the presence of PVA on the surface of the particles is expected to prolong their blood circulation time. A significantly lower tmax (2.00 min) was also observed with SLN-14 compared with the oral suspension of Valdoxan® (15.67 min; P=0.001), which can be attributed to the faster systemic absorption from the nasal route than the oral administration.
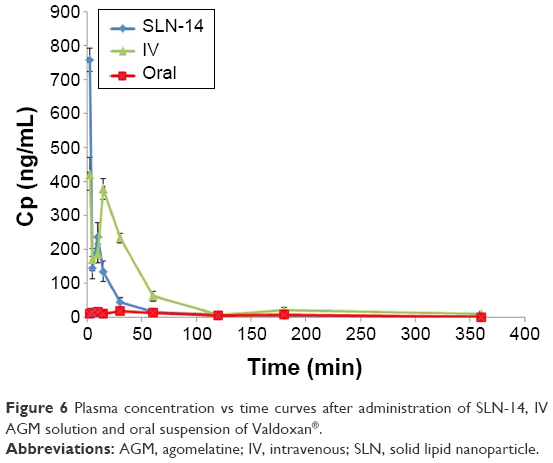 | Figure 6 Plasma concentration vs time curves after administration of SLN-14, IV AGM solution and oral suspension of Valdoxan®. |
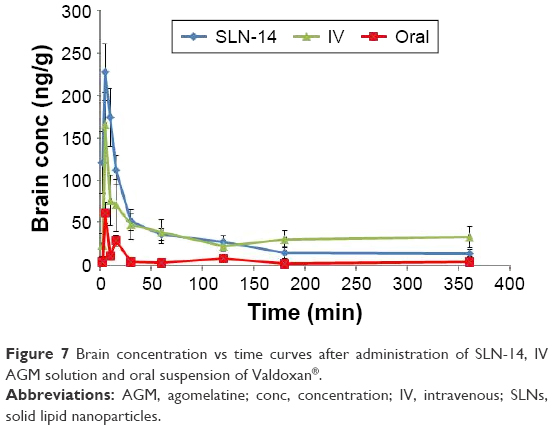 | Figure 7 Brain concentration vs time curves after administration of SLN-14, IV AGM solution and oral suspension of Valdoxan®. |
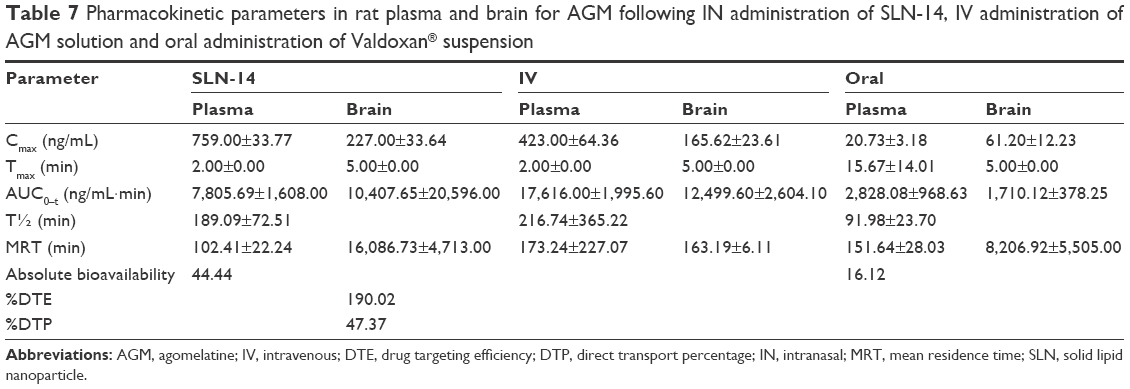 | Table 7 Pharmacokinetic parameters in rat plasma and brain for AGM following IN administration of SLN-14, IV administration of AGM solution and oral administration of Valdoxan® suspension |
For the pharmacokinetic parameters in the brain, SLN-14 was found to have significantly higher Cmax and AUC(0–360 min) (227.00 ng/mL and 10,407.65 ng·min/mL, respectively) than the oral suspension (61.20 ng/mL and 1,710.12 ng·min/mL, respectively; P<0.0001). The enhanced brain drug delivery can be explained based on several factors. First, the enhanced absolute bioavailability and the higher AGM plasma concentrations with the intranasal SLNs can create a higher driving force for the diffusion across the BBB due to the increased concentration gradient from the systemic circulation to the brain. Second, drug-loaded SLNs are known to have preferential BBB permeation compared with the free drug form. The enhanced BBB permeation of the SLNs can be related to the lipid content of the SLNs, which can enhance the transcellular diffusion across the BBB. Also, the surfactants associated with the SLNs such as SDC and PVA can act as absorption enhancers, decrease nanoparticle clearance by the reticuloendothelial system and inhibit the efflux system, especially P-glycoprotein enhancing the transport across BBB.25,26 The associated surfactants can also increase brain uptake via transient opening of the brain endothelial tight junctions.27 The SLNs can also be endocytosed by the BBB endothelial cells28 from where they can be transcytosed into the brain29 or release AGM within the endothelial cells that diffuses into the brain.
Third, the preferential brain delivery of the SLNs can be attributed to the direct transport from the nose to the brain where AGM-loaded SLNs or the free AGM can reach the brain using the olfactory or trigeminal nerves that extend from the nasal cavity into the brain away from the BBB. It was also found that SLN-14 has significantly longer brain MRT (16,086.73 min) than the oral suspension of Valdoxan® (8,206.92 min; P<0.05).
The %DTE was calculated for SLN-14 to assess the success of achieving brain targeting by this formula. SLN-14 yielded %DTE of 190.02 that indicated, as being >100, a superior drug delivery to the brain with the intranasal SLN-14 compared with the systemic administration. The favored brain drug delivery can be related to the route of administration, where the formula given by the intranasal route has the ability to be directly transported to the brain using the direct pathways of the olfactory and trigeminal nerves without having to cross the BBB, whereas the intravenously administrated AGM does not have any direct pathway to the brain and must cross the BBB to reach it. Another reason for the better brain delivery can be attributed to the form of AGM where the intranasal formula contained loaded AGM within SLNs, whereas the intravenous formula contained AGM in the free form. As previously mentioned, the SLNs are expected to have better BBB uptake than the free drug.
Drug uptake into the brain from the nasal mucosa occurs mainly via two different pathways. One is the systemic pathway by which some of AGM is absorbed into the systemic circulation and subsequently reaches the brain by crossing the BBB. The other is the olfactory and the trigeminal neural pathway by which AGM directly travels from the nasal cavity to the cerebrospinal fluid and/or brain tissue. To determine the relative contribution of the direct pathway in the overall brain delivery, the % DTP of SLN-14 was calculated. Negative % DTP indicates more efficient drug delivery to the brain through BBB permeation than direct nose-to-brain routes, whereas the positive values of the % DTE indicate a significant contribution of the direct routes to the overall brain delivery. SLN-14 yielded % DTP of 47.37, which revealed a significant role of the direct pathway in the brain delivery.
Conclusion
The formulation of AGM as SLNs and the intranasal administration of the prepared formulation have effectively enhanced both the absolute bioavailability and the brain delivery of AGM probably due to the avoidance of the first-pass metabolism, the favored BBB uptake of the SLNs and the direct transport from the nose to the brain through the olfactory and trigeminal nerves.
Disclosure
The authors report no conflicts of interest in this work.
References
Millan MJ, Gobert A, Lejeune F, et al. The novel melatonin agonist agomelatine (S20098) is an antagonist at 5-hydroxytryptamine2C receptors, blockade of which enhances the activity of frontocortical dopaminergic and adrenergic pathways. J Pharmacol Exp Ther. 2003;306(3):954–964. | ||
Audinot V, Mailliet F, Lahaye-Brasseur C, et al. New selective ligands of human cloned melatonin MT1 and MT2 receptors. Naunyn Schmiedebergs Arch Pharmacol. 2003;367(6):553–561. | ||
Yous S, Andrieux J, Howell H, et al. Novel naphthalenic ligands with high affinity for the melatonin receptor. J Med Chem. 1992;35(8):1484–1486. | ||
Zupancic M, Guilleminault C. Agomelatine. CNS Drugs. 2006;20(12):981–992. | ||
Haque S, Md S, Sahni JK, Ali J, Baboota S. Development and evaluation of brain targeted intranasal alginate nanoparticles for treatment of depression. J Psychiatr Res. 2014;48(1):1–12. | ||
Alsarra IA, Hamed AY, Alanazi FK, El Maghraby GM. Vesicular systems for intranasal drug delivery. Drug Delivery to the Central Nervous System. New York: Springer; 2010:175–203. | ||
Soane R, Hinchcliffe M, Davis S, Illum L. Clearance characteristics of chitosan based formulations in the sheep nasal cavity. Int J Pharm. 2001;217(1):183–191. | ||
Dhuria SV, Hanson LR, Frey WH. Intranasal delivery to the central nervous system: mechanisms and experimental considerations. J Pharm Sci. 2010;99(4):1654–1673. | ||
Kozlovskaya L, Abou-Kaoud M, Stepensky D. Quantitative analysis of drug delivery to the brain via nasal route. J Control Release. 2014;189:133–140. | ||
Johnson PH, Quay SC. Advances in nasal drug delivery through tight junction technology. Expert Opin Drug Deliv. 2005;2(2):281–298. | ||
Battaglia L, Gallarate M. Lipid nanoparticles: state of the art, new preparation methods and challenges in drug delivery. Expert Opinion Drug Deliv. 2012;9(5):497–508. | ||
Gastaldi L, Battaglia L, Peira E, et al. Solid lipid nanoparticles as vehicles of drugs to the brain: current state of the art. Eur J Pharm Biopharm. 2014;87(3):433–444. | ||
Lasa-Saracibar B, Estella-Hermoso de Mendoza A, Guada M, Dios-Vieitez C, Blanco-Prieto MJ. Lipid nanoparticles for cancer therapy: state of the art and future prospects. Expert Opin Drug Deliv. 2012;9(10):1245–1261. | ||
Kaur IP, Bhandari R, Bhandari S, Kakkar V. Potential of solid lipid nanoparticles in brain targeting. J Controlled Release. 2008; 127(2):97–109. | ||
Garcia-Fuentes M, Torres D, Alonso M. Design of lipid nanoparticles for the oral delivery of hydrophilic macromolecules. Colloids Surf B. 2003;27(2):159–168. | ||
Montgomery DC. Design and Analysis of Experiments. Vol 7. New York: Wiley; 1984. | ||
Pramanick S. Excipient selection in injectable/parenteral formulation development. Pharma Times. 2013;45(3):65–77. | ||
CDER U. Guidance for industry: estimating the maximum safe starting dose in initial clinical trials for therapeutics in adult healthy volunteers. US Department of Health and Human Services Food and Drug Administration Center for Drug Evaluation and Research. Rockville, MD; 2005. | ||
Muller RH, Mader K, Gohla S. Solid lipid nanoparticles (SLN) for controlled drug delivery–a review of the state of the art. Eur J Pharm Biopharm. 2000;50(1):161–177. | ||
Müller RH, Peters K. Nanosuspensions for the formulation of poorly soluble drugs: I. Preparation by a size-reduction technique. Int J Pharm. 1998;160(2):229–237. | ||
Phipps L. The High Pressure dairy homogenizer. Vol 6. Reading: National Institute for Research in Dairying; 1985. | ||
Mehnert W, Mäder K. Solid lipid nanoparticles: production, characterization and applications. Adv Drug Deliv Rev. 2001;47(2):165–196. | ||
Romeo VD, deMeireles JC, Gries WJ, et al. Optimization of systemic nasal drug delivery with pharmaceutical excipients. Adv Drug Deliv Rev. 1998;29(1–2):117–133. | ||
Moghimi SM, Porter CJ, Muir IS, Illum L, Davis SS. Non-phagocytic uptake of intravenously injected microspheres in rat spleen: influence of particle size and hydrophilic coating. Biochem Biophys Res Commun. 1991;177(2):861–866. | ||
Esposito E, Fantin M, Marti M, et al. Solid lipid nanoparticles as delivery systems for bromocriptine. Pharm Res. 2008;25(7):1521–1530. | ||
Tsai MJ, Wu C, Huang YB, et al. Baicalein loaded in tocol nanostructured lipid carriers (tocol NLCs) for enhanced stability and brain targeting. Int J Pharm. 2012;423(2):461–470. | ||
Alam MI, Beg S, Samad A, et al. Strategy for effective brain drug delivery. Eur J Pharm Sci. 2010;40(5):385–403. | ||
Kreuter J. Application of nanoparticles for the delivery of drugs to the brain. Int Congress Ser. 2005;1277:85–94. | ||
Dehouck B, Fenart L, Dehouck MP, Pierce A, Torpier G, Cecchelli R. A new function for the LDL receptor: transcytosis of LDL across the blood–brain barrier. J Cell Biol. 1997;138(4):877–889. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.