Back to Journals » OncoTargets and Therapy » Volume 11
Interleukin-33 enhanced the migration and invasiveness of human lung cancer cells
Authors Yang Z, Gao X, Wang J, Xu L ![]() , Zheng Y, Xu Y
, Zheng Y, Xu Y ![]()
Received 2 November 2017
Accepted for publication 15 December 2017
Published 16 February 2018 Volume 2018:11 Pages 843—849
DOI https://doi.org/10.2147/OTT.S155905
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Yao Dai
Zhiping Yang,1 Xin Gao,2 Jingyu Wang,3 Longsheng Xu,4 Ying Zheng,4 Yufen Xu1
1Department of Oncology (04-F-14), The First Affiliated Hospital of Jiaxing University, Jiaxing, 2Department of Oncology, Suzhou Municipal Hospital of Nanjing Medical University, Suzhou, 3Department of Pathology, 4Department of Central Laboratory, The First Affiliated Hospital of Jiaxing University, Jiaxing, People’s Republic of China
Aim: Interleukin-33 (IL-33), belonging to IL-1 family cytokines, has been reported to participate in cancer growth and metastasis. The clinical values of IL-33 in lung cancer have been previously investigated. We aimed to elucidate the probable role of IL-33 in the migration and invasion of lung cancer cells.
Methods: Cell migration and invasiveness were tested by Transwell assay. Western blotting analysis was performed to detect protein expression.
Results: We found that IL-33 treatment in human lung A549 cells dose-dependently enhanced their migratory and invasive ability, accompanied by elevated expression of matrix metalloproteinase (MMP) 2 and MMP9. Meanwhile, IL-33-induced cell migration and invasion were significantly abolished by small interfering RNA-targeting ST2, the specific receptor of IL-33. Furthermore, IL-33 exposure induced the phosphorylation of AKT. Pretreatment with an AKT inhibitor LY294002 markedly attenuated IL-33-induced cell migration and invasion.
Conclusion: IL-33/ST2 promoted the migration and invasiveness of lung cancer cells through AKT pathway. Our findings strongly suggest that IL-33 may serve as a promising therapeutic strategy for lung cancer.
Keywords: ST2, AKT, migration, invasion
Introduction
Lung cancer is the principal cause of cancer-associated death and accounts for ~1.4 million deaths every year in the world.1 More than 80% of primary lung cancers are identified as non-small-cell lung carcinoma (NSCLC).2 Lung cancer is highly malignant and rapidly metastasizing, which is the main reason for the poor 5-year survival rate of lung cancer patients. Increasing evidence suggests that genetic and epigenetic alterations, which cause DNA damage, can transform normal lung epithelial cells into lung cancer cells, thus causing lung carcinogenesis.3
Interleukin-33 (IL-33), a specific ligand of ST2, belongs to the IL-1 family cytokines. Once secreted, IL-33 binds to ST2, activates mitogen-activated protein kinase (MAPK), nuclear factor (NF)-κB, or AKT,4,5 and then initiates downstream effector responses.6 The functions of IL-33 have been widely studied in a variety of inflammatory diseases, such as asthma, arthritis, and inflammatory bowel disease.7–10 Recent data have showed the implication of IL-33/ST2 axis in tumor development and metastasis.11–13 By using 4T1 breast cancer model, Jovanovic et al11 showed that IL-33/ST2 axis could increase the intratumoral accumulation of immunosuppressive cells and then enhance the progression of breast cancer. Liu et al12 demonstrated that ectopic expression of IL-33 in colon cancer cells enhanced their growth and metastasis in nude mice. A subsequent study showed the promoting effects of IL-33 on the invasiveness and migration of gastric cancer cells.13 The clinical values of IL-33 in lung cancer have been studied.14,15 Hu et al14 reported that NSCLC patients had higher levels of serum IL-33 than the healthy control and patients with benign lung diseases. They proposed that serum IL-33 was an independent prognostic indicator for NSCLC. A study based on male patients who were current smokers or ex-smokers showed that the plasma level of IL-33 was increased in Stage I of lung cancer in comparison to that in normal healthy control but was evidently reduced in Stages III and IV of lung cancer.15 However, the roles of IL-33 in the metastasis of lung cancer still need to be elucidated.
In this study, we have investigated the biological functions of IL-33 in the migration and invasiveness of lung cancer cells. The results showed that IL-33 significantly promoted cell invasion and migration and induced the expression of matrix metalloproteinase (MMP) 2 and MMP9 via ST2 and AKT pathway.
Methods
Cell culture
Human lung cancer cell lines, A549 and NCI-H1299 (Cell Bank of the Chinese Academy of Sciences, Shanghai, China), were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM; Thermo Fisher Scientific, Waltham, MA, USA) containing 10% fetal bovine serum (FBS; Hyclone, Logan, UT, USA). The cells were maintained at 37°C in an atmosphere of 5% CO2 and 95% air.
Small interfering RNA (siRNA) transfection
A549 cells at 70%–80% confluence were transfected with 2 μg/mL ST2 siRNA or Ctrl siRNA using Lipofectamine 2000 (Thermo Fisher Scientific) following the manufacturer’s instruction. The knockdown efficiency was analyzed by Western blot analysis at 48 h posttransfection. The sequences of ST2 siRNAs are as follows: siST2-1, 5′-GCGAAUGUCACCAUAUAUA-3′; siST2-2, 5′-GCACCUCUUGAGUGGUUUA-3′; siST2-3, 5′-GCACUUUGUUCACCAGAUU-3′.
Experimental grouping
To study the effects of IL-33, A549 and NCI-H1299 cells were treated with 0, 20, or 50 ng/mL IL-33 (R&D Systems, Inc., Minneapolis, MN, USA). To investigate the involvement of ST2, IL-33 siRNA (siST2) or Ctrl siRNA was transfected into A549 cells. At 24 h following transfection, A549 cells were treated with 0 or 50 ng/mL IL-33. To explore the role of AKT, A549 cells were pretreated with LY294002 (10 μM; Merck Millipore, Billerica, MA, USA) or vehicle ( dimethylsulfoxide) for 1 h and then with 0 or 50 ng/mL IL-33. Cell migration, invasion, and protein expression were assessed at 24 h after treatment.
Transwell assay
Non-Matrigel-coated and Matrigel-coated Boyden chambers (BD Biosciences, San Jose, CA, USA) were used to detect the migration and invasive abilities of A549 and NCI-H1299 cells, respectively. After starvation overnight in serum-free DMEM, the cells were harvested by trypsinization. Equal numbers of cells (1×105/well) in serum-free medium were plated onto the upper chamber, and DMEM with 10% FBS was added to the lower chamber. After 24 h, the nonmigrated cells were carefully scraped from the upper chamber with cotton tips. The migrated cells were subjected to 4% formaldehyde fixation and then 0.5% crystal violet staining. The migrated and invaded cells from at least five random microscopic fields (200×) were counted under a light microscope.
Western blotting
A549 cells were lysed in ice-cold radioimmunoprecipitation assay buffer with fresh-added protease inhibitor cocktail (Hoffman-La Roche Ltd., Basel, Switzerland) at 4°C. Bicinchoninic acid assay (Pierce, Rockford, IL, USA) was used to measure protein concentration. A total of 30 μg of protein was resolved by sodium dodecyl sulfate-polyacrylamide gels and electronically transferred onto nitrocellulose membranes (EMD Millipore, Billerica, MA, USA). The membranes were blocked with 5% nonfat milk in Tris-buffered saline/Tween 20 (TBST) and then probed with primary antibodies according to the manufacturers’ protocols. After washing three times with TBST, the membranes were incubated with the corresponding horseradish peroxidase-conjugated secondary antibodies (Beyotime, Shanghai, China) at room temperature for 1 h. Enhanced chemiluminescent (ECL) (Pierce) was used for the detection of signals. The sources of primary antibodies are as follows: anti-ST2 was purchased from Santa Cruz Biotechnology Inc. (Dallas, TX, USA); anti-MMP2 and anti-MMP9 were purchased from Abcam (Cambridge, MA, USA); and antibodies against phosphorylation of AKT (p-AKT) (S473), AKT, and GAPDH were obtained from Cell Signaling Technology (Danvers, MA, USA). GAPDH was used as loading Ctrl. The intensity of the Western blot signals was analyzed using the ImageJ software (http://rsb.info.nih.gov/ij/, Bethesda, MD, USA).
Statistical analysis
The experiments were performed three times independently. Data presented are mean ± standard deviation (SD). Statistics was calculated with GraphPad Prism Version 6 (GraphPad Software, Inc., La Jolla, CA, USA). One-way analysis of variance (ANOVA) followed by Tukey’s test was used for statistical comparisons. A P-value of <0.05 was defined as statistically significant.
Results
IL-33 exposure leads to an increase in cell migration and invasion of lung cancer cell lines
To determine whether the IL-33 affects the migration and invasion of lung cancer cells, Transwell assays were conducted on A549 and NCI-H1299 cells exposed to various concentrations of IL-33 (0, 20, or 50 ng/mL). As shown in Figure 1A and B, stimulation with IL-33 greatly increased the number of cells that migrated across the non-Matrigel-coated membranes or Matrigel-coated membranes in a dose-dependent manner. Similar results were achieved in NCI-H1299 cells. These abovementioned results suggested that exposing lung cancer cells to IL-33 significantly enhanced cell migration and invasiveness.
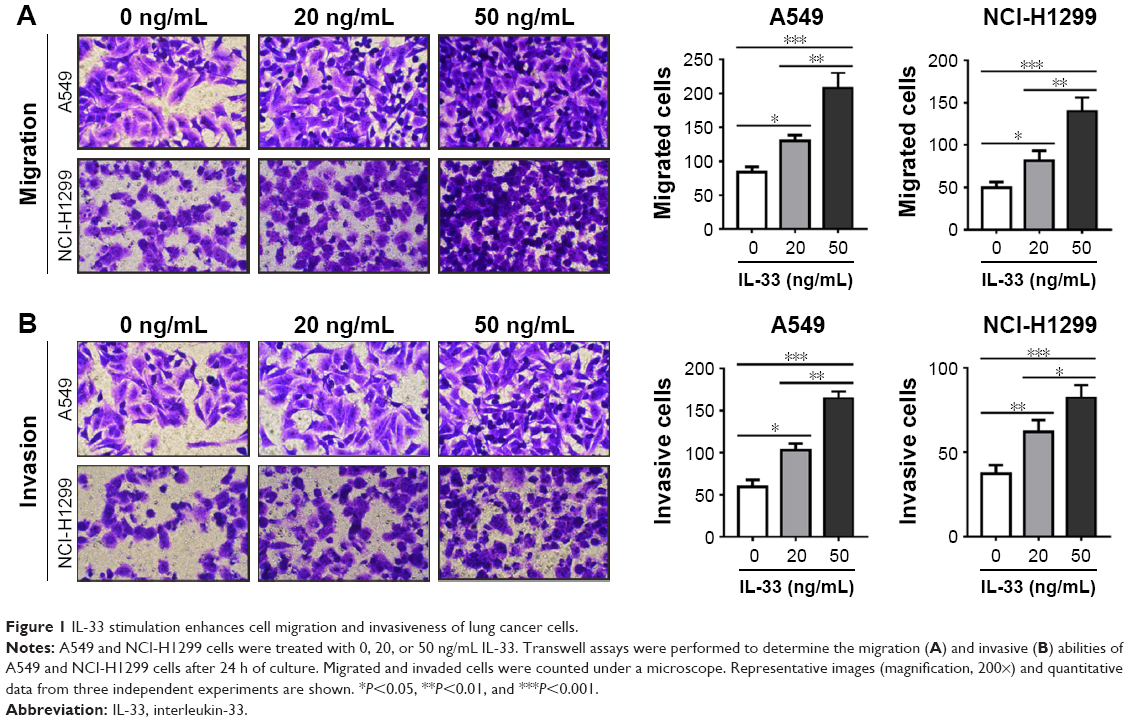 | Figure 1 IL-33 stimulation enhances cell migration and invasiveness of lung cancer cells. |
IL-33 upregulates the levels of MMP2, MMP9 and p-AKT
AKT, MMP2, and MMP916,17 have been proven to function in tumor metastasis; thus, p-AKT and the expression of MMP2 and MMP9 were examined to further investigate the mechanisms of how IL-33 affects migration and invasion. The results showed that the levels of MMP2, MMP9, and p-AKT were upregulated in A549 cells exposed to 20 or 50 ng/mL of IL-33 when compared with that in cells without IL-33 treatment (Figure 2A and B).
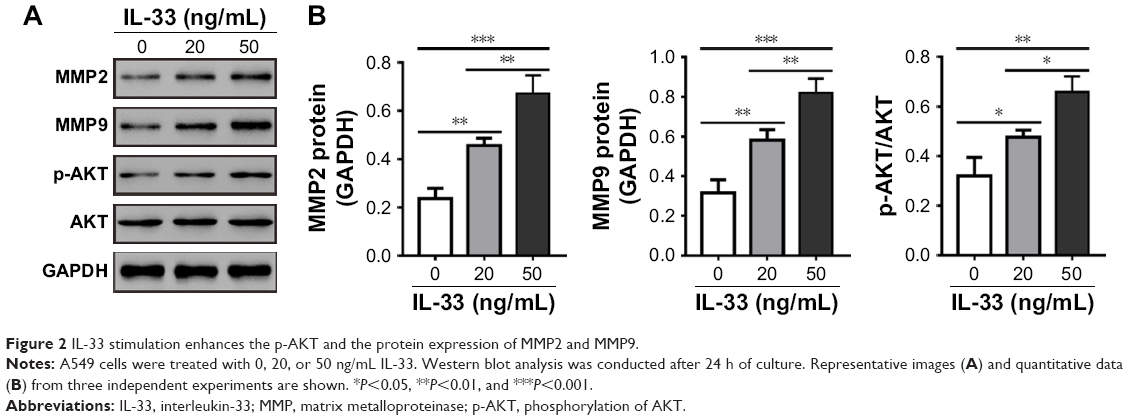 | Figure 2 IL-33 stimulation enhances the p-AKT and the protein expression of MMP2 and MMP9. |
Knockdown of ST2 mitigates the effects of IL-33 on A549 cells
IL-33 exerts functions via its specific receptor, ST2. To investigate the involvement of ST2 in IL-33-stimulated migration and invasiveness of A549 cells, ST2 expression was knocked down by siRNA transfection in A549 cells. Western blot analysis showed that A549 cells transfected with ST2 siRNAs (siST2-1, -, and -3) expressed a significantly lower level of ST2 when compared with A549 cells transfected with Ctrl siRNA or wild-type (WT) A549 cells (Figure 3A). siST2-1 showed the best knockdown efficiency and was used in the subsequent studies.
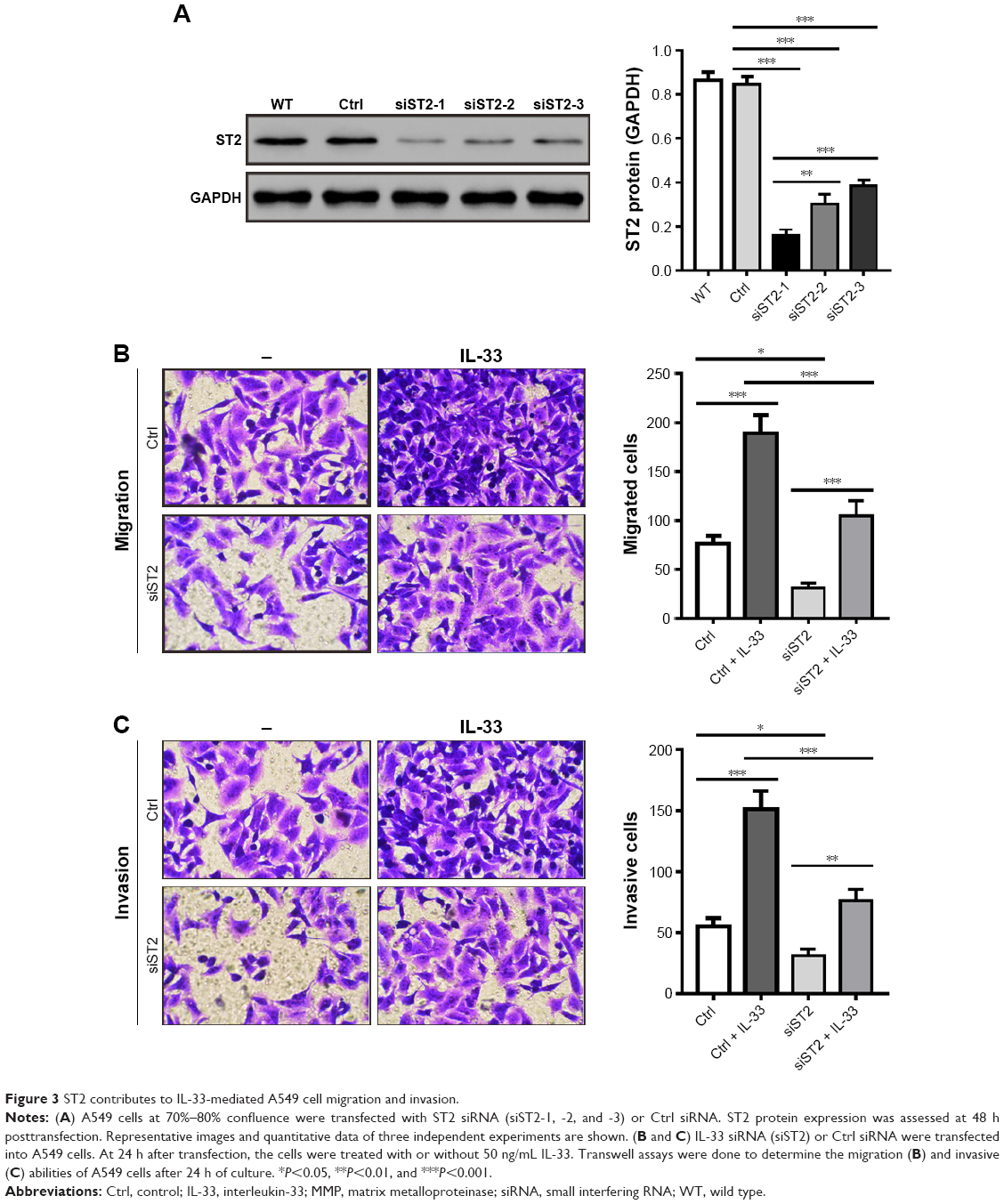 | Figure 3 ST2 contributes to IL-33-mediated A549 cell migration and invasion. |
Next, A549 cells were transfected with siST2-1 or Ctrl siRNA and then treated with 50 ng/mL of IL-33. We found that IL-33-stimulated migration and invasion were notably attenuated in siST2-transfected cells as compared to Ctrl-transfected cells (Figure 3B and C). Additionally, as shown in Figure 4, knockdown of ST2 reduced the expression of MMP2, MMP9, and p-AKT induced by IL-33. These abovementioned results suggested that the stimulation effects of IL-33 on the migration and invasion, as well as the levels of MMP2, MMP9, and p-AKT in A549 cells, were dependent on ST2.
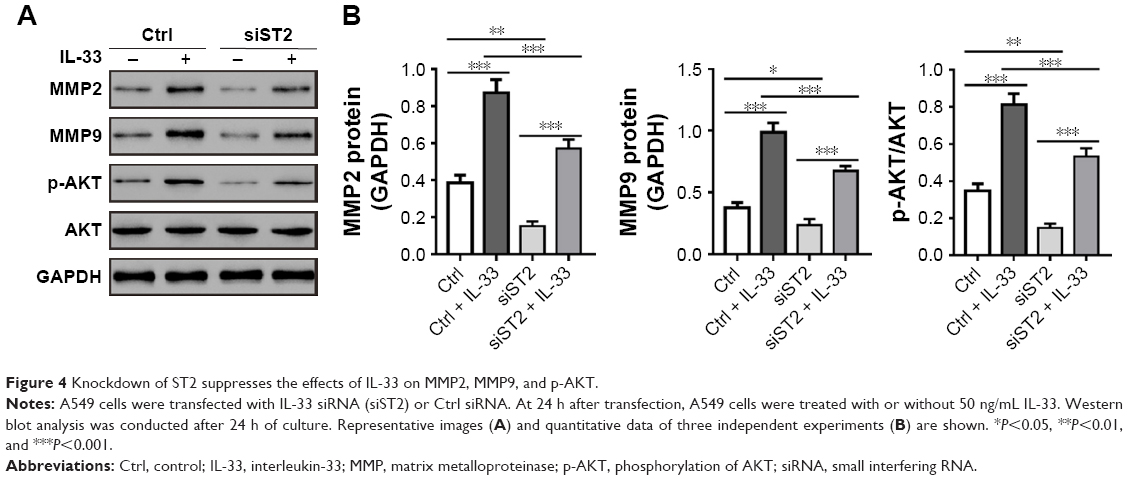 | Figure 4 Knockdown of ST2 suppresses the effects of IL-33 on MMP2, MMP9, and p-AKT. |
Inhibition of AKT pathway suppresses IL-33-mediated migration and invasion
To further clarify the role of AKT during IL-33-mediated migration and invasion, AKT activity was inhibited by LY294002 in A549 cells. As illustrated in Figure 5, LY294002 treatment significantly suppressed the protein levels of MMP2, MMP9, and p-AKT, as well as the migration and invasion of A549 cells with or without IL-33 treatment. These data indicated that IL-33 promoted cell migration and invasion through AKT pathway.
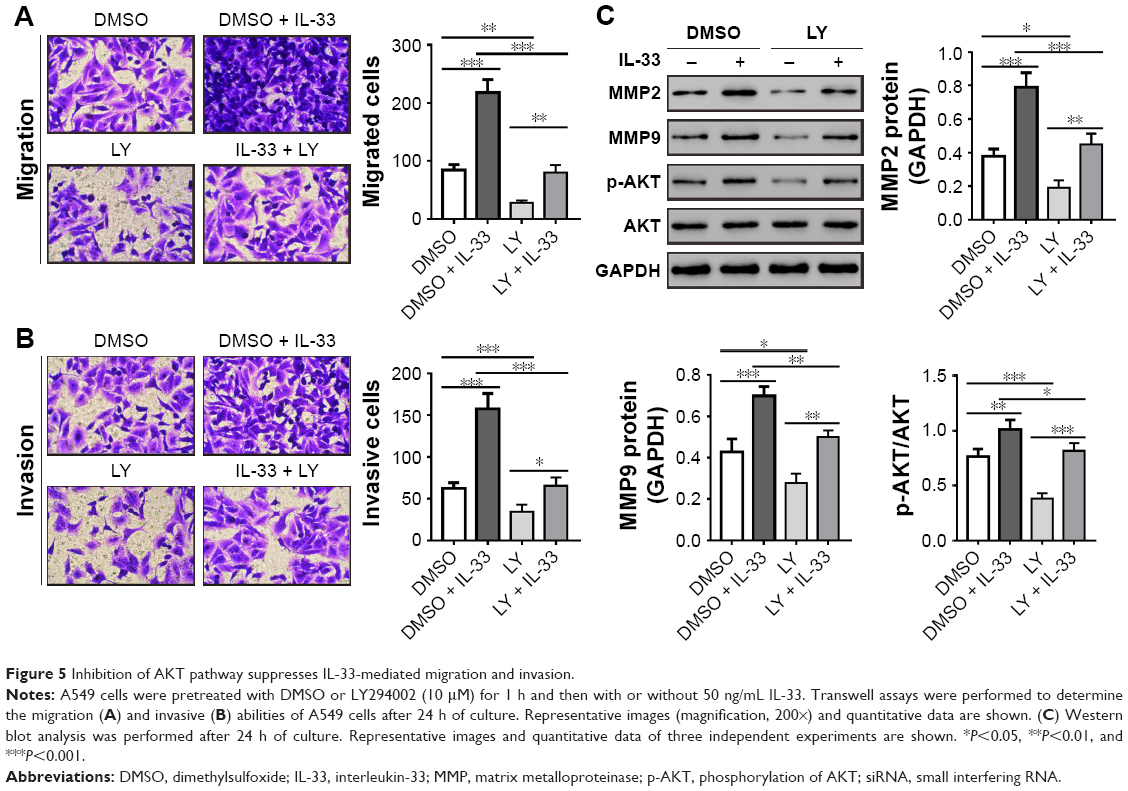 | Figure 5 Inhibition of AKT pathway suppresses IL-33-mediated migration and invasion. |
Discussion
Accumulating evidence has supported that IL-33, an IL-1 family cytokine, plays an essential role in various inflammatory diseases7–10 and cancer growth and metastasis.11–13 Recent studies have revealed the clinical values of IL-33 in lung cancer.14,15 In this study, we investigated the probable role of IL-33 in the migration and invasion of lung cancer cells and characterized the signaling pathway affected by IL-33 by the stimulation of A549 cells with recombinant IL-33. We found that stimulation with IL-33 significantly promoted the migration and invasion of A549 cells. The present study also demonstrated that, 1) IL-33 exposure was associated with AKT activation and the increased expression of MMP2 and MMP9 in A549 cells; 2) IL-33 treatment caused a remarkable increase in cell migration and invasion in an ST2-dependent manner; and 3) AKT took part in mediating the effect of IL-33 on cell migration and invasiveness. Our data suggest that IL-33 regulates the migration and invasion of lung cancer cells via ST2 and activation of AKT pathway.
High invasive potential is one of the main hallmarks of lung cancer.3 Previous studies have demonstrated that IL-33 promotes the metastasis of colorectal cancer and the migration and invasion of gastric cancer cells.12,13 Consistent with these findings, we showed that IL-33 stimulation significantly enhanced the migration and invasion of A549 cells. MMPs, a family of zinc-dependent endopeptidases, are key players in the degradation of extracellular matrix (ECM). Considerable evidence has implicated MMPs in cancer cell invasion, the metastatic process, and angiogenesis.16,18 In particular, the clinical values of MMP2 and MMP9, two type IV collagenases, have been reported in lung cancer.19,20 Here, Western blot analysis revealed a much higher level of MMP2 and MMP9 in IL-33-treated cells than that in the untreated cells, further suggesting the role of IL-33 in the migration and invasion of A549 cells. IL-33 binds to its specific receptor, ST2 and then initiates downstream effector responses.6 In the present study, our results demonstrated that transfection with ST2-targeted siRNA significantly repressed the cell migration and invasion and the expression of MMP2 and MMP9 induced by IL-33, suggesting that IL-33 induced cell migration and invasion in an ST2-dependent manner.
PI3K/AKT has been found to be activated in several cancers,21–25 including pancreatic, thyroid, ovarian, and lung cancers. IL-33 activates AKT phosphorylation in immune cells.4 However, it remains unknown whether IL-33 treatment is associated with the activation of AKT pathway in lung cancer cells. Our data showed that AKT phosphorylation was dose-dependently increased by IL-33 exposure. ST2 played a critical role in the induction effects of IL-33 on AKT phosphorylation. Moreover, blocking of AKT with LY294002 attenuated IL-33-induced cell migration and invasion. Thus, we suggest that IL-33/ST2 activates AKT in lung cancer cells, which subsequently enhances cellular migration and invasion. Previous reports have shown that AKT can promote cell migration and invasion via inducing MMP proteins.26 Here, we found that increased AKT phosphorylation was associated with the increased levels of MMP2 and MMP9. Thus, our findings suggest that IL-33/ST2 axis exerted the migration- and invasion-promoting effect in A549 cells via a cellular signaling pathway involving AKT, MMP2, and MMP9.
Conclusion
Our findings in the present study indicated that IL-33 treatment in A549 cells promoted cell migration and invasiveness and increased the expression of MMP2 and MMP9 in an ST2-dependent manner. Furthermore, our results suggested that AKT signaling pathway was involved in this process.
Acknowledgment
This study was funded by the National Natural Science Foundation of China (81650012) and the Jiaxing Innovation Team of early diagnosis and comprehensive therapy for lung cancer.
Disclosure
The authors report no conflicts of interest in this work.
References
Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61(2):69–90. | ||
Koudelakova V, Kneblova M, Trojanec R, Drabek J, Hajduch M. Non-small cell lung cancer – genetic predictors. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub. 2013;157(2):125–136. | ||
Cooper WA, Lam DC, O’Toole SA, Minna JD. Molecular biology of lung cancer. J Thorac Dis. 2013;5(Suppl 5):S479–S490. | ||
Salmond RJ, Mirchandani AS, Besnard AG, Bain CC, Thomson NC, Liew FY. IL-33 induces innate lymphoid cell-mediated airway inflammation by activating mammalian target of rapamycin. J Allergy Clin Immunol. 2012;130(5):1159.e6–1166.e6. | ||
Castellani ML, Kempuraj D, Salini V, et al. The latest interleukin: IL-33 the novel IL-1-family member is a potent mast cell activator. J Biol Regul Homeost Agents. 2009;23(1):11–14. | ||
Liew FY, Pitman NI, McInnes IB. Disease-associated functions of IL-33: the new kid in the IL-1 family. Nat Rev Immunol. 2010;10(2):103–110. | ||
Xi H, Katschke KJ Jr, Li Y, et al. IL-33 amplifies an innate immune response in the degenerating retina. J Exp Med. 2016;213(2):189–207. | ||
Lloyd CM. IL-33 family members and asthma – bridging innate and adaptive immune responses. Curr Opin Immunol. 2010;22(6):800–806. | ||
Talabot-Ayer D, McKee T, Gindre P, et al. Distinct serum and synovial fluid interleukin (IL)-33 levels in rheumatoid arthritis, psoriatic arthritis and osteoarthritis. Joint Bone Spine. 2012;79(1):32–37. | ||
Beltran CJ, Nunez LE, Diaz-Jimenez D, et al. Characterization of the novel ST2/IL-33 system in patients with inflammatory bowel disease. Inflamm Bowel Dis. 2010;16(7):1097–1107. | ||
Jovanovic IP, Pejnovic NN, Radosavljevic GD, et al. Interleukin-33/ST2 axis promotes breast cancer growth and metastases by facilitating intratumoral accumulation of immunosuppressive and innate lymphoid cells. Int J Cancer. 2014;134(7):1669–1682. | ||
Liu X, Zhu L, Lu X, et al. IL-33/ST2 pathway contributes to metastasis of human colorectal cancer. Biochem Biophys Res Commun. 2014;453(3):486–492. | ||
Yu XX, Hu Z, Shen X, Dong LY, Zhou WZ, Hu WH. IL-33 promotes gastric cancer cell invasion and migration via ST2-ERK1/2 pathway. Dig Dis Sci. 2015;60(5):1265–1272. | ||
Hu LA, Fu Y, Zhang DN, Zhang J. Serum IL-33 as a diagnostic and prognostic marker in non-small cell lung cancer. Asian Pac J Cancer Prev. 2013;14(4):2563–2566. | ||
Kim MS, Kim E, Heo JS, et al. Circulating IL-33 level is associated with the progression of lung cancer. Lung Cancer. 2015;90(2):346–351. | ||
Egeblad M, Werb Z. New functions for the matrix metalloproteinases in cancer progression. Nat Rev Cancer. 2002;2(3):161–174. | ||
Hennessy BT, Smith DL, Ram PT, Lu Y, Mills GB. Exploiting the PI3K/AKT pathway for cancer drug discovery. Nat Rev Drug Discov. 2005;4(12):988–1004. | ||
Mott JD, Werb Z. Regulation of matrix biology by matrix metalloproteinases. Curr Opin Cell Biol. 2004;16(5):558–564. | ||
Ylisirnio S, Hoyhtya M, Turpeenniemi-Hujanen T. Serum matrix metalloproteinases-2, −9 and tissue inhibitors of metalloproteinases-1, −2 in lung cancer – TIMP-1 as a prognostic marker. Anticancer Res. 2000;20(2b):1311–1316. | ||
Yu C, Pan K, Xing D, et al. Correlation between a single nucleotide polymorphism in the matrix metalloproteinase-2 promoter and risk of lung cancer. Cancer Res. 2002;62(22):6430–6433. | ||
Brognard J, Clark AS, Ni Y, Dennis PA. Akt/protein kinase B is constitutively active in non-small cell lung cancer cells and promotes cellular survival and resistance to chemotherapy and radiation. Cancer Res. 2001;61(10):3986–3997. | ||
Cheng JQ, Ruggeri B, Klein WM, et al. Amplification of AKT2 in human pancreatic cells and inhibition of AKT2 expression and tumorigenicity by antisense RNA. Proc Natl Acad Sci U S A. 1996;93(8):3636–3641. | ||
Vasko V, Saji M, Hardy E, et al. Akt activation and localisation correlate with tumour invasion and oncogene expression in thyroid cancer. J Med Genet. 2004;41(3):161–170. | ||
Yuan ZQ, Sun M, Feldman RI, et al. Frequent activation of AKT2 and induction of apoptosis by inhibition of phosphoinositide-3-OH kinase/Akt pathway in human ovarian cancer. Oncogene. 2000;19(19):2324–2330. | ||
Lee SH, Kim HS, Park WS, et al. Non-small cell lung cancers frequently express phosphorylated Akt; an immunohistochemical study. APMIS. 2002;110(7–8):587–592. | ||
Chin YR, Toker A. Function of Akt/PKB signaling to cell motility, invasion and the tumor stroma in cancer. Cell Signall. 2009;21(4):470–476. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.