Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 15
Interleukin-17A Deficiency Attenuated Emphysema and Bone Loss in Mice Exposed to Cigarette Smoke
Authors Xiong J, Tian J, Zhou L, Le Y, Sun Y
Received 20 October 2019
Accepted for publication 22 January 2020
Published 10 February 2020 Volume 2020:15 Pages 301—310
DOI https://doi.org/10.2147/COPD.S235384
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Chunxue Bai
Jing Xiong,1 Jieyu Tian,2 Lu Zhou,1 Yanqing Le,1 Yongchang Sun1
1Department of Respiratory and Critical Care Medicine, Peking University Third Hospital, Beijing, 100191, People’s Republic of China; 2Hematology Oncology Center, Beijing Children’s Hospital, Capital Medical University, Beijing 100045, People’s Republic of China
Correspondence: Yongchang Sun
Department of Respiratory and Critical Care Medicine, Peking University Third Hospital, 49 North Garden Road, Haidian District, Beijing 100191, People’s Republic of China
Tel +86 156 1196 3697
Email [email protected]
Background and Purpose: Chronic obstructive pulmonary disease (COPD) is a common chronic inflammatory disease, which is associated with various comorbidities including osteoporosis. Interleukin(IL)-17 has been reported to play important roles in the pathogenesis of COPD and also associated with bone destruction in inflammatory diseases. However, the role of IL-17A in COPD-related osteoporosis is yet unknown. The purpose of our study was to investigate the potential contribution of IL-17A in COPD-related bone loss.
Materials and Methods: We examined the bone mass and bone microarchitecture in wild-type and IL-17A-/- mice exposed to long-term cigarette smoke (CS). Osteoclast activities and the expression of receptor activator of nuclear factor-κB ligand (RANKL) in bone tissues were assessed, and the blood levels of inflammatory cytokines were measured.
Results: Less bone loss as well as attenuated emphysema were shown in IL-17A-/- mice compared with wild-type mice. CS-exposed IL-17A-/- mice had decreased TRAP+ osteoclast numbers and lower RANKL expression compared with CS-exposed wild-type mice. Inflammatory cytokines including IL-6 and IL-1β in circulation were decreased in IL-17A-/- mice exposed to CS compared with wild-type mice.
Conclusion: This study indicates that IL-17A is involved in CS-induced bone loss and may be a common link between COPD and osteoporosis.
Keywords: chronic obstructive pulmonary disease, osteoporosis, interleukin 17, receptor activator of nuclear factor-κB ligand
Introduction
Chronic obstructive pulmonary disease (COPD) is a globally prevalent airway disease characterized by persistent inflammation and progressive airflow limitation caused by noxious particles or gases, particularly cigarette smoke (CS),1 which is also commonly associated with extra-pulmonary comorbidities such as cardiovascular diseases, muscle wasting and osteoporosis.2,3 As one of the major systemic comorbidities of COPD, osteoporosis is associated with poor health status and prognosis of the disease.4,5 However, the underlying mechanistic links between COPD and osteoporosis remain elusive, although there is evidence suggesting that systemic inflammation, presumably resulting from “overspill” of inflammatory mediators from the lungs, may play key roles, among other factors, in the pathogenesis of COPD-related osteoporosis.6,7
Studies have shown that bone and immune cells, particularly activated T cells including Th17 cells, are functionally connected.8,9 For example, CD4+ T cells produce both proinflammatory cytokines such as IL-17, and the osteoclastogenic cytokine RANKL, which was found to be dysregulated in COPD in our previous study.10 IL-17, mainly produced by Th17 cells, has been found to play important roles in the pathogenesis of COPD.11,12 Expression of IL-17A was increased in airways of COPD patients and correlated with lung function decline.13,14 Besides, data from animal models have suggested a critical role for IL-17A in CS-induced lung inflammation and emphysema.15,16
Interestingly, IL-17 is also a key molecule in osteoimmunology. As a potent osteoclastogenic cytokine, IL-17A was found to increase RANKL expression in osteoblasts and synovial fibroblasts, enhancing the formation and activation of osteoclasts and bone destruction in rheumatoid arthritis.17,18
While the involvement of IL-17 in bone metabolism has been demonstrated in inflammatory diseases,19 and long-term CS exposure is associated with bone loss,20,21 the role of IL-17 in COPD-related osteoporosis is yet unknown. Therefore, in this study, we employed a long-term CS exposure mouse model to investigate the potential role of IL-17A in COPD-related osteoporosis. The results revealed that long-term CS exposure induced emphysema as well as bone loss in mice, and IL-17A deficiency was associated with attenuated emphysema and less bone loss in this model, suggesting that IL-17A may be a common mechanistic link between CS-induced COPD and osteoporosis.
Materials and Methods
Animals
Female wild-type (WT) C57BL/6 mice (6–8 weeks of age, 20–25g body weight) were purchased from Beijing Vital River Experimental Animal Company (Beijing, China). IL-17A-/- mice on the C57BL/6 background (generated by Dr. Yoichiro Iwakura, and provided by Dr. Huanzhong Shi, Capital Medical University) were bred in house. All mice were housed in sterilized cages under a 12-h light-dark cycle, with free access to sterilized food and water. All experimental protocols and procedures complied with the guidelines of the Committee of Peking University on the care and use of laboratory animals, and were approved by the Ethical Committee for Animal Research of Peking University Third Hospital.
Cigarette Smoke Exposure
Wild-type mice and IL-17A-/- mice were, respectively, divided randomly into two groups: control groups and CS-exposed groups. Mice were exposed to active smoke generated by commercially available cigarettes (Baisha cigarettes with filter purchased from Hunan, China) using a nose-only smoke exposure system (SG-300; SIBATA, Tokyo, Japan), as described previously in our study.22 Briefly, each mouse was restrained in an individual exposure chamber and exposed nose-only to diluted cigarette smoke. A computer-controlled suction was generated, with 20mL CS per 8 seconds into the inhalation tower. The optimal smoke: air ratio of 1:9 was obtained. Animals were exposed twice a day for 50 mins, 5 days per week for 24 weeks. The control animals were exposed only to filtered air in the same duration.
Lung Function Measurements
At the end of the 24 weeks CS exposure, each mouse was deeply anesthetized with intraperitoneally administered pentobarbital sodium. Tracheostomy was performed, and the trachea cannulated. After intubation, the tracheal cannula was attached to a Flexivent ventilator (Scireq, Montreal, QC, Canada), and lung mechanics were measured following the protocol described previously.22 The ventilation rate was set at 150 breaths per min with a tidal volume of 10 mL/kg and a positive end-expiratory pressure of 2cm H2O. Total lung capacity (TLC), airway resistance (R) and lung dynamic compliance were measured and recorded.
Lung Histology and Measurement of Emphysema
Mouse lung tissues were obtained and fixed with 4% paraformaldehyde for 24 hrs, and then embedded in paraffin, cut into sections of 4 μm thickness, followed by H&E staining. Airspace enlargement was measured by the mean linear intercept (Lm) and destruction of alveolar walls was quantified by the destructive index (DI) according to described protocols.23
Microcomputed Tomography of Bones
Mouse femurs were dissected and fixed in 4% paraformaldehyde. Distal femurs were scanned using micro-computed tomography (SkyScan 1172; Kontich, Belgium) with a 5µm voxel size. X-ray source was set at a voltage of 45 kV with a 0.5 mm aluminium filter. Scanning angular rotation was 180° and images were captured every 0.4°. Three-dimensional (3D) reconstruction was carried out using SkyScan NRecon software and data were analyzed using CT-Analyzer software. For trabecular measurement, region of interest (ROI) was drawn starting from 0.5mm proximally to the growth plate over 2 mm in the proximal direction, and the cortical ROI was drawn from 3.25 mm proximally to the growth plate with 0.5 mm of thickness as described previously.24 Bone morphometric parameters were measured by CT-Analyzer software, including trabecular bone volume (BV/TV), trabecular thickness (Tb.Th), trabecular separation (Tb.Sp), structure model index (SMI). BMD values were calculated from calcium hydroxyapatite micro-CT phantoms and were averaged for the volumes of interest.
Histochemistry and Immunohistochemistry of Bone Tissues
At the time of sacrifice, mouse femurs were fixed in 4% paraformaldehyde for 24 hrs, decalcified in 10% EDTA for 21 days and embedded in paraffin. The osteoclast marker tartrate-resistant acidic phosphatase (TRAP) was detected using TRAP staining kit (Servicebio, Wuhan, China). Briefly, paraffin sections were routinely dewaxed in water and incubated with the TRAP activity mix for 1 h at 37°C. For immunohistochemistry of RANKL, bone tissue on slides was treated with 0.3% H2O2 for 10 min to block endogenous peroxidase. Antibodies against RANKL (1:150, Santa Cruz Biotechnology, CA) were added and incubated at 4°C overnight. After incubation with peroxidase-conjugated secondary antibody, signals were visualized with a DAB peroxidase substrate kit. Cancellous bone was assessed starting from the lower edge of the growth plate and extending proximally by 4 mm. Images were captured using Olympus BX51 microscope, and cells per bone perimeter (B.Pm) or per bone marrow area was measured to evaluate the number of positive cells with Image Pro Plus 6.0 software (Media Cybernetics, MD, USA) as previously described.25
Cytokine Measurements
Serum levels of IL-6 (BMS603/2 eBioscience, USA) were determined using commercial ELISA kits. Serum levels of IL-1β, TNF-α and IL-17A were measured by Procarta cytokine assay kit (Affymetrix eBioscience, San Diego, CA, USA) according to the manufacturer’s protocol.
Statistical Analysis
All statistical analyses were performed using the SPSS20.0 software (IBM Corporation, Armonk, USA). Data are presented as mean ± SD. Statistical analysis was performed by one-way ANOVA test (equal variances assumed) or Tamhane’s T2 signed-rank (equal variances not assumed). Differences were considered significant at p-value <0.05.
Results
IL-17A Contributed to CS-Induced Airspace Enlargement and Lung Inflammatory Response
Following 24 weeks CS exposure, mouse lung function and morphometric assessment of lung sections were performed. Wild-type mice receiving chronic CS exposure exhibited impaired pulmonary function, a significant increase in total lung capacity and airway resistance (Figure 1A–B), as compared to those exposed to air only. Histological analysis of lung sections showed alveolar enlargement as well as increased inflammatory cell infiltration in the lung parenchyma and interstitial spaces (Figure 1C–F), indicating that COPD-like responses were successfully induced in CS-exposed mice.
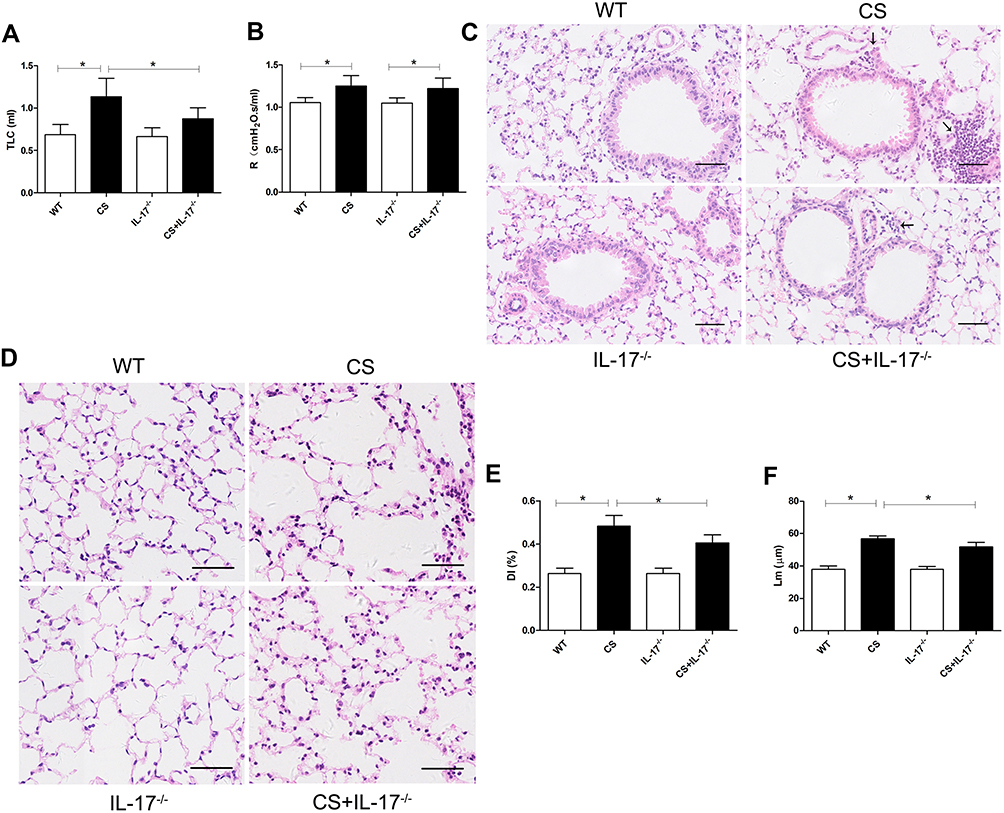 |
Figure 1 Mouse lung function and histology of lung tissue. (A) Total lung capacity (TLC) and (B) airway resistance (R) were measured in mice (n=6). (C–D) Histology of lung tissue of mice from each group by hematoxylin and eosin staining. Arrows indicate inflammatory cell infiltration. Scale bar=50 µm. Representative (E) destructive index (DI) and (F) average linear intercept (Lm) of alveoli were assessed. Data presented as mean ± SD (n=6 per group). * P<0.05. |
In contrast, in IL-17A-deficient (IL-17A-/-) mice exposed to CS, the changes in total lung capacity (Figure 1A) and emphysema were partially ameliorated, as demonstrated by a statistically decreased DI and Lm (Figure 1E–F), as well as decreased inflammatory cell infiltration in lung tissues (Figure 1C). These results indicated that IL-17A contributed to COPD-like changes in our murine model.
Deletion of IL-17A Was Associated with Attenuated Bone Loss in Mice with Chronic CS Exposure
To explore the potential role of IL-17A in bone loss in mice exposed to CS, bone microarchitecture of the femur was assessed using high-resolution micro-computed tomography (microCT). Three-dimensional reconstruction images of trabecular bones and cortical bones showed that the bone microarchitecture was significantly altered in CS-exposed wild-type mice, with thinner and less dense trabeculae, as well as reduced connectivity, which were ameliorated in IL-17A-/- mice with CS exposure (Figure 2A). Moreover, quantitative analyses showed that in wild-type mice exposed to CS, as compared to those exposed to air, a significant decrease in trabecular thickness (Tb.Th) as well as bone mineral density (BMD) of the trabeculae and cortical bone were observed, whereas in IL-17A-/- mice with CS exposure, these changes were significantly attenuated (Figure 2B–D). Bone volume (BV/TV) was also significantly decreased in wild-type mice exposed to CS (P<0.05), but showed a trend toward increase in IL-17A-/- mice with CS exposure (P=0.081) (Figure 2E). However, structural model index (SMI) and trabecular spacing (Tb.sp) showed no significant change (Figure 2F–G). Taken together, these results indicated that IL-17A was also involved in bone loss in CS-exposed mice.
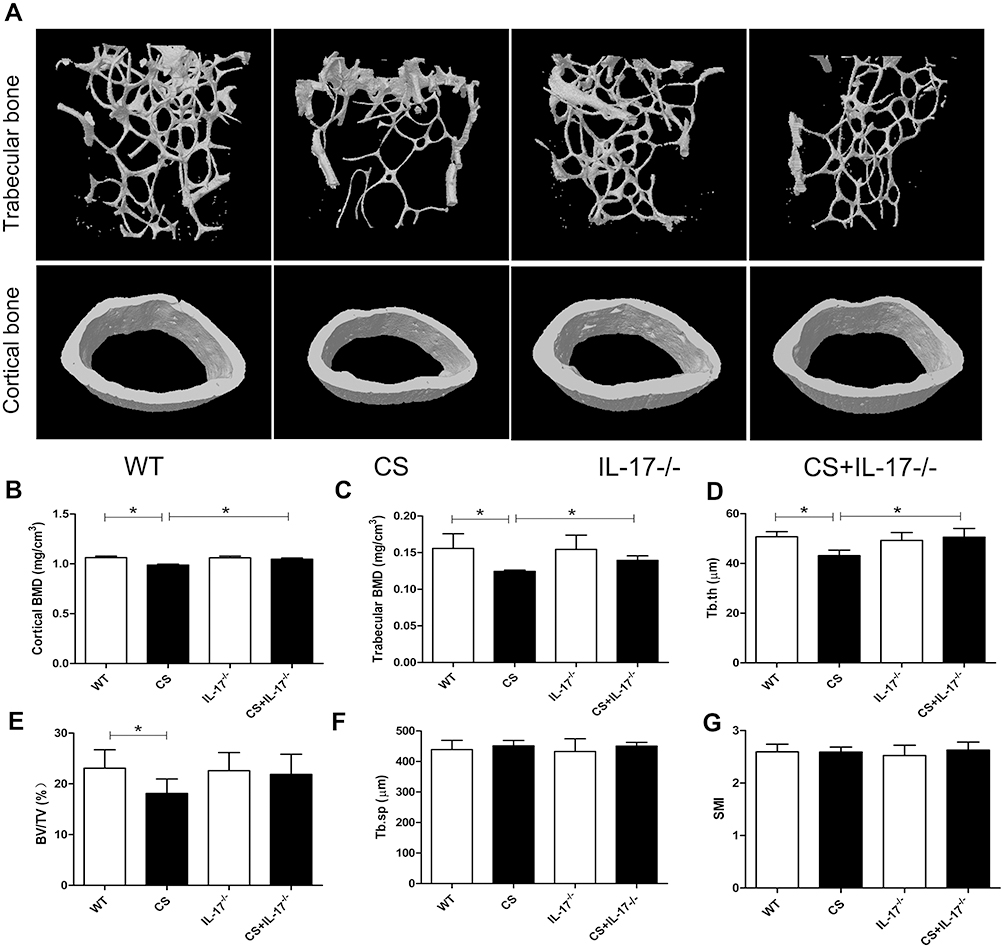 |
Figure 2 Deletion of IL-17A had a protective effect on bone mass of CS-exposed mice. (A) Representative microCT image reconstruction of trabecular bones (upper) and cortical bones (lower) towards the distal side of the femur. (B) Quantitative analysis of BMD of cortical bone and (C) trabecular bone. Quantitative analysis of trabecular parameters including (D) trabecular thickness (Tb.Th), (E) bone volume density (BV/TV), (F) trabecular spacing (Tb.sp), (G) structural model index (SMI). Data presented as mean ± SD (n=6 per group). * P<0.05. |
Osteoclastogenesis and RANKL Expression Were Downregulated in Bone Tissues of CS-Exposed Mice with IL-17A Deletion
As osteoclasts were the only cells definitively shown to degrade bone,26 we therefore performed TRAP staining on paraffin-embedded femur sections to examine osteoclastic activity in bone of CS-exposed mice. Marked TRAP+ osteoclast cells on the surface of bone were seen in wild-type mice with CS exposure, whereas a significantly decreased number of TRAP+ osteoclast cells were found in IL-17A-/- mice with CS exposure (Figure 3A–B).
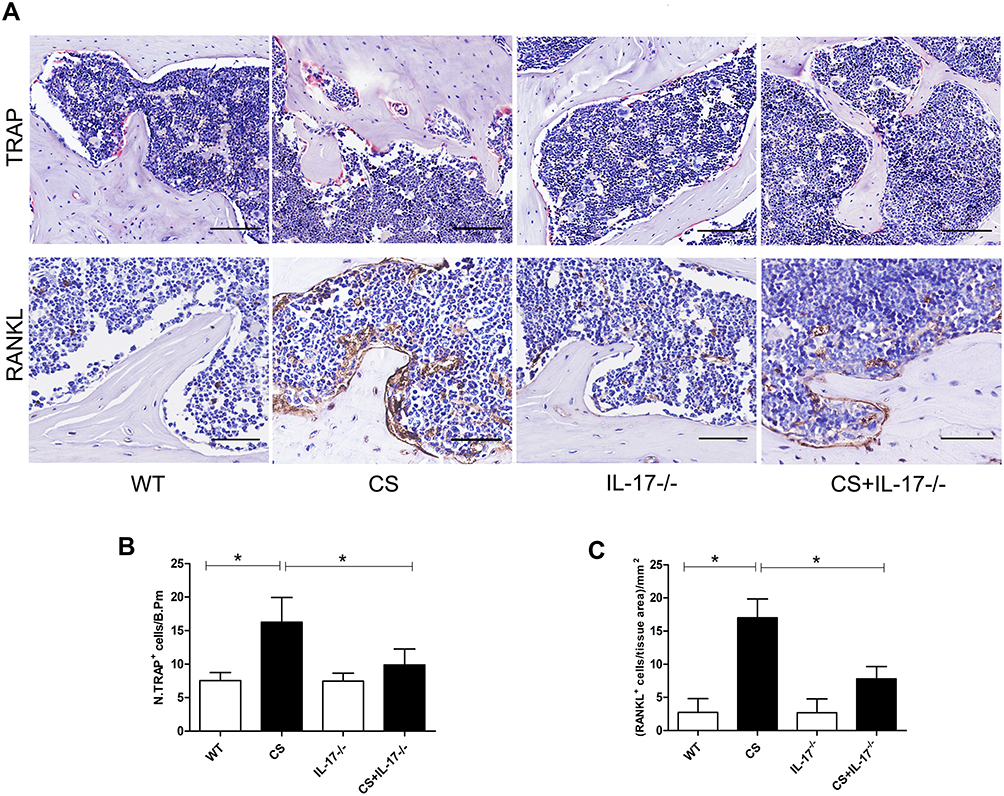 |
Figure 3 TRAP staining and RANKL expression in femur sections. (A) TRAP staining (upper) and immunohistochemical staining of RANKL expression (lower) in bone along the distal femur. TRAP-positive osteoclasts are stained red purple on the surface of bone. Scale bar=100 µm. Brown cells are RANKL expression positive cells. Magnification ×200. Scale bar=50 µm. (B) Numbers of TRAP-positive osteoclasts on the bone surface, measured as cells per millimeter of perimeter(/B.Pm). (C) Quantitative analysis of RANKL-positive cells per bone marrow area (mm2), Data presented as mean ± SD (n=5 per group). * P<0.05. |
As RANKL was a canonical mediator in osteoclast differentiation and bone destruction,27 and in our earlier study circulating RANKL was found increased in COPD patients with lower BMD,28 we, therefore, performed immunohistochemistry for RANKL in bone tissues. We found marked RANKL staining in wild-type mice exposed to CS as compared to those exposed to air, but its expression was decreased in IL-17A-/- mice with CS exposure (Figure 3A and C). These data suggest that IL-17A may be involved in osteoclastogenesis via enhancing local expression of RANKL in bone tissues in this model.
IL-17A Deficiency Was Associated with Downregulation of Pro-Osteoclastic Inflammatory Cytokines in Circulation
Several proinflammatory cytokines have been reported to be responsible for bone destruction.29 Therefore, we examined the serum levels of cytokines IL-1β, IL-6, and TNF-α, which were potentially associated with both COPD and bone loss. Our results showed that chronic CS exposure was associated with upregulation of IL-1β, IL-6, and TNF-α, as well as IL-17A (Figure 4A–D). By contrast, the levels of IL-6 and IL-1β were decreased in CS-exposed IL-17A-/- mice as compared with wild-type mice exposed to CS (Figure 4B–C), whereas the levels of TNF-α remained unaffected (Figure 4D), suggesting that IL-17A may also promote bone loss indirectly by modulating some of the pro-osteoclastic cytokines.
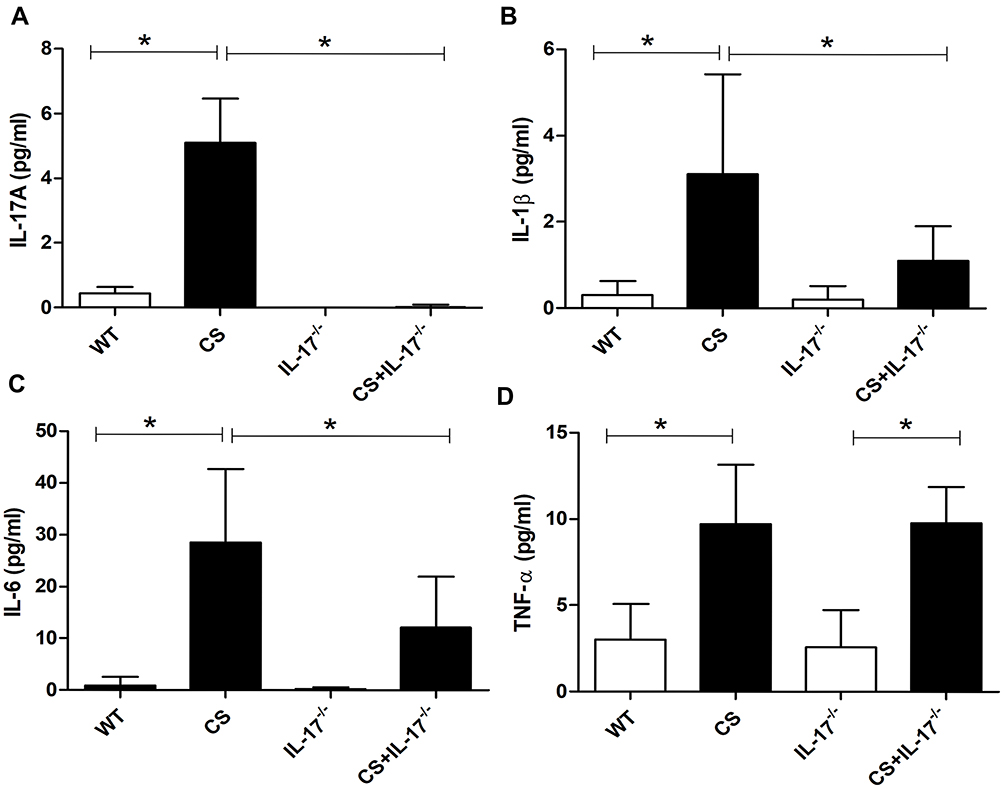 |
Figure 4 Concentration of IL-17A, IL-1β, IL-6, and TNF-α in serum. Levels of serum (A) IL-17A, (B) IL-1β, (C) IL-6 and (D) TNF-α were determined by Procarta cytokine profiling assay or enzyme-linked immunosorbent assay (ELISA). Data presented as mean ± SD (n=6 per group). * P<0.05. |
Discussion
While accumulating evidence has implicated IL-17 in the pathogenesis of both COPD30,31 and osteoclastogenesis associated with inflammatory diseases,19 its role in osteoporosis of COPD has not been established. In this study, for the first time to our knowledge, we demonstrated that IL-17A contributed to bone loss induced by long-term CS exposure in a well-established mouse model of COPD, probably through its proinflammatory effects and upregulation of RANKL expression in bone tissues.
Osteoporosis is a major comorbidity of COPD. In a systemic review by Graat-Verboom et al, the overall mean prevalence of osteoporosis in COPD was 35.1% (272 of 775 patients), although a large variation was observed because of differences in patients enrolled, methods and definitions used for osteoporosis.32 Osteoporosis may cause fractures, impair mobility, increase morbidity and mortality, and therefore carries a poorer outcome of COPD.5 Understanding of the pathogenesis of osteoporosis associated with COPD is imperative for prevention and targeted interventions. However, the mechanisms underlying exaggerated bone loss in COPD remain speculative.33 For example, it has been hypothesized that inflammation in the lung results in “overspill” into the circulation causing systemic inflammation which triggers osteoporosis.6 It is also interesting to note that in smokers, radiographic emphysema was a strong, independent predictor of low BMD, and this relationship suggested that there may be common mechanistic links between emphysema and osteoporosis.34 In a most recent study, Tsukamoto et al observed systemic bone loss in a mouse model of elastase-induced pulmonary emphysema, strongly suggesting a potential link between emphysema and osteoporosis.35 However, in CS-exposed mouse models, the effect of CS on bone was inconsistent, depending on the routes and duration of CS exposure. In a recent study, Sasaki et al found, in a nose-only CS exposure mouse model, that only prolonged (20–40weeks) exposure led to bone resorption.20 In the current study, we used a same exposure system, and found that 24 weeks CS exposure caused bone loss, as well as emphysema consistent with COPD. In agreement with previous studies,15 we confirmed the critical role of IL-17A in development of lung inflammation and alveolar destruction in this model, and more importantly, we made the novel finding that deficiency of IL-17A attenuated bone loss together with attenuated emphysema. These results indicated that IL-17A might be one of the common links between emphysema and osteoporosis associated with chronic cigarette smoking.
Bone is a dynamic organ undergoing continuous remodeling via bone resorption and formation. At the cellular level, the occurrence of osteoporosis mainly results from excessive bone resorption by osteoclasts.36 Although the activities of osteoclasts are reported to be regulated by several factors, including the RANKL-RANK-OPG pathway, estradiol and various cytokines, only RANKL is indispensable for osteoclast differentiation and activation.37–39 In our previous studies, we found increased RANKL levels in peripheral blood of COPD patients with osteoporosis.10,28 Thus, in this animal study, we further investigated osteoclast activities and RANKL expression in bone tissues, and as expected, our data demonstrated that long-term CS exposure was associated with significantly higher numbers of TRAP+ osteoclasts, and local upregulation of RANKL expression. Interestingly, we also observed that IL-17A deficiency reduced the numbers of osteoclasts as well as RANKL expression in bone tissues of CS-exposed mice, suggesting involvement of IL-17A/RANKL axis in regulating bone resorption in this CS-exposed model. Although the direct effects of IL-17A on differentiation of osteoclast precursors into osteoclasts remain controversial,40,41 there is evidence suggesting that IL-17A induces osteoclast formation via RANKL expression from other cells. For example, one in vitro study revealed that IL-17 induced differentiation of osteoclast progenitors into mature osteoclasts by stimulating RANKL expression in osteoblasts.17 In collagen-induced arthritis mice, overexpression of IL-17 in the knee joint promoted osteoclastic bone destruction by enhancing RANKL expression in the synovium.42 Taken together, in our animal model, long-term CS-induced boss loss was dependent, at least partially, on the IL-17A/RANKL pathway.
As our previous study in COPD patients revealed an association of lower BMD with higher serum levels of inflammatory cytokines including TNF-α, IL-1β, and IL-6,28 which are well-known inducers of osteoclasts leading to increased bone resorption,43 we further explored whether these systemic inflammatory mediators were dysregulated and associated with IL-17A in this model. The results showed that CS-exposed mice exhibited elevated proinflammatory cytokines, including IL-17A, TNF-α, IL-1β, and IL-6, in circulation. It is worth noting that mice deficient of IL-17A had lower IL-1β and IL-6 expression. Consistently, experimental studies by others had shown that IL-17A was able to promote the production of IL-1β, IL-6, TNF-α by macrophages,44 and in collagen-induced arthritis mice, IL-17A increased the synovial expression of IL-1β and IL-6.45 Our findings here suggest a regulatory role of IL-17A in production of proinflammatory cytokines, which are probably involved in CS-induced osteoclastogenesis in this model.
One limitation of our study is the lack of evidence to demonstrate whether bone loss is consequent on lung disease, as cigarette smoking was associated with lower BMD independently of COPD,34 and certain components in cigarettes could directly induce osteoclastic bone resorption.46 Although the critical role of IL-17 in CS-induced emphysema is clear in our mouse model, its independent role in bone loss cannot be established. Considering the complexity of pathogenesis of COPD and its comorbid conditions, a causal relationship is technically difficult to demonstrate. Deleting IL-17 in specific organ systems may be useful to reveal its role in certain comorbidities of COPD in future studies.
Conclusion
Taken together, our study revealed the critical role of IL-17A in long-term CS exposure-induced emphysema and bone loss in mice, supporting IL-17A as a common mechanistic link between CS-induced COPD and osteoporosis. These results provide novel insights into the mechanisms underlying COPD and its major comorbidities.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (No.81470239 and No.81770040). Jing Xiong and Jieyu Tian are co-first authors for this study.
Author Contributions
All authors contributed to data analysis, drafting and revising the article, gave final approval of the version to be published, and agreed to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Vogelmeier CF, Criner GJ, Martinez FJ, et al. Global strategy for the diagnosis, management, and prevention of chronic obstructive lung disease 2017 report. GOLD executive summary. Am J Respir Crit Care Med. 2017;195(5):557–582. doi:10.1164/rccm.201701-0218PP
2. Biskobing DM. COPD and osteoporosis. Chest. 2002;121(2):609–620. doi:10.1378/chest.121.2.609
3. Hillas G, Perlikos F, Tsiligianni I, Tzanakis N. Managing comorbidities in COPD. Int J Chron Obstruct Pulmon Dis. 2015;10:95–109. doi:10.2147/COPD.S54473
4. Smith MC, Wrobel JP. Epidemiology and clinical impact of major comorbidities in patients with COPD. Int J Chron Obstruct Pulmon Dis. 2014;9:871–888. doi:10.2147/COPD
5. Nussbaumer-Ochsner Y, Rabe KF. Systemic manifestations of COPD. Chest. 2011;139(1):165–173. doi:10.1378/chest.10-1252
6. Sinden NJ, Stockley RA, LEE J-H. Systemic inflammation and comorbidity in COPD: a result of ‘overspill’ of inflammatory mediators from the lungs? Review of the evidence. Thorax. 2010;65(10):930–936. doi:10.1136/thx.2009.130260
7. Barnes PJ. Inflammatory mechanisms in patients with chronic obstructive pulmonary disease. J Allergy Clin Immunol. 2016;138(1):16–27. doi:10.1016/j.jaci.2016.05.011
8. Kong YY, Feige U, Sarosi I, et al. Activated T cells regulate bone loss and joint destruction in adjuvant arthritis through osteoprotegerin ligand. Nature. 1999;402(6759):304–309. doi:10.1038/46303
9. Sato K, Suematsu A, Okamoto K, et al. Th17 functions as an osteoclastogenic helper T cell subset that links T cell activation and bone destruction. J Exp Med. 2006;203(12):2673–2682. doi:10.1084/jem.20061775
10. Chen Y, Bai P, Liu L, Han J, Zeng H, Sun Y. Increased RANKL expression in peripheral T cells is associated with decreased bone mineral density in patients with COPD. Int J Mol Med. 2016;38(2):585–593. doi:10.3892/ijmm.2016.2629
11. Duan MC, Zhang JQ, Liang Y, et al. Infiltration of IL-17-producing T cells and treg cells in a mouse model of smoke-induced emphysema. Inflammation. 2016;39(4):1334–1344. doi:10.1007/s10753-016-0365-8
12. Chen K, Pociask DA, McAleer JP, et al. IL-17RA is required for CCL2 expression, macrophage recruitment, and emphysema in response to cigarette smoke. PLoS One. 2011;6(5):e20333. doi:10.1371/journal.pone.0020333
13. Di Stefano A, Caramori G, Gnemmi I, et al. T helper type 17-related cytokine expression is increased in the bronchial mucosa of stable chronic obstructive pulmonary disease patients. Clin Exp Immunol. 2009;157(2):316–324. doi:10.1111/cei.2009.157.issue-2
14. Eustace A, Smyth L, Mitchell L, Williamson K, Plumb J, Singh D. Identification of cells expressing IL-17A and IL-17F in the lungs of patients with COPD. Chest. 2011;139(5):1089–1100. doi:10.1378/chest.10-0779
15. Chang Y, Al-Alwan L, Audusseau S, et al. Genetic deletion of IL-17A reduces cigarette smoke-induced inflammation and alveolar type II cell apoptosis. Am J Physiol Lung Cell Mol Physiol. 2014;306(2):L132–L143. doi:10.1152/ajplung.00111.2013
16. Shan M, Yuan X, Song LZ, et al. Cigarette smoke induction of osteopontin (SPP1) mediates T(H)17 inflammation in human and experimental emphysema. Sci Transl Med. 2012;4(117):117–119. doi:10.1126/scitranslmed.3003041
17. Kotake S, Udagawa N, Takahashi N, et al. IL-17 in synovial fluids from patients with rheumatoid arthritis is a potent stimulator of osteoclastogenesis. J Clin Invest. 1999;103(9):1345–1352. doi:10.1172/JCI5703
18. Milanova V, Ivanovska N, Dimitrova P. TLR2 elicits IL-17-mediated RANKL expression, IL-17, and OPG production in neutrophils from arthritic mice. Mediators Inflamm. 2014;2014:643406. doi:10.1155/2014/643406
19. Lee Y. The role of interleukin-17 in bone metabolism and inflammatory skeletal diseases. BMB Rep. 2013;46(10):479–483. doi:10.5483/BMBRep.2013.46.10.141
20. Sasaki M, Chubachi S, Kameyama N, et al. Effects of long-term cigarette smoke exposure on bone metabolism, structure, and quality in a mouse model of emphysema. PLoS One. 2018;13(1):e191611. doi:10.1371/journal.pone.0191611
21. Ko CH, Chan RL, Siu WS, et al. Deteriorating effect on bone metabolism and microstructure by passive cigarette smoking through dual actions on osteoblast and osteoclast. Calcif Tissue Int. 2015;96(5):389–400. doi:10.1007/s00223-015-9966-8
22. Zhou L, Le Y, Tian J, et al. Cigarette smoke-induced RANKL expression enhances MMP-9 production by alveolar macrophages. Int J Chron Obstruct Pulmon Dis. 2019;14:81–91. doi:10.2147/COPD.S190023
23. Bracke KR, D’Hulst AI, Maes T, et al. Cigarette smoke-induced pulmonary inflammation and emphysema are attenuated in CCR6-deficient mice. J Immunol. 2006;177(7):4350–4359. doi:10.4049/jimmunol.177.7.4350
24. Sreenivasan D, Tu PT, Dickinson M, et al. Computer modelling integrated with micro-CT and material testing provides additional insight to evaluate bone treatments: application to a beta-glycan derived whey protein mice model. Comput Biol Med. 2016;68:9–20. doi:10.1016/j.compbiomed.2015.10.017
25. Zhen G, Wen C, Jia X, et al. Inhibition of TGF-beta signaling in mesenchymal stem cells of subchondral bone attenuates osteoarthritis. Nat Med. 2013;19(6):704–712. doi:10.1038/nm.3143
26. Tanaka Y, Nakayamada S, Okada Y. Osteoblasts and osteoclasts in bone remodeling and inflammation. Curr Drug Targets Inflamm Allergy. 2005;4(3):325–328. doi:10.2174/1568010054022015
27. Tanaka S, Tanaka Y, Ishiguro N, Yamanaka H, Takeuchi T. RANKL: a therapeutic target for bone destruction in rheumatoid arthritis. Mod Rheumatol. 2018;28(1):9–16. doi:10.1080/14397595.2017.1369491
28. Bai P, Sun Y, Jin J, et al. Disturbance of the OPG/RANK/RANKL pathway and systemic inflammation in COPD patients with emphysema and osteoporosis. Respir Res. 2011;12:157. doi:10.1186/1465-9921-12-157
29. Ginaldi L, Di Benedetto MC, De Martinis M. Osteoporosis, inflammation and ageing. Immun Ageing. 2005;2:14. doi:10.1186/1742-4933-2-14
30. Roos AB, Sanden C, Mori M, Bjermer L, Stampfli MR, Erjefalt JS. IL-17A Is elevated in end-stage chronic obstructive pulmonary disease and contributes to cigarette smoke-induced lymphoid neogenesis. Am J Respir Crit Care Med. 2015;191(11):1232–1241.
31. Roos AB, Stampfli MR. Targeting interleukin-17 signalling in cigarette smoke-induced lung disease: mechanistic concepts and therapeutic opportunities. Pharmacol Therapeut. 2017;178:123–131. doi:10.1016/j.pharmthera.2017.04.001
32. Graat-Verboom L, Wouters EF, Smeenk FW, van den Borne BE, Lunde R, Spruit MA. Current status of research on osteoporosis in COPD: a systematic review. Eur Respir J. 2009;34(1):209–218. doi:10.1183/09031936.50130408
33. Lehouck A, Boonen S, Decramer M, Janssens W. COPD, bone metabolism, and osteoporosis. Chest. 2011;139(3):648–657. doi:10.1378/chest.10-1427
34. Bon J, Fuhrman CR, Weissfeld JL, et al. Radiographic emphysema predicts low bone mineral density in a tobacco-exposed cohort. Am J Respir Crit Care Med. 2011;183(7):885–890. doi:10.1164/rccm.201004-0666OC
35. Tsukamoto M, Mori T, Wang KY, et al. Systemic bone loss, impaired osteogenic activity and type I muscle fiber atrophy in mice with elastase-induced pulmonary emphysema: establishment of a COPD-related osteoporosis mouse model. Bone. 2019;120:114–124. doi:10.1016/j.bone.2018.10.017
36. Rachner TD, Khosla S, Hofbauer LC. Osteoporosis: now and the future. Lancet. 2011;377(9773):1276–1287. doi:10.1016/S0140-6736(10)62349-5
37. Yoon V, Maalouf NM, Sakhaee K. The effects of smoking on bone metabolism. Osteoporos Int. 2012;23(8):2081–2092. doi:10.1007/s00198-012-1940-y
38. Vega D, Maalouf NM, Sakhaee K. Clinical review #: the role of receptor activator of nuclear factor-kappaB (RANK)/RANK ligand/osteoprotegerin: clinical implications. J Clin Endocrinol Metab. 2007;92(12):4514–4521. doi:10.1210/jc.2007-0646
39. Leibbrandt A, Penninger JM. RANK/RANKL: regulators of immune responses and bone physiology. Ann N Y Acad Sci. 2008;1143:123–150. doi:10.1196/nyas.2008.1143.issue-1
40. Kitami S, Tanaka H, Kawato T, et al. IL-17A suppresses the expression of bone resorption-related proteinases and osteoclast differentiation via IL-17RA or IL-17RC receptors in RAW264.7 cells. Biochimie. 2010;92(4):398–404. doi:10.1016/j.biochi.2009.12.011
41. Adamopoulos IE, Chao CC, Geissler R, et al. Interleukin-17A upregulates receptor activator of NF-kappaB on osteoclast precursors. Arthritis Res Ther. 2010;12(1):R29. doi:10.1186/ar2936
42. Lubberts E, Koenders MI, Oppers-Walgreen B, et al. Treatment with a neutralizing anti-murine interleukin-17 antibody after the onset of collagen-induced arthritis reduces joint inflammation, cartilage destruction, and bone erosion. Arthritis Rheum. 2004;50(2):650–659. doi:10.1002/(ISSN)1529-0131
43. Braun T, Schett G. Pathways for bone loss in inflammatory disease. Curr Osteoporos Rep. 2012;10(2):101–108. doi:10.1007/s11914-012-0104-5
44. Geesala R, Dhoke NR, Das A. Cox-2 inhibition potentiates mouse bone marrow stem cell engraftment and differentiation-mediated wound repair. Cytotherapy. 2017;19(6):756–770. doi:10.1016/j.jcyt.2017.03.072
45. Lee JH, Cho ML, Kim JI, et al. Interleukin 17 (IL-17) increases the expression of Toll-like receptor-2, 4, and 9 by increasing IL-1beta and IL-6 production in autoimmune arthritis. J Rheumatol. 2009;36(4):684–692. doi:10.3899/jrheum.080169
46. Iqbal J, Sun L, Cao J, et al. Smoke carcinogens cause bone loss through the aryl hydrocarbon receptor and induction of Cyp1 enzymes. Proc Natl Acad Sci U S A. 2013;110(27):11115–11120. doi:10.1073/pnas.1220919110
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.