Back to Journals » Open Access Rheumatology: Research and Reviews » Volume 11
Interstitial Lung Disease in Systemic Sclerosis: Focus on Early Detection and Intervention
Authors Fischer A, Patel NM, Volkmann ER ![]()
Received 10 August 2019
Accepted for publication 9 November 2019
Published 9 December 2019 Volume 2019:11 Pages 283—307
DOI https://doi.org/10.2147/OARRR.S226695
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Chuan-Ju Liu
Aryeh Fischer,1 Nina M Patel,2 Elizabeth R Volkmann3
1Division of Rheumatology, Division of Pulmonary Sciences and Critical Care Medicine, Department of Medicine, University of Colorado School of Medicine, Denver, CO, USA; 2Division of Pulmonary, Allergy and Critical Care Medicine, Columbia University Irving Medical Center, New York, NY, USA; 3Division of Rheumatology, Department of Medicine, David Geffen School of Medicine, University of California, Los Angeles, CA, USA
Correspondence: Elizabeth R Volkmann
Division of Rheumatology, Department of Medicine, David Geffen School of Medicine, University of California, 1000 Veteran Avenue, Suite 32-59, Los Angeles, CA 90095, USA
Tel +1 310-825-2448
Fax +1 310-206-6553
Email [email protected]
Abstract: Systemic sclerosis (SSc) is a progressive and often devastating disease characterized by autoimmune dysfunction, vasculopathy, and fibrosis. Interstitial lung disease (ILD) is identified in the majority of patients with SSc and is the leading cause of SSc-related mortality. Although clinical manifestations and ILD severity vary among patients, lung function typically declines to the greatest extent during the first 3–4 years after disease onset. We aim to provide an overview of SSc-associated ILD (SSc-ILD) with a focus on current and emerging tools for early diagnosis of ILD and current and novel treatments under investigation. Early detection of ILD provides the opportunity for early therapeutic intervention, which could improve patient outcomes. Thoracic high-resolution computed tomography is the most effective method of identifying ILD in patients with SSc; it enables detection of mild lung abnormalities and plays an important role in monitoring disease progression. Cyclophosphamide and mycophenolate mofetil are the most commonly prescribed treatments for SSc-ILD. Recently, nintedanib (an antifibrotic) was approved by the Food and Drug Administration for patients with SSc-ILD; it is indicated for slowing the rate of decline in pulmonary function. However, there is a need for additional effective and well-tolerated disease-modifying therapy. Ongoing studies are evaluating other antifibrotics and novel agents. We envision that early detection of lung involvement, combined with the emergence and integration of novel therapies, will lead to improved outcomes in patients with SSc-ILD.
Keywords: systemic sclerosis, interstitial lung diseases, early diagnosis, disease progression, treatment outcome
Plain Language Summary
Systemic sclerosis (SSc) is a rare condition characterized by immunologic abnormalities, organ fibrosis and vasculopathy. Interstitial lung disease (ILD), also called pulmonary fibrosis, is a common manifestation of SSc. ILD in SSc is often associated with a decline in lung function within the first several years of lung disease onset. Effective screening to improve early diagnosis of patients with SSc with associated ILD (SSc-ILD) is of paramount importance. We analyzed the SSc-ILD medical literature to look at available and emerging tools for the early diagnosis of ILD, current treatments, and novel agents under study. Several methods are available to diagnose ILD, including high-resolution computed tomography, the “gold standard” method for detecting SSc-ILD, and lung function tests. Cyclophosphamide and mycophenolate are recommended for the treatment of SSc-ILD based on data from the Scleroderma Lung Studies I and II. In addition, the FDA recently approved nintedanib to slow the decline of lung function in patients with progressive fibrotic SSc-ILD. There remains a need to identify additional, more effective therapies for SSc-ILD. We hope that early diagnosis of lung involvement and the development of safe and more effective medicines will lead to improved outcomes in SSc-ILD.
Introduction
Systemic sclerosis (SSc) is a clinically heterogeneous disease characterized by a complex interplay between autoimmunity, vasculopathy, and fibrosis. This condition affects multiple organ systems, including the skin, gastrointestinal tract, lungs, kidneys, and heart.1–3 The most common pulmonary manifestations of SSc, interstitial lung disease (ILD) and pulmonary arterial hypertension (PAH), are the leading causes of death and account for up to 60% of the SSc-associated mortality.4,5 In a meta-analysis, patients with SSc with associated ILD (SSc-ILD) were found to have a mortality risk nearly three times greater than SSc patients without ILD.6
When examined using high-resolution computed tomography (HRCT), ILD in patients with SSc is typically characterized by bilateral, lower-lobe predominant reticulations, ground-glass opacities, and in some cases, honeycombing.7 The initial clinical presentation of SSc-ILD, however, varies, which can make diagnosis challenging. Patients with mild ILD can be asymptomatic in the early stages of disease and, therefore, may not undergo pulmonary function testing or diagnostic radiology until they experience symptoms such as dyspnea on exertion and an increasingly persistent cough. Despite recent improvements in the overall survival rates of patients with SSc, current therapies do not curtail disease-related inflammation or fibrosis consistently.8–10 Clinical trials have demonstrated that immunosuppressant therapy can provide modest benefits in patients with SSc-ILD, and some patients experience ILD progression despite receiving such treatment.11
Administration of treatment early in the course of SSc-ILD may lead to improved clinical outcomes.12 This was demonstrated in a retrospective study comparing the use of cyclophosphamide (CYC) with other drugs and no treatment in patients with SSc-ILD.13 Irrespective of the drug used, the factor that predicted significant improvement in lung function was the initiation of treatment at an early stage in the disease process.13 In line with this, rigorous screening programs to facilitate early diagnosis of SSc-ILD and, hopefully, early initiation of treatment are of paramount importance.14
In this review, we aim to provide an overview of SSc-ILD with a focus on current and emerging tools for early diagnosis of ILD, as well as novel treatments currently under investigation. Relevant articles were identified by screening the literature using the PubMed search engine, with various combinations of the following search terms: “systemic sclerosis” OR “scleroderma”; “interstitial lung disease”; “pathology”; “epidemiology”; “treatment” OR “therapy”; and “detection” OR “diagnosis”. The contents of the retrieved articles were reviewed to identify those of relevance. We were particularly interested in literature that discussed early detection and interventions that were published within the last 10 years; however, strict limits on time from publication were not imposed. We also searched clinicaltrials.gov to identify relevant clinical trials on SSc-ILD.
Epidemiology and Clinical Outcomes of SSc-ILD
For the majority (70–90%) of patients with SSc who develop ILD, ILD is observed within the first 3 years of SSc diagnosis; 40–75% of the patients with SSc have reduced pulmonary function.15,16 SSc-ILD tends to occur with increased severity and/or increased risk of progression in patients with Scl-70 (anti-topoisomerase I) antibodies,5,17 male sex,18 and African–American ethnicity.19 SSc-ILD is most often associated with a histological pattern of non-specific interstitial pneumonia, while usual interstitial pneumonia and other histologic patterns are less commonly observed.20–22 Differences in histological patterns do not appear to have prognostic significance in SSc-ILD.20
When evaluating SSc-ILD, a variety of parameters should be considered as relevant to the patient’s prognosis. The presence of Scl-70 antibodies is a key risk factor for disease severity and mortality.5,17,23 Diffusing capacity of the lung for carbon monoxide (DLco) at baseline has been shown to be an independent predictor of mortality in SSc-ILD including in early SSc-ILD, while the extent of disease on baseline HRCT independently predicts ILD progression, as well as mortality.24 Patients with cardiac involvement are more likely to have worse lung function.19 Increased cutaneous sclerosis is associated with decreased lung function,19,25 and the extent of skin disease appears to be an independent predictor of mortality.26 Decline in forced vital capacity (FVC) and DLco over 2 years, and increased age are also independent predictors of mortality.26
Considering the heterogeneity of SSc-ILD, the potential absence of symptoms in early or mild disease, and the potential impact of ILD on outcomes in patients with SSc, it is prudent to consider the above parameters when assessing and monitoring newly diagnosed patients. Patients may be diagnosed early and more efficiently through multidisciplinary evaluation and collaboration; this is expected to help optimize care and lead to improved clinical outcomes.
Current Diagnostic Tools for ILD in SSc
The most frequently used methods to diagnose ILD in patients with SSc are HRCT and pulmonary function tests (PFTs).27,28 HRCT is considered the “gold standard” method for diagnosing SSc-ILD, as well as other types of ILD.29–31 Despite the established utility of PFTs in assessing the progression of ILD, these tests should not be used in isolation to screen for, or diagnose, SSc-ILD.32 PFT results suggesting restricted lung function can be caused by SSc without ILD due to severe skin involvement, while results outside the normal range may not be apparent in patients with early ILD. These points are discussed in more detail below.
High-Resolution Computed Tomography
For the diagnosis of SSc-ILD, HRCT is more sensitive than conventional chest CT or PFTs and allows recognition of mild abnormalities.28,33,34 The high sensitivity of HRCT can help identify mild or early interstitial abnormalities that are of unknown clinical significance,34 which should prompt heightened surveillance for signs of progression. HRCT is well suited to detecting mild or early SSc-ILD; a scan of a patient with mild ILD compared with a patient with severe ILD is presented in Figure 1. As a non-invasive diagnostic tool, HRCT is used not only to detect ILD but also to predict ILD progression and decline in pulmonary function. In one study, 68% of the patients with SSc with areas of ground-glass opacities on their initial HRCT scan showed progressive lung fibrosis in a second scan that was conducted 2–5 years later.35 This suggests that ground-glass opacities may precede lung fibrosis in SSc and that early intervention with immunosuppressive therapy should be considered.35 HRCT can not only be used to detect ILD, predict the development of fibrosis, ILD progression and decline in pulmonary function,36 but it can also be used to identify patients with SSc with low risk of developing ILD. Of 108 patients with SSc and no signs of lung fibrosis at baseline, none exhibited fibrosis upon follow-up HRCT assessment a mean of 3.1 years later.36
 |
Figure 1 HRCT scans of lungs from patients with SSc-ILD of different severities. Notes: Patient with mild severity: coronal view (A) and axial view (C); patient with severe disease: coronal view (B) and axial view (D). Both patients participated in the SLS II trial. Abbreviations: HRCT, high-resolution computed tomography; SLS, Scleroderma Lung Study; SSc-ILD, systemic sclerosis with associated interstitial lung disease. |
As alluded to earlier, baseline HRCT findings can be used to predict the likelihood of future progression, decline in FVC, or response to therapy,36–38 and serial HRCT scans can be performed to check whether a patient has developed ILD or to monitor the progression of existing disease. HRCT also enables the detection of increased esophageal diameter, which has been associated with, and is an important predictor of, severe ILD and low DLco in patients with SSc.34,39,40
Radiation exposure is considered a potential limiting factor in the use of HRCT.41 Recent findings highlight that the required HRCT slice number can be reduced, thereby limiting the radiation dose to the patient without affecting the assessment of lung fibrosis (severity or extent) in comparison to standard protocols.42,43 This approach may be most useful in patients with established ILD, in whom HRCT may be used regularly to monitor progression. Nevertheless, it has been reported that sequential reduced 9-slice HRCT could reliably detect mild ILD42 and discriminate lung fibrosis at a threshold of 20% with high sensitivity and specificity.43 It may, therefore, be useful in detecting early disease progression in patients with SSc. However, further validation in additional cohorts of patients with SSc is required.
Quantitative imaging is emerging as a powerful tool for measuring the extent of lung fibrosis and for assessing disease progression more objectively than visual assessment.44,45 In this method, computer-generated algorithms are applied to quantify the extent of features such as fibrosis, ground-glass opacities, and honeycombing, in a uniform and standard manner. Currently, quantitative imaging is being used primarily in SSc-ILD clinical trials, but may become established in daily clinical practice in due course.44–46 Semi-quantitative imaging may also be used for diagnosis and quantification of lung involvement in SSc-ILD; however, this method does not eliminate subjectivity or variability from the assessor.47,48
Pulmonary Function Tests
FVC and DLco are important pulmonary function parameters for assessing lung involvement in SSc-ILD.27 A recent systematic review of 219 SSc-ILD studies reported that FVC% predicted was the primary endpoint in 70.4% of the studies, and DLco was the primary endpoint in 11.3% of the studies.29 Despite official American Thoracic Society guidelines which define ILD by a reduction of total lung capacity, this parameter remains rarely used as an outcome measure in SSc-ILD studies.29
Data from the Pittsburgh Scleroderma Databank demonstrated that the greatest declines in FVC often occur early, within the first 3 years of SSc onset, even in asymptomatic patients.3,15 This was supported by the results of the Scleroderma Lung Study (SLS) I study. In SLS I, patients with SSc-ILD (n=77) who were randomly assigned to receive placebo had a 4.2% decline in unadjusted FVC% predicted and an 8.2% decline in DLco% predicted over 12 months. The unadjusted decline in FVC% predicted and DLco% predicted were similar regardless of the disease duration (0–7 years) group, although an association between the extent of fibrosis (assessed via HRCT) at baseline and decline in FVC was most evident during the first 2 years after disease onset.37
FVC% or DLco% predicted values >80% are considered to be “normal” in patients without respiratory symptoms, but the threshold for “normal” varies between individual patients, potentially affecting interpretation of test results. In our experience and in line with the literature, a decline from baseline of 5–10% in FVC and 10–15% in DLco in a patient with SSc-ILD should be further evaluated, as it may be indicative of disease progression, even if absolute values remain above the 80% threshold.32 Goh et al showed that a decline of ≥10% in FVC, or a decline of 5–9% in FVC together with a decline of ≥15% in DLco, was associated with a higher risk of subsequent mortality in patients with SSc-ILD.49 This finding was attributed to disease progression in patients with extensive lung fibrosis. To illustrate this point, a patient with early SSc-ILD with an FVC of 90% would usually be considered normal. However, if this patient had an FVC of 110% a year earlier, the value of 90% represents a significant decline from their baseline, and, potentially, disease progression. It should be noted that the rate of decline in pulmonary function from baseline during the first 12 months has been shown to have strong prognostic significance.
The clinical course of FVC decline in SSc is highly variable between patients, with the possibility of stability or improvement even in patients with low baseline FVC. Man et al identified the following groups of patients within SSc, based on baseline values and changes over the subsequent 72 months in FVC: (1) very low – slow decline (5.5% of the overall population of SSc patients); (2) very low – improve (13.8%); (3) low – fast decline (9.5%); low – stable (19.7%); (4) low-normal – improve (31.1%); (5) normal – improve (16.1%); and normal – stable (4.3%).50 Patients with longer disease duration (>4 years) are less likely to experience a future decline in lung function compared with those who have early stage disease.37
Values for both FVC and DLco can be influenced by SSc disease processes other than ILD, and this should be a consideration during ILD assessment.29 For example, a disproportionate decline in DLco relative to FVC may be related to the development of PAH.51 FVC correlates poorly with the quantitative extent of fibrosis, while DLco has been shown to be the best predictor of HRCT-measured ILD.52 The reliability of lung function as a predictor of SSc-ILD remains controversial. In a recent study involving 256 patients with SSc, among patients with radiographic ILD (n=188; 71%), 59 patients (31%) had a normal FVC (≥80% predicted). Of the 256 patients, 151 had accurate DLco measurements available; among these patients, 65 (43%) had a DLco ≥60% predicted.33 These data emphasize limitations of PFTs and show that normal FVC and DLco measurements in patients with SSc do not exclude the presence of ILD or obviate the need to pursue HRCT imaging to definitively assess for ILD. Due to the variability arising from technical factors, diurnal or seasonal changes, and patient-related factors, PFTs alone are insufficient for determining the true extent of ILD and changes in PFT parameters do not mirror changes in the extent of radiographic fibrosis consistently.53
Combined Data from HRCT and PFTs
Considering the aforementioned limitations of HRCT and PFTs, combined use of these tests can provide more informative and reliable prognostic information than either component used alone.54,55 We suggest that, in addition to assessing and quantifying dyspnea and cough, and performing PFTs in patients with newly diagnosed SSc, HRCT should be used to assess the presence of ILD. Depending on the clinical scenario, appropriate follow-up investigations might include DLco for PAH evolution or serial FVC, DLco, and HRCT assessments in patients with established ILD. In order to increase the likelihood of diagnosing ILD at an early stage, yearly PFTs with a low threshold are recommended in patients with SSc and no known ILD. HRCT should be pursued if there is a decline in FVC and/or DLco, or a change in the clinical scenario (e.g., increased dyspnea or audible crackles on chest auscultation).
Other Diagnostic Tools
Lung Ultrasound and Other Imaging Modalities
One drawback of HRCT is radiation exposure,41 and this casts concern over the frequent use of this method to monitor for SSc-ILD disease progression. The past few years have seen extensive evaluation of lung ultrasound (LUS) as a non-invasive and non-ionizing imaging method for detecting ILD.56 Discrete vertical hyperechoic reverberation artifacts arising from the pleural line and extending to the bottom of the screen without fading when the lung parenchyma air content is decreased or when the interstitial space is expanded, known as B-lines, have been observed in 51% of the patients with SSc.57 Several studies have reported a significant linear correlation between the number of B-lines and HRCT score.58–60 Pleural irregularity (PI; loss of normal hyperechoic linear pleural contouring) represents another potential LUS assessment criterion. A recent study reported significantly higher PI scores in patients with SSc with ILD versus without ILD, and in patients with extensive versus limited disease. In addition, the PI scores correlated with the Wells scores and with DLco measurements.61 Thus, the PI score could be valuable in diagnosing, classifying, and assessing SSc-ILD.
LUS may also represent a useful tool for the detection of ILD at an early stage in patients with SSc.62 In a recent study, 58 patients with SSc were evaluated for signs of ILD with HRCT and then LUS. Using HRCT, evidence of ILD was confirmed in 88% of the patients with SSc and in 41% of the patients with early stage SSc. When subsequently analyzed with LUS, there was a significant difference in the number of B-lines in patients with or without evidence of ILD on HRCT (57±53 vs 9±9; p<0.0001), with a concordance rate of 83%.62
Overall, LUS seems to be an attractive and promising technique, with the potential to be integrated with HRCT and PFTs in the screening and evaluation of ILD in patients with SSc. However, at this stage, LUS remains a relatively experimental and unproven technique for ILD diagnosis. Validation and protocol standardization across institutions are needed before LUS can be implemented in routine clinical practice.
Further imaging modalities with potential application in early SSc-ILD include magnetic resonance imaging (MRI), positron emission tomography (PET), and single-photon emission computed tomography (SPECT).63 Such modalities have been used to obtain diagnostic information with regard to disease activity and treatment response in cardiac disease, neurodegenerative disease, and oncology, and are being investigated more frequently in pulmonary diseases including SSc-ILD.63 The use of MRI to image pulmonary fibrosis in SSc-ILD has been investigated. For instance, the capacity of MRI to determine the presence and degree of ILD was assessed retrospectively in 18 patients with SSc who underwent MRI and HRCT of the chest within a 12-month period. MRI could detect ILD with high sensitivity and specificity in patients with greater than 0.5% parenchymal involvement.63,64 However, in comparison with HRCT, MRI was found to be less sensitive and under-measured disease extent.64 MRI has been investigated in terms of its capacity to detect early functional changes with treatment in various diseases; however, further research is needed to confirm if MRI could be used to detect early changes in SSc-ILD.
Molecular probes can be combined with various imaging modalities, such as MRI, SPECT, or PET to help visualize and quantify the expression of specific molecular targets. For instance, probes have been designed to target molecular components involved in the onset of inflammation following epithelial injury or the development of pulmonary fibrosis.65 Several molecular probes have been utilized in animal models of pulmonary fibrosis or in humans with pulmonary fibrosis including SSc-ILD; further research and development in this area may lay foundations for early and more personalized precise diagnostic and treatment approaches.63,65 At present, however, these methods are investigational in relation to the diagnosis and assessment of SSc-ILD and not commonly used currently in clinical practice.63,66
Bronchoalveolar Lavage
Currently, bronchoalveolar lavage (BAL) is most often performed when there is suspicion of an underlying infection. It is seldom used for diagnosis or screening of SSc-ILD due to inconclusive study outcomes,67 as well as being an invasive procedure with potential complications. In a follow-up study from SLS I, evaluation of BAL inflammation did not improve prediction of disease progression or treatment response, in comparison with physiologic and HRCT findings.68 However, in a later study, BAL was used in conjunction with comprehensive clinical information and HRCT findings, and this approach provided useful information for the diagnostic evaluation of patients with suspected ILD.69 Overall, the role of BAL as a tool for early diagnosis of ILD in patients with SSc is controversial and not recommended.
Surgical Lung Biopsy
Obtaining lung tissue samples is an invasive procedure that requires the use of anesthesia and carries the potential risk of significant complications, including acute exacerbation of ILD, prolonged air leak, and respiratory failure.70–72 Given the availability of the methods described earlier and the unclear implications of defining histologic subsets in SSc-ILD, lung biopsy is not clinically necessary in most patients.20,73,74 However, it can be diagnostically useful in certain circumstances, such as when there are contradictions between clinical and radiological findings.75
Serum Biomarkers
Although techniques such as HRCT and PFTs can be used to detect early development or progression of SSc-ILD, it is necessary to find additional reliable and easily obtained markers of disease progression. Biomarkers may have a role in this respect since they can be readily obtained, are informative, and can be monitored over time.76 Elevated serum levels of the glycoprotein Krebs von den Lungen-6 (KL-6) at first visit have been associated with lung involvement in patients with SSc.77 Elevated serum KL-6 has been linked to the degree of inflammation and fibrosis, and future decline in lung function.78–83 Thus, KL-6 is emerging as a promising biomarker for the diagnosis of SSc-ILD, with potential for predicting lung involvement and disease progression. The detection of high KL-6 levels at baseline could help to identify patients in need of therapy or more frequent clinical monitoring.78,81,84
High levels of the cytokines, chemokine (C-C motif) ligand 285 and CCL18,86 are predictive of ILD progression82,83 and poor survival in patients with early SSc-ILD.82 However, a recent study highlighted that, although CCL18 levels are higher in patients with SSc-ILD, they do not predict progression of ILD based on FVC decline.81 Serum levels of surfactant protein D (SP-D) are also elevated in patients with SSc-ILD,83 and longitudinal measurement of SP-D levels may help to monitor fibrosis. However, there is some uncertainty regarding the interpretation of SP-D results and validation is required.77 Several other biomarkers, including interleukin-687 and CXC chemokine ligand 4,88,89 may also be important in SSc-ILD. High levels of certain serum biomarkers could potentially serve to prompt physicians to undertake further pulmonary imaging and PFTs. For most biomarkers, the available evidence is insufficient to justify utilizing them in clinical practice; however, further research may shed light on the most important biomarkers for ILD screening in patients with SSc. The key strengths and limitations of each technique discussed in this section are summarized in Table 1.
 |  |  |
Table 1 Diagnostic Tests for ILD: Summary of Available Methods, and Their Main Strengths and Limitations |
Monitoring and Predicting the Progression of ILD in Patients with SSc-ILD
Considering the potentially progressive fibrosing phenotype of SSc-ILD and the risk of rapid deterioration particularly evident in the first 3 years of disease onset, effective and early disease monitoring is of utmost importance.14 In addition to this, monitoring treatment response is important. Disease progression and response to treatment are usually assessed through the evolution of respiratory symptoms, exercise tolerance, HRCT findings, and PFTs. In SLS I, patients with any cough at baseline displayed increased ILD severity.90 Treatment of SSc-ILD patients with CYC and mycophenolate (MMF)91 was shown to produce significant reductions in cough,92 along with improvements in measures of ILD severity over 2 years.91
Not only is it important to detect and monitor SSc-ILD, it is essential to identify those who are at high risk of disease progression at an early stage. Recently, studies have been undertaken to investigate the possibility of using predictive models for this purpose.50,93–95 Ryerson et al showed that the modified du Bois index (parameters: age, respiratory-related hospitalization in the past 6 months, and predicted baseline and 24-week change in FVC) was slightly superior to the composite physiologic index, ILD-gender, age, physiology index, and the du Bois index in predicting 1-year mortality in SSc-ILD.94 Additionally, the FVC and 6 min walk test distance (6MWT) were identified as independent predictors of mortality.94 The 6MWT is frequently used as a measure of exercise tolerance in patients with SSc-ILD.96 However, although it is a straightforward and non-invasive assessment of exercise capacity, its usefulness and accuracy remain unclear in patients with SSc-ILD due to potential concomitant neuromuscular effects that can introduce bias.96
Despite recommendations to perform these assessments (as well as 6MWT) at regular intervals in patients with idiopathic pulmonary fibrosis (IPF),97 there are no formal guidelines for monitoring progression in SSc-ILD. Most clinicians would perform PFTs every 3–6 months,30 whereas opinions on the optimal interval between HRCT assessments are variable. Table 2 shows our suggestions for monitoring patients with SSc-ILD, including those with early disease.
 |
Table 2 Monitoring the Progression of SSc-ILD in Different Clinical Scenarios: Suggested Frequencies for Assessmenta |
Current Treatments for SSc-ILD
Due to the complexity and heterogeneous nature of SSc-ILD, treatment approaches should be tailored to the individual, with consideration of disease stage and organ-specific complications.98 While early treatment of ILD in SSc is of high importance to reduce the rate of lung deterioration and mortality,13 not every patient with SSc-ILD requires treatment for their ILD ─ treatment is often reserved for patients with progressive disease, ILD-related clinical impairment, or those patients at high risk for ILD progression. Table 3 provides a summary of treatments currently being investigated for the treatment of SSc-ILD, and Figure 2 presents a diagrammatic summary of the mechanism of action of these drugs.
 |
Table 3 Current and Potential Future Therapeutic Agents: Supporting Evidence and Ongoing Studies |
 |
Figure 2 Key pathogenic pathways involved in SSc-ILD and purported targets of existing and potential therapeutic agents. Notes: Khanna D, Tashkin DP, Denton CP, et al, Ongoing clinical trials and treatment options for patients with systemic sclerosis–associated interstitial lung disease, Rheumatology 2019; 58 (4): 567–579, doi:10.1093/rheumatology/key151. Reprinted by permission of Oxford University Press on behalf of the British Society for Rheumatology. © The Author(s) 2018. All rights reserved. For permissions, please email: [email protected].7 aMultiple growth factor receptors. Abbreviations: ADCC, antibody-dependent cell-mediated toxicity; CD, cluster of differentiation; CMC, complement-mediated cytotoxicity; DNA, deoxyribonucleic acid; FGF, fibroblast growth factor; IL, interleukin; LPA, lysophosphatidic acid; MMF, mycophenolate mofetil; PDGF, platelet-derived growth factor; SSc-ILD, systemic sclerosis with associated interstitial lung disease; TGF, tumor growth factor; VEGF, vascular endothelial growth factor. |
Immunosuppressive Therapy
CYC and MMF are the most broadly prescribed therapies for SSc-ILD. A retrospective study from the European Scleroderma Trials and Research group involving over 3000 patients with SSc-ILD recently reported that non-selective immunosuppressive therapy was prescribed to 71% (2681/3778) of patients.99 Also, a survey from the Scleroderma Clinical Trial Consortium and the Canadian Scleroderma Research group (n=170) revealed that 69% of the experts surveyed would use CYC, MMF, or rituximab (RTX) for induction therapy, and MMF for maintenance therapy.100 However, CYC and MMF are not consistently disease-modifying for all patients, as some patients experience ILD progression despite treatment with these agents.
Cyclophosphamide
In the early 1990s, CYC was found to improve pulmonary function in patients with SSc-ILD when administrated with low-dose prednisone.101 It was subsequently shown that CYC alone improved lung function and survival outcomes.102 A retrospective study provided evidence supporting the role of CYC in SSc-ILD, particularly for patients with early SSc-ILD (<4 years), compared with high-dose prednisolone, immunosuppressive other than CYC, D-penicillamine, and no drug. The CYC-treated group showed significantly more improvement in FVC than the other groups.13 The randomized, controlled trial SLS I provided robust evidence of CYC efficacy compared with placebo; after 12 months of CYC therapy, modest but significant improvements were reported in FVC decline, total lung capacity, dyspnea, skin thickening, and health-related quality of life.90 Follow-up data from the SLS I study showed that the beneficial effects of 12 months of CYC therapy persisted for a few months after cessation of therapy, although there was no difference in FVC between the two study groups at 24 months post-treatment.103 Quantitative analysis based on HRCT findings from SLS I showed decreased quantitative lung fibrosis scores in the CYC group compared with placebo at 12 months.44 Subsequent studies of CYC in SSc-ILD have yielded conflicting results. Nannini et al conducted a systematic review and meta-analysis of randomized, controlled trials and prospective observational studies in patients with SSc-ILD.104 The meta-analysis showed that CYC treatment does not result in clinically significant improvement of pulmonary function.104
Despite the conflicting efficacy data, CYC remains the most widely used treatment for SSc-ILD in the world.99 However, CYC therapy is associated with toxicities, including an increased risk of leukopenia and neutropenia.90,105 Consequently, CYC is unsuitable for use in some patients. Additionally, a recent study found that 1 year of treatment with CYC did not improve long-term survival compared with placebo in patients who participated in SLS I.26
Mycophenolate Mofetil
MMF has been suggested as an alternative to CYC and was shown to stabilize lung function in patients with SSc-ILD in a retrospective study.106 Additionally, a pilot study assessed the safety and efficacy of anti-thymocyte globulin given for 5 days, followed by MMF for 12 months, in patients with recent-onset diffuse SSc. It was shown that systemic disease was stabilized as measured by various assessments including FVC and that MMF was well tolerated.107 In a prospective, observational 1-year study of 14 SSc-ILD patients treated with MMF, six patients reported at least a 10% improvement in their FVC and five had stable pulmonary function.108 In SLS II, 2 years of treatment with MMF was compared with 1 year of treatment with CYC followed by 1 year of placebo in patients with SSc-ILD. The two treatments produced similar improvements in FVC% predicted at 24 months; MMF and CYC treatment led to an overall improvement in the FVC% predicted with an absolute change from baseline in FVC% predicted of 3.3%±1.1 and 3.0%±1.2, respectively. However, MMF showed a more favorable safety and tolerability profile.10 A recent analysis of data from the SLS I and SLS II studies was performed to compare MMF with placebo, with adjustments for disease severity. Patients treated with MMF showed a significant improvement in FVC% predicted, DLco% predicted, and dyspnea compared with the placebo group.109 Together, these data support the continued use of MMF to treat SSc-ILD.
Nintedanib
Nintedanib is a tyrosine kinase inhibitor (TKI) that acts on the platelet-derived growth factor, fibroblast growth factor, and vascular endothelial growth factor receptor signaling pathways. As a result, it inhibits cytokine-induced activation of fibroblasts and has demonstrated potent antifibrotic effects in mouse models of SSc.110 Recently, nintedanib became the first Food and Drug Administration-approved treatment for SSc-ILD; it is indicated for slowing the rate of decline in pulmonary function in patients with SSc-ILD111 based on the results of the phase III, randomized, double-blind, placebo-controlled Safety and Efficacy of Nintedanib in Systemic Sclerosis (SENSCIS) trial (NCT02597933).112 In the SENSCIS trial, a broad population of patients with SSc-ILD were selected, including those with limited and diffuse cutaneous SSc, those receiving mycophenolate at a stable dose for 6 months at baseline, and those with several lung function impairments. In the primary endpoint analysis, there was significant difference in the adjusted annual rate of change in FVC between nintedanib and placebo (difference 41.0 mL/year; 95% confidence interval [CI] 2.9–79.0; p=0.04) over a 1-year period in the total study population. The differences in change from baseline in the modified Rodnan skin score (mRSS) and total score on the St George’s Respiratory Questionnaire were not significant. Adverse events (AEs) were similar between the nintedanib and placebo groups. In line with the known safety profile of nintedanib in IPF, diarrhea was the most common AE. Among nintedanib-treated patients who experienced diarrhea AE, diarrhea events which were at worst mild or moderate in severity were reported in 49.5% and 45% of the patients, respectively.112 An open-label extension trial to assess whether there is sustained efficacy with nintedanib, as well as its long-term safety (NCT03313180), is ongoing.
Azathioprine
A retrospective case series study suggested that azathioprine (AZA) given in combination with low-dose prednisone may play a role in stabilizing pulmonary function in patients with SSc.113 A recent case study reported that AZA combined with prednisone improved pulmonary function.114 However, no significant improvements in FVC and DLco were observed compared with placebo in patients with SSc-ILD who received CYC and low-dose prednisolone followed by oral AZA in a randomized, controlled trial.115 In an open-label prospective study conducted in 13 patients with early diffuse SSc (nine of whom had lung involvement), patients who completed a year of treatment with low-dose intravenous pulse CYC received 1 year of treatment with AZA (50 mg/day for the first 20 days and 100 mg/day thereafter).116 The results showed that the improvement following a year of CYC therapy was maintained by AZA treatment; no outcome measures deteriorated, including FVC and DLco. This study, therefore, may suggest a role of AZA in maintaining the improvement induced by low-dose pulse CYC in early diffuse SSc; however, these findings require confirmation in controlled studies.116 Therefore, the clinical value of AZA in SSc-ILD remains unclear and, to the best of our knowledge, this drug is not currently being investigated further in SSc-ILD.
Adjunctive Therapies
The primary aim of treatment for SSc-ILD is to optimize patients’ quality of life as early as possible. Pharmacologic therapy is the primary focus, but adjunctive supportive measures can lessen the symptom burden.117 The primary adjunctive measures to consider in the care of patients with SSc-ILD are similar to those used for other types of ILD. These include smoking cessation, appropriate vaccinations, supplemental oxygen (if oxygen saturation is below 89% at rest or upon exertion), and pulmonary rehabilitation. Additionally, the association between gastroesophageal reflux disease (GERD) and ILD necessitates appropriate management of GERD in patients with SSc-ILD.117
Other Potential Future Therapeutic Agents
Several treatments are being investigated in SSc-ILD. This review focuses on treatment options for SSc-ILD with the most published evidence supporting their use in this condition. Additional agents under investigation for SSc-ILD that are not included here are bortezomib and abatacept.
Rituximab
RTX is a CD20-targeting chimeric monoclonal antibody that causes rapid B-cell depletion and immunosuppression. It is licensed for the treatment of non-Hodgkin lymphoma, chronic lymphocytic leukemia, rheumatoid arthritis, and granulomatosis with polyangiitis.118 In a 1-year, proof-of-principle study in SSc-ILD, a 10% increase in FVC% predicted versus baseline was observed with RTX compared with a 5% decrease in the control group (p=0.002).119 A retrospective analysis of data from the European League Against Rheumatism Scleroderma Trials and Research Group database showed that over a mean follow-up period of 2 years, RTX use was associated with an acceptable safety profile, and significant improvement was observed in skin fibrosis, but not in lung function.120 RTX is currently being compared with CYC in a randomized, controlled trial, as a first-line treatment in patients with SSc-ILD and other connective tissue diseases with associated ILDs.121
Antifibrotic Agents
Antifibrotic agents slow lung function decline in IPF and were approved for the treatment of this lung disease in 2014.122 Due to the clinical and mechanistic parallels between IPF and SSc-ILD, including lung parenchymal injury, transforming growth factor (TGF)-β-mediated fibroblast activation, and myofibroblast accumulation,123 antifibrotics are being studied as potential treatment options in SSc-ILD. The exact mechanism of action of the antifibrotic agent pirfenidone is currently under investigation and is not yet fully understood, but it has been shown to reduce TGF-β-stimulated fibrosis.124,125 A small study of five patients with SSc-ILD reported improved FVC upon treatment with pirfenidone at doses ranging from 1200 to 1800 mg/day.126 Subsequently, in the randomized, open-label, phase II Safety and Tolerability of Pirfenidone in Patients With Systemic Sclerosis−Related Interstitial Lung Disease (LOTUSS) study (NCT01933334), pirfenidone showed an acceptable safety and tolerability profile.127 The ongoing SLS III trial (NCT03221257) will compare pirfenidone plus MMF versus placebo plus MMF in patients with SSc-ILD.
TKIs other than nintedanib are also being assessed in SSc-ILD. Imatinib was first investigated in a phase I/IIa, open-label, pilot study (NCT00512902) in patients with SSc-ILD and showed trends towards improvement in FVC and DLco over 1 year.128 However, treatment-related toxicities were observed with the 600 mg/day dose.128 A subsequent phase IIa study assessed imatinib 400 mg/day in patients with diffuse cutaneous SSc, 53% of whom had SSc-ILD; most patients tolerated the treatment, and sustained FVC improvements were observed.129 Conversely, in a small phase I/II study (NCT00506831) conducted in seven patients with SSc, imatinib 300 mg/day produced no improvement in pulmonary function. A subsequent phase II pilot study reported that imatinib 200 mg/day stabilized lung function and improved HRCT scan patterns in patients with SSc-ILD who were unresponsive to CYC (NCT00573326).130
Dasatinib and nilotinib are examples of second-generation TKIs; these compounds block several receptor and non-receptor kinases, many of which have been shown to be activated in SSc. In a single-arm, open-label, phase IIa study of dasatinib in SSc-ILD (NCT00764309), the safety profile was acceptable.131 No significant changes at Day 169 versus baseline were observed in mRSS or PFTs, but comparison with a historical, placebo-treated control group indicated that dasatinib may have reduced the rate of deterioration of lung fibrosis. An open-label pilot study of nilotinib was, therefore, performed in patients with SSc, three (30%) of whom had ILD. Nilotinib was well tolerated, and the principal safety finding was mild QTc-prolongation. The study showed significant improvements in skin parameters in response to nilotinib, but these findings should not be considered as definitive because of the open-label study design and the small number of patients included.132 Further investigations are warranted.
Dabigatran
The thrombin inhibitor dabigatran directly prevents fibrin formation and, therefore, could have therapeutic potential in fibrosing ILD. Preliminary data from a phase I, open-label clinical trial (NCT02426229), which investigated the safety and efficacy of dabigatran in patients with SSc-ILD, suggested that individuals with high baseline plasma/BAL fluid thrombin activity were more likely to benefit from dabigatran.133
Tocilizumab
Tocilizumab is a monoclonal antibody that targets interleukin-6, a cytokine that has been implicated in the pathogenesis of SSc. Two major placebo-controlled studies have investigated the safety and efficacy of tocilizumab in patients with SSc. These studies were focused on patients with an early, active progressive diffuse cutaneous SSc (progression defined by increase in mRSS), but not specifically those diagnosed with associated ILD, as in the SLS and SENSCIS trials; nevertheless, both tocilizumab trials included lung function assessment. In the phase II Safety And Efficacy Of Subcutaneous Tocilizumab In Adults with SSc (faSScinate) trial, the percentage of patients with SSc showing a decline in FVC at 48 weeks was significantly lower in the tocilizumab group than in the placebo group (p=0.0373).134 This benefit was maintained in a 48-week, open-label extension of the study. Tocilizumab treatment was associated with benefits in skin fibrosis, lung fibrosis, and physical function in patients with SSc; however, it was also associated with increased risk of serious infections. Taken together, the results of the faSScinate trial and its associated open-label extension period may indicate a role for tocilizumab for the treatment of patients with progressive SSc who have few treatment options.135 The phase III Efficacy and Safety of Tocilizumab in Participants With SSc (focuSSced) trial reported a 167 mL (95% CI 83–250 mL) difference in favor of tocilizumab versus placebo in the mean change from baseline in FVC at Week 48.136 The primary endpoints in the focuSSced and faSScinate clinical trials, which were change in mRSS at 48 and 24 weeks, respectively, were not met.
Additional Therapeutic Options
Stem Cell Transplantation
For over 2 decades, hematopoietic stem cell transplantation (HSCT) has been used in the treatment of autoimmune disorders that are refractory to conventional immunosuppressive therapy.137 HSCT has been studied in three randomized, controlled trials in patients with SSc; a summary of each of these studies is provided in Table 4. There is evidence that HSCT may prevent organ deterioration and improve skin thickening, pulmonary function, and long-term survival in patients with SSc.137 In addition, follow-up assessment of participants in the Scleroderma: Cyclophosphamide or Transplantation trial (summarized in Table 4) showed that HSCT results in significant correction of molecular signatures associated with SSc.138 However, HSCT has been associated with treatment-related mortality in the early, post-transplant period, and some patients who demonstrate an initial response may subsequently experience progression of SSc-related symptoms. Overall, HSCT is likely to be beneficial in patients with early, diffuse skin disease who are at risk of rapid progression, although candidates need to be selected carefully, with particular consideration of cardiac disease.139
 | 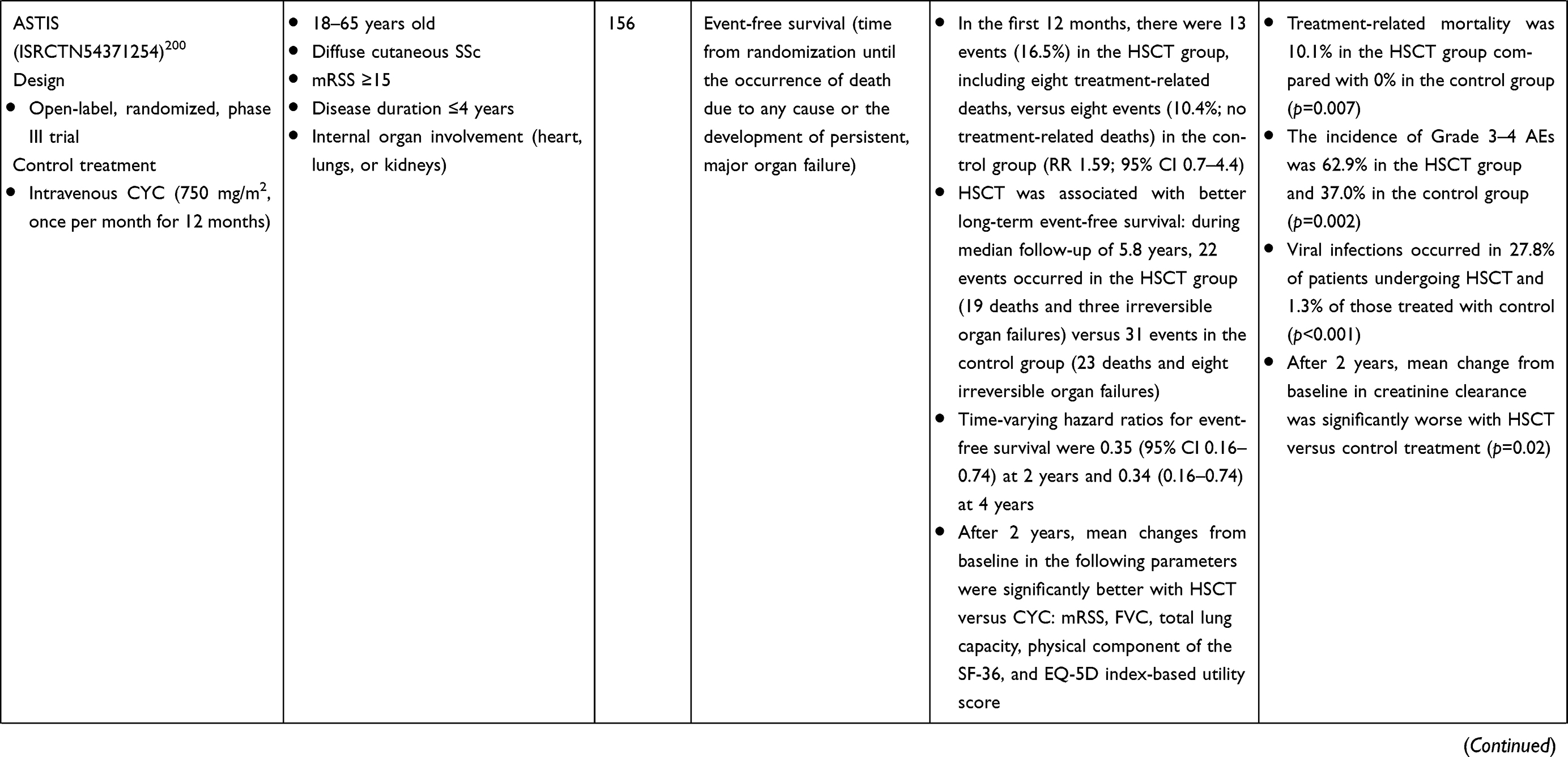 | 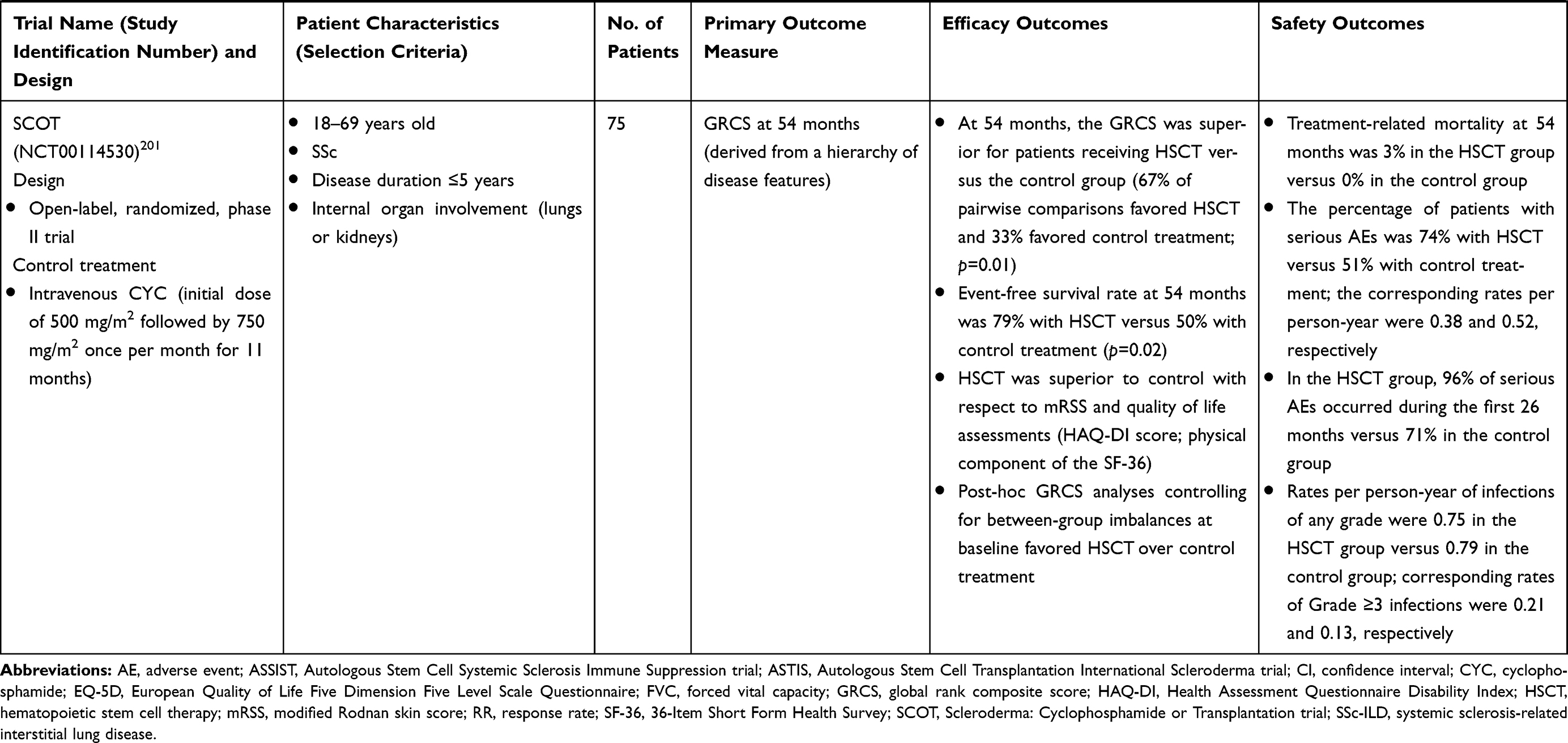 |
Table 4 Randomized Controlled Trials of HSCT in SSc-ILD |
Lung Transplantation
Lung transplantation represents a life-saving option for selected patients with progressive SSc-ILD who are at risk of respiratory failure.140 Short- and intermediate-term survival rates after lung transplantation appear similar in carefully selected patients with SSc-ILD compared with PAH and other connective tissue disease-ILDs.141–143 A retrospective, single-center study also showed that outcomes were similar in patients with or without SSc who underwent lung transplant; and that esophageal dysfunction – present in 60% of the patients with SSc – only rarely precluded or affected the listing of the outcome of lung transplantation.144 Overall, patients with advanced and refractory SSc-ILD may be candidates for lung transplantation, although careful consideration of age and comorbidities (e.g., esophageal dysfunction) is essential.141
Conclusion
As ILD is the leading cause of death in SSc, screening for its presence is essential – even in the absence of symptoms. HRCT is the most effective means of identifying ILD in patients with SSc, as it enables the detection of mild lung abnormalities; it can also play an important role in monitoring disease progression. Early detection of ILD provides the opportunity for early therapeutic intervention, which could improve patient outcomes. Due to multi-organ involvement and the complex nature of SSc-ILD, interdisciplinary engagement is key to provide optimal care to patients. Currently, immunosuppressive treatment with CYC or MMF is most commonly prescribed for SSc-ILD. However, these drugs may not provide consistent and sustained disease-modifying effects, and may not improve long-term survival; toxicity is also a concern (particularly with CYC). New therapies for SSc-ILD are urgently needed, and ongoing studies are evaluating antifibrotics and other novel agents.
Acknowledgments
The authors meet criteria for authorship as recommended by the International Committee of Medical Journal Editors (ICMJE). The authors received no direct compensation related to the development of the manuscript. Writing assistance was provided by Leon Newman, PhD, Emily Ruban-Fell, PhD, and Ken Sutor, BSc, of GeoMed, an Ashfield company, part of UDG Healthcare plc, which was contracted and funded by Boehringer Ingelheim Pharmaceuticals, Inc. (BIPI). BIPI was given the opportunity to review the manuscript for medical and scientific accuracy, as well as intellectual property considerations.
Author Contributions
All authors contributed to data analysis, drafting or revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
AF reports receiving grants and personal fees from Boehringer Ingelheim Pharmaceuticals, Inc. during the development of the submitted work and personal fees from Genentech Inc./F. Hoffmann-La Roche Ltd, Pfizer Inc. and Bristol-Myers Squibb outside the submitted work. AF is currently an employee of Bristol-Myers Squibb Company, Princeton, NJ. NMP and ERV both report receiving personal fees from Boehringer Ingelheim Pharmaceuticals, Inc. outside the submitted work. The authors report no other conflicts of interest in this work.
References
1. Hinchcliff M, Varga J. Systemic sclerosis/scleroderma: a treatable multisystem disease. Am Fam Physician. 2008;78(8):961–968.
2. Muangchan C; Canadian Scleroderma Research Group, Baron M, et al. The 15% rule in scleroderma: the frequency of severe organ complications in systemic sclerosis. A systematic review. J Rheumatol. 2013;40(9):1545–1556. doi:10.3899/jrheum.121380
3. Steen VD, Medsger TA
4. Steen VD, Medsger TA. Changes in causes of death in systemic sclerosis, 1972–2002. Ann Rheum Dis. 2007;66(7):940–944. doi:10.1136/ard.2006.066068
5. Tyndall AJ, Bannert B, Vonk M, et al. Causes and risk factors for death in systemic sclerosis: a study from the EULAR Scleroderma Trials and Research (EUSTAR) database. Ann Rheum Dis. 2010;69(10):1809–1815. doi:10.1136/ard.2009.114264
6. Rubio-Rivas M, Royo C, Simeon CP, Corbella X, Fonollosa V. Mortality and survival in systemic sclerosis: systematic review and meta-analysis. Semin Arthritis Rheum. 2014;44(2):208–219. doi:10.1016/j.semarthrit.2014.05.010
7. Khanna D, Tashkin DP, Denton CP, Lubell MW, Vazquez-Mateo C, Wax S. Ongoing clinical trials and treatment options for patients with systemic sclerosis-associated interstitial lung disease. Rheumatology (Oxford). 2018;58(4):567–579. doi:10.1093/rheumatology/key151
8. Glasser SW, Hagood JS, Wong S, Taype CA, Madala SK, Hardie WD. Mechanisms of lung fibrosis resolution. Am J Pathol. 2016;186(5):1066–1077. doi:10.1016/j.ajpath.2016.01.018
9. Poudel DR, Derk CT. Mortality and survival in systemic sclerosis: a review of recent literature. Curr Opin Rheumatol. 2018;30(6):588–593. doi:10.1097/BOR.0000000000000551
10. Tashkin DP, Roth MD, Clements PJ, et al. Mycophenolate mofetil versus oral cyclophosphamide in scleroderma-related interstitial lung disease (SLS II): a randomised controlled, double-blind, parallel group trial. Lancet Respir Med. 2016;4(9):708–719. doi:10.1016/S2213-2600(16)30152-7
11. Volkmann ER, Tashkin DP. Treatment of systemic sclerosis-related interstitial lung disease: a review of existing and emerging therapies. Ann Am Thorac Soc. 2016;13(11):2045–2056. doi:10.1513/AnnalsATS.201606-426FR
12. Schoenfeld SR, Castelino FV. Evaluation and management approaches for scleroderma lung disease. Ther Adv Respir Dis. 2017;11(8):327–340. doi:10.1177/1753465817713680
13. Steen VD, Lanz JK
14. Suliman S, Al Harash A, Roberts WN, Perez RL, Roman J. Scleroderma-related interstitial lung disease. Respir Med Case Rep. 2017;22:109–112. doi:10.1016/j.rmcr.2017.07.007
15. Steen VD, Conte C, Owens GR, Medsger TA
16. Steen VD, Owens GR, Fino GJ, Rodnan GP, Medsger TA
17. Walker UA, Tyndall A, Czirjak L, et al. Clinical risk assessment of organ manifestations in systemic sclerosis: a report from the EULAR scleroderma trials and research group database. Ann Rheum Dis. 2007;66(6):754–763. doi:10.1136/ard.2006.062901
18. Peoples C, Medsger TA
19. McNearney TA, Reveille JD, Fischbach M, et al. Pulmonary involvement in systemic sclerosis: associations with genetic, serologic, sociodemographic, and behavioral factors. Arthritis Rheum. 2007;57(2):318–326. doi:10.1002/(ISSN)1529-0131
20. Bouros D, Wells AU, Nicholson AG, et al. Histopathologic subsets of fibrosing alveolitis in patients with systemic sclerosis and their relationship to outcome. Am J Respir Crit Care Med. 2002;165(12):1581–1586. doi:10.1164/rccm.2106012
21. Savarino E, Bazzica M, Zentilin P, et al. Gastroesophageal reflux and pulmonary fibrosis in scleroderma: a study using pH-impedance monitoring. Am J Respir Crit Care Med. 2009;179(5):408–413. doi:10.1164/rccm.200808-1359OC
22. Sharp C, Dodds N, Mayers L, Millar AB, Gunawardena H, Adamali H. The role of biologics in treatment of connective tissue disease-associated interstitial lung disease. QJM. 2015;108(9):683–688. doi:10.1093/qjmed/hcv007
23. Ho KT, Reveille JD. The clinical relevance of autoantibodies in scleroderma. Arthritis Res Ther. 2003;5(2):80–93. doi:10.1186/ar628
24. Winstone TA, Assayag D, Wilcox PG, et al. Predictors of mortality and progression in scleroderma-associated interstitial lung disease: a systematic review. Chest. 2014;146(2):422–436. doi:10.1378/chest.13-2626
25. Jaeger VK, Wirz EG, Allanore Y, et al. Incidences and risk factors of organ manifestations in the early course of systemic sclerosis: a longitudinal EUSTAR study. PLoS One. 2016;11(10):e0163894. doi:10.1371/journal.pone.0163894
26. Volkmann ER, Tashkin DP, Sim M, et al. Short-term progression of interstitial lung disease in systemic sclerosis predicts long-term survival in two independent clinical trial cohorts. Ann Rheum Dis. 2019;78:122–130. doi:10.1136/annrheumdis-2018-213708
27. Behr J, Furst DE. Pulmonary function tests. Rheumatology (Oxford). 2008;47(Suppl 5):v65–67. doi:10.1093/rheumatology/ken313
28. Raghu G. Interstital lung disease. In: Goldman L, Schafer AI, editors. Goldman-Cecil Medicine. Philadelphia, USA: Elsevier Science; 2016:575–588.
29. Caron M, Hoa S, Hudson M, Schwartzman K, Steele R. Pulmonary function tests as outcomes for systemic sclerosis interstitial lung disease. Eur Respir Rev. 2018;27:148. doi:10.1183/16000617.0102-2017
30. Chowaniec M, Skoczynska M, Sokolik R, Wiland P. Interstitial lung disease in systemic sclerosis: challenges in early diagnosis and management. Reumatologia. 2018;56(4):249–254. doi:10.5114/reum.2018.77977
31. Elicker BM, Kallianos KG, Henry TS. The role of high-resolution computed tomography in the follow-up of diffuse lung disease: number 2 in the series “Radiology” edited by Nicola Sverzellati and Sujal Desai. Eur Respir Rev. 2017;26(144):
32. Suliman YA, Dobrota R, Huscher D, et al. Brief report: pulmonary function tests: high rate of false-negative results in the early detection and screening of scleroderma-related interstitial lung disease. Arthritis Rheumatol. 2015;67(12):3256–3261. doi:10.1002/art.39405
33. Showalter K, Hoffmann A, Rouleau G, et al. Performance of forced vital capacity and lung diffusion cutpoints for associated radiographic interstitial lung disease in systemic sclerosis. J Rheumatol. 2018;45(11):1572–1576. doi:10.3899/jrheum.171362
34. Strollo D, Goldin J. Imaging lung disease in systemic sclerosis. Curr Rheumatol Rep. 2010;12(2):156–161. doi:10.1007/s11926-010-0095-0
35. Launay D, Remy-Jardin M, Michon-Pasturel U, et al. High resolution computed tomography in fibrosing alveolitis associated with systemic sclerosis. J Rheumatol. 2006;33(9):1789–1801.
36. Hoffmann-Vold AM, Aalokken TM, Lund MB, et al. Predictive value of serial high-resolution computed tomography analyses and concurrent lung function tests in systemic sclerosis. Arthritis Rheumatol. 2015;67(8):2205–2212. doi:10.1002/art.39166
37. Khanna D, Tseng CH, Farmani N, et al. Clinical course of lung physiology in patients with scleroderma and interstitial lung disease: analysis of the Scleroderma Lung Study Placebo Group. Arthritis Rheum. 2011;63(10):3078–3085. doi:10.1002/art.30467
38. Roth MD, Tseng CH, Clements PJ, et al. Predicting treatment outcomes and responder subsets in scleroderma-related interstitial lung disease. Arthritis Rheum. 2011;63(9):2797–2808. doi:10.1002/art.30438
39. Richardson C, Agrawal R, Lee J, et al. Esophageal dilatation and interstitial lung disease in systemic sclerosis: a cross-sectional study. Semin Arthritis Rheum. 2016;46(1):109–114. doi:10.1016/j.semarthrit.2016.02.004
40. Salaffi F, Di Carlo M, Carotti M, Fraticelli P, Gabrielli A, Giovagnoni A. Relationship between interstitial lung disease and oesophageal dilatation on chest high-resolution computed tomography in patients with systemic sclerosis: a cross-sectional study. Radiol Med. 2018;123(9):655–663. doi:10.1007/s11547-018-0894-3
41. Picano E, Matucci-Cerinic M. Unnecessary radiation exposure from medical imaging in the rheumatology patient. Rheumatology (Oxford). 2011;50(9):1537–1539. doi:10.1093/rheumatology/keq412
42. Frauenfelder T, Winklehner A, Nguyen TD, et al. Screening for interstitial lung disease in systemic sclerosis: performance of high-resolution CT with limited number of slices: a prospective study. Ann Rheum Dis. 2014;73(12):2069–2073. doi:10.1136/annrheumdis-2014-205637
43. Nguyen-Kim TDL, Maurer B, Suliman YA, Morsbach F, Distler O, Frauenfelder T. The impact of slice-reduced computed tomography on histogram-based densitometry assessment of lung fibrosis in patients with systemic sclerosis. J Thorac Dis. 2018;10(4):2142–2152. doi:10.21037/jtd.2018.04.39
44. Kim HJ, Brown MS, Elashoff R, et al. Quantitative texture-based assessment of one-year changes in fibrotic reticular patterns on HRCT in scleroderma lung disease treated with oral cyclophosphamide. Eur Radiol. 2011;21(12):2455–2465. doi:10.1007/s00330-011-2223-2
45. Salaffi F, Carotti M, Bosello S, et al. Computer-aided quantification of interstitial lung disease from high resolution computed tomography images in systemic sclerosis: correlation with visual reader-based score and physiologic tests. Biomed Res Int. 2015;2015:834262. doi:10.1155/2015/834262
46. Salaffi F, Carotti M, Di Donato E, Di Carlo M, Ceccarelli L, Giuseppetti G. Computer-aided tomographic analysis of interstitial lung disease (ILD) in patients with systemic sclerosis (SSc). Correlation with pulmonary physiologic tests and patient-centred measures of perceived dyspnea and functional disability. PLoS One. 2016;11(3):e0149240. doi:10.1371/journal.pone.0149240
47. Khanna D, Nagaraja V, Tseng CH, et al. Predictors of lung function decline in scleroderma-related interstitial lung disease based on high-resolution computed tomography: implications for cohort enrichment in systemic sclerosis-associated interstitial lung disease trials. Arthritis Res Ther. 2015;17:372. doi:10.1186/s13075-015-0872-2
48. Moore OA, Goh N, Corte T, et al. Extent of disease on high-resolution computed tomography lung is a predictor of decline and mortality in systemic sclerosis-related interstitial lung disease. Rheumatology (Oxford). 2013;52(1):155–160. doi:10.1093/rheumatology/kes289
49. Goh NS, Hoyles RK, Denton CP, et al. Short-term pulmonary function trends are predictive of mortality in interstitial lung disease associated with systemic sclerosis. Arthritis Rheumatol. 2017;69(8):1670–1678. doi:10.1002/art.40130
50. Man A, Davidyock T, Ferguson LT, Ieong M, Zhang Y, Simms RW. Changes in forced vital capacity over time in systemic sclerosis: application of group-based trajectory modelling. Rheumatology (Oxford). 2015;54(8):1464–1471. doi:10.1093/rheumatology/kev016
51. Steen VD, Graham G, Conte C, Owens G, Medsger TA
52. Tashkin DP, Volkmann ER, Tseng CH, et al. Relationship between quantitative radiographic assessments of interstitial lung disease and physiological and clinical features of systemic sclerosis. Ann Rheum Dis. 2016;75(2):374–381. doi:10.1136/annrheumdis-2014-206076
53. Wells AU. Pulmonary function tests in connective tissue disease. Semin Respir Crit Care Med. 2007;28(4):379–388. doi:10.1055/s-2007-985610
54. Goh NS, Desai SR, Veeraraghavan S, et al. Interstitial lung disease in systemic sclerosis: a simple staging system. Am J Respir Crit Care Med. 2008;177(11):1248–1254. doi:10.1164/rccm.200706-877OC
55. Hax V, Bredemeier M, Didonet Moro AL, et al. Clinical algorithms for the diagnosis and prognosis of interstitial lung disease in systemic sclerosis. Semin Arthritis Rheum. 2017;47(2):228–234. doi:10.1016/j.semarthrit.2017.03.019
56. Delle Sedie A, Doveri M, Frassi F, et al. Ultrasound lung comets in systemic sclerosis: a useful tool to detect lung interstitial fibrosis. Clin Exp Rheumatol. 2010;28(5 Suppl 62):S54.
57. Gargani L, Doveri M, D’Errico L, et al. Ultrasound lung comets in systemic sclerosis: a chest sonography hallmark of pulmonary interstitial fibrosis. Rheumatology (Oxford). 2009;48(11):1382–1387. doi:10.1093/rheumatology/kep263
58. Gigante A, Rossi Fanelli F, Lucci S, et al. Lung ultrasound in systemic sclerosis: correlation with high-resolution computed tomography, pulmonary function tests and clinical variables of disease. Intern Emerg Med. 2016;11(2):213–217. doi:10.1007/s11739-015-1329-y
59. Tardella M, Di Carlo M, Carotti M, Filippucci E, Grassi W, Salaffi F. Ultrasound B-lines in the evaluation of interstitial lung disease in patients with systemic sclerosis: cut-off point definition for the presence of significant pulmonary fibrosis. Medicine (Baltimore). 2018;97(18):e0566. doi:10.1097/MD.0000000000010566
60. Vizioli L, Ciccarese F, Forti P, et al. Integrated use of lung ultrasound and chest X-ray in the detection of interstitial lung disease. Respiration. 2017;93(1):15–22. doi:10.1159/000452225
61. Pinal-Fernandez I, Pallisa-Nunez E, Selva-O’Callaghan A, et al. Pleural irregularity, a new ultrasound sign for the study of interstitial lung disease in systemic sclerosis and antisynthetase syndrome. Clin Exp Rheumatol. 2015;33(4 Suppl 91):S136–141.
62. Barskova T, Gargani L, Guiducci S, et al. Lung ultrasound for the screening of interstitial lung disease in very early systemic sclerosis. Ann Rheum Dis. 2013;72(3):390–395. doi:10.1136/annrheumdis-2011-201072
63. Montesi SB, Caravan P. Novel imaging approaches in systemic sclerosis-associated interstitial lung disease. Curr Rheumatol Rep. 2019;21(6):25. doi:10.1007/s11926-019-0826-9
64. Pinal-Fernandez I, Pineda-Sanchez V, Pallisa-Nunez E, et al. Fast 1.5 T chest MRI for the assessment of interstitial lung disease extent secondary to systemic sclerosis. Clin Rheumatol. 2016;35(9):2339–2345. doi:10.1007/s10067-016-3267-0
65. Montesi SB, Desogere P, Fuchs BC, Caravan P. Molecular imaging of fibrosis: recent advances and future directions. J Clin Invest. 2019;129(1):24–33. doi:10.1172/JCI122132
66. Walsh SLF, Devaraj A, Enghelmayer JI, et al. Role of imaging in progressive-fibrosing interstitial lung diseases. Eur Respir Rev. 2018;27:150. doi:10.1183/16000617.0073-2018
67. Goh NS, Veeraraghavan S, Desai SR, et al. Bronchoalveolar lavage cellular profiles in patients with systemic sclerosis-associated interstitial lung disease are not predictive of disease progression. Arthritis Rheum. 2007;56(6):2005–2012. doi:10.1002/art.22696
68. Strange C, Bolster MB, Roth MD, et al. Bronchoalveolar lavage and response to cyclophosphamide in scleroderma interstitial lung disease. Am J Respir Crit Care Med. 2008;177(1):91–98. doi:10.1164/rccm.200705-655OC
69. Meyer KC, Raghu G, Baughman RP, et al. An official American Thoracic Society clinical practice guideline: the clinical utility of bronchoalveolar lavage cellular analysis in interstitial lung disease. Am J Respir Crit Care Med. 2012;185(9):1004–1014. doi:10.1164/rccm.201202-0320ST
70. Qureshi RA, Ahmed TA, Grayson AD, Soorae AS, Drakeley MJ, Page RD. Does lung biopsy help patients with interstitial lung disease? Eur J Cardiothorac Surg. 2002;21(4):621–626. doi:10.1016/S1010-7940(02)00021-0
71. Manhire A, Charig M, Clelland C, et al. Guidelines for radiologically guided lung biopsy. Thorax. 2003;58(11):920–936. doi:10.1136/thorax.58.11.920
72. Cottin V. Lung biopsy in interstitial lung disease: balancing the risk of surgery and diagnostic uncertainty. Eur Respir J. 2016;48(5):1274–1277. doi:10.1183/13993003.01633-2016
73. Solomon JJ, Olson AL, Fischer A, Bull T, Brown KK, Raghu G. Scleroderma lung disease. Eur Respir Rev. 2013;22(127):6–19. doi:10.1183/09059180.00005512
74. Cottin V, Brown KK. Interstitial lung disease associated with systemic sclerosis (SSc-ILD). Respir Res. 2019;20(1):13. doi:10.1186/s12931-019-0980-7
75. Branley HM, Lake FR. 2008 British guidelines on the management of interstitial lung diseases. What are the key new messages for clinical practice? Pol Arch Med Wewn. 2009;119(3):112–114.
76. Yu C, Song S, Chen J. Peripheral blood biomarkers in systemic sclerosis-associated interstitial lung diseases. Int J Clin Rheumatol. 2018;13(1):11–19. doi:10.4172/1758-4272
77. Yanaba K, Hasegawa M, Takehara K, Sato S. Comparative study of serum surfactant protein-D and KL-6 concentrations in patients with systemic sclerosis as markers for monitoring the activity of pulmonary fibrosis. J Rheumatol. 2004;31(6):1112–1120.
78. Benyamine A, Heim X, Resseguier N, et al. Elevated serum Krebs von den Lungen-6 in systemic sclerosis: a marker of lung fibrosis and severity of the disease. Rheumatol Int. 2018;38(5):813–819. doi:10.1007/s00296-018-3987-3
79. Bonella F, Volpe A, Caramaschi P, et al. Surfactant protein D and KL-6 serum levels in systemic sclerosis: correlation with lung and systemic involvement. Sarcoidosis Vasc Diffuse Lung Dis. 2011;28(1):27–33.
80. Kumanovics G, Gorbe E, Minier T, Simon D, Berki T, Czirjak L. Follow-up of serum KL-6 lung fibrosis biomarker levels in 173 patients with systemic sclerosis. Clin Exp Rheumatol. 2014;32(6 Suppl 86):S138–144.
81. Salazar GA, Kuwana M, Wu M, et al. KL-6 but not CCL-18 is a predictor of early progression in systemic sclerosis-related interstitial lung disease. J Rheumatol. 2018;45(8):1153–1158. doi:10.3899/jrheum.170518
82. Volkmann ER, Tashkin DP, Kuwana M, et al. Pneumoproteins KL-6 and CCL-18 predict progression of interstitial lung disease in systemic sclerosis. Arthritis Rheumatol. 2019. doi:10.1002/art.41020
83. Elhai M, Hoffmann-Vold AM, Avouac J, et al. Performance of candidate serum biomarkers for systemic sclerosis-associated interstitial lung disease. Arthritis Rheumatol. 2019;71(6):972–982. doi:10.1002/art.40815
84. Kuwana M, Shirai Y, Takeuchi T. Elevated serum Krebs von den Lungen-6 in early disease predicts subsequent deterioration of pulmonary function in patients with systemic sclerosis and interstitial lung disease. J Rheumatol. 2016;43(10):1825–1831. doi:10.3899/jrheum.160339
85. Wu M, Baron M, Pedroza C, et al. CCL2 in the circulation predicts long-term progression of interstitial lung disease in patients with early systemic sclerosis: data from two independent cohorts. Arthritis Rheumatol. 2017;69(9):1871–1878. doi:10.1002/art.40171
86. Hoffmann-Vold AM, Tennoe AH, Garen T, et al. High level of Chemokine CCL18 is associated with pulmonary function deterioration, lung fibrosis progression, and reduced survival in systemic sclerosis. Chest. 2016;150(2):299–306. doi:10.1016/j.chest.2016.03.004
87. De Lauretis A, Sestini P, Pantelidis P, et al. Serum interleukin 6 is predictive of early functional decline and mortality in interstitial lung disease associated with systemic sclerosis. J Rheumatol. 2013;40(4):435–446. doi:10.3899/jrheum.120725
88. van Bon L, Affandi AJ, Broen J, et al. Proteome-wide analysis and CXCL4 as a biomarker in systemic sclerosis. N Engl J Med. 2014;370(5):433–443. doi:10.1056/NEJMoa1114576
89. Volkmann ER, Tashkin DP, Roth MD, et al. Changes in plasma CXCL4 levels are associated with improvements in lung function in patients receiving immunosuppressive therapy for systemic sclerosis-related interstitial lung disease. Arthritis Res Ther. 2016;18(1):305. doi:10.1186/s13075-016-1203-y
90. Tashkin DP, Elashoff R, Clements PJ, et al. Cyclophosphamide versus placebo in scleroderma lung disease. N Engl J Med. 2006;354(25):2655–2666. doi:10.1056/NEJMoa055120
91. Tashkin DP, Volkmann ER, Tseng CH, et al. Improved cough and cough-specific quality of life in patients treated for scleroderma-related interstitial lung disease: results of Scleroderma Lung Study II. Chest. 2017;151(4):813–820. doi:10.1016/j.chest.2016.11.052
92. Theodore AC, Tseng CH, Li N, Elashoff RM, Tashkin DP. Correlation of cough with disease activity and treatment with cyclophosphamide in scleroderma interstitial lung disease: findings from the Scleroderma Lung Study. Chest. 2012;142(3):614–621. doi:10.1378/chest.11-0801
93. Morisset J, Vittinghoff E, Elicker BM, et al. Mortality risk prediction in scleroderma-related interstitial lung disease: the SADL Model. Chest. 2017;152(5):999–1007. doi:10.1016/j.chest.2017.06.009
94. Ryerson CJ, O’Connor D, Dunne JV, et al. Predicting mortality in systemic sclerosis-associated interstitial lung disease using risk prediction models derived from idiopathic pulmonary fibrosis. Chest. 2015;148(5):1268–1275. doi:10.1378/chest.15-0003
95. Wu W, Jordan S, Becker MO, et al. Prediction of progression of interstitial lung disease in patients with systemic sclerosis: the SPAR model. Ann Rheum Dis. 2018;77(9):1326–1332. doi:10.1136/annrheumdis-2018-213201
96. Rizzi M, Sarzi-Puttini P, Airoldi A, Antivalle M, Battellino M, Atzeni F. Performance capacity evaluated using the 6-minute walk test: 5-year results in patients with diffuse systemic sclerosis and initial interstitial lung disease. Clin Exp Rheumatol. 2015;33(4 Suppl 91):S142–147.
97. National Institute for Health and Care Excellence (NICE). Idiopathic pulmonary fibrosis in adults: diagnosis and management. Clinical guideline [CG163]. 2017. Available from: https://www.nice.org.uk/guidance/CG163/chapter/1-Recommendations#review-and-follow-up-2.
98. Cossu M, Beretta L, Mosterman P, de Hair MJH, Radstake T. Unmet needs in systemic sclerosis understanding and treatment: the knowledge gaps from a scientist’s, clinician’s, and patient’s perspective. Clin Rev Allergy Immunol. 2018;55(3):312–331. doi:10.1007/s12016-017-8636-1
99. Adler S, Huscher D, Siegert E, et al. Systemic sclerosis associated interstitial lung disease - individualized immunosuppressive therapy and course of lung function: results of the EUSTAR group. Arthritis Res Ther. 2018;20(1):17. doi:10.1186/s13075-018-1517-z
100. Fernandez-Codina A, Walker KM, Pope JE, Scleroderma Algorithm Group. Treatment algorithms for systemic sclerosis according to experts. Arthritis Rheumatol. 2018;70(11):1820–1828. doi:10.1002/art.40560
101. Silver RM, Warrick JH, Kinsella MB, Staudt LS, Baumann MH, Strange C. Cyclophosphamide and low-dose prednisone therapy in patients with systemic sclerosis (scleroderma) with interstitial lung disease. J Rheumatol. 1993;20(5):838–844.
102. White B, Moore WC, Wigley FM, Xiao HQ, Wise RA. Cyclophosphamide is associated with pulmonary function and survival benefit in patients with scleroderma and alveolitis. Ann Intern Med. 2000;132(12):947–954. doi:10.7326/0003-4819-132-12-200006200-00004
103. Tashkin DP, Elashoff R, Clements PJ, et al. Effects of 1-year treatment with cyclophosphamide on outcomes at 2 years in scleroderma lung disease. Am J Respir Crit Care Med. 2007;176(10):1026–1034. doi:10.1164/rccm.200702-326OC
104. Nannini C, West CP, Erwin PJ, Matteson EL. Effects of cyclophosphamide on pulmonary function in patients with scleroderma and interstitial lung disease: a systematic review and meta-analysis of randomized controlled trials and observational prospective cohort studies. Arthritis Res Ther. 2008;10(5):R124. doi:10.1186/ar2534
105. Berezne A, Valeyre D, Ranque B, Guillevin L, Mouthon L. Interstitial lung disease associated with systemic sclerosis: what is the evidence for efficacy of cyclophosphamide? Ann N Y Acad Sci. 2007;1110:271–284. doi:10.1196/annals.1423.029
106. Koutroumpas A, Ziogas A, Alexiou I, Barouta G, Sakkas LI. Mycophenolate mofetil in systemic sclerosis-associated interstitial lung disease. Clin Rheumatol. 2010;29(10):1167–1168.
107. Stratton RJ, Wilson H, Black CM. Pilot study of anti-thymocyte globulin plus mycophenolate mofetil in recent-onset diffuse scleroderma. Rheumatology (Oxford). 2001;40(1):84–88. doi:10.1093/rheumatology/40.1.84
108. Simeon-Aznar CP, Fonollosa-Pla V, Tolosa-Vilella C, Selva-O’Callaghan A, Solans-Laque R, Vilardell-Tarres M. Effect of mycophenolate sodium in scleroderma-related interstitial lung disease. Clin Rheumatol. 2011;30(11):1393–1398. doi:10.1007/s10067-011-1823-1
109. Volkmann ER, Tashkin DP, Li N, et al. Mycophenolate mofetil versus placebo for systemic sclerosis-related interstitial lung disease: an analysis of scleroderma lung studies I and II. Arthritis Rheumatol. 2017;69(7):1451–1460. doi:10.1002/art.v69.7
110. Huang J, Beyer C, Palumbo-Zerr K, et al. Nintedanib inhibits fibroblast activation and ameliorates fibrosis in preclinical models of systemic sclerosis. Ann Rheum Dis. 2016;75(5):883–890. doi:10.1136/annrheumdis-2014-207109
111. Boehringher Ingelheim. OFEV (nintedanib) US Prescribing Information. September 2019.
112. Distler O, Highland KB, Gahlemann M, et al. Nintedanib for systemic sclerosis-associated interstitial lung disease. N Engl J Med. 2019;380(26):2518–2528. doi:10.1056/NEJMoa1903076
113. Dheda K, Lalloo UG, Cassim B, Mody GM. Experience with azathioprine in systemic sclerosis associated with interstitial lung disease. Clin Rheumatol. 2004;23(4):306–309. doi:10.1007/s10067-004-0906-7
114. Kotani T, Takeuchi T, Makino S, Hanafusa T. Successful treatment of long-term severe progressive interstitial pneumonia with low-dose corticosteroid and azathioprine in a patient with diffuse systemic sclerosis. Case Rep Rheumatol. 2012;2012:143927.
115. Hoyles RK, Ellis RW, Wellsbury J, et al. A multicenter, prospective, randomized, double-blind, placebo-controlled trial of corticosteroids and intravenous cyclophosphamide followed by oral azathioprine for the treatment of pulmonary fibrosis in scleroderma. Arthritis Rheum. 2006;54(12):3962–3970. doi:10.1002/(ISSN)1529-0131
116. Paone C, Chiarolanza I, Cuomo G, et al. Twelve-month azathioprine as maintenance therapy in early diffuse systemic sclerosis patients treated for 1-year with low dose cyclophosphamide pulse therapy. Clin Exp Rheumatol. 2007;25(4):613–616.
117. Silver KC, Silver RM. Management of systemic-sclerosis-associated interstitial lung disease. Rheum Dis Clin North Am. 2015;41(3):439–457. doi:10.1016/j.rdc.2015.04.006
118. Biogen Idec Inc, Genentech Inc. Rituximab prescribing information: FDA. 2010. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2010/103705s5311lbl.pdf.
119. Daoussis D, Liossis SN, Tsamandas AC, et al. Experience with rituximab in scleroderma: results from a 1-year, proof-of-principle study. Rheumatology (Oxford). 2010;49(2):271–280. doi:10.1093/rheumatology/kep093
120. Elhai M, Boubaya M, Distler O, et al. Outcomes of patients with systemic sclerosis treated with rituximab in contemporary practice: a prospective cohort study. Ann Rheum Dis. 2019;78(7):979–987. doi:10.1136/annrheumdis-2018-214816
121. Saunders P, Tsipouri V, Keir GJ, et al. Rituximab versus cyclophosphamide for the treatment of connective tissue disease-associated interstitial lung disease (RECITAL): study protocol for a randomised controlled trial. Trials. 2017;18(1):275. doi:10.1186/s13063-017-2016-2
122. Raghu G, Selman M. Nintedanib and pirfenidone. New antifibrotic treatments indicated for idiopathic pulmonary fibrosis offer hopes and raises questions. Am J Respir Crit Care Med. 2015;191(3):252–254. doi:10.1164/rccm.201411-2044ED
123. Kreuter M, Walscher J, Behr J. Antifibrotic drugs as treatment of nonidiopathic pulmonary fibrosis interstitial pneumonias: the time is now (?). Curr Opin Pulm Med. 2017;23(5):418–425. doi:10.1097/MCP.0000000000000408
124. Nakayama S, Mukae H, Sakamoto N, et al. Pirfenidone inhibits the expression of HSP47 in TGF-beta1-stimulated human lung fibroblasts. Life Sci. 2008;82(3–4):210–217. doi:10.1016/j.lfs.2007.11.003
125. Yu W, Guo F, Song X. Effects and mechanisms of pirfenidone, prednisone and acetylcysteine on pulmonary fibrosis in rat idiopathic pulmonary fibrosis models. Pharm Biol. 2017;55(1):450–455. doi:10.1080/13880209.2016.1247879
126. Miura Y, Saito T, Fujita K, et al. Clinical experience with pirfenidone in five patients with scleroderma-related interstitial lung disease. Sarcoidosis Vasc Diffuse Lung Dis. 2014;31(3):235–238.
127. Khanna D, Albera C, Fischer A, et al. An open-label, phase II study of the safety and tolerability of pirfenidone in patients with scleroderma-associated interstitial lung disease: the LOTUSS Trial. J Rheumatol. 2016;43(9):1672–1679. doi:10.3899/jrheum.151322
128. Khanna D, Saggar R, Mayes MD, et al. A one-year, phase I/IIa, open-label pilot trial of imatinib mesylate in the treatment of systemic sclerosis-associated active interstitial lung disease. Arthritis Rheum. 2011;63(11):3540–3546. doi:10.1002/art.v63.11
129. Spiera RF, Gordon JK, Mersten JN, et al. Imatinib mesylate (gleevec) in the treatment of diffuse cutaneous systemic sclerosis: results of a 1-year, phase IIa, single-arm, open-label clinical trial. Ann Rheum Dis. 2011;70(6):1003–1009. doi:10.1136/ard.2010.143974
130. Fraticelli P, Gabrielli B, Pomponio G, et al. Low-dose oral imatinib in the treatment of systemic sclerosis interstitial lung disease unresponsive to cyclophosphamide: a phase II pilot study. Arthritis Res Ther. 2014;16(4):R144.
131. Martyanov V, Kim GJ, Hayes W, et al. Novel lung imaging biomarkers and skin gene expression subsetting in dasatinib treatment of systemic sclerosis-associated interstitial lung disease. PLoS One. 2017;12(11):e0187580. doi:10.1371/journal.pone.0187580
132. Gordon JK, Martyanov V, Magro C, et al. Nilotinib (tasigna) in the treatment of early diffuse systemic sclerosis: an open-label, pilot clinical trial. Arthritis Res Ther. 2015;17:213.
133. Silver R, Atanelishvili I, Akter T, et al. Safety and suitability of a direct thrombin inhibitor, dabigatran etexilate, in scleroderma-associated interstitial lung disease (SSc-ILD) patients (abstract). Am J Respir Crit Care Med. 2018;197:A1055.
134. Khanna D, Denton CP, Jahreis A, et al. Safety and efficacy of subcutaneous tocilizumab in adults with systemic sclerosis (faSScinate): a phase 2, randomised, controlled trial. Lancet. 2016;387(10038):2630–2640. doi:10.1016/S0140-6736(16)00232-4
135. Khanna D, Denton CP, Lin CJF, et al. Safety and efficacy of subcutaneous tocilizumab in systemic sclerosis: results from the open-label period of a phase II randomised controlled trial (faSScinate). Ann Rheum Dis. 2018;77(2):212–220. doi:10.1136/annrheumdis-2017-211682
136. Khanna D, Lin CJF, Kuwana M, et al. Efficacy and safety of tocilizumab for the treatment of systemic sclerosis: results from a phase 3 randomized controlled trial (abstract). Arthritis Rheumatol. 2018;70:A898.
137. Walker UA, Saketkoo LA, Distler O. Haematopoietic stem cell transplantation in systemic sclerosis. RMD Open. 2018;4(1):e000533. doi:10.1136/rmdopen-2017-000533
138. Assassi S, Wang X, Chen G, et al. Myeloablation followed by autologous stem cell transplantation normalises systemic sclerosis molecular signatures. Ann Rheum Dis. 2019;78(10):1371–1378. doi:10.1136/annrheumdis-2019-215770
139. Burt RK, Oliveira MC, Shah SJ, et al. Cardiac involvement and treatment-related mortality after non-myeloablative haemopoietic stem-cell transplantation with unselected autologous peripheral blood for patients with systemic sclerosis: a retrospective analysis. Lancet. 2013;381(9872):1116–1124. doi:10.1016/S0140-6736(12)62114-X
140. Bernstein EJ, Peterson ER, Sell JL, et al. Survival of adults with systemic sclerosis following lung transplantation: a nationwide cohort study. Arthritis Rheumatol. 2015;67(5):1314–1322. doi:10.1002/art.39021
141. Crespo MM, Bermudez CA, Dew MA, et al. Lung transplant in patients with scleroderma compared with pulmonary fibrosis. Short- and long-term outcomes. Ann Am Thorac Soc. 2016;13(6):784–792. doi:10.1513/AnnalsATS.201503-177OC
142. Khan IY, Singer LG, de Perrot M, et al. Survival after lung transplantation in systemic sclerosis. A systematic review. Respir Med. 2013;107(12):2081–2087. doi:10.1016/j.rmed.2013.09.015
143. Sottile PD, Iturbe D, Katsumoto TR, et al. Outcomes in systemic sclerosis-related lung disease after lung transplantation. Transplantation. 2013;95(7):975–980. doi:10.1097/TP.0b013e3182845f23
144. Miele CH, Schwab K, Saggar R, et al. Lung transplant outcomes in systemic sclerosis with significant esophageal dysfunction. A comprehensive single-center experience. Ann Am Thorac Soc. 2016;13(6):793–802. doi:10.1513/AnnalsATS.201512-806OC
145. Kang M, Deoghuria D, Varma S, Gupta D, Bhatia A, Khandelwal N. Role of HRCT in detection and characterization of pulmonary abnormalities in patients with febrile neutropenia. Lung India. 2013;30(2):124–130. doi:10.4103/0970-2113.110420
146. Rosas IO, Yao J, Avila NA, Chow CK, Gahl WA, Gochuico BR. Automated quantification of high-resolution CT scan findings in individuals at risk for pulmonary fibrosis. Chest. 2011;140(6):1590–1597. doi:10.1378/chest.10-2545
147. Padley SP, Hansell DM, Flower CD, Jennings P. Comparative accuracy of high resolution computed tomography and chest radiography in the diagnosis of chronic diffuse infiltrative lung disease. Clin Radiol. 1991;44(4):222–226. doi:10.1016/S0009-9260(05)80183-7
148. Bradley B, Branley HM, Egan JJ, et al. Interstitial lung disease guideline: the British Thoracic Society in collaboration with the Thoracic Society of Australia and New Zealand and the Irish Thoracic Society. Thorax. 2008;63(Suppl 5):v1–58. doi:10.1136/thx.2008.101691
149. Tovar Perez M, Rodriguez Mondejar MR. Acquired heart disease in adults: what can a chest X-ray tell us? Radiologia. 2017;59(5):446–459. doi:10.1016/j.rx.2017.06.006
150. Ghane MR, Saburi A, Javadzadeh HR. A recommended method in order to interpret chest x-rays for diagnosing small size pneumothorax. Int J Crit Illn Inj Sci. 2013;3(1):36–39. doi:10.4103/2229-5151.109417
151. Meyer KC. Diagnosis and management of interstitial lung disease. Transl Respir Med. 2014;2:4. doi:10.1186/2213-0802-2-4
152. Gargani L, Picano E. The risk of cumulative radiation exposure in chest imaging and the advantage of bedside ultrasound. Crit Ultrasound J. 2015;7:4.
153. Kolodziej M, de Veer MJ, Cholewa M, Egan GF, Thompson BR. Lung function imaging methods in cystic fibrosis pulmonary disease. Respir Res. 2017;18(1):96. doi:10.1186/s12931-017-0578-x
154. Manolescu D, Davidescu L, Traila D, Oancea C, Tudorache V. The reliability of lung ultrasound in assessment of idiopathic pulmonary fibrosis. Clin Interv Aging. 2018;13:437–449. doi:10.2147/CIA
155. Gargani L, Volpicelli G. How I do it: lung ultrasound. Cardiovasc Ultrasound. 2014;12:25. doi:10.1186/1476-7120-12-25
156. Falcetta A, Leccardi S, Testa E, Papaleo F, Fenoglio L, Melchio R. The role of lung ultrasound in the diagnosis of interstitial lung disease. Shanghai Chest. 2018;2(5). doi:10.21037/shc.2018.04.10
157. Romei C, Turturici L, Tavanti L, et al. The use of chest magnetic resonance imaging in interstitial lung disease: a systematic review. Eur Respir Rev. 2018;27:150. doi:10.1183/16000617.0062-2018
158. Metcalfe P, Liney GP, Holloway L, et al. The potential for an enhanced role for MRI in radiation-therapy treatment planning. Technol Cancer Res Treat. 2013;12(5):429–446. doi:10.7785/tcrt.2012.500342
159. Guimaraes MD, Hochhegger B, Santos MK, et al. Magnetic resonance imaging of the chest in the evaluation of cancer patients: state of the art. Radiol Bras. 2015;48(1):33–42. doi:10.1590/0100-3984.2013.1921
160. Biederer J, Mirsadraee S, Beer M, et al. MRI of the lung (3/3)-current applications and future perspectives. Insights Imaging. 2012;3(4):373–386.
161. Wielputz M, Kauczor HU. MRI of the lung: state of the art. Diagn Interv Radiol. 2012;18(4):344–353. doi:10.4261/1305-3825.DIR.5365-11.0
162. Biederer J, Beer M, Hirsch W, et al. MRI of the lung (2/3). Why … when … how? Insights Imaging. 2012;3(4):355–371. doi:10.1007/s13244-011-0146-8
163. Mills GH, Wild JM, Eberle B, Van Beek EJ. Functional magnetic resonance imaging of the lung. Br J Anaesth. 2003;91(1):16–30. doi:10.1093/bja/aeg149
164. Yang CM. The impact of global budget on the diffusion of innovations: the example of positron emission tomography in Taiwan. BMC Health Serv Res. 2018;18(1):905. doi:10.1186/s12913-018-3731-4
165. Jame SZ, Sari AA, Majdzadeh R, Rashidian A, Arab M, Rahmani H. The extent of inappropriate use of magnetic resonance imaging in low back pain and its contributory factors. Int J Prev Med. 2014;5(8):1029–1036.
166. Russo RJ, Costa HS, Silva PD, et al. Assessing the risks associated with MRI in patients with a pacemaker or defibrillator. N Engl J Med. 2017;376(8):755–764. doi:10.1056/NEJMoa1603265
167. Sammet S. Magnetic resonance safety. Abdom Radiol (NY). 2016;41(3):444–451. doi:10.1007/s00261-016-0680-4
168. Evans R, Taylor S, Janes S, et al. Patient experience and perceived acceptability of whole-body magnetic resonance imaging for staging colorectal and lung cancer compared with current staging scans: a qualitative study. BMJ Open. 2017;7(9):e016391. doi:10.1136/bmjopen-2017-016391
169. Dewey M, Schink T, Dewey CF. Claustrophobia during magnetic resonance imaging: cohort study in over 55,000 patients. J Magn Reson Imaging. 2007;26(5):1322–1327. doi:10.1002/(ISSN)1522-2586
170. Müller CS, Warszawiak D, Paiva EDS, Escuissato DL. Pulmonary magnetic resonance imaging is similar to chest tomography in detecting inflammation in patients with systemic sclerosis. Rev Bras Reumatol Engl Ed. 2017;57(5):419–424. doi:10.1016/j.rbr.2016.12.003
171. Vaquero JJ, Kinahan P. Positron emission tomography: current challenges and opportunities for technological advances in clinical and preclinical imaging systems. Annu Rev Biomed Eng. 2015;17:385–414. doi:10.1146/annurev-bioeng-071114-040723
172. Maurer AH. Combined imaging modalities: PET/CT and SPECT/CT. Health Phys. 2008;95(5):571–576. doi:10.1097/01.HP.0000334064.46217.20
173. Capitanio S, Nordin AJ, Noraini AR, Rossetti C. PET/CT in nononcological lung diseases: current applications and future perspectives. Eur Respir Rev. 2016;25(141):247–258. doi:10.1183/16000617.0051-2016
174. Mostard RL, Verschakelen JA, van Kroonenburgh MJ, et al. Severity of pulmonary involvement and (18)F-FDG PET activity in sarcoidosis. Respir Med. 2013;107(3):439–447. doi:10.1016/j.rmed.2012.11.011
175. Cherry SR, Jones T, Karp JS, et al. maximizing sensitivity to create new opportunities for clinical research and patient care. J Nucl Med. 2018;59(1):3–12. doi:10.2967/jnumed.116.184028
176. Foster B, Bagci U, Mansoor A, Xu Z, Mollura DJ. A review on segmentation of positron emission tomography images. Comput Biol Med. 2014;50:76–96. doi:10.1016/j.compbiomed.2014.04.014
177. Peelen DM, Zwezerijnen B, Nossent EJ, et al. FRI0642 The potential value of positron emission tomography (PET)-scan in systemic sclerosis for the quantitative assessment of interstitial lung disease. Ann Rheum Dis. 2017;76:732–733.
178. Guckel B, Gatidis S, Enck P, et al. Patient comfort during positron emission tomography/magnetic resonance and positron emission tomography/computed tomography examinations: subjective assessments with visual analog scales. Invest Radiol. 2015;50(10):726–732. doi:10.1097/RLI.0000000000000177
179. Petersson J, Sanchez-Crespo A, Larsson SA, Mure M. Physiological imaging of the lung: single-photon-emission computed tomography (SPECT). J Appl Physiol (1985). 2007;102(1):468–476. doi:10.1152/japplphysiol.00732.2006
180. Schniering J, Guo L, Brunner M, et al. Evaluation of (99m)Tc-rhAnnexin V-128 SPECT/CT as a diagnostic tool for early stages of interstitial lung disease associated with systemic sclerosis. Arthritis Res Ther. 2018;20(1):183. doi:10.1186/s13075-018-1681-1
181. Bybel B, Brunken RC, DiFilippo FP, Neumann DR, Wu G, Cerqueira MD. SPECT/CT imaging: clinical utility of an emerging technology. Radiographics. 2008;28(4):1097–1113. doi:10.1148/rg.284075203
182. Zhou Y, Chen H, Ambalavanan N, et al. Noninvasive imaging of experimental lung fibrosis. Am J Respir Cell Mol Biol. 2015;53(1):8–13.
183. Shaffer TM, Drain CM, Grimm J. Optical imaging of ionizing radiation from clinical sources. J Nucl Med. 2016;57(11):1661–1666. doi:10.2967/jnumed.116.178624
184. Roper J, Bowsher J, Yin FF. On-board SPECT for localizing functional targets: a simulation study. Med Phys. 2009;36(5):1727–1735. doi:10.1118/1.3113902
185. Radha S, Afroz T, Prasad S, Ravindra N. Diagnostic utility of bronchoalveolar lavage. J Cytol. 2014;31(3):136–138. doi:10.4103/0970-9371.145636
186. Patel PH, Bhimji SS; National Center for Biotechnology Information (NCBI). Bronchoalveolar Lavage. 2018. Available from: https://www.ncbi.nlm.nih.gov/books/NBK430762/#_NBK430762_dtls.
187. Chockalingam A, Duraiswamy R, Jagadeesan M. Bronchoalveolar lavage cellular analyses in conjunction with high-resolution computed tomography imaging as a diagnostic intervention for patients with suspected interstitial lung disease. Lung India. 2016;33(3):287–291. doi:10.4103/0970-2113.180806
188. Collins AM, Rylance J, Wootton DG, et al. Bronchoalveolar lavage (BAL) for research; obtaining adequate sample yield. J Vis Exp. 2014;85:e4345.
189. Nguyen W, Meyer KC. Surgical lung biopsy for the diagnosis of interstitial lung disease: a review of the literature and recommendations for optimizing safety and efficacy. Sarcoidosis Vasc Diffuse Lung Dis. 2013;30(1):3–16.
190. Kaarteenaho R. The current position of surgical lung biopsy in the diagnosis of idiopathic pulmonary fibrosis. Respir Res. 2013;14:43. doi:10.1186/1465-9921-14-43
191. Keeratichananont W, Thanadetsuntorn C, Keeratichananont S. Value of preoperative 6-minute walk test for predicting postoperative pulmonary complications. Ther Adv Respir Dis. 2016;10(1):18–25. doi:10.1177/1753465815615509
192. Garin MC, Highland KB, Silver RM, Strange C. Limitations to the 6-minute walk test in interstitial lung disease and pulmonary hypertension in scleroderma. J Rheumatol. 2009;36(2):330–336. doi:10.3899/jrheum.080447
193. Keir GJ, Silver RM, Wells AU. Clinical assessment of lung disease. In: Varga J, Denton CP, Wigley FM, editors. Scleroderma: From Pathogenesis to Comprehensive Management. New York, NY, USA: Springer; 2012:403–414.
194. Deuschle K, Weinert K, Becker MO, Backhaus M, Huscher D, Riemekasten G. Six-minute walk distance as a marker for disability and complaints in patients with systemic sclerosis. Clin Exp Rheumatol. 2011;29(2 Suppl 65):S53–59.
195. Lota HK, Renzoni EA. Circulating biomarkers of interstitial lung disease in systemic sclerosis. Int J Rheumatol. 2012;2012:121439. doi:10.1155/2012/121439
196. Tzouvelekis A, Kouliatsis G, Anevlavis S, Bouros D. Serum biomarkers in interstitial lung diseases. Respir Res. 2005;6:78. doi:10.1186/1465-9921-6-78
197. Le Gouellec N, Duhamel A, Perez T, et al. Predictors of lung function test severity and outcome in systemic sclerosis-associated interstitial lung disease. PLoS One. 2017;12(8):e0181692. doi:10.1371/journal.pone.0181692
198. Moore OA, Proudman SM, Goh N, et al. Quantifying change in pulmonary function as a prognostic marker in systemic sclerosis-related interstitial lung disease. Clin Exp Rheumatol. 2015;33(4 Suppl 91):S111–116.
199. Burt RK, Shah SJ, Dill K, et al. Autologous non-myeloablative haemopoietic stem-cell transplantation compared with pulse cyclophosphamide once per month for systemic sclerosis (ASSIST): an open-label, randomised phase 2 trial. Lancet. 2011;378(9790):498–506. doi:10.1016/S0140-6736(11)60982-3
200. van Laar JM, Farge D, Sont JK, et al. Autologous hematopoietic stem cell transplantation vs intravenous pulse cyclophosphamide in diffuse cutaneous systemic sclerosis: a randomized clinical trial. JAMA. 2014;311(24):2490–2498. doi:10.1001/jama.2014.6368
201. Sullivan KM, Goldmuntz EA, Keyes-Elstein L, et al. Myeloablative autologous stem-cell transplantation for severe scleroderma. N Engl J Med. 2018;378(1):35–47.
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.