Back to Journals » Clinical, Cosmetic and Investigational Dermatology » Volume 12
Interferon Kappa Is Up-Regulated in Psoriasis and It Up-Regulates Psoriasis-Associated Cytokines in vivo
Authors Li Y, Song Y ![]() , Zhu L, Wang X, Yang B
, Zhu L, Wang X, Yang B ![]() , Lu P
, Lu P ![]() , Chen Q, Bin L
, Chen Q, Bin L ![]() , Deng L
, Deng L
Received 4 June 2019
Accepted for publication 6 November 2019
Published 29 November 2019 Volume 2019:12 Pages 865—873
DOI https://doi.org/10.2147/CCID.S218243
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Jeffrey Weinberg
Yuanyuan Li,1,* Yueqi Song,1,* Leqing Zhu,1,* Xiao Wang,1 Bin Yang,2 Ping Lu,2 Quan Chen,3 Lianghua Bin,4 Liehua Deng5
1Biomedical Translational Research Institute, The First Affiliated Hospital, Jinan University, Guangzhou, Guangdong Province, People’s Republic of China; 2Department of Dermatology, Dermatology Hospital of Southern Medical University, Guangzhou, Guangdong Province, People’s Republic of China; 3Division of Research Informatics Services, Department of Medicine, National Jewish Health, Denver, CO, USA; 4Department of Pediatrics, National Jewish Health, Denver, CO, USA; 5Department of Dermatology, The First Affiliated Hospital, Jinan University, Guangzhou, Guangdong Province, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Lianghua Bin
Department of Pediatrics, National Jewish Health, 1400 Jackson Street, Denver, CO 80206, USA
Tel +1-303-270-2055
Email [email protected]
Liehua Deng
Department of Dermatology, The First Affiliated Hospital, Jinan University, West Huangpu Dadao, No 605, Tianhe Qu, Guangzhou, Guangdong Province, People’s Republic of China
Tel +86-137-25356728
Email [email protected]
Purpose: There is increased type I interferon signature in psoriasis patients. Interferon-kappa (IFN-κ) is a member of type I interferon family that is constitutively expressed by keratinocytes. In this study, we investigate whether IFN-κ is involved in psoriasis etiology.
Patients and methods: Twenty healthy individuals, 20 psoriasis vulgaris patients and 10 atopic dermatitis (AD) were included for this study. Immunohistochemistry staining, normal human epidermal keratinocytes (NHEK) culture, Ca2Cl-induced differentiation, quantitative reverse transcription (qRT-PCR), ELISA and murine experiments were performed.
Results: We found IFN-κ protein expression was extremely low in the epidermis of normal skin, but it was significantly increased in the suprabasal layers of epidermal keratinocytes in psoriatic skin lesions. However, its expression in the skin lesions of AD was similar to normal skin. Additionally, IFN-κ protein was detected in sera from psoriasis patients, but not in sera from normal subjects and AD. We further investigated the regulation of IFNk gene expression in NHEK. We found that IFNk was significantly induced by types of nucleic acid pathogen recognition receptor (PRR) agonists in NHEK. While its expression was significantly induced by itself and IFN-γ, it was inhibited by type 2 immunity cytokines IL4 and IL13; other inflammatory cytokines including IL1 super-family members and IL17A did not alter its expression. Addition of recombinant IFN-κ did not affect keratinocytes differentiation. Using the murine experimental model, we demonstrated that subcutaneous administration of recombinant IFN-κ did not increase skin thickness, but significantly increased the transcription of TNFA and IL17A in mice skin.
Conclusion: Increased IFN-κ in psoriasis may be caused by injured cells-released nucleic acids, increased IFN-γ and self-activation. Its enhancement may contribute to the etiology of the disease by enhancing TNFA and IL17A gene expression.
Keywords: IFN-κ, psoriasis, TNFA, IL17A, atopic dermatitis
Introduction
Psoriasis is a chronic inflammatory skin disease affecting approximately 2% to 3% of the Caucasian population and 0.1% to 0.5% of Asians.1–5 Epidermal hyperplasia, angiogenesis, infiltration of T cells and neutrophils are the key pathological characteristics of the disease. Current understanding of the disease etiology has revealed that it is a complicated immune system disorder involving the dynamic interplay of innate and adaptive immunity.6 Cytokines including TNFα, IL-17, and IL-23 are critical signals to psoriasis pathogenesis, which are proved by the effective disease control when systemically use therapeutic blockers of these cytokines.7–9 In addition to TNFα, IL-17, and IL-23, overexpression of IFN-regulated genes has been seen in psoriasis skin, and type I IFN has been proposed as the potential therapeutic target for psoriasis.5,10–12 However, sources of type I IFNs in the skin of psoriasis are not completely clear.
IFN-κ is a member of type I IFN family. It consists of 207 amino acids including a 27 amino acid signal peptide and a series of cysteine conserved in type I IFN, and has about 30% homology to other family members.13,14 IFN-κ is expressed by limited cell sources, including keratinocytes.14 It activates the same set of genes as other family members by using receptors of IFNRA1/IFNRA2. Recently, IFN-κ has been found to associate with autoimmune disease such as cutaneous lupus erythematous: its expression is increased in keratinocytes from cutaneous lupus erythematous, and its function has been suggested to promote inflammatory responses.15–17
More than a decade ago, a research group investigated IFN-κ gene expression in skin chronic inflammatory diseases that only included 5 subjects of both AD and psoriasis.18 Based on results from these few cases, they concluded that down-regulation of IFN-κ in AD and psoriasis.18 Given the accumulated evidence of enhanced IFN signals in the epidermis of psoriasis, we think it is necessary to re-evaluate the role of IFN-κ in psoriasis, as it is a keratinocyte-specific type I IFN. In this study, we investigated IFN-κ gene expression in psoriasis skin lesions compared to normal skin and skin lesions of AD. We also investigated the regulation of IFNk gene by an array of pathogen recognition receptors’ agonists and cytokines associated with skin inflammation, as well as its effect on keratinocyte’s differentiation. Finally, we investigated the impact of IFN-κ to skin inflammation in vivo by using a murine subcutaneous injection model. Our results suggest that IFN-κ may contribute to psoriasis pathogenesis by promoting gene expression of TNFA and IL17A in skin tissue.
Materials and Methods
Human Subjects
Human skin biopsies and serum were obtained from normal human subjects, psoriasis patients and AD at the First Affiliated Hospital of Jinan University and Dermatology Hospital of Southern Medical University at Guangzhou, China. This study was approved by the Ethics Committee of the First Affiliated Hospital of Jinan University and Dermatology Hospital of Southern Medical University and conducted according to the Declaration of Helsinki. Written informed consents were obtained from all participating human subjects. Subjects included 20 healthy individuals with no history of other skin diseases (10 males and 10 females, mean age 35.9 ± 17.5), 20 patients with psoriasis vulgaris (12 males and 8 females, mean age 47.68 ± 18.35) (but only 17 psoriasis subjects and 17 normal subjects provided serum samples) and 10 patients with AD (5 females and 5 males, mean age 38.7 ± 10.5). None of the subjects had received therapy for at least 7 days before biopsies and blood donation.
Immunohistochemical Staining
Rabbit polyclonal anti-IFN-κ (ab168119) raised by full length of the protein (1–207aa) (used as 25 µg/mL) was purchased from Abcam (Cambridge, MA). After a serial of de-paraffinization, antigen retrieval, and inactivation of endogenous peroxidase with 0.3% H2O2, Paraffin-embedded skin sections (4 µm) were blocked with 5% BSA, then incubated with primary antibody at 4°C overnight. The following day, skin sections were washed by 1x PBS, incubated with secondary antibody conjugated HRP for 30 mins, and then developed with 3,3ʹ-diaminobenzidine at room temperature. Sections were counterstained with hematoxylin solution. Pictures were acquired using a Leica DM6000 microscope system. Optical density scores of IFN-κ positive signals (in brown color) per the entire epidermis area of each sample were determined using the Image-Pro PLUS program (Media Cybernetics, Inc., Shanghai, China), we designated the optical density scores as staining intensity of IFN-κ.
Enzyme-Linked Immunosorbent Assay (ELISA)
Human IFN-κ ELISA kit was purchased from CUSABIO (Wuhan, Hubei, China). The experiments were carried out as standardized ELISA protocol.
NHEK Cell Culture and Treatment
Primary human neonatal foreskin keratinocytes were purchased from Thermo Fisher Scientific and maintained in EpiLife Medium containing 0.06 mM CaCl2 and S7 supplemental reagent in 5% of CO2 and 37°C. For NHEK differentiation, cells were seeded in 24 well dishes at 2x105/well to form a confluent monolayer. The following day, the cells were subjected to differentiation by increasing CaCl2 to1.3mM in the culture media for 2 days, followed by treatment with cytokines or various PRR agonists for additional 24 hrs.
PRR Agonists and Cytokines
Pathogen recognition receptor (PRR) agonists lipopeptides Pam2csk4, FSC-1, Poly(I:C) HMW/naked, Poly(I:C) LMW/naked, HKLM, Poly(dA:dT)/LyoVec, LPS, Imiquimod, CpG (ODN2006), FLA, ss40, Poly(I:C)HMW/LyoVec and Poly(I:C)LMW/LyoVec were purchased from InvivoGen (San Diego, CA). Human recombinant cytokines of IL36α, IL36β, IL36γ, IL1α, IL1β, TNFα, IL18, IL33, IL17A, IL-4, IL13, IFN-κ and IFN-γ were purchased from R&D Biosystems.
Total RNA Extraction and qRT-PCR
Total RNA was extracted from NHEK cells and mice skin tissues using RNeasy mini kit according to the manufacture’s guideline (QIAGEN, MD). RNA was then reverse transcribed into cDNA using superScript® III reverse transcriptase from Invitrogen (Portland, OR) and analyzed by real-time RT-PCR using an ABI Prism 7000 sequence detector (Applied Biosystems, Foster City, CA). Primers and probes for human HPRT1 (Hs02800696_m1), IFNk (Hs00737883_m1), KRT1 (Hs00196158_m1), KRT10 (Hs00166289_m1), KRT5 (Hs00361185_m1), KRT14 (Hs00265033_m1), FLG (Hs00856927_g1), LOR (Hs1894962_s1), MKI67 (Hs04260396_g1), CCL20 (Hs00171125_m1), OAS 1(Hs00242943_m1), MX1 (Hs00182073_m1), mTNFA (Mm00443258_m1), mIL17A (Mm00439618_m1) and mHPRT (Mm03024075) were purchased from Applied Biosystems (Foster City, CA). Quantities of all target genes in test samples were normalized to the corresponding HPRT1 or mHPRT levels.
Animal Experiments
Balb/c mice were purchased from Guangdong experimental animal institute (Guangzhou, China). All animals were maintained under specific pathogen-free conditions and used for experiments at 8–11 weeks of age. The Institutional Animal Care and Use Committee at Jinan University approved the experiments, and they were performed according to the committee’s approved protocol. The rmIFN-κ (1µg in 20 µL of saline) or saline (20 µL) were administrated subcutaneously to the ear once the other day. The loci of the injection were marked. Ear thickness was measured daily using a caliper. Erythema and scaling of the ear skin were scored using an adapted version of the clinical Psoriasis Area and Severity Index. After 10 days, tissues from the injection sites were taken for RNA extraction and qRT-PCR measurement of IL17A and TNFA; or fixed in 10% PBS-buffered formalin.
Statistical Analysis
We used GraphPad prism software (version 5.03, San Diego, CA) and SAS (version 9.4) for statistical analyses. Comparisons of expression levels were performed using one-way ANOVA analyses or two-tailed unpaired t-tests as appropriate. We used Wilcoxon rank-sum test to analyze IFN-κ ELISA data because the values of IFN-κ in both normal controls and AD subjects were undetectable. Differences were considered significant at P<0.05. ns, not significant; *p<0.05; **p<0.01; ***p<0.001.
Results
IFN-κ Is Significantly Increased in the Epidermis of Psoriatic Skin Lesions and Blood Sera
Transcriptomic analyses revealed evidence that type I IFN signal pathway was significantly elevated in psoriatic skin lesions,5 we therefore investigated IFN-κ expression in skin lesions of psoriasis patients. AD is another common chronic skin inflammatory disease which is often associated with T helper 2 immunity.19 We included AD as controls to psoriasis in this study. As shown in Figure 1A, IFN-κ protein-positive staining was significantly increased in the suprabasal layers of KC in psoriasis skin lesions as compared to normal skin epidermis and AD skin lesions. Semi-quantitative analyses of IFN-κ positive signals showed significantly increased staining intensity scores in psoriatic skin lesions (n=20) than normal epidermis (n=20) as well as AD skin lesions (n=10) (Figure 1B). We also investigated the protein levels of other IFNs, include IFNα1, IFNβ and IFNλ1. None of these IFNs’ proteins were found in the epidermis of psoriasis skin lesions (data not shown).
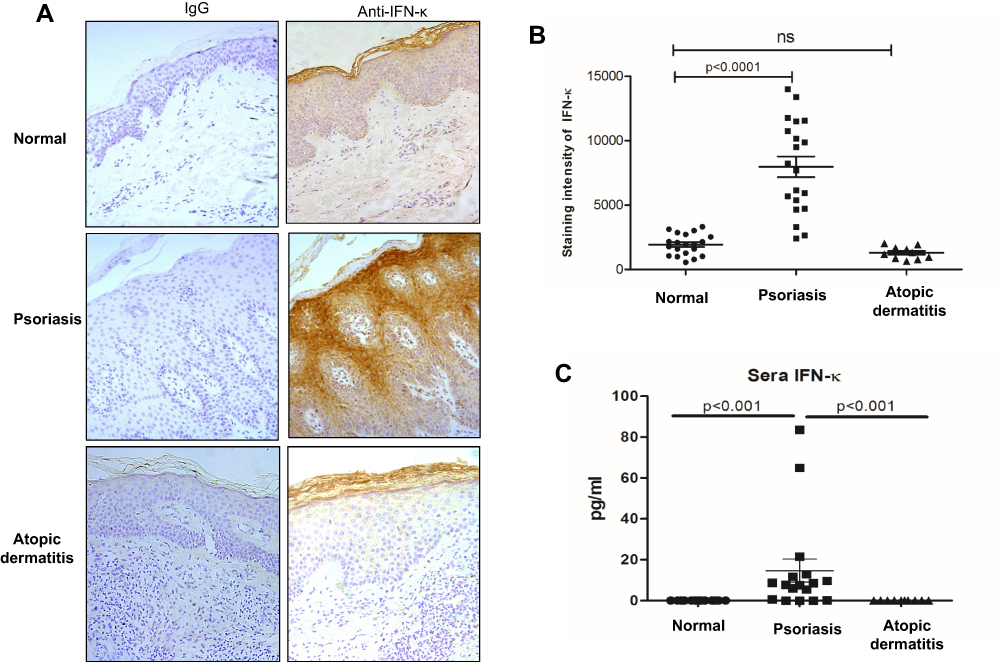 |
Figure 1 IFN-κ protein expression is increased in psoriasis lesions and sera. (A) Representative pictures of IFN-κ staining in normal skin, psoriasis lesions and AD lesions. IFN-κ protein is stained as brown color. Magnification x 200. (B) Semi-quantification analyses of IFN-κ protein staining intensity in normal skin (n=20), psoriasis lesions (n=20) and AD lesions (n=10). (C) IFN-κ in normal (n=17), psoriasis (n=17) and AD (n=10) sera. Statistical analyses were Wilcoxon rank-sum test for (C). |
We also investigated IFN-κ protein levels in blood serum from psoriasis patients, AD and normal subjects using ELISA method. As shown in Figure 1C, IFN-κ is undetectable in serum of both AD and normal subjects, but it was detected in the serum of the majority of psoriasis patients. Although the serum levels of IFN-κ were significantly increased in psoriasis patients compared to normal and AD subjects, the amounts of IFN-κ protein were fairly low in the blood of psoriasis patients.
IFN-κ Is Up-Regulated by Nucleic Acid Types of PRR Agonists and IFNs
Previously IFN-κ was reported to be up-regulated in proliferating NHEK cells by poly(I:C) stimulation.20 Since IFN-κ is predominately expresses in differentiated KC in psoriasis lesions (Figure 1A), we therefore investigated a panel of PRR’s agonists on their effects of inducing IFNk in differentiated NHEK. We set up 2-fold change as the threshold of significant increase, As shown in Figure 2A, poly(I:C)HMW/LyoVec, poly(I:C)LMW/LyoVec and poly(dA:dT)/LyoVec could significantly up-regulate IFNk mRNA expression in differentiated NHEK, while other PRR agonists did not significantly alter IFNk. Poly(I:C)HMW/naked and poly(I:C)LMW/naked activate Toll-like receptor (TLR) 3 signal pathway,21 transfected poly(I:C)HMW/LyoVec and poly(I:C)LMW/LyoVec activate RIG-1/MDA5 signaling pathways that recognize cytoplasmic RNA,22 and transfected poly(dA:dT)/LyoVec activates signaling pathways that recognize cytoplasmic DNA,23 and imiquimod and ss40 activate TLR7 and TLR8 signaling pathways,24 respectively. Our results suggest that IFNk gene expression is activated by cytoplasm RNA and DNA sensors-mediated signaling pathways in differentiated NHEK cells.
 |
Figure 2 IFNk gene expression is regulated by nucleic acids-type of PRR agonists and IFNs. NHEK cells were induced by 1.3 mM CaCl2 for 2 days, 13 indicated PRR agonists (A) and 13 different cytokines (B) were added to the cells and incubated for an additional 24 hrs. The cells were then harvested for qRT-PCR to evaluate IFNk gene expression. (C) Undifferentiated NHEK cells were stimulated with indicated reagents for 24 hrs, the cells were then harvested for qRT-PCR to evaluate IFNk gene expression. One of the three independent experiments is presented. Data presented as mean ± SEM. The two-tailed unpaired t-test was used to analyze Media treatment and indicated treatment. ns, not significant; *p<0.05; **p<0.01; ***p<0.001. |
Since there are increased inflammatory cytokines in psoriasis lesion, such as IL36, TNFα, IL17A, IFN-γ;25,26 we therefore investigated whether these cytokines regulate IFNk gene expression. As shown in Figure 2B, IFNk gene expression is significantly up-regulated by IFN-γ and itself, and significantly inhibited by type 2 cytokines IL-13 and IL-4. But other cytokines, include IL1 family members, IL17A and TNFα have no effect on IFNk gene expression in differentiated NHEK.
We also investigated whether the PRR agonists and cytokines could activate IFNk gene expression in undifferentiated NHEK. As shown in Figure 2C, poly(dA:dT)/LyoVec, poly(I:C) LMW/LyoVec, poly(I:C) HMW/LyoVec, IFN-γ and itself could significantly up-regulate IFNk gene expression in undifferentiated NHEK, while IL-4 inhibited its expression.
IFN-κ Increases Gene Expression of MX1 and OAS1, but Does Not Regulate Gene Expression of NHEK Differentiation Markers
In order to investigate whether increased IFN-κ in psoriasis contributes to the disease etiology, we examined whether IFN-κ up-regulates type I IFN targets as well as NHEK differentiation markers. We found the addition of recombinant IFN-κ to NHEK significantly up-regulated MX1 and OAS1 gene expression, the two representative genes induced by type 1 IFNs (Figure 3A and B). A panel of NHEK differentiation markers were also evaluated. These markers included basal layer markers of keratin 5 (KRT5) and keratin 14 (KRT14), spinous layer markers of keratin 1 (KRT1) and keratin 10 (KRT10), and granular layer markers of filaggrin (FLG) and loricrin (LOR).27 As shown in Figure 3C–H, addition of IFN-κ did not change gene expression of KRT5, KRT14, KRT1, KRT10, FLG and LOR. We also investigated MKI67, an indicator of cell proliferation. We found that differentiated NHEK had significantly decreased MKI67 gene expression compared to undifferentiated NHEK, addition of IFN-κ led to decreased gene expression of MKI67 in both undifferentiated and differentiated NHEK cells (Figure 3I), suggesting that IFN-κ inhibits the proliferation of keratinocytes. Addition of IFN-κ did not change CCL20 and TNFA gene expression in both undifferentiated and differentiated NHEK cells (Figure 3J and K).
 |
Figure 3 IFN-κ significantly up-regulates MX1 and OSA1, but does not regulate KC differentiation markers. 10ng/mL of human recombinant IFN-κ protein was added to undifferentiated (Undiff-) and differentiated (Diff-) NHEK for 24 hrs of incubation. MX1 (A), OAS1 (B), KRT5 (C), KRT14 (D), KRT1 (E), KRT10 (F), FLG (G), LOR (H), MKI67 (I), CCL20 (J) and TNFA (K). One of the three independent experiments is presented. Data presented as mean ± SEM. ns, not significant; ***p<0.001. |
Murine Recombinant IFN-κ Protein (mrIFN-κ) Significantly Increases TNFA and IL17A mRNA Levels in Skin in vivo
In order to test whether IFN-κ can induce skin inflammation in vivo, we subcutaneously administrated mrIFN-κ to Balb/c mice ear, marked the injection loci, and repeated injection every other day for 10 days. Injection of mrIFN-κ protein did not induce obvious skin inflammation lesions (Figure 4A), nor increase mice ear thickness (Figure 4B), however, injection of mrIFN-κ protein-induced increased mRNA levels of TNFA and IL17A in skin as compared to injection of saline (Figure 4C and D).
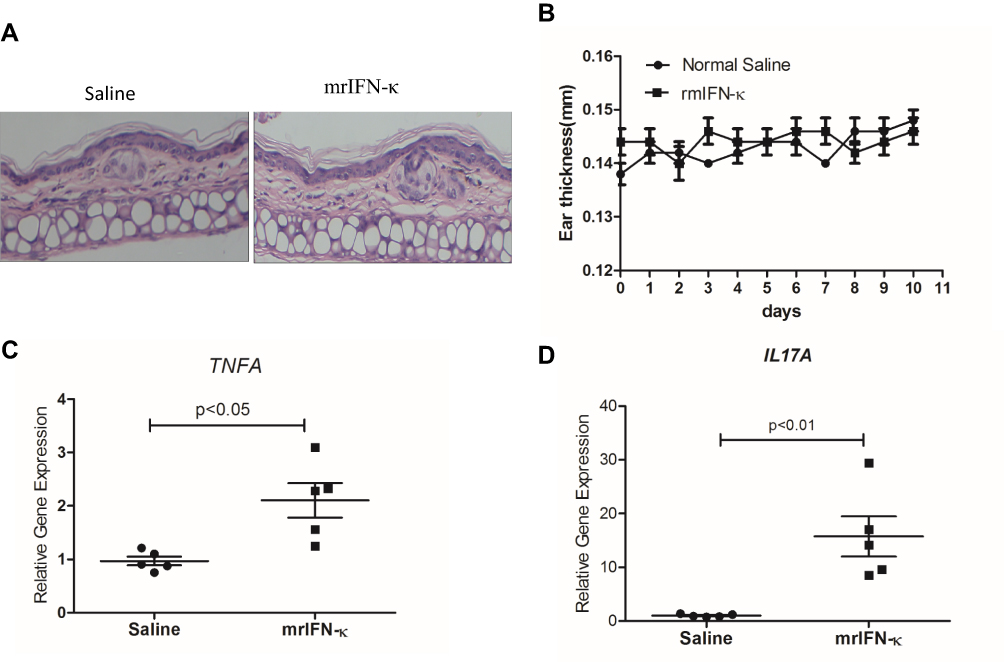 |
Figure 4 mrIFN-κ induces up-regulation of TNFA and IL17A transcription in murine skin. (A) Representative pictures of skin HE staining (magnification x 200) for each experimental groups. (B) Ear thickness measures (mm) for each experimental groups (n=5) at indicated days of treatment. (C and D) After 10 days of indicated treatment, mice were sacrificed and skin tissues of treatment loci were taken for RNA extraction and qRT-PCR evaluation of TNFA and IL17A transcripts (n=5). Data presented as mean ± SEM. |
Discussion
Type I IFN cytokines are normally produced in response to viral infections and play a critical role in host defense mechanisms.28,29 They induce the expression of anti-viral genes to inhibit viral replication in host cells and eradicate infected cells. In addition, they stimulate the maturation of dendritic cells and involve in activation of the adaptive immune system. To date, 13 IFN-α subtypes, IFN-β, -κ, -ε, -σ, -ω and -δ in type I IFN family are identified with differences in sources, receptor affinities and biological activities.28,29
Elevated type I IFN-inducible signals in skin lesions of psoriasis patients have been reported by different investigators.5,11,12,30 In addition, treatment with IFNs can induce psoriasis in non-psoriatic patients or exacerbate psoriasis.31–33 The direct association of psoriasis and type I IFN signals is further supported by a xenograft murine model of human psoriasis. In this model, nonlesional skin from psoriatic patients developed into psoriatic lesions when transplanted into immunocompromised mice, but inhibition of type I IFN signaling prevented the development of psoriasis disease.34 Although the evidence of type I IFN signals in psoriasis is solid, the sources of type I IFN cytokines in the skin of psoriatic lesions are not completely clear. Previous studies attempted to find the sources of type I IFNs in psoriasis skin, only weak IFN-α mRNA were detected by using in situ hybridization method, no IFN-α protein and IFN-β mRNA/protein were detected.11,35 However, recently, Zheng et al showed IFN-β positive staining in freshly prepared frozen sections of skin biopsies from psoriasis lesions, but did not specify how many subjects they investigated.36 Yao et al found IFN-κ mRNA were increased in psoriasis skin lesional tissues, but could not tell which cell type produced it.5 In this study, we demonstrated that IFN-κ protein was significantly increased in the epidermis of psoriasis lesions, and it is also increased in a subset of psoriasis patients’ peripheral blood serum (Figure 1). We found IFN-κ in psoriatic lesions was located in suprabasal layers of KC in the epidermis, which matched with protein expression of type I IFNs-induced STAT1 and ISG15 shown by the study of Yao et al.5 Our results suggest that IFN-κ of epidermal keratinocytes is a source for increased type I IFN induced signatures in psoriasis. We were not able to detect other types of IFNs in our samples, the discrepancy may be caused by different approaches of preparing staining samples. For example, we used paraffin-embedded tissue sections, while Zhang et al used OTC-embedded fresh tissue sections.36
Since IFN-κ is increased in the suprabasal layers of KC in psoriasis lesions, we therefore investigated the regulation of IFNk gene expression in differentiated human normal KC. We found nucleic acid types of PRR agonists, especially the transfected double-stranded RNA and DNA agonists could significantly up-regulate IFNk (Figure 2A). In psoriasis lesions, rapid turnover of KC and dead neutrophils would provide abundant sources of double-stranded RNA and DNA fragments, these could be the nature triggers for induction of IFNk. We also found IFNk mRNA could be up-regulated significantly by type II IFN-γ, suggesting infiltrated T help 1 cells are another possible trigger for increased IFN-κ in psoriasis lesions.37 Additionally, IFN-κ itself can positively reinforce its expression in KC of psoriasis lesions. In the undifferentiated keratinocytes, IFNk gene expression could also be significantly up-regulated by the same set of stimulates as its induction in differentiated keratinocytes. Stimulation of NHEK with IFN-κ in vitro could significantly up-regulate MX1 and OAS1 in both undifferentiated and differentiated KC, the two type I IFN-inducible genes, which are often over-expressed in psoriasis lesions.5,30 However, gene expression of KRT5, KRT15, KRT1, KRT10, FLG and LOR in differentiated NHEK was not altered by IFN-κ stimulation. We did see that the addition of IFN-κ inhibits the gene expression of the proliferation marker MKI67, suggesting that IFN-κ has proliferation inhibition function as other IFNs.
Although subcutaneous injection of recombinant IFN-κ did not induce psoriatic skin lesions, we observed the up-regulation of TNFA and IL17A mRNA in the injection sites of mice skin biopsies (Figure 4C and D). The sources of IL17A are Th17 cells and ILC3 cells.38 This result suggests IFN-κ may promote the development of Th17. Both TNFA and IL17A are important for psoriasis pathogenesis, the reason that subcutaneous injection of recombinant IFN-κ did not induce psoriatic lesions may be caused by insufficient dose and bioactivity of recombinant IFN-κ. As the addition of IFN-κ directly to the cultured human NHEK did not alter gene expression of inflammatory cytokines such as CCL20 and TNFA, indicating that increased TNFA mRNA in mice model was most likely caused by systemic effect triggered by mrIFN-κ.
One limitation of this study is that the sample size of human subjects is too small, that led to not having enough power for the correlation analysis between IFN-κ and psoriasis severity. Another limitation of this study is lack of a transgenic mice model. Transgenic mice with epidermis specific over-expression of IFN-κ would elucidate better the role of IFN-κ in psoriasis pathogenesis in vivo.
Currently, there is no application of using antagonistic strategies of IFNk to treat psoriasis patients. Our study opens a new avenue to investigate whether IFNk is a compelling target for psoriasis treatment.
Conclusion
IFN-κ is significantly increased in the epidermis of psoriasis skin lesions, it is a source for increased type I IFN gene signatures in psoriasis. The increased IFN-κ in psoriasis may be caused by dead cells-released double-stranded RNA and DNA fragments, increased IFN-γ and self-promotion. IFN-κ may up-regulate TNFA and IL17A to promote inflammation responses in psoriasis. Anti-IFN-κ may be a potential target for psoriasis treatment.
Abbreviations
AD, atopic dermatitis; ELISA, enzyme-linked immunosorbent assay; FLG, filaggrin; HPRT1, hypoxanthine phosphoribosyltransferase 1; IFN, interferon; KC, keratinocyte; KRT1, keratin 1; KRT10, keratin 10; LOR, loricrin; mHPRT, Mus musculus hypoxanthine phosphoribosyltransferase; MX1, MX dynamin-like GTPase 1; NHEK, normal human epidermal keratinocytes; PRR, pathogen recognition receptor; qRT-PCR, quantitative reverse transcription-polymerase chain reaction; OAS1, 2ʹ,5ʹ-oligoadenylate synthetase 1.
Acknowledgment
This work is supported by National Natural Science Foundation of China (No. 81371716).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Boehncke WH, Schon MP. Psoriasis. Lancet. 2015;386(9997):983–994. doi:10.1016/S0140-6736(14)61909-7
2. Kaufman BP, Alexis AF. Psoriasis in skin of color: insights into the epidemiology, clinical presentation, genetics, quality-of-life impact, and treatment of psoriasis in non-white racial/ethnic groups. Am J Clin Dermatol. 2018;19(3):405–423. doi:10.1007/s40257-017-0332-7
3. Lebwohl M. Psoriasis. Lancet. 2003;361(9364):1197–1204. doi:10.1016/S0140-6736(03)12954-6
4. Parisi R, Symmons DP, Griffiths CE, et al. Global epidemiology of psoriasis: a systematic review of incidence and prevalence. J Invest Dermatol. 2013;133(2):377–385. doi:10.1038/jid.2012.339
5. Yao Y, Richman L, Morehouse C, et al. Type I interferon: potential therapeutic target for psoriasis? PLoS One. 2008;3(7):e2737. doi:10.1371/journal.pone.0002737
6. Georgescu SR, Tampa M, Caruntu C, et al. Advances in understanding the immunological pathways in psoriasis. Int J Mol Sci. 2019;20:3. doi:10.3390/ijms20030739
7. Dong J, Goldenberg G. New biologics in psoriasis: an update on IL-23 and IL-17 inhibitors. Cutis. 2017;99(2):123–127.
8. Leonardi CL, Powers JL, Matheson RT, et al. Etanercept as monotherapy in patients with psoriasis. N Engl J Med. 2003;349(21):2014–2022. doi:10.1056/NEJMoa030409
9. Menter A, Strober BE, Kaplan DH, et al. Joint AAD-NPF guidelines of care for the management and treatment of psoriasis with biologics. J Am Acad Dermatol. 2019;80(4):1029–1072. doi:10.1016/j.jaad.2018.11.057
10. Baldwin HM, Pallas K, King V, et al. Microarray analyses demonstrate the involvement of type I interferons in psoriasiform pathology development in D6-deficient mice. J Biol Chem. 2013;288(51):36473–36483. doi:10.1074/jbc.M113.491563
11. Schmid P, Itin P, Cox D, McMaster GK, Horisberger MA. The type I interferon system is locally activated in psoriatic lesions. J Interferon Res. 1994;14(5):229–234. doi:10.1089/jir.1994.14.229
12. van der Fits L, van der Wel LI, Laman JD, Prens EP, Verschuren MC. In psoriasis lesional skin the type I interferon signaling pathway is activated, whereas interferon-alpha sensitivity is unaltered. J Invest Dermatol. 2004;122(1):51–60. doi:10.1046/j.0022-202X.2003.22113.x
13. LaFleur DW, Nardelli B, Tsareva T, et al. Interferon-kappa, a novel type I interferon expressed in human keratinocytes. J Biol Chem. 2001;276(43):39765–39771. doi:10.1074/jbc.M102502200
14. Nardelli B, Zaritskaya L, Semenuk M, et al. Regulatory effect of IFN-kappa, a novel type I IFN, on cytokine production by cells of the innate immune system. J Immunol. 2002;169(9):4822–4830. doi:10.4049/jimmunol.169.9.4822
15. Harley IT, Niewold TB, Stormont RM, et al. The role of genetic variation near interferon-kappa in systemic lupus erythematosus. J Biomed Biotechnol. 2010;2010.
16. Sarkar MK, Hile GA, Tsoi LC, et al. Photosensitivity and type I IFN responses in cutaneous lupus are driven by epidermal-derived interferon kappa. Ann Rheum Dis. 2018;77(11):1653–1664. doi:10.1136/annrheumdis-2018-213197
17. Stannard JN, Reed TJ, Myers E, et al. Lupus skin is primed for IL-6 inflammatory responses through a keratinocyte-mediated autocrine type I interferon loop. J Invest Dermatol. 2017;137(1):115–122. doi:10.1016/j.jid.2016.09.008
18. Scarponi C, Nardelli B, Lafleur DW, et al. Analysis of IFN-kappa expression in pathologic skin conditions: downregulation in psoriasis and atopic dermatitis. J Interferon Cytokine Res. 2006;26(3):133–140. doi:10.1089/jir.2006.26.133
19. Czarnowicki T, He H, Krueger JG, Guttman-Yassky E. Atopic dermatitis endotypes and implications for targeted therapeutics. J Allergy Clin Immunol. 2019;143(1):1–11. doi:10.1016/j.jaci.2018.10.032
20. Reiser J, Hurst J, Voges M, et al. High-risk human papillomaviruses repress constitutive kappa interferon transcription via E6 to prevent pathogen recognition receptor and antiviral-gene expression. J Virol. 2011;85(21):11372–11380. doi:10.1128/JVI.05279-11
21. Alexopoulou L, Holt AC, Medzhitov R, Flavell RA. Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature. 2001;413(6857):732–738. doi:10.1038/35099560
22. Takeuchi O, Akira S. MDA5/RIG-I and virus recognition. Curr Opin Immunol. 2008;20(1):17–22. doi:10.1016/j.coi.2008.01.002
23. Zhong B, Zhang L, Lei C, et al. The ubiquitin ligase RNF5 regulates antiviral responses by mediating degradation of the adaptor protein MITA. Immunity. 2009;30(3):397–407. doi:10.1016/j.immuni.2009.01.008
24. Saha B, Kodys K, Adejumo A, Szabo G. Circulating and exosome-packaged hepatitis C single-stranded RNA induce monocyte differentiation via TLR7/8 to polarized macrophages and fibrocytes. J Immunol. 2017;198(5):1974–1984. doi:10.4049/jimmunol.1600797
25. Hawkes JE, Chan TC, Krueger JG. Psoriasis pathogenesis and the development of novel targeted immune therapies. J Allergy Clin Immunol. 2017;140(3):645–653. doi:10.1016/j.jaci.2017.07.004
26. Keermann M, Koks S, Reimann E, Prans E, Abram K, Kingo K. Transcriptional landscape of psoriasis identifies the involvement of IL36 and IL36RN. BMC Genomics. 2015;16:322. doi:10.1186/s12864-015-1508-2
27. Candi E, Schmidt R, Melino G. The cornified envelope: a model of cell death in the skin. Nat Rev Mol Cell Biol. 2005;6(4):328–340. doi:10.1038/nrm1619
28. McNab F, Mayer-Barber K, Sher A, Wack A, O’Garra A. Type I interferons in infectious disease. Nat Rev Immunol. 2015;15(2):87–103. doi:10.1038/nri3787
29. Schreiber G. The molecular basis for differential type I interferon signaling. J Biol Chem. 2017;292(18):7285–7294. doi:10.1074/jbc.R116.774562
30. Ruano J, Suarez-Farinas M, Shemer A, Oliva M, Guttman-Yassky E, Krueger JG. Molecular and cellular profiling of scalp psoriasis reveals differences and similarities compared to skin psoriasis. PLoS One. 2016;11(2):e0148450. doi:10.1371/journal.pone.0148450
31. Afshar M, Martinez AD, Gallo RL, Hata TR. Induction and exacerbation of psoriasis with interferon-alpha therapy for hepatitis C: a review and analysis of 36 cases. J Eur Acad Dermatol Venereol. 2013;27(6):771–778. doi:10.1111/jdv.2013.27.issue-6
32. Tas F, Atsu N. Exacerbation of psoriasis induced by interferon-alpha treatment for melanoma. Cutan Ocul Toxicol. 2016;35(1):83–84. doi:10.3109/15569527.2015.1020545
33. Wu MC, Lee JY. Generalized flare of pustular psoriasis induced by PEGylated interferon-alpha2b therapy for chronic hepatitis C. Australas J Dermatol. 2012;53(4):e69–72. doi:10.1111/j.1440-0960.2011.00767.x
34. Nestle FO, Conrad C, Tun-Kyi A, et al. Plasmacytoid predendritic cells initiate psoriasis through interferon-alpha production. J Exp Med. 2005;202(1):135–143. doi:10.1084/jem.20050500
35. Livden JK, Nilsen R, Bjerke JR, Matre R. In situ localization of interferons in psoriatic lesions. Arch Dermatol Res. 1989;281(6):392–397. doi:10.1007/BF00455323
36. Zhang LJ, Sen GL, Ward NL, et al. Antimicrobial peptide LL37 and MAVS signaling drive interferon-beta production by epidermal keratinocytes during skin injury. Immunity. 2016;45(1):119–130. doi:10.1016/j.immuni.2016.06.021
37. Furiati SC, Catarino JS, Silva MV, et al. Th1, Th17, and treg responses are differently modulated by TNF-alpha inhibitors and methotrexate in psoriasis patients. Sci Rep. 2019;9(1):7526. doi:10.1038/s41598-019-43899-9
38. Keijsers RR, Joosten I, van Erp PE, Koenen HJ, van de Kerkhof PC. Cellular sources of IL-17 in psoriasis: a paradigm shift? Exp Dermatol. 2014;23(11):799–803. doi:10.1111/exd.12487
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.