Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 10 » Issue 1
Inter-alpha-trypsin inhibitor heavy chain 4: a novel biomarker for environmental exposure to particulate air pollution in patients with chronic obstructive pulmonary disease
Authors Lee K, Feng P, Ho S, Chuang K, Chen T, Su C, Liu W ![]() , Chuang H
, Chuang H
Received 26 January 2015
Accepted for publication 10 March 2015
Published 24 April 2015 Volume 2015:10(1) Pages 831—841
DOI https://doi.org/10.2147/COPD.S81611
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Richard Russell
Kang-Yun Lee,1–3 Po-Hao Feng,1,2 Shu-Chuan Ho,4 Kai-Jen Chuang,5,6 Tzu-Tao Chen,2,3 Chien-Ling Su,2,4 Wen-Te Liu,2,4 Hsiao-Chi Chuang2,4
1Department of Internal Medicine, School of Medicine, College of Medicine, Taipei Medical University, 2Division of Pulmonary Medicine, Department of Internal Medicine, Shuang Ho Hospital, Taipei Medical University, 3Graduate Institute of Clinical Medicine, College of Medicine, Taipei Medical University, 4School of Respiratory Therapy, College of Medicine, Taipei Medical University, 5Department of Public Health, School of Medicine, College of Medicine, Taipei Medical University, 6School of Public Health, College of Public Health and Nutrition, Taipei Medical University, Taipei, Taiwan
Abstract: Chronic obstructive pulmonary disease (COPD) is a chronic inflammatory disease that is correlated with environmental stress. Particulate matter ≤10 µm (PM10) is considered to be a risk factor for COPD development; however, the effects of PM10 on the protein levels in COPD remain unclear. Fifty subjects with COPD and 15 healthy controls were recruited. Gene ontology analysis of differentially expressed proteins identified immune system process and binding as the most important biological process and molecular function, respectively, in the responses of PM10-exposed patients with COPD. Biomarkers for PM10 in COPD were identified and compared with the same in healthy controls and included proteoglycan 4 (PRG4), inter-alpha-trypsin inhibitor heavy chain 4 (ITIH4), and apolipoprotein F (APOF). PRG4 and ITIH4 were associated with a past 3-year PM10 exposure level. The receiver operating characteristic curve analysis showed that ITIH4 is a sensitive and specific biomarker for PM10 exposure (area under the curve [AUC] =0.690, P=0.015) compared with PRG4 (AUC =0.636, P=0.083), APOF (AUC =0.523, P=0.766), 8-isoprostane (AUC =0.563, P=0.405), and C-reactive protein (CRP; AUC =0.634, P=0.086). ITIH4 levels were correlated with CRP (r=0.353, P=0.005), suggesting that ITIH4 may be involved in an inflammatory mechanism. In summary, serum ITIH4 may be a PM10-specific biomarker in COPD and may be related to inflammation.
Keywords: air pollution, apolipoprotein F, C-reactive protein, inflammation, proteoglycan 4, oxidative stress
Introduction
Epidemiological and toxicological research has identified adverse human health effects due to particulate air pollution exposure. Particulate matter ≤10 μm in aerodynamic diameter (PM10) is associated with increased hospital admissions and mortality due to pulmonary and cardiovascular diseases.1 Air pollutants participate in the development of pulmonary disease by interfering with both nonspecific and specific lung defenses.2 Chronic obstructive pulmonary disease (COPD), for instance, is recognized as an environmentally related pulmonary disease that is associated with exposure to air pollution.3 Schikowski et al4 in 2014, provided evidence that the chronic effects of air pollution affect the prevalence and incidence of COPD among adults. Kumar et al5 (2013) noted the association between acute COPD exacerbation and particulate air pollution, finding that the risk of acute COPD exacerbation increased by 2.3% following a unit increase in PM2.5 (≤2.5 μm). However, the effects of PM10 on protein expression in COPD are not well understood.
A protein biomarker is capable of objectively and quantitatively acting as an indicator to identify normal or pathological processes and the corresponding exposure to environmental stresses.6 For example, 8-isoprostane and C-reactive protein (CRP), the biomarkers for oxidative stress and inflammation, respectively, have been widely used to evaluate the cardiopulmonary toxicity of PM10.7,8 However, they are nonspecific PM10 biomarkers for oxidative inflammatory reactions. Thus, an increasing number of studies have attempted to identify more specific biomarkers for the assessment of specific pollutant exposures. For example, urinary p-nitrophenol has been reported to be a specific biomarker for occupational exposure to methyl parathion,9 and DNA modifications, such as F2RL3 methylation, have been suggested as biomarkers for tobacco exposure.10 Ideally, a quality biomarker can be used for treatment and intervention decisions. However, biomarker measurement may also vary according to exposure sources and susceptible populations.
A useful biomarker not only differentiates stable disease from exacerbations but also predicts the severity of pollutant exposure. The objective of this study was to identify biomarkers for PM10 exposure in patients with COPD. This study identified and assessed the use of serum biomarkers in the evaluation of PM10 compared with the existing biomarkers of inflammation, namely 8-isoprostane and CRP. We used a proteomics approach to comprehensively screen protein profiles in healthy subjects and patients with COPD exposed to low and high levels of PM10 over the past 3 years. The identified protein candidates were determined in serum samples obtained from 50 patients with COPD and 15 subjects without COPD. The specific and sensitive values of 8-isoprostane, CRP, and identified biomarkers were compared.
Materials and methods
Study population
We recruited 50 patients with COPD and 15 subjects without COPD (nonsmokers and smokers) in a medical center in Taiwan between January 2013 and August 2014. All subjects were between 40 years and 80 years of age at the time of inclusion. Patients identified with current cancer or active inflammatory disease or who had an exacerbation during the 4 weeks before the study were excluded. All subjects with COPD were informed of diagnosis and exhibited postbronchodilation forced expiratory volume in 1 second (FEV1)/forced vital capacity (FVC) ratios of <70%. Subjects without COPD exhibited an FEV1/FVC ratio of ≥75% and FEV1 ≥80% of predicted value. The Ethics Committees of Taipei Medical University-Joint Institutional Review Board approved the study protocol (number 201310027). The Chinese Clinical Trial Register number is ChiCTR-OCC-13004025. All subjects received written and oral information before inclusion and provided informed consent.
Data collection
Before recruitment into the study, a physical examination was performed, as was a clinical interview regarding smoking habits, comorbidities, and medicinal use. All subjects with COPD continued with a stable regimen of medications throughout the study. Lung function parameters were assessed at the time of recruitment using a Vitalograph Spirotac V™. Postbronchodilation measurements for FEV1 and FVC were taken, and FEV1/FVC was calculated. Serum samples were obtained and stored at −80°C for analysis.
PM10 exposure
PM10 data were obtained from 25 monitoring stations (operated by the Taiwan Environmental Protection Administration, Taiwan) throughout northern Taiwan. The daily concentrations of PM10 were collected, corresponding to subject exposure. If a subject resided within 10 km of multiple monitoring stations, the weighted average was used to calculate the PM10 levels. The PM10 data were used to estimate the 1-year, 2-year, and 3-year effects of PM10 on COPD.
Protein digestion
To determine the effects of PM10 on protein expression in COPD, the five subjects with the highest average 3-year PM10 values were classified as the High Ambient Particles (HAP) group (exposure range =63.2–64.5 μg/m3), and the five subjects with the lowest average 3-year PM10 served as the Low Ambient Particles (LAP) group (exposure range =35.4–43.9 μg/m3). Serum samples from healthy controls, HAP, and LAP were collected and pooled together following protein digestion.11 After protein depletion to remove albumin and immunoglobulin, the serum samples were diluted and denatured with 8 M urea/10 mM ammonium bicarbonate for 1 hour; they were then alkylated with 50 mM iodoacetamide for 30 minutes, tryptically digested in a solution of 50 mM ammonium bicarbonate at 37°C for 18 hours, and desalted on C18 columns.
Mass spectrometry and protein identification
Tryptic samples were analyzed using a mass spectrometer (LTQ Orbitrap XL; Thermo Fisher Scientific, Bremen, Germany) coupled with an Ultimate 3000 RSLC system (Dionex, Sunnyvale, CA, USA) using previously documented protocols.12 Briefly, peptides were separated using a 150 mm length ×100 mm inner diameter self-pulled spray tip (5 μm tip opening) and packed with C18 material (ReproSil; 3 μm; Dr Maisch, Ammerbuch, Germany). The linear gradient was from 5% to 10% of mobile phase B (mobile phase A: 0.5% acetic acid, mobile phase B: 80% acetonitrile/0.5% acetic acid) for 5 minutes, 10% to 40% of mobile phase B for 30 minutes, and 40% to 99% of mobile phase B for 5 minutes, with a total of 75 minutes’ separation time. Five ranges of full mass spectrometry (MS) scans (m/z 350–2,000, m/z 350–600, m/z 600–800, m/z 800–1,200, and m/z 1,200–2,000) were followed by the selection of the ten most intense ions for MS/MS scans. Data were analyzed using the Proteome Discoverer 1.4 program to perform a Mascot database search of the National Center for Biotechnology Information and UniProt databases both for prediction as well as a literature search. The maximum mass tolerance was set to 10 ppm for precursor ions and 0.05 Da for fragment ions. Variable modifications for deamidation (NQ), oxidation (M), and fixed modification for carbamidomethyl (C) were selected for the search parameters.
Protein functional analyses
Unique proteins expressed in the HAP and LAP groups were analyzed using the Protein ANalysis THrough Evolutionary Relationships (PANTHER) classification system.13,14 The UniProt accession database was used to relate the expressed proteins identified in this study to related biological processes and molecular functions.
Enzyme-linked immunosorbent assay
Enzyme-linked immunosorbent assays (ELISAs) were used to determine the serum levels of 8-isoprostane (Cayman, Ann Arbor, MI, USA), CRP (CRP; R&D Systems, Minneapolis, MN, USA), proteoglycan 4 (PRG4), inter-alpha-trypsin inhibitor heavy chain 4 (ITIH4), and apolipoprotein F (APOF; MyBioSource, San Diego, CA, USA) according to the manufacturer’s instructions.
Statistical analysis
All ELISA experiments were performed at least three times. Data are expressed as the mean ± standard deviation. For multiple comparison analysis, one-way analysis of variance with Tukey’s post hoc was used. Spearman’s rank correlation coefficient was used to examine the correlation of quintiles of PM10 with PRG4, ITIH4, and APOF, as well as the correlation of 8-isoprostane and CRP with PRG4, ITIH4, and APOF. Receiver operating characteristic (ROC) curve analysis was used to determine the specificity and sensitivity of PRG4, ITIH4, and APOF to PM10 (≥50 μg/m3 and <50 μg/m3) in COPD. Statistical analyses were performed using GraphPad Version 5 (La Jolla, CA, USA) for Windows. The level of significance was set to P<0.05.
Results
Characteristics of study subjects
Fifty subjects with COPD, ten healthy controls, and five healthy smoking volunteers were enrolled. Detailed baseline characteristics of the 65 subjects in the study population are presented in Table 1. The ages of healthy controls, healthy smokers, and subjects with COPD were 67.8±6.2 years, 59.8±14.6 years, and 70.7±8.4 years, respectively. The majority of the study population consisted of men with body mass indexes that ranged between 22.5 kg/m2 and 24.8 kg/m2. The current smoking percentage among the subjects with COPD was 9.1%. The FEV1 percentages of predicted values for the healthy controls, healthy smokers, and subjects with COPD were 88.0%±11.5%, 85.5%±14.9%, and 42.6%±12.4%, respectively. The levels of 8-isoprostane and CRP for subjects with COPD were 13.4±3.0 pg/mL and 5.6±6.4 mg/L, respectively. The average PM10 concentrations for the study subjects were estimated, as shown in Table 2, and were 46.0±8.4 μg/m3 for the 1-year average, 48.5±7.2 μg/m3 for the 2-year average, and 50.8±7.0 μg/m3 for the 3-year average.
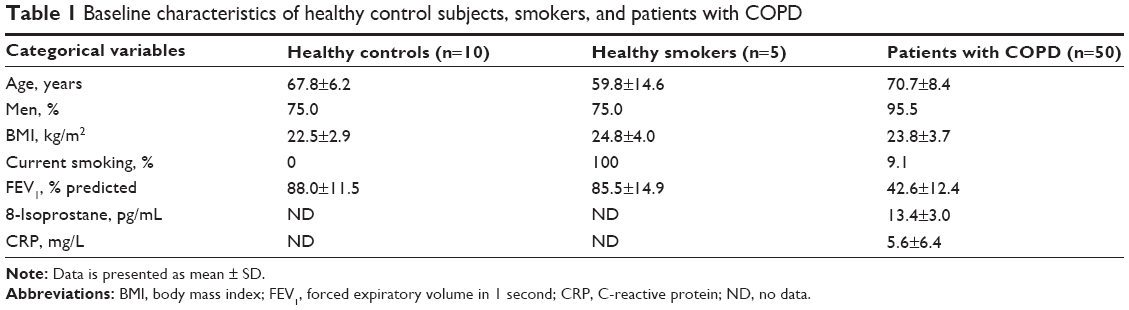 | Table 1 Baseline characteristics of healthy control subjects, smokers, and patients with COPD |
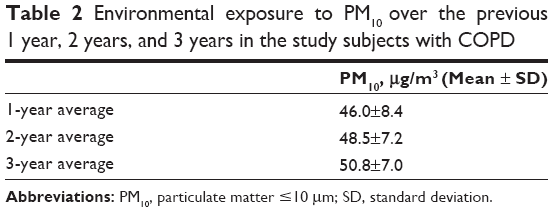 | Table 2 Environmental exposure to PM10 over the previous 1 year, 2 years, and 3 years in the study subjects with COPD |
Selection of potential biomarkers in patients with COPD
Overlaps in protein profiles between healthy controls and the LAP- and HAP-exposed subjects with COPD were compared to investigate protein expression in COPD (Figure 1A). A total of 179 proteins were identified in the healthy controls, and 178 and 187 proteins were identified in the LAP- and HAP-exposed subjects with COPD, respectively (Table S1). A total of 14 and 23 proteins were unique to LAP- and HAP-exposed subjects with COPD, respectively. Six serum proteins were common to both LAP- and HAP-exposed subjects with COPD: PRG4, ITIH4, APOF, Ig kappa chain V-I region DEE, type I cytoskeletal 16 keratin, and type II cytoskeletal 6A keratin.
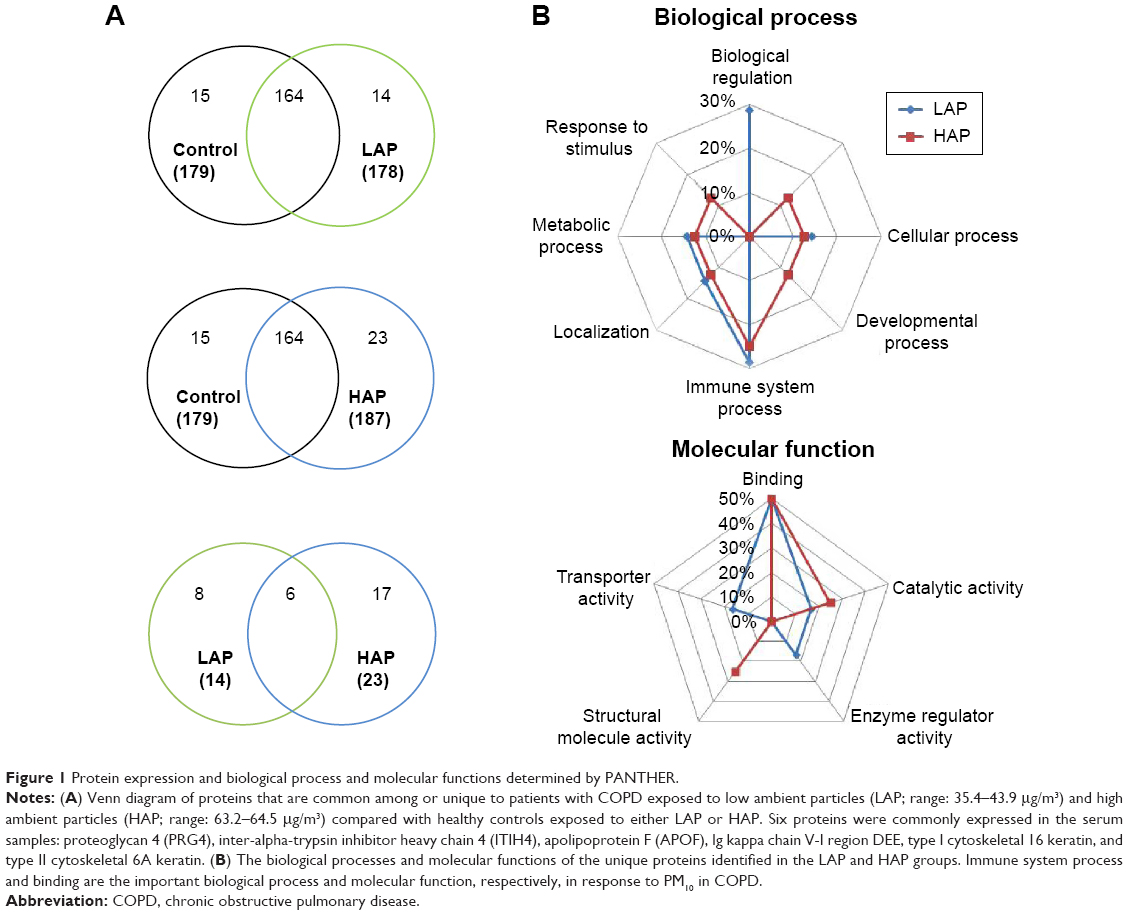 | Figure 1 Protein expression and biological process and molecular functions determined by PANTHER. |
PANTHER analysis was conducted on the proteins unique to LAP- and HAP-exposed subjects with COPD to understand their biological context (Figure 1B). The common biological processes identified in the LAP and HAP groups included biological regulation (29% for LAP), cellular component organization or biogenesis (13% for HAP), cellular process (14% for LAP and 13% for HAP), developmental process (13% for HAP), immune system process (29% for LAP and 25% for HAP), localization (14% for LAP and 13% for HAP), metabolic process (14% for LAP and 13% for HAP), and response to stimulus (13% for HAP), with immune system process terms being the most common. The common molecular functions identified between the LAP and HAP groups included binding (50% for LAP and 50% for HAP), catalytic activity (17% for LAP and 25% for HAP), enzyme regulator activity (17% for LAP), structural molecule activity (25% for HAP), and transporter activity (17% for LAP), with binding terms being the most common.
Validation of PRG4, ITIH4, and APOF in patients with COPD and control subjects
The levels of PRG4, ITIH4, and APOF in healthy controls, smokers, and subjects with COPD are presented in Figure 2. There were no differences in the PRG4, ITIH4, and APOF levels between healthy controls and healthy smokers. The PRG4 levels in the subjects with COPD were significantly higher than those in healthy controls or smokers (P<0.05). The ITIH4 levels in subjects with COPD were lower than those in healthy controls (P<0.05) but not those of healthy smokers. There was no significant difference in the APOF levels across subjects with COPD, healthy controls, or smokers.
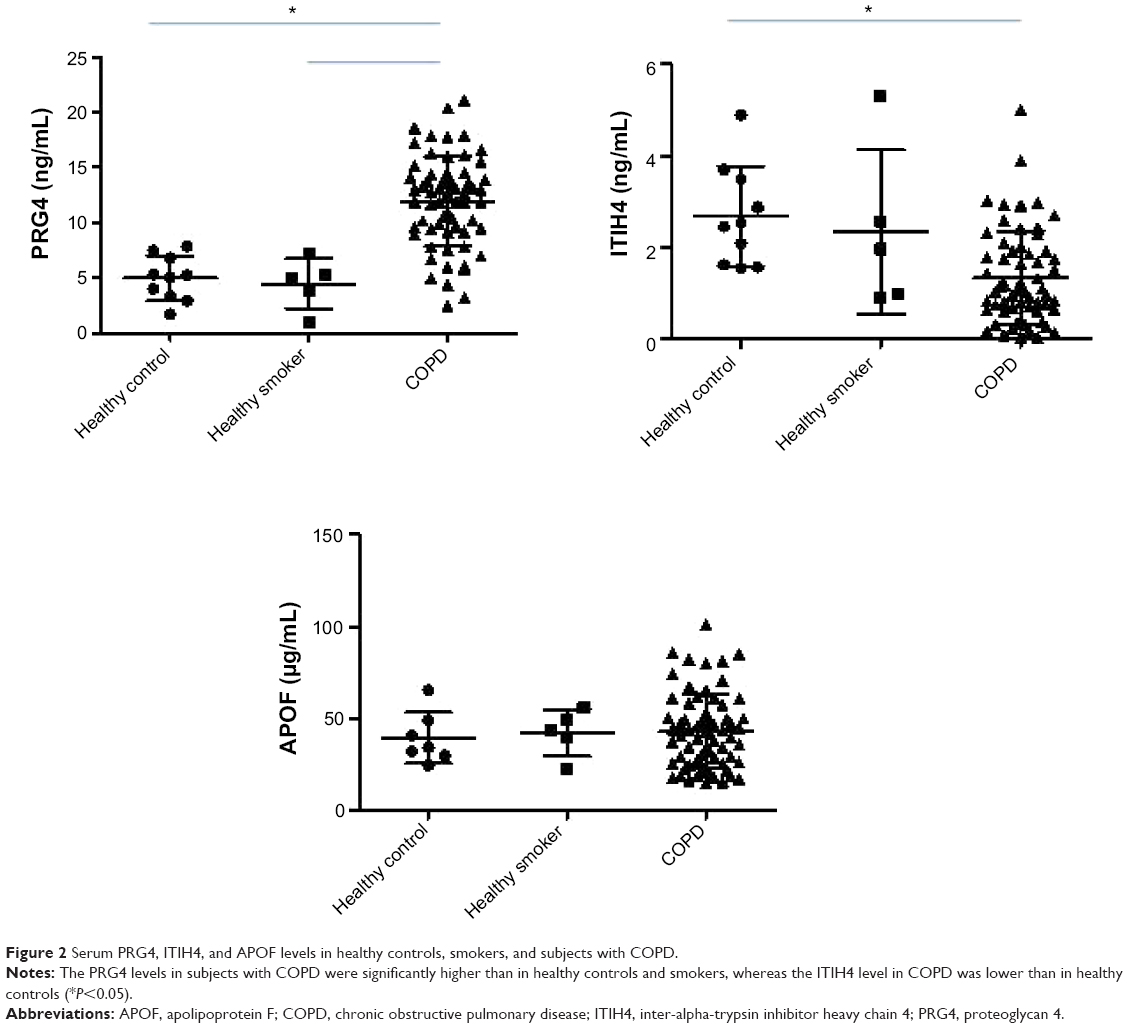 | Figure 2 Serum PRG4, ITIH4, and APOF levels in healthy controls, smokers, and subjects with COPD. |
Correlation of PM10 with PRG4, ITIH4, and APOF
The changes in PM10 for the 1-year, 2-year, and 3-year averages in quintiles were related to PRG4, ITIH4, and APOF (Figure 3). The PRG4 levels were correlated with 2-year (r=0.278, P=0.033) and 3-year (r=0.349, P=0.007) PM10 averages, whereas the ITIH4 levels were correlated with the 1-year (r=−0.325, P=0.012), 2-year (r=−0.401, P=0.002), and 3-year PM10 averages (r=−0.342, P=0.008). There was no correlation between APOF and PM10 levels over the previous 3 years.
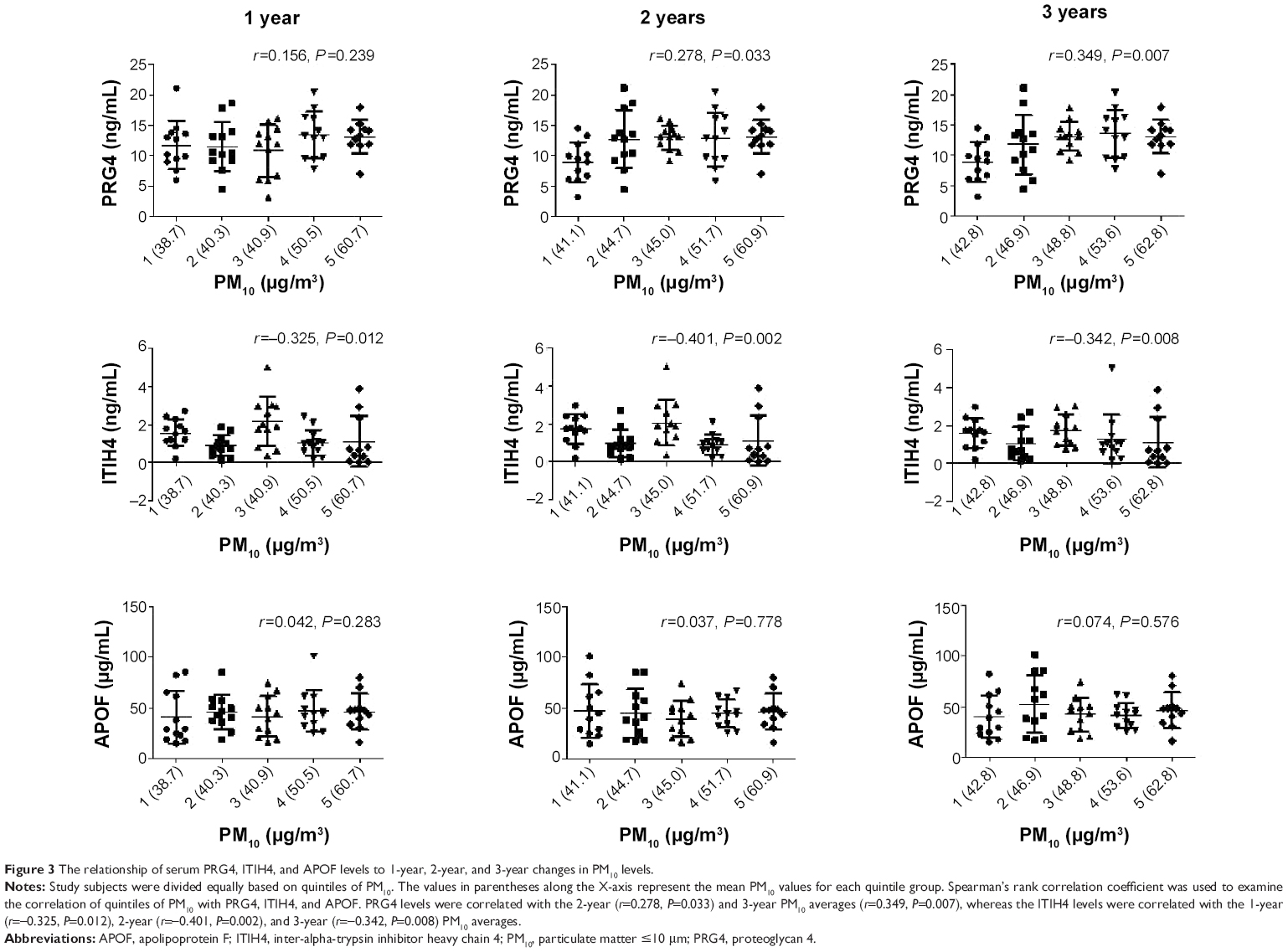 | Figure 3 The relationship of serum PRG4, ITIH4, and APOF levels to 1-year, 2-year, and 3-year changes in PM10 levels. |
Figure 4 shows the ROC curve of the sensitivity and specificity obtained for the PRG4, ITIH4, and APOF levels when discriminating between the ≥50 μg/m3 and <50 μg/m3 average 3-year PM10 levels in patients with COPD. The area under the curve (AUC) value was 0.563 (95% confidence interval [CI] =0.413–0.713, P=0.405) for 8-isoprostane, 0.634 (95% CI =0.486–0.782, P=0.086) for CRP, 0.636 (95% CI =0.492–0.780, P=0.083) for PRG4, 0.690 (95% CI =0.545–0.836, P=0.015) for ITIH4, and 0.523 (95% CI =0.376–0.671, P=0.766) for APOF.
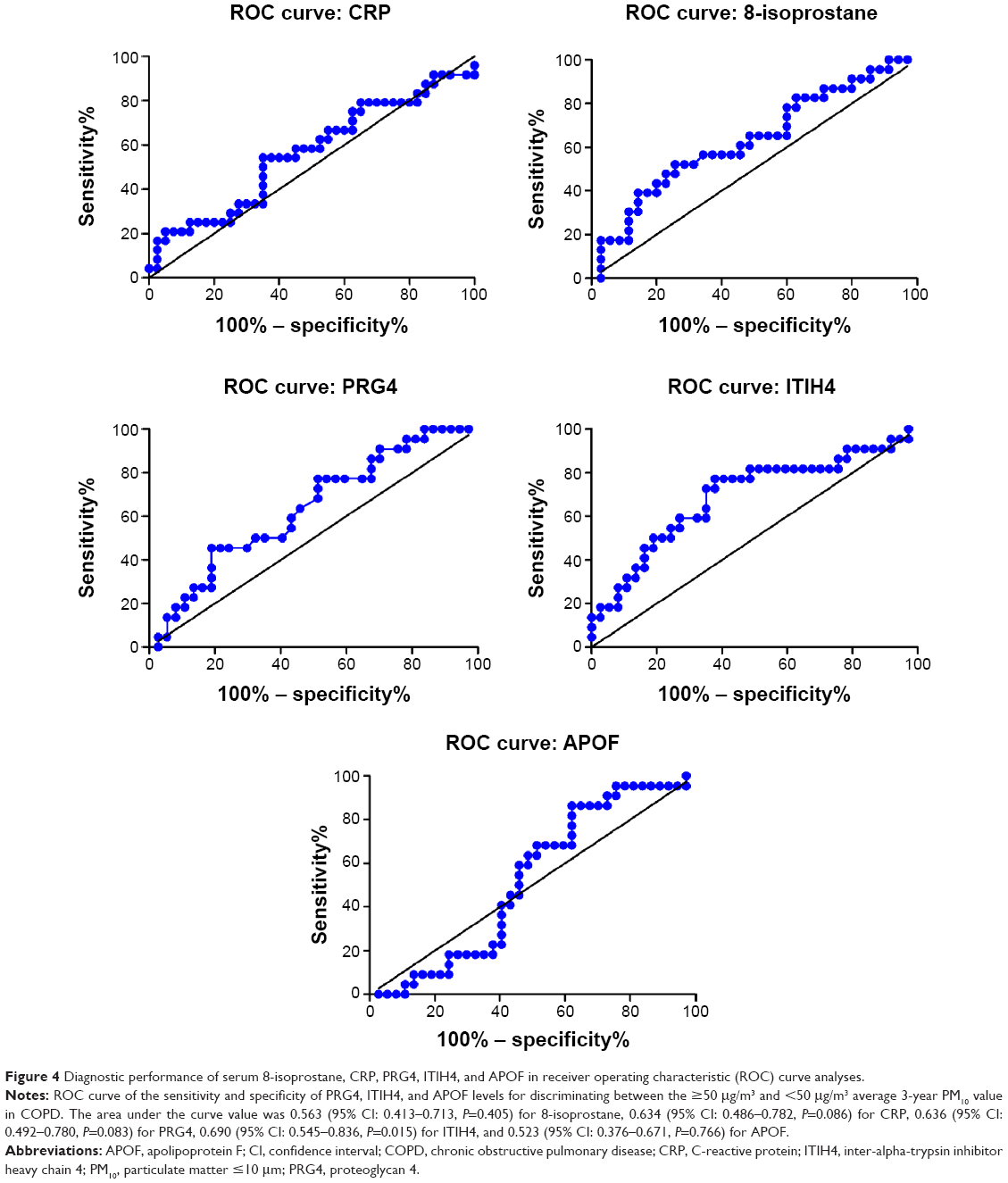 | Figure 4 Diagnostic performance of serum 8-isoprostane, CRP, PRG4, ITIH4, and APOF in receiver operating characteristic (ROC) curve analyses. |
Correlation of 8-isoprostane and CRP with PRG4, ITIH4, and APOF
Correlations between 8-isoprostane and CRP with PRG4, ITIH4, and APOF for COPD were determined (Figure 5). There were no statistical associations of 8-isoprostane and CRP to PRG4 and APOF in patients with COPD. Only ITIH4 was correlated with the CRP levels (r=0.353, P=0.005).
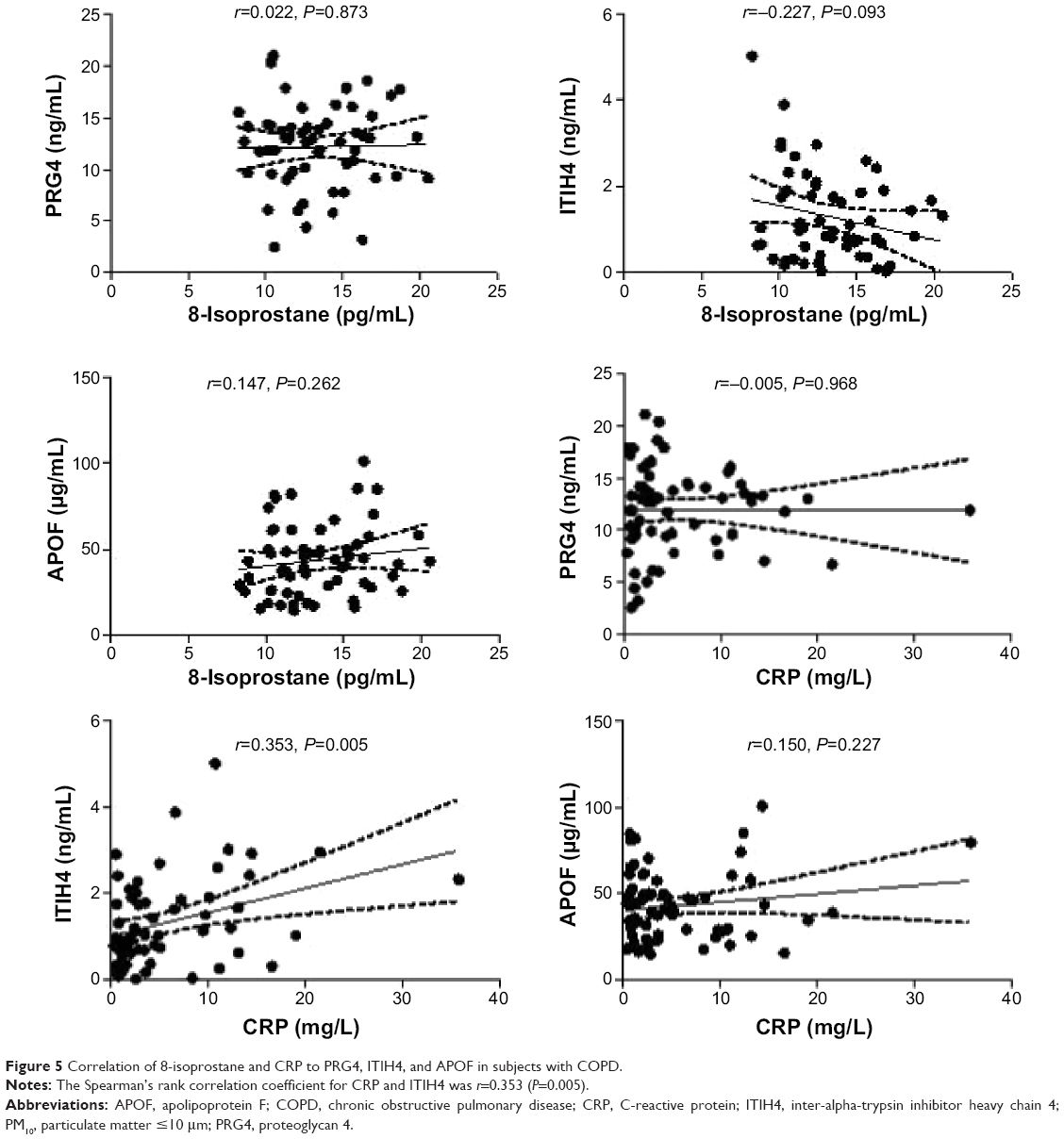 | Figure 5 Correlation of 8-isoprostane and CRP to PRG4, ITIH4, and APOF in subjects with COPD. |
Discussion
COPD, a chronic inflammatory disease, is recognized by irreversible airflow obstruction and by structural modifications in the proximal and peripheral airways.15 An abnormal inflammatory response of the lung to PM10 appears to play a central role in the pathogenesis of COPD;16,17 however, few studies have investigated protein profiles in patients with COPD residing in polluted urban areas. The present study discovered differences in protein expression in patients with COPD who were exposed to low and high levels of PM10 (LAP and HAP). Three major findings are reported in the present study: 1) immune system process and binding are the most important biological process and molecular function, respectively, in the response to PM10 in patients with COPD, 2) ITIH4 is a biomarker for PM10 in COPD, and 3) ITIH4 is associated with inflammatory response.
Oxidative stress and inflammatory reactions are considered common biological responses that result from the inhalation of PM10. Toxicology exposure experiments in animals and humans have identified several biological mechanisms that may be affected by the inhalation of PM10, including oxidative stress and systemic inflammation.18 The present study identified the immune system process as an important biological process in the response to PM10 in COPD and the binding terms as important molecular functions. Immune response is considered an important determinant for the exacerbation of COPD,19,20 and immune disorder commonly associated with PM10 contributes to lung function impairment and mortality. Systemic inflammation is considered a common link between COPD and immune response.21 Furthermore, a link between binding and COPD has been reported.22 For example, a heavy subunit with a regulatory light subunit can modulate glutamate cysteine ligase activity.23 Notably, a key common feature between PM10 exposure and COPD is “inflammatory response.” Oxidative inflammatory reactions may be a result of exposure-related variances in protein regulation; therefore, an investigation of the effect of PM10 on protein profiling in COPD is important.
Discovering biomarkers for susceptible populations assists with human environmental exposure assessment and risk evaluation. Despite increased efforts to identify COPD biomarkers, however, the gap between clinical and environmental medicine is substantial. We identified six biomarker candidates from healthy controls and from LAP- and HAP-exposed patients with COPD using a proteomics approach on serum samples: PRG4, ITIH4, APOF, Ig kappa chain V-I region DEE, type I cytoskeletal 16 keratin, and type II cytoskeletal 6A keratin. Type I cytoskeletal 16 keratin and type II cytoskeletal 6A keratin are associated with epithelial tissues from nail beds, esophagus, tongue, and hair follicles,24,25 which are less likely related to the mechanisms of PM10 and COPD. Thus, we next examined PRG4, ITIH4, and APOF as candidate biomarkers. We observed that the PRG4 levels in subjects with COPD were significantly higher than those in healthy controls and smokers, whereas the ITIH4 levels were significantly lower than those in healthy controls. These results suggest PRG4 and ITIH4 could be the potential biomarkers for COPD and that APOF is not sensitive as a potential biomarker for COPD and PM10 exposure. We then correlated the PM10 levels for the previous 1-year, 2-year, and 3-year averages with these three biomarker candidates. The correlations showed that PRG4 and ITIH4 may be biomarkers for alteration in PM10 levels, particularly ITIH4. Previously, 8-isoprostane and CRP were characterized as biomarkers of oxidative stress and inflammation, respectively, in response to particulate air pollution.7,26 Therefore, we used them for the comparison with PRG4, ITIH4, and APOF in COPD. The US Environmental Protection Agency’s health-based national air quality standard for PM10 (annual average of 50 μg/m3) was used as the cutoff level for the ROC analysis. We discovered that ITIH4 is the only significant biomarker for PM10 in COPD; no significance was observed for 8-isoprostane, CRP, PRG4, and APOF. This finding suggests that ITIH4 is a good biomarker for evaluating PM10 exposure in subjects with COPD.
To understand the potential mechanisms underlying PM10-regulated COPD in the present study, 8-isoprostane and CRP were correlated with PRG4, ITIH4, and APOF. Statistical significance was observed between CRP and ITIH4 in COPD, suggesting that ITIH4 may be involved in inflammatory responses. ITIH4 is a type II acute phase protein and has been associated with inflammatory responses.27,28 Our findings are consistent with those of a previous study that observed correlation between the ITIH4 and CRP levels.27,28 However, the correlations between PM10 and ITIH4 are negatively associated with increasing PM10 concentrations. CRP is commonly used in the assessment of several acute clinical conditions, such as COPD.29 Notably, CRP levels are also elevated in many other conditions, including infective, inflammatory, and neoplastic pathologies.30–32 Because of CRP’s involvement in multiple pathologies, ITIH4 may be a more specific biomarker for PM10 in COPD than CRP.
Conclusion
In conclusion, circulating ITIH4 is an important biomarker to assess PM10 exposure in patients with COPD and could be involved in postexposure inflammatory responses. Furthermore, metabolic process and catalytic activity are the important biological process and molecular function, respectively, in response to PM10 exposure in COPD. The limitation of the present study is the sex distribution in the study subjects. The sex difference may associated with the biomarker identification for PM10 exposure. Future studies are required to investigate the potential mechanisms underlying PM10 exposure and to determine whether ITIH4 can provide a specific index for evaluating PM10 exposure in COPD.
Acknowledgments
The authors thank Yi-Syuan Lin and Ching-Hsuan Law for their technical assistance in this research. This study was funded by the Ministry of Science and Technology of Taiwan (MOST103-2314-B-038-018 and MOST102-2314-B-038-055-MY2), Taipei Medical University (TMU103-AE1-B12), and the Taipei Medical University-Shuang Ho Hospital (103TMU-SHH-01-1).
Author contributions
All authors have contributed substantially to the concept and design of the study, the drafting of the article, and the critical revision of the manuscript for important intellectual content. All authors have read and approved the final version of the manuscript for publication.
Disclosure
The authors report no conflicts of interest in this work.
References
Dominici F, Peng RD, Bell ML, et al. Fine particulate air pollution and hospital admission for cardiovascular and respiratory diseases. JAMA. 2006;295(10):1127–1134. | ||
Olivieri D, Scoditti E. Impact of environmental factors on lung defences. Eur Respir Rev. 2005;14(95):51–56. | ||
Yorifuji T, Kashima S, Tsuda T, et al. Long-term exposure to traffic-related air pollution and the risk of death from hemorrhagic stroke and lung cancer in Shizuoka, Japan. Sci Total Environ. 2012;443C:397–402. | ||
Schikowski T, Mills IC, Anderson HR, et al. Ambient air pollution – a cause for COPD? Eur Respir J. 2014;43(1):250–263. | ||
Kumar N, Liang D, Comellas A, Chu AD, Abrams T. Satellite-based PM concentrations and their application to COPD in Cleveland, OH. J Expo Sci Environ Epidemiol. 2013;23(6):637–646. | ||
Schrohl AS, Würtz S, Kohn E, et al. Banking of biological fluids for studies of disease-associated protein biomarkers. Mol Cell Proteomics. 2008;7(10):2061–2066. | ||
Hennig F, Fuks K, Moebus S, et al; Heinz Nixdorf Recall Study Investigative Group. Association between source-specific particulate matter air pollution and hs-CRP: local traffic and industrial emissions. Environ Health Perspect. 2014;122(7):703–710. | ||
Zhang J, Zhu T, Kipen H, et al; HEI Health Review Committee. Cardiorespiratory biomarker responses in healthy young adults to drastic air quality changes surrounding the 2008 Beijing Olympics. Res Rep Health Eff Inst. 2013;(174):5–174. | ||
Hryhorczuk DO, Moomey M, Burton A, et al. Urinary p-nitrophenol as a biomarker of household exposure to methyl parathion. Environ Health Perspect. 2002;110(suppl 6):1041–1046. | ||
Zhang Y, Yang R, Burwinkel B, Breitling LP, Brenner H. F2RL3 methylation as a biomarker of current and lifetime smoking exposures. Environ Health Perspect. 2014;122(2):131–137. | ||
Yu CJ, Wang CL, Wang CI, et al. Comprehensive proteome analysis of malignant pleural effusion for lung cancer biomarker discovery by using multidimensional protein identification technology. J Proteome Res. 2011;10(10):4671–4682. | ||
Pan CH, Liu WT, Bien MY, et al. Effects of size and surface of zinc oxide and aluminum-doped zinc oxide nanoparticles on cell viability inferred by proteomic analyses. Int J Nanomedicine. 2014;9:3631–3643. | ||
Huang DW, Sherman BT, Tan Q, et al. DAVID bioinformatics resources: expanded annotation database and novel algorithms to better extract biology from large gene lists. Nucleic Acids Res. 2007;35:W169–W175. | ||
Su CL, Chen TT, Chang CC, et al; Taiwan CardioPulmonary Research Group (T-CPR). Comparative proteomics of inhaled silver nanoparticles in healthy and allergen provoked mice. Int J Nanomedicine. 2013;8:2783–2799. | ||
Vestbo J, Hurd SS, Agustí AG, et al. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2013;187(4):347–365. | ||
Wood AM, Harrison RM, Semple S, Ayres JG, Stockley RA. Outdoor air pollution is associated with rapid decline of lung function in alpha-1-antitrypsin deficiency. Occup Environ Med. 2010;67(8):556–561. | ||
Kurmi OP, Semple S, Simkhada P, Smith WC, Ayres JG. COPD and chronic bronchitis risk of indoor air pollution from solid fuel: a systematic review and meta-analysis. Thorax. 2010;65(3):221–228. | ||
Brookes PS, Yoon Y, Robotham JL, Anders MW, Sheu SS. Calcium, ATP, and ROS: a mitochondrial love-hate triangle. Am J Physiol Cell Physiol. 2004;287(4):C817–C833. | ||
Watz H, Waschki B, Kirsten A, et al. The metabolic syndrome in patients with chronic bronchitis and COPD: frequency and associated consequences for systemic inflammation and physical inactivity. Chest. 2009;136(4):1039–1046. | ||
Clini E, Crisafulli E, Radaeli A, Malerba M. COPD and the metabolic syndrome: an intriguing association. Intern Emerg Med. 2013;8(4):283–289. | ||
Rovina N, Koutsoukou A, Koulouris NG. Inflammation and immune response in COPD: where do we stand? Mediators Inflamm. 2013; 2013:413735. | ||
Haenen S, Clynen E, Nemery B, Hoet PHM, Vanoirbeek JAJ. Biomarker discovery in asthma and COPD: application of proteomics techniques in human and mice. EuPA Open Proteom. 2014;4(0):101–112. | ||
Rahman I, Adcock IM. Oxidative stress and redox regulation of lung inflammation in COPD. Eur Respir J. 2006;28(1):219–242. | ||
Rosenberg M, Fuchs E, Le Beau MM, Eddy RL, Shows TB. Three epidermal and one simple epithelial type II keratin genes map to human chromosome 12. Cytogenet Cell Genet. 1991;57(1):33–38. | ||
Schweizer J, Bowden PE, Coulombe PA, et al. New consensus nomenclature for mammalian keratins. J Cell Biol. 2006;174(2):169–174. | ||
MacNee W, Donaldson K. Mechanism of lung injury caused by PM10 and ultrafine particles with special reference to COPD. Eur Respir J Suppl. 2003;40:47s–51s. | ||
Xu H, Shang Q, Chen H, et al. ITIH4: a new potential biomarker of “Toxin Syndrome” in coronary heart disease patient identified with proteomic method. Evid Based Complement Alternat Med. 2013;2013: 360149. | ||
Piñeiro M, Alava MA, González-Ramón N, et al. ITIH4 serum concentration increases during acute-phase processes in human patients and is up-regulated by interleukin-6 in hepatocarcinoma HepG2 cells. Biochem Biophys Res Commun. 1999;263(1):224–229. | ||
Thomsen M, Ingebrigtsen TS, Marott JL, et al. Inflammatory biomarkers and exacerbations in chronic obstructive pulmonary disease. JAMA. 2013;309(22):2353–2361. | ||
Agassandian M, Shurin GV, Ma Y, Shurin MR. C-reactive protein and lung diseases. Int J Biochem Cell Biol. 2014;53C:77–88. | ||
Palaniyar N, Nadesalingam J, Reid KB. Pulmonary innate immune proteins and receptors that interact with gram-positive bacterial ligands. Immunobiology. 2002;205(4–5):575–594. | ||
Zbroja-Sontag W. Defense proteins and immune complexes in the blood serum of women with inflammatory and neoplastic lesions of the ovary. Am J Reprod Immunol. 1983;4(1):11–20. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.