Back to Journals » Clinical, Cosmetic and Investigational Dermatology » Volume 19
Integrative Transcriptomic and Genetic Analysis Prioritizes SLC1A5 as a Programmed Cell Death-Associated Candidate Risk Gene in Vitiligo
Authors Yao J, Chen H, Zhou X, Hu T, Guo H, Ma L, Liu M, Liu F, Zhang R, Li W
Received 15 April 2026
Accepted for publication 30 May 2026
Published 18 July 2026 Volume 2026:19 617044
DOI https://doi.org/10.2147/CCID.S617044
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Monica K. Li
Jizhao Yao, Honghui Chen, Xiulian Zhou, Ting Hu, Hengshan Guo, Lili Ma, Minhong Liu, Fangyan Liu, Ruiyang Zhang, Weiquan Li
Department of Dermatology, Yuebei People’s Hospital, Shaoguan, Guangdong, 512026, People’s Republic of China
Correspondence: Weiquan Li, Department of Dermatology, Yuebei People’s Hospital, No. 133, South Huimin Road, Wujiang District, Shaoguan, Guangdong, 512026, People’s Republic of China, Email [email protected]
Background: Programmed cell death (PCD) has been implicated in various autoimmune disorders, but its role in vitiligo remains poorly understood. This study aimed to identify PCD-related genes and elucidate their potential contribution to vitiligo pathogenesis through integrative bioinformatics analysis.
Methods: Three GEO datasets (GSE65127, GSE53146, GSE75819) were merged to obtain a combined cohort of 40 controls and 30 vitiligo samples. Differentially expressed genes (DEGs) were identified using limma. GSVA was applied to assess 11 PCD pathways. Summary-data-based Mendelian randomization (SMR) and HEIDI testing integrated eQTL data with vitiligo to pinpoint causal genes. Bayesian colocalization and immune infiltration analyses were further performed.
Results: A total of 922 DEGs were identified, with pyroptosis and cuproptosis signatures upregulated in vitiligo whereas overall autophagy- and lysosome-dependent cell death–related gene expression was decreased. In contrast, pathway-level GSVA using a broader autophagy-related gene set indicated upregulated autophagy signaling, highlighting the context dependence of autophagy-related signatures. Overlapping DEGs with PCD gene sets yielded 75 differentially expressed PCD-related genes. SMR analysis prioritized 602 genes associated with vitiligo risk, and intersection with PCD genes highlighted PARK7 and SLC1A5 as key candidates. Bayesian colocalization analysis provided strong genetic support for SLC1A5 as a candidate gene, with lead SNP rs8105903 showing consistent eQTL and GWAS signals. GSVA revealed downregulated melanogenesis and tyrosine metabolism alongside upregulated autophagy and NOD-like receptor signaling in vitiligo. Single-gene enrichment linked SLC1A5 to glycosphingolipid biosynthesis and melanogenesis. Immune infiltration analysis showed elevated aDC, T helper, and Th2 cells but reduced NK CD56bright cells in vitiligo. SLC1A5 was significantly downregulated in vitiligo samples and demonstrated moderate diagnostic value.
Conclusion: This study identifies SLC1A5 as a genetically anchored PCD-associated gene potentially involved in vitiligo through metabolic reprogramming and immune modulation, and provides strong genetic evidence supporting SLC1A5 as a candidate for further mechanistic and translational investigation, while recognizing that functional studies are required before it can be considered a therapeutic target.
Keywords: vitiligo, programmed cell death, transcriptomics, immune cell infiltration, genetic susceptibility, SMR analysis
Introduction
Vitiligo is a complex autoimmune disorder characterized by progressive destruction of melanocytes, resulting in patchy depigmentation of the skin.1,2 It affects approximately 0.5–2.0% of the global population, across all ethnic groups, and can develop at any age.2,3 Beyond the visible depigmentation, vitiligo is associated with substantial psychological and social burden, including stigmatization, diminished quality of life, and increased rates of anxiety and depression.4,5 The chronic course, frequent resistance to therapy, and high relapse rates further underscore its clinical and public health importance.6 Despite its prevalence, the underlying molecular and genetic mechanisms contributing to vitiligo pathogenesis remain poorly understood. Current evidence suggests that immune dysregulation plays a pivotal role, and transcriptomic studies have highlighted altered immune responses, oxidative stress, and apoptosis as significant contributors to melanocyte degeneration.7–9 Furthermore, programmed cell death (PCD) mechanisms, including apoptosis, pyroptosis, ferroptosis, necroptosis, autophagy-related cell death, and lysosome-dependent cell death, have emerged as critical pathways in cellular homeostasis and autoimmune disorders, yet their specific roles and interactions in vitiligo remain unexplored.10–12
Genetic studies have made significant progress in identifying risk variants associated with vitiligo susceptibility. Genome-wide association studies (GWAS) have localized several candidate loci, including variants within or near HLA class I and II regions, CD80, CCR6, CD44, TYR, TYRP1, and MC1R, which implicate both immune regulatory and melanocyte-specific pathways in disease risk.13,14 However, connecting these findings to functional genes and pathways relevant to melanocyte biology has proven challenging, particularly because many risk variants reside in noncoding regions and may exert their effects through context-specific regulation of gene expression. Integrative approaches utilizing transcriptomic, epigenetic, and genetic data are thus essential to fully elucidate the molecular landscape of vitiligo.
Among the genes and pathways implicated in immune regulation and cellular stress responses, amino acid transporters and glutamine metabolism have gained increasing attention. Solute carrier family members, including SLC1A5, play key roles in glutamine uptake, metabolic reprogramming, redox balance, and activation of immune cells.15,16 SLC1A5, a high-affinity neutral amino acid transporter, has been shown to regulate T cell function, cytokine production, and susceptibility to various forms of PCD in other disease contexts.17,18 However, its potential involvement in melanocyte biology, cutaneous immunity, and vitiligo pathogenesis has not been systematically investigated. Given that glutamine availability and amino acid transport can influence oxidative stress responses, immune activation, and cell survival, SLC1A5 represents a biologically plausible candidate linking genetic susceptibility, metabolic regulation, and PCD in vitiligo.
The present study aims to address existing gaps by comprehensively investigating PCD-related pathways, genetic susceptibility, and immune dysfunction in vitiligo, with a particular focus on SLC1A5 and amino acid transport. Utilizing transcriptomic and genomic datasets, we hypothesize that specific genes within PCD pathways act as central mediators linking immune dysregulation and genetic susceptibility. To test this hypothesis, we combined differential expression analysis, Summary-data-based Mendelian randomization (SMR), regional colocalization, and pathway enrichment methods to identify key genes and pathways.19,20 More specifically, this study aimed to: (1) identify differentially expressed PCD-related genes in vitiligo; (2) prioritize genetically supported candidate genes using SMR and colocalization analysis; (3) explore functional pathways associated with the leading candidate gene, with emphasis on SLC1A5 and glutamine metabolism; and (4) assess its relationship with immune infiltration and diagnostic performance. This multidisciplinary approach offers new insights into molecular drivers of autoimmune dysfunction and melanocyte destruction in vitiligo, providing direction for future therapeutic development.
Methods
Data Acquisition and Preprocessing
The gene expression datasets GSE65127, GSE53146, and GSE75819 were downloaded from the Gene Expression Omnibus (GEO) database (https://www.ncbi.nlm.nih.gov/geo/). Raw data in CEL format were processed using R and Bioconductor packages. Preprocessing involved background correction via the robust multi-array average (RMA) method, normalization, and imputation of missing values using the “affy” and “impute” packages. Probe-to-gene annotation was performed by mapping probes to gene symbols using annotation files provided by the array manufacturers. Duplicate probe sets corresponding to the same gene were resolved by calculating the median expression value, and probes without matching gene symbols were excluded. Sample characteristics are summarised in Table S1. Briefly, GSE65127 contributed ten non-lesional epidermis samples and ten healthy controls, plus ten lesional epidermis samples. No further clinical details were available for the healthy controls in this dataset. GSE53146 included five lesional epidermis samples and five healthy controls; the control skin was obtained from age- and site-matched excision tips without pathology. GSE75819 provided fifteen lesional and fifteen non-lesional epidermis samples, without healthy controls. Across the three datasets, no information on treatment status, disease activity, or vitiligo subtype could be retrieved. Controls were not uniformly matched by age, sex, or anatomical site due to inconsistent reporting, nor were all platforms identical (GPL570, GPL14951, GPL6884). After merging, the integrated matrix contained 40 control samples (non-lesional or healthy) and 30 vitiligo lesional samples. Batch effects were assessed and corrected with the sva package.
Differential Expression Analysis
DEGs between vitiligo and control groups were identified using the “limma” package. Statistical significance was determined using an adjusted p-value < 0.05, with |log2 fold change| > 1 set as the differential expression threshold. Volcano plots illustrating DEGs were generated using the “EnhancedVolcano” package, while heatmaps of the top 50 DEGs were constructed using the “ComplexHeatmap” package for visualization.
GSVA Analysis of PCD Pathways
To investigate PCD mechanisms, 11 gene sets related to cell death modalities, including apoptosis, pyroptosis, ferroptosis, necroptosis, and others, were curated from literature sources.21 The pathway activity levels of these PCD gene sets were evaluated using GSVA with the “GSVA” package. Pathway scores were compared between vitiligo and control groups using a Wilcoxon test, and results were visualized using “ggplot2”.
Summary-Data-Based Mendelian Randomization (SMR) Analysis
To identify putatively causal genes whose expression levels influence vitiligo susceptibility, we performed a SMR analysis.22 Vitiligo GWAS summary statistics (95 cases and 337,064 controls of European ancestry) were sourced from the IEU OpenGWAS Project platform (https://gwas.mrcieu.ac.uk/, accession ukb-a-115).23 Genetic instruments for gene expression were defined using cis-expression quantitative trait loci (cis-eQTL) data. For each gene, the most significantly associated single nucleotide polymorphism (SNP) within a ±1000 kb window of the gene’s transcription start site was selected as the instrumental variable, provided its GWAS p-value met a genome-wide significance threshold of 5×10−8. An initial gene-level association with vitiligo risk was assessed under a liberal pSMR threshold of 0.05. To distinguish causality from confounding due to linkage disequilibrium (LD), we applied the heterogeneity in dependent instruments (HEIDI) test. Associations with a pHEIDI value < 0.05 were considered evidence of pleiotropy/LD and were subsequently excluded from the final set of candidate causal genes.24 The SMR and HEIDI analyses were performed using SMR software (version 1.3.1).
Bayesian Colocalization Analysis
To assess whether SLC1A5 expression and vitiligo risk share the same causal genetic variant, we performed Bayesian colocalization analysis using the coloc package (version 5.2.3, https://github.com/chr1swallace/coloc) in R (version 4.2.0). Summary statistics for the vitiligo GWAS (ukb-a-115) and SLC1A5 expression quantitative trait loci (eQTL) were integrated via the ieugwasr_to_coloc function. We defined a symmetric genomic window centered on SLC1A5, corresponding to ±250 kb around the gene locus, and included all SNPs present in both the GWAS and eQTL datasets within this region. For each region, we report the total number of overlapping SNPs used in the colocalization analysis. The coloc.abf function was then applied with default prior probabilities (p1 = 1.0×10−4 for association with vitiligo, p2 = 1.0×10−4 for association with SLC1A5 expression, and p12 = 1.0×10−5 for a shared causal variant), to estimate the posterior probability for each of the five hypotheses: H0 (no association with either trait), H1 (association with vitiligo only), H2 (association with SLC1A5 expression only), H3 (both traits associated but with distinct causal variants), and H4 (both traits associated and sharing a common causal variant). We report the full posterior probability distribution (PP.H0–PP.H4), rather than PP.H4 alone, to distinguish between shared and distinct causal signals. A PP.H4 > 80% was considered strong evidence for a shared causal variant, whereas a high PP.H3 was interpreted as evidence for distinct causal variants in the same region. Regional plots and locus-specific visualizations were generated using the locuscompare function from the locuscompare package (https://github.com/boxiangliu/locuscompare), allowing direct comparison of GWAS and eQTL association signals across the genomic region of SLC1A5.
Gene Set Variation Analysis (GSVA) and Single-Gene Enrichment Analysis
To evaluate pathway activity differences between control and vitiligo samples, GSVA was performed using the GSVA package. Gene sets were obtained from the Molecular Signatures Database (MSigDB, https://www.gsea-msigdb.org/gsea/msigdb/). GSVA was run with default parameters, using the normalized expression matrix. The resulting enrichment scores were compared between groups to identify differentially activated pathways. For single-gene enrichment analysis of SLC1A5, we employed a gene set enrichment analysis (GSEA)-like approach. Samples were stratified based on SLC1A5 expression levels, and differential expression analysis was performed between high- and low-expression groups using the limma package. GSEA was conducted using the clusterProfiler package with the GSEA function, using 1000 permutations and default parameters. Enrichment scores (ES) and nominal P-values were computed for each gene set. Visualization of leading edge pathways was performed using the enrichplot package.
Immune Cell Infiltration Analysis
Immune cell infiltration analysis was conducted to assess the immunological landscape of vitiligo using expression data from the integrated datasets. Single-sample Gene Set Enrichment Analysis (ssGSEA) was performed with the “GSVA” R package to quantify infiltration levels of various immune cell subsets based on predefined immune-related gene sets obtained from publicly available databases and literature.25 Correlation analysis between SLC1A5 expression levels and the abundance of each immune cell type was conducted using Spearman’s rank correlation test, implemented with the cor.test function in R. The results of the correlation analysis were visualized as lollipop plots using the ggplot2 package.
Results
Identification of DEGs and PCD Pathways in Vitiligo
Figure 1 shows the flowchart for this study. To correct for technical batch effects across the three GEO datasets (GSE65127, GSE53146, GSE75819), we applied the ComBat method. As shown in Figure S1, the UMAP plot after batch correction revealed substantial intermingling of samples from the three datasets, indicating effective removal of platform- and study-specific biases while preserving biological variability. To characterize the transcriptional landscape of vitiligo, we first performed differential expression analysis on the merged dataset, identifying a total of 922 DEGs (Figure 2A). Among these, the top 50 DEGs were selected for hierarchical clustering, and the resulting heatmap demonstrated clear segregation between control and vitiligo samples, reflecting substantial disease-associated transcriptomic shifts (Figure 2B). To further explore the involvement of regulated cell death mechanisms, we applied GSVA to evaluate eleven common PCD pathways; notably, pyroptosis and cuproptosis signatures were enriched in vitiligo samples, whereas autophagy and lysosome-dependent cell death pathways were downregulated relative to controls (Figure 2C). Integration of DEG analysis with PCD-related gene sets enabled the identification of 75 DEGs directly associated with programmed cell death, representing a critical subset of genes potentially driving pathogenic processes in vitiligo (Figure 2D). These findings provide insight into the interplay between PCD mechanisms and vitiligo pathogenesis, highlighting avenues for targeted research and therapeutic exploration.
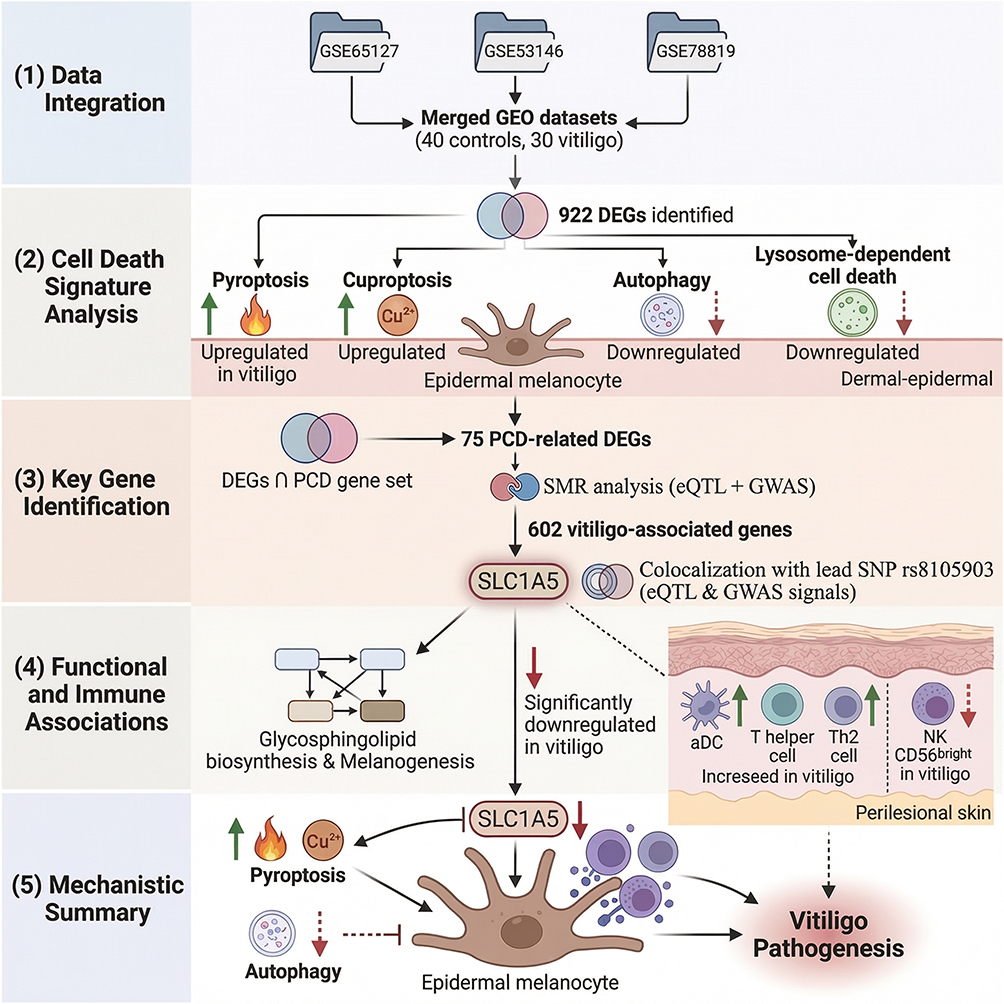 |
Figure 1 Flowchart of this study. An upward arrow indicates an increase, whilst a downward arrow indicates a decrease. |
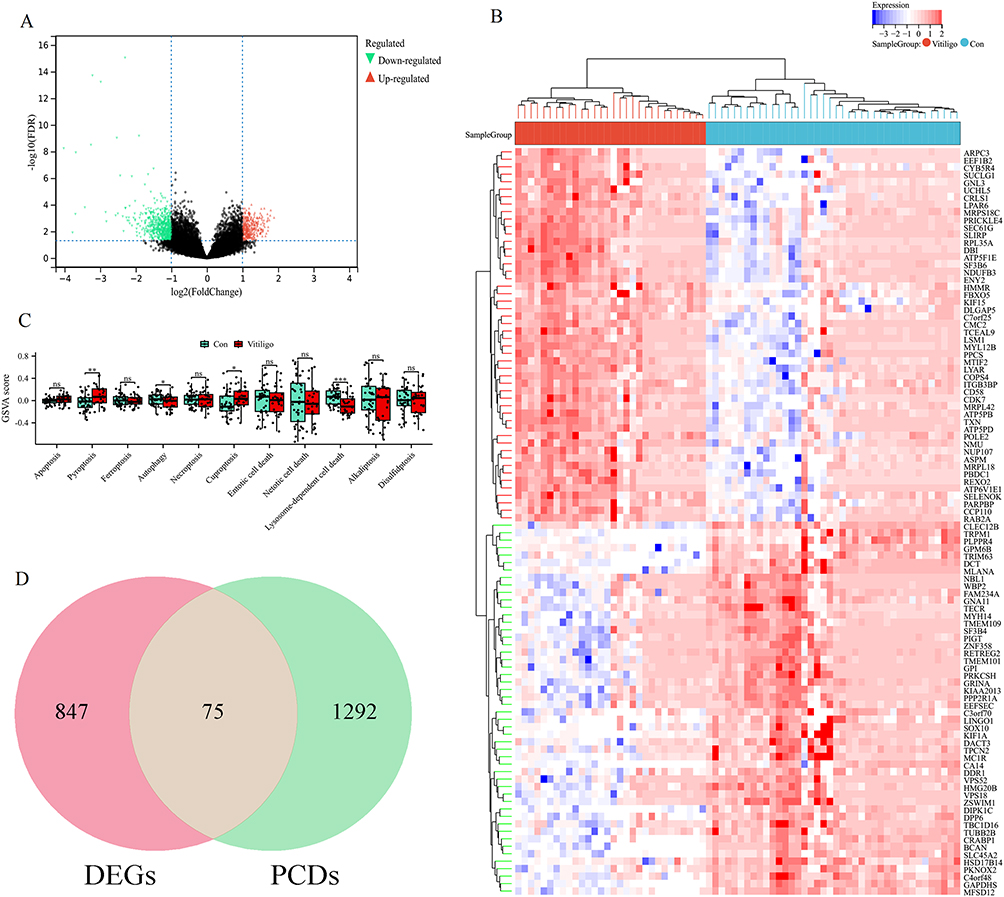 |
Figure 2 Comprehensive analysis of DEGs and PCD pathways in vitiligo. (A) Volcano plot illustrating DEGs identified from the integrated vitiligo dataset based on differential expression analysis. (B) Heatmap displaying hierarchical clustering of the top 50 DEGs, highlighting distinct expression patterns between vitiligo and control samples. (C) GSVA of 11 common programmed cell death pathways comparing vitiligo and control groups. ns indicates no statistically significant difference, *p < 0.05, **p < 0.01, ***p < 0.001. (D) Venn diagram demonstrating the intersection between DEGs and programmed cell death-related genes. |
Identification of Putative Causal Genes Linking Genetic Variants to Vitiligo
To identify putative causal genes linking genetic variants to vitiligo pathogenesis, we performed a SMR analysis integrating cis-eQTL data with large-scale GWAS summary statistics, followed by HEIDI testing to prioritize likely causal associations over those driven by linkage (Figure 3A and Table S2). Because none of the tested genes remained significant after FDR correction (FDR < 0.05), we adopted an exploratory threshold of nominal pSMR < 0.05 combined with HEIDI P-value > 0.05, in line with previously published SMR studies.26 Using this criterion, this integrative genomics approach identified 602 genes whose expression levels were nominally associated with vitiligo risk (Figure 3B). Overlapping these risk-associated genes with the previously identified PCD-related gene set yielded a refined list, and further intersection analysis pinpointed two key PCD-associated genes (PARK7 and SLC1A5) as potential core targets linking genetic susceptibility to cell death dysregulation in vitiligo (Figure 3C and D).
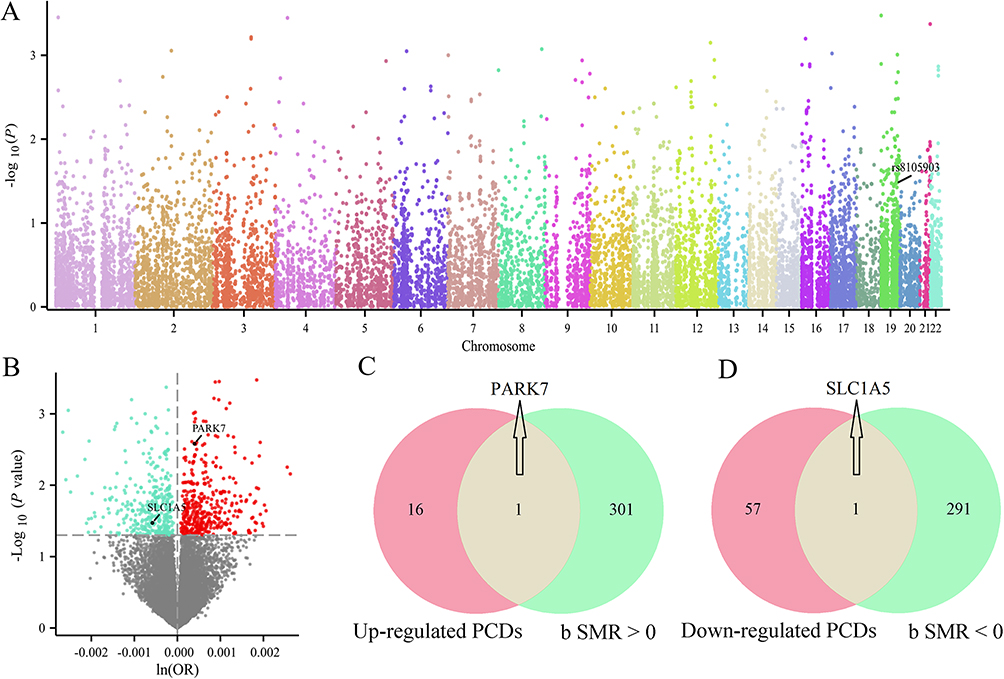 |
Figure 3 Integration of eQTL and GWAS data to identify genetically associated PCDs in vitiligo. (A) Manhattan plot displaying genome-wide SMR analysis results for cis-eQTLs against vitiligo risk using GWAS summary data. Each dot represents a single SNP-gene association; color-coding reflects chromosomal position. (B) Volcano plot of SMR p values versus ln(OR) estimates for gene-level associations with vitiligo risk. Venn diagrams showing the overlap between SMR-identified vitiligo-associated genes and (C) up-regulated PCDs-related DEGs, and (D) down-regulated PCDs-related DEGs. |
Regional Association and Colocalization Analysis
As shown in Table 1, colocalization analysis was further performed to assess whether the two identified PCD-related genes shared causal variants with vitiligo risk. For SLC1A5, the lead SNP rs8105903 yielded an odds ratio of 0.998 (95% CI: 0.998–0.999) with a SMR P-value of 0.034 and a HEIDI P-value of 0.476, indicating no evidence of pleiotropy, and the posterior probability of colocalization (PP.H4) reached 1.000, providing very strong statistical support that the GWAS and eQTL signals are consistent with a shared causal variant. For PARK7, the top SNP rs35675666 showed an OR of 1.000 (95% CI: 1.000–1.001), a SMR P-value of 0.003, a HEIDI P-value of 0.588, and a PP.H4 of 0.538, suggesting a moderate probability of colocalization. Together, these results support SLC1A5 as a genetically prioritized PCD-related candidate gene potentially involved in vitiligo pathogenesis, while acknowledging that colocalization alone does not establish biological causality or therapeutic actionability. To further illustrate the genetic colocalization at the SLC1A5 locus, we performed a detailed regional association analysis. Figure 4A presents a scatter plot comparing the strength of association (-log10(P-value)) for SNPs in the SLC1A5 region between the vitiligo GWAS and the eQTL data. The high correlation (r2) between the two trait associations, visually represented by the color gradient of the data points, suggests a shared genetic signal. The lead variant, rs8105903, is prominently positioned within this correlated cluster. This observation is further substantiated in Figure 4B, which shows the coordinated peak of association for both the vitiligo GWAS (ukb-a-115) and the SLC1A5 eQTL (eqtl-a-ENSG00000105281) precisely at the genomic position of rs8105903 on chromosome 19. The aligned peaks, coupled with a low local recombination rate in this interval, are consistent with the hypothesis that the same variant influences both SLC1A5 expression and vitiligo susceptibility, in line with the high posterior probability for colocalization, but should be interpreted as evidence for a shared genetic signal rather than definitive proof that SLC1A5 is the causal gene or a validated therapeutic target.
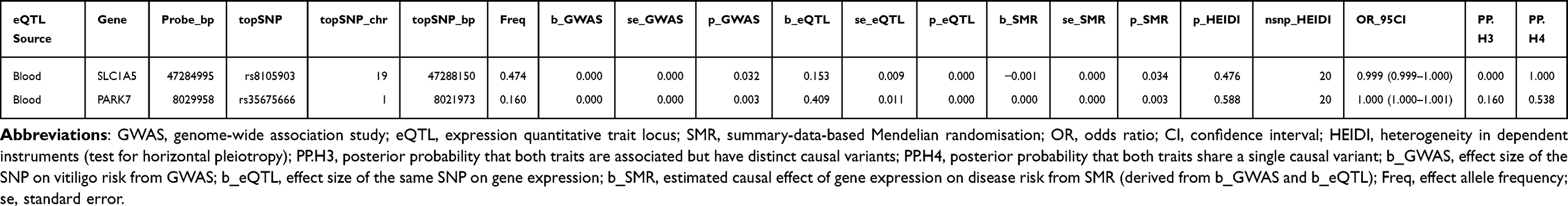 |
Table 1 Causal Associations and Colocalization Analysis of Potential Therapeutic Targets in Vitiligo |
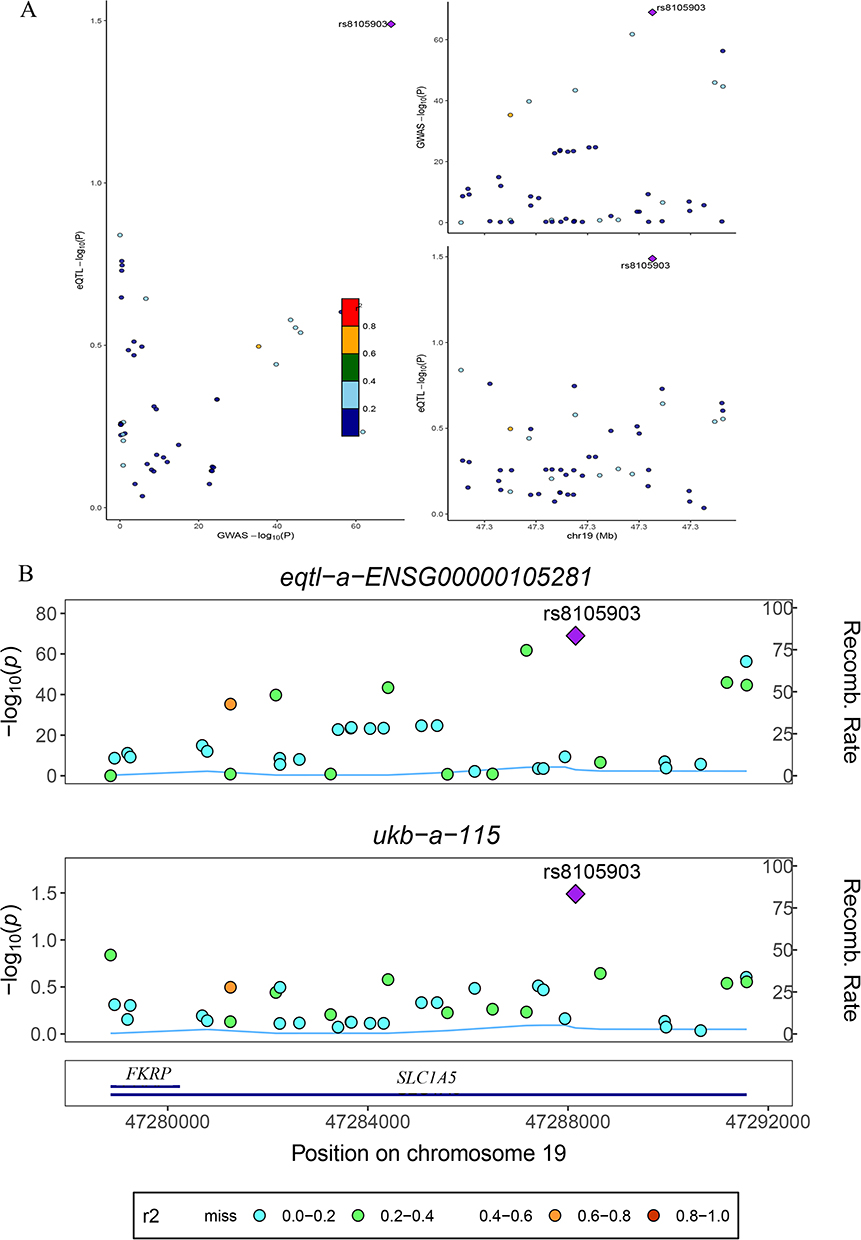 |
Figure 4 Regional association and colocalization analysis at the SLC1A5 locus. (A) Left: Scatter plot of the association strength (-log10(P-value)) for genetic variants in the SLC1A5 region, comparing the vitiligo GWAS (x-axis) and the SLC1A5 cis-eQTL (y-axis). Points are colored by their linkage disequilibrium (r2) with the sentinel variant rs8105903 (purple diamond), according to the color key. Right: Regional association plot for the vitiligo GWAS on chromosome 19. Each circle represents a variant, colored by LD with rs8105903; the purple diamond highlights the sentinel variant’s position. (B) Regional association plots for the SLC1A5 cis-eQTL study (eqtl-a-ENSG00000105281; top) and the vitiligo GWAS (ukb-a-115; bottom) across the same genomic region. In each plot, the left y-axis shows the –log10(P-value) for association, the right y-axis shows the local recombination rate, and points are colored by LD with rs8105903. The genomic position (Mb) is indicated on the x-axis. The bottom track displays the location and orientation of the SLC1A5 gene. The shared sentinel variant, rs8105903, is marked by a purple diamond in both plots. The color key for linkage disequilibrium (r2) is provided below the plots. |
Pathway Activity Profiling and Functional Enrichment Analysis of SLC1A5
To further investigate the functional impact of SLC1A5, the prioritized causal gene from our genetic analysis, we conducted pathway activity and single-gene enrichment analyses. As shown in Figure 5A, GSVA revealed distinct pathway activity profiles between Con and Vitiligo samples. Notably, pathways directly relevant to melanocyte biology and cellular homeostasis were dysregulated. The MELANOGENESIS and TYROSINE METABOLISM pathways were significantly downregulated, aligning with the core pathology of pigment loss. In our earlier analysis of eleven curated PCD gene sets, we observed that the AUTOPHAGY and LYSOSOME-DEPENDENT_CELL_DEATH pathways were downregulated in vitiligo (Figure 2C). By contrast, in the broader GO-based GSVA shown in Figure 5A, the REGULATION_OF_AUTOPHAGY pathway appeared upregulated. This apparent discrepancy reflects differences in gene set composition and analytic focus: the PCD-specific GSVA captures a decrease in autophagy/lysosomal cell death–related effector genes, whereas the REGULATION_OF_AUTOPHAGY gene set is enriched for upstream regulatory and stress-response components that may be activated despite reduced downstream autophagic execution. Concurrently, pathways associated with cellular stress and programmed cell death, including REGULATION_OF_AUTOPHAGY and NOD_LIKE_RECEPTOR_SIGNALING_PATHWAY, were upregulated, suggesting an activated state of pro-death signaling. Furthermore, a broad upregulation was observed in core metabolic and biosynthetic pathways such as RIBOSOME, OXIDATIVE_PHOSPHORYLATION, and CELL_CYCLE, indicating potential cellular dysfunction or metabolic reprogramming. Subsequently, single-gene enrichment analysis for SLC1A5 revealed that this gene positively correlated with several pathways, most notably glycosphingolipid biosynthesis with an enrichment score (ES) of 0.6092 and nominal P-value (NP) of 0.0000, followed by fructose and mannose metabolism (ES=0.5937, NP=0.0082), glycosaminoglycan degradation (ES=0.5135, NP=0.0388), tyrosine metabolism (ES=0.4525, NP=0.0220), amino sugar and nucleotide sugar metabolism (ES=0.4271, NP=0.0099), melanogenesis (ES=0.4008, NP=0.0060), and arginine and proline metabolism (ES=0.3662, NP=0.0414) (Figure 5B). These findings implicate SLC1A5 in the dysregulation of glycosphingolipid metabolism and melanin biosynthesis pathways, potentially linking genetic susceptibility to metabolic reprogramming in vitiligo.
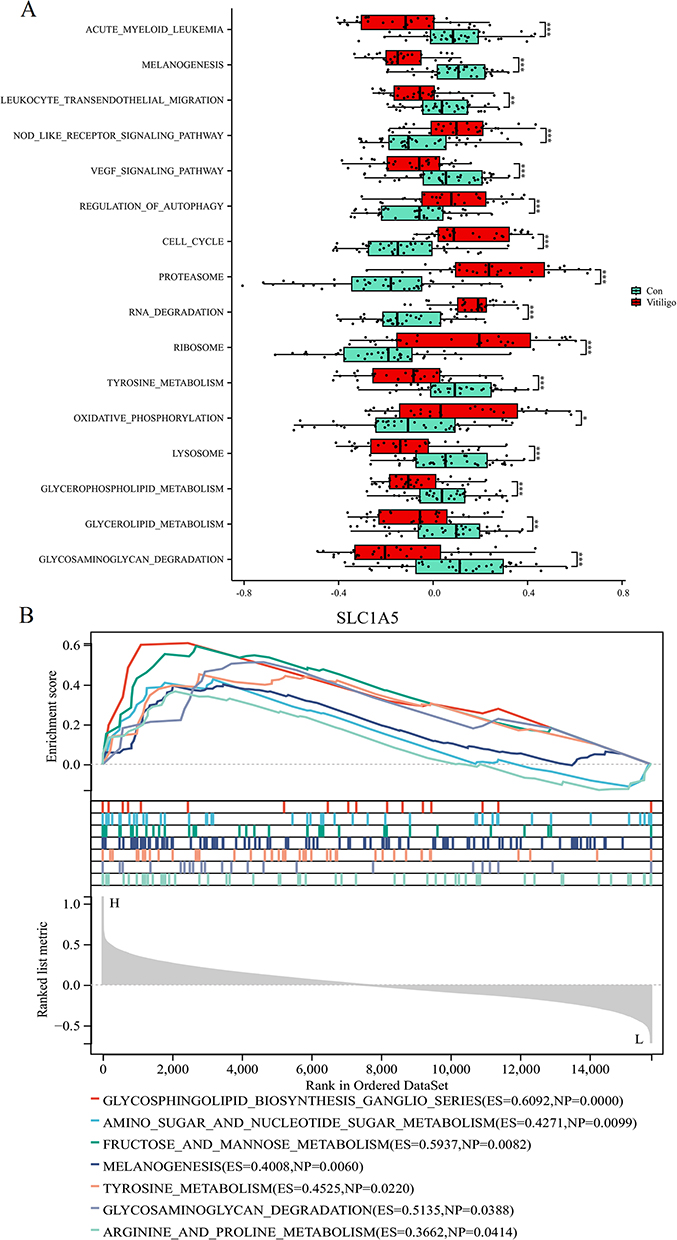 |
Figure 5 Pathway activity profiling and functional enrichment analysis of SLC1A5. (A) Box plots depicting the GSVA enrichment scores for selected biological pathways between control and vitiligo samples. *p < 0.05, **p < 0.01, ***p < 0.001. (B) Single-gene enrichment analysis for SLC1A5, showing enrichment scores and nominal P-values for the top correlated pathways. |
Immune Cell Infiltration Analysis and SLC1A5 Expression in Vitiligo
The immune cell infiltration analysis between vitiligo and control samples (Figure 6A) revealed significant differences in several immune cell subsets. Specifically, aDC, T cells, T helper cells, Tcm, and Th2 cells exhibited higher infiltration levels in vitiligo, while NK CD56bright cells, NK CD56dim cell levels, and Th17 cells appeared relatively reduced. Given that Th17-related pathways are frequently implicated in autoimmune diseases, the observed decrease in Th17 cells in this dataset should be interpreted with caution. This pattern may reflect characteristics of the included cohorts (eg, lesion site, disease stage, or treatment status), limitations of the deconvolution algorithm and gene signatures used to infer cell populations, or the possibility that Th17 involvement varies across different phases or subtypes of vitiligo Therefore, the Th17 result is considered exploratory rather than definitive. Correlation analysis between SLC1A5 expression and immune cell infiltration (Figure 6B) demonstrated that SLC1A5 is positively correlated with NK CD56bright cells, iDCs, and NK CD56dim cells, indicating its potential role in regulating immune microenvironments. However, negative associations were observed with certain T cell subsets such as Th2 cells and Tcm. Furthermore, differential expression analysis (Figure 6C) revealed a significant downregulation of SLC1A5 in vitiligo samples compared to controls. The ROC curve (Figure 6D) validated the diagnostic potential of SLC1A5 for distinguishing vitiligo from controls, with an AUC of 0.733, indicating moderate predictive accuracy. These findings suggest that SLC1A5 may play an essential role in immune regulation associated with vitiligo pathogenesis, although the inferred immune-cell alterations, require validation in independent cohorts and with orthogonal experimental methods.
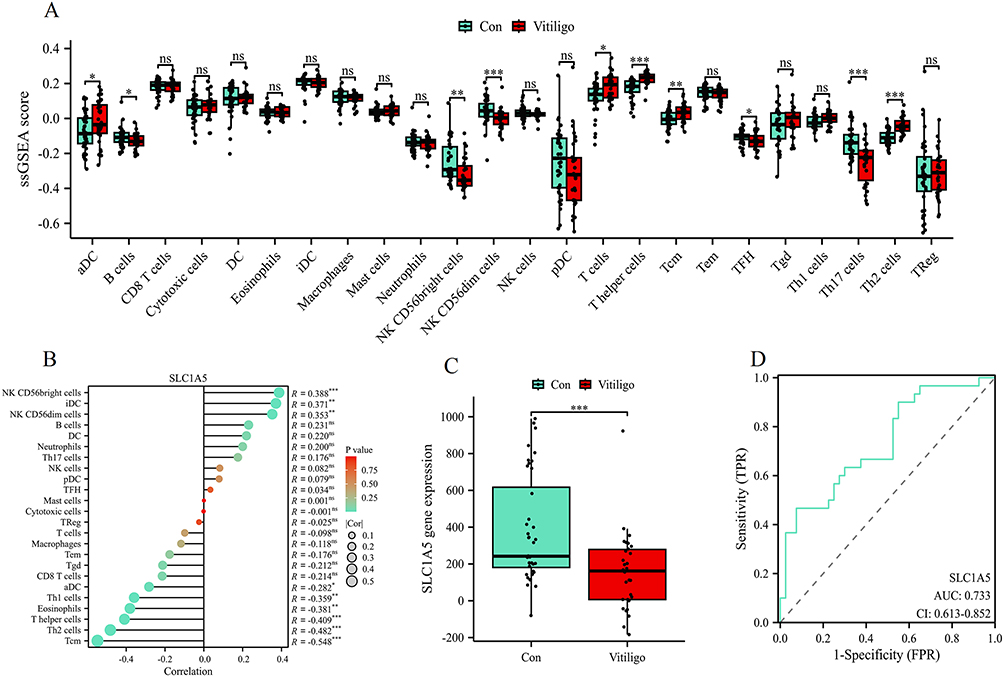 |
Figure 6 Immune cell infiltration analysis and SLC1A5 expression in vitiligo. (A) Immune cell infiltration levels in control and vitiligo groups analyzed using ssGSEA. ns indicates no statistically significant difference, *p < 0.05, **p < 0.01, ***p < 0.001. (B) Correlation between SLC1A5 expression and immune cell infiltration levels. (C) Expression analysis of SLC1A5 in control and vitiligo groups using the combined dataset. ***p < 0.001. (D) ROC curve evaluating the diagnostic performance of SLC1A5 in distinguishing vitiligo samples from controls. |
Discussion
This comprehensive bioinformatics analysis reveals a previously underappreciated role of programmed cell death mechanisms in vitiligo pathogenesis, with particular emphasis on the genetic and functional prioritization of SLC1A5. Our integrative approach, combining transcriptomic analysis, MR, and colocalization studies, provides computational evidence that dysregulated PCD pathways contribute to vitiligo susceptibility through genetically-anchored mechanisms involving amino acid transport and cellular metabolism.
The identification of 75 PCD-related DEGs in vitiligo skin samples, coupled with the enrichment of pyroptosis and cuproptosis signatures, suggests that inflammatory cell death pathways are aberrantly activated in this autoimmune condition.27 Particularly noteworthy is the concurrent downregulation of protective autophagy and lysosome-dependent clearance mechanisms, which may create a cellular environment conducive to melanocyte dysfunction and death. Through rigorous genetic analysis integrating cis-eQTL data with large-scale GWAS statistics, we prioritized SLC1A5 as a candidate gene linking genetic susceptibility to PCD dysregulation, supported by strong colocalization evidence at the rs8105903 variant. However, it is important to emphasize that colocalization indicates a shared genetic signal between SLC1A5 expression and vitiligo risk, and does not, by itself, prove that SLC1A5 is the causal gene or that it is therapeutically actionable.
Our findings align with and extend previous observations regarding the role of oxidative stress and cellular dysfunction in vitiligo pathogenesis. The enrichment of pyroptosis, a highly inflammatory form of cell death, is consistent with recent studies demonstrating elevated levels of inflammasome activation and IL-1β signaling in vitiligo lesions.8,28,29 The identification of cuproptosis enrichment represents a novel finding, suggesting that copper-mediated cell death mechanisms may contribute to melanocyte loss, potentially through disruption of mitochondrial metabolism and protein aggregation pathways. The downregulation of autophagy pathways observed in our analysis corroborates earlier reports indicating impaired autophagic clearance in vitiligo melanocytes.30,31 This dysfunction likely compromises the cellular capacity to manage oxidative stress and protein aggregation, thereby sensitizing melanocytes to various death stimuli. Our genetic prioritization of SLC1A5, encoding the neutral amino acid transporter ASCT2, provides a hypothesis-generating link between these cellular processes and inherited susceptibility factors.32 SLC1A5 has been extensively studied in cancer biology due to its role in glutamine transport and metabolic reprogramming.33 However, its involvement in autoimmune skin disorders represents a relatively unexplored area. Our functional enrichment analysis revealing SLC1A5’s positive correlation with glycosphingolipid biosynthesis and melanogenesis pathways suggests that this transporter may influence melanocyte survival through metabolic mechanisms rather than direct effects on pigment production. These inferences are based on bulk-tissue transcriptomic associations and should be interpreted as hypotheses requiring experimental validation in melanocyte and immune-cell models.
We propose several interconnected mechanisms through which SLC1A5 dysfunction might contribute to vitiligo pathogenesis. First, impaired glutamine transport could compromise cellular energy metabolism and antioxidant defense systems, as glutamine serves as a critical substrate for glutathione synthesis and the tricarboxylic acid cycle.34,35 This metabolic dysfunction may render melanocytes more susceptible to oxidative stress-induced cell death, particularly in the context of inflammatory immune responses.36 This scenario remains speculative at present and will need to be tested in vitro using melanocyte cultures and ex vivo patient samples. Second, our observation that SLC1A5 positively correlates with NK cell populations while showing negative associations with certain T helper cell subsets suggests a complex immunomodulatory role. Glutamine availability is crucial for T cell activation and proliferation,16 and SLC1A5 dysregulation may skew immune responses toward melanocyte-targeting effector functions. Nevertheless, these correlations are derived from deconvolution of bulk-tissue data and may be influenced by cell-composition changes; therefore, they should be viewed as indicative rather than definitive. The reduced SLC1A5 expression in vitiligo samples could represent either a consequence of chronic inflammation or a predisposing factor that alters local immune homeostasis. The strong correlation between SLC1A5 and glycosphingolipid biosynthesis pathways is particularly intriguing, as these lipid species play crucial roles in membrane stability, cellular signaling, and immune recognition.37 Dysregulated glycosphingolipid metabolism has been implicated in various autoimmune conditions,38,39 and our findings suggest that SLC1A5-mediated metabolic alterations may influence melanocyte membrane composition and susceptibility to immune-mediated destruction. Furthermore, the enrichment of cuproptosis signatures in vitiligo samples, combined with SLC1A5’s role in amino acid transport, raises the possibility that altered copper homeostasis contributes to disease pathogenesis. Copper is essential for tyrosinase activity and melanin synthesis, and dysregulated copper metabolism could simultaneously impair pigment production while promoting copper-dependent cell death pathways.40,41 All of these proposed links—glutamine metabolism, glycosphingolipid biosynthesis, copper homeostasis, and cuproptosis—should be considered working hypotheses generated by integrative analyses, which require confirmation in targeted functional assays and in vivo models.
An important alternative explanation for our observations is bulk-tissue confounding. The skin biopsies analyzed here contain mixtures of melanocytes, keratinocytes, fibroblasts, endothelial cells, and infiltrating immune cells. Reduced SLC1A5 expression in vitiligo lesions could therefore reflect a lower proportion of SLC1A5-expressing melanocytes or other cell types, rather than true downregulation within a specific population. Similarly, the inferred immune-cell differences, including changes in Th17, NK, and dendritic cell signatures, may partly arise from shifts in cellular composition or limitations of the gene-signature–based deconvolution method. Future studies integrating single-cell or spatial transcriptomics, along with cell-type–specific validation, will be necessary to disentangle cell-intrinsic expression changes from alterations in tissue architecture.
The moderate diagnostic accuracy of SLC1A5 expression suggests potential utility as a biomarker, though additional validation in larger cohorts is warranted. At this stage, SLC1A5 should be viewed as a promising candidate biomarker and mechanistic gene rather than a proven therapeutic target. More importantly, our identification of specific PCD pathways and their genetic determinants opens new avenues for therapeutic intervention. Targeting pyroptosis through NLRP3 inflammasome inhibitors or modulating copper homeostasis could represent novel treatment strategies for vitiligo.42,43 The metabolic dependencies revealed by our SLC1A5 analysis also suggest that nutritional interventions or metabolic modulators might influence disease progression.44 However, these potential interventions are speculative and remain to be tested in appropriate experimental systems, including melanocyte cultures, immune-cell models, patient-derived tissues, and animal models. Given the central role of glutamine in immune cell function, therapeutic approaches aimed at selectively modulating amino acid availability in skin microenvironments could potentially restore immune tolerance to melanocytes,15,16 but this concept requires systematic preclinical evaluation before any clinical application is considered.
Our study benefits from several methodological strengths, including the integration of multiple analytical approaches and the use of well-validated statistical methods for genetic colocalization. The GSVA-based pathway analysis provides robust assessment of functional themes, while the SMR approach with HEIDI testing effectively distinguishes causal associations from confounding linkage effects. However, several limitations should be acknowledged. First, the merged transcriptomic cohort is relatively small (40 controls, 30 lesional samples), and we did not have access to an independent external validation cohort. This limited sample size and lack of replication may reduce statistical power and increase the risk that some findings are dataset-specific. The cross-sectional nature of transcriptomic data limits causal inference regarding temporal relationships between gene expression changes and disease progression. Second, no experimental validation was performed in this study. We did not confirm SLC1A5 or other candidate genes at the mRNA or protein level using qPCR, Western blotting, or immunohistochemistry, nor did we assess their functional roles in melanocytes or immune cells through in vitro or in vivo assays. Sample metadata are incomplete across the three GEO datasets. Control matching for age, sex, and anatomical site was only reported for GSE53146; the other two datasets did not provide such matching. Moreover, the three datasets used different platforms (GPL570, GPL14951, GPL6884), introducing potential technical heterogeneity despite batch correction. No information on treatment status, disease activity, or vitiligo subtype could be retrieved from the original entries. Fourth, because our analyses were based on bulk-tissue transcriptomes, the observed changes in SLC1A5 expression and immune-cell signatures may partly reflect shifts in cell-type composition rather than genuine within-cell expression changes. No information on treatment status, disease activity, or vitiligo subtype could be retrieved from the original entries. Fifth, clinical metadata were limited: key variables such as disease stage, lesion site, treatment history, and vitiligo subtype were unavailable for most samples, precluding stratified analyses and potentially introducing uncontrolled clinical heterogeneity. Sixth, the GSVA, immune infiltration, and single-gene enrichment outputs should be interpreted as hypothesis-generating rather than definitive, given the multiple comparisons involved. The reliance on skin biopsy samples, while clinically relevant, may not fully capture the systemic nature of vitiligo pathogenesis or the heterogeneity of disease presentations across different anatomical sites. Seventh, the PCD gene sets and pathway definitions used in this study were curated from general databases and are not specifically tailored to skin or vitiligo. As a result, they may omit skin-specific regulators or include genes less relevant to melanocyte biology. Future work should refine these gene sets with tissue- and disease-specific information. Future studies incorporating single-cell RNA sequencing approaches could provide more detailed insights into cell type-specific expression patterns and intercellular communication networks. This work establishes a foundation for understanding the genetic architecture of PCD dysregulation in vitiligo and highlights SLC1A5 as a promising therapeutic target. Future investigations should focus on experimental validation of the proposed mechanisms through functional studies in melanocyte culture systems and animal models. Additionally, longitudinal studies examining the temporal dynamics of SLC1A5 expression and PCD pathway activation during disease progression could inform optimal timing for therapeutic interventions.
Conclusions
In conclusion, this study establishes a robust, genetics-informed framework for understanding the role of PCD in vitiligo. We have identified SLC1A5 as a genetically supported, PCD-associated candidate gene that may contribute to linking genetic predisposition to the metabolic and immunologic aberrations that drive vitiligo pathogenesis. The findings suggest a potential connection between genetic susceptibility, metabolic regulation, immune infiltration, and melanocyte dysfunction, but do not demonstrate direct causality. Rather than redefining vitiligo as a primary metabolic fragility of the melanocyte, our results generate the hypothesis that altered amino acid transport and associated metabolic pathways might modulate melanocyte vulnerability and local immune responses. This hypothesis-generating perspective opens new avenues for therapeutic exploration, while emphasizing that strategies aimed at modulating SLC1A5 activity or correcting downstream metabolic dysregulation will require rigorous experimental and clinical validation before their mechanistic or therapeutic relevance in vitiligo can be established.
Data Sharing Statement
The datasets analyzed in this study were obtained from publicly accessible databases, specifically the GEO database, with the following accession numbers: GSE65127, GSE53146, and GSE75819.
Ethics Approval and Consent to Participate
The present study made use of publicly available GWAS and GEO datasets. According to the Declaration of Helsinki (2024 revision) and China’s Measures for the Ethical Review of Life Science and Medical Research Involving Humans (2023), research that relies solely on fully anonymised and openly accessible data is exempt from further ethical review.
Funding
There is no funding to report.
Disclosure
All authors declare no conflicts of interest in this work.
References
1. Bergqvist C, Ezzedine K. Vitiligo: a review. Dermatology. 2020;236(6):571–16. doi:10.1159/000506103
2. Ezzedine K, Eleftheriadou V, Whitton M, van Geel N. Vitiligo. Lancet. 2015;386(9988):74–84. doi:10.1016/S0140-6736(14)60763-7
3. Krüger C, Schallreuter KU. A review of the worldwide prevalence of vitiligo in children/adolescents and adults. Int J Dermatol. 2012;51(10):1206–1212. doi:10.1111/j.1365-4632.2011.05377.x
4. Gauthier Y, Benzekri L. Non-cultured epidermal suspension in vitiligo: from laboratory to clinic. Indian J Dermatol Venereol Leprol. 2012;78(1):59–63. doi:10.4103/0378-6323.90947
5. Kapur S, Goyal E, Kumar A, Puria A, Raj R. Psychiatric morbidity among patients suffering from Vitiligo. Ind Psychiatry J. 2023;32(Suppl 1):S131–s135. doi:10.4103/ipj.ipj_220_23
6. Rork JF, Rashighi M, Harris JE. Understanding autoimmunity of vitiligo and alopecia areata. Curr Opin Pediatr. 2016;28(4):463–469. doi:10.1097/MOP.0000000000000375
7. Yang P, Luan M, Li W, et al. Single-cell transcriptomics reveals peripheral immune responses in non-segmental vitiligo. Front Immunol. 2023;14:1221260. doi:10.3389/fimmu.2023.1221260
8. Białczyk A, Wełniak A, Kamińska B, Czajkowski R. Oxidative stress and potential antioxidant therapies in vitiligo: a narrative review. Mol Diagn Ther. 2023;27(6):723–739. doi:10.1007/s40291-023-00672-z
9. Srivastava N, Gupta S, Parsad D. Melanocyte adhesion and apoptosis in vitiligo: linking puzzle blocks. Curr Mol Med. 2023;23(8):709–711. doi:10.2174/1566524022666220621125552
10. Sun J, Huang L, Wang J, Hu Y, Wang W, Zhu H. Programmed cell death in autoimmune diseases: ferroptosis. Ann Biol Clin. 2024;82(1):33–42. doi:10.1684/abc.2024.1866
11. Tang D, Kang R, Berghe TV, Vandenabeele P, Kroemer G. The molecular machinery of regulated cell death. Cell Res. 2019;29(5):347–364. doi:10.1038/s41422-019-0164-5
12. Gibellini L, Moro L. Programmed Cell Death in Health and Disease. Cells. 2021;10(7):1765. doi:10.3390/cells10071765
13. Jin Y, Birlea SA, Fain PR, et al. Genome-wide association analyses identify 13 new susceptibility loci for generalized vitiligo. Nat Genet. 2012;44(6):676–680. doi:10.1038/ng.2272
14. Quan C, Ren YQ, Xiang LH, et al. Genome-wide association study for vitiligo identifies susceptibility loci at 6q27 and the MHC. Nat Genet. 2010;42(7):614–618. doi:10.1038/ng.603
15. Sinclair LV, Rolf J, Emslie E, Shi YB, Taylor PM, Cantrell DA. Control of amino-acid transport by antigen receptors coordinates the metabolic reprogramming essential for T cell differentiation. Nat Immunol. 2013;14(5):500–508. doi:10.1038/ni.2556
16. Nakaya M, Xiao Y, Zhou X, et al. Inflammatory T cell responses rely on amino acid transporter ASCT2 facilitation of glutamine uptake and mTORC1 kinase activation. Immunity. 2014;40(5):692–705. doi:10.1016/j.immuni.2014.04.007
17. Ren W, Rajendran R, Zhao Y, et al. Amino acids as mediators of metabolic cross talk between host and pathogen. Front Immunol. 2018;9:319. doi:10.3389/fimmu.2018.00319
18. Ye J, Kumanova M, Hart LS, et al. The GCN2-ATF4 pathway is critical for tumour cell survival and proliferation in response to nutrient deprivation. EMBO J. 2010;29(12):2082–2096. doi:10.1038/emboj.2010.81
19. Zhang PA, Wang JL, Dong MH, et al. Genetic influence of the brain imaging phenotypes, brain and cerebrospinal fluid metabolites and brain genes on migraine subtypes: a Mendelian randomization and multi-omics study. J Headache Pain. 2025;26(1):124. doi:10.1186/s10194-025-02063-7
20. Xia L, Wang J, Gao J, et al. Integrative multi-omics analysis identifies key genes and colocalized signals associated with colorectal cancer risk. BMC Cancer. 2025;25(1):1372. doi:10.1186/s12885-025-14798-2
21. Wei Q, Jiang X, Miao X, Zhang Y, Chen F, Zhang P. Molecular subtypes of lung adenocarcinoma patients for prognosis and therapeutic response prediction with machine learning on 13 programmed cell death patterns. J Cancer Res Clin Oncol. 2023;149(13):11351–11368. doi:10.1007/s00432-023-05000-w
22. Zhu Z, Zhang F, Hu H, et al. Integration of summary data from GWAS and eQTL studies predicts complex trait gene targets. Nat Genet. 2016;48(5):481–487. doi:10.1038/ng.3538
23. Kurki MI, Karjalainen J, Palta P, et al. FinnGen provides genetic insights from a well-phenotyped isolated population. Nature. 2023;613(7944):508–518. doi:10.1038/s41586-022-05473-8
24. Song ZQ, Xu YP, Chen YQ, et al. Proteome-wide Mendelian randomization and colocalization analyses identify novel protein targets for cardiac conduction disorders. J Cardiol. 2026;87(2):180–189. doi:10.1016/j.jjcc.2025.07.005
25. Bindea G, Mlecnik B, Tosolini M, et al. Spatiotemporal dynamics of intratumoral immune cells reveal the immune landscape in human cancer. Immunity. 2013;39(4):782–795. doi:10.1016/j.immuni.2013.10.003
26. Yu Z, Meng X, Zong Z, Song Q, Huo Y, Chen H. Exploring the causal biological association between mitochondrial genes and carotid plaques: a multiomics Mendelian randomization study. Atherosclerosis. 2026;413:120570. doi:10.1016/j.atherosclerosis.2025.120570
27. Sasaki K, Himeno A, Nakagawa T, Sasaki Y, Kiyonari H, Iwai K. Modulation of autoimmune pathogenesis by T cell-triggered inflammatory cell death. Nat Commun. 2019;10(1):3878. doi:10.1038/s41467-019-11858-7
28. Geng Y, Huang P, Cui T. Mechanisms of pyroptosis in vitiligo. Tissue Cell. 2025;97:103081. doi:10.1016/j.tice.2025.103081
29. Zhang D, Li Y, Du C, et al. Evidence of pyroptosis and ferroptosis extensively involved in autoimmune diseases at the single-cell transcriptome level. J Transl Med. 2022;20(1):363. doi:10.1186/s12967-022-03566-6
30. Zeng K, Zhu Y, Han Z, et al. NLRP3 autophagic degradation disruption in melanocytes contributes to vitiligo development. Cell Death Differ. 2026;33(2):343–357. doi:10.1038/s41418-025-01578-5
31. Bastonini E, Kovacs D, Raffa S, et al. A protective role for autophagy in vitiligo. Cell Death Dis. 2021;12(4):318. doi:10.1038/s41419-021-03592-0
32. Scalise M, Pochini L, Console L, Losso MA, Indiveri C. The Human SLC1A5 (ASCT2) Amino Acid Transporter: from Function to Structure and Role in Cell Biology. Front Cell Dev Biol. 2018;6:96. doi:10.3389/fcell.2018.00096
33. Yoo HC, Park SJ, Nam M, et al. A variant of SLC1A5 is a mitochondrial glutamine transporter for metabolic reprogramming in cancer cells. Cell Metab. 2020;31(2):267–283.e212. doi:10.1016/j.cmet.2019.11.020
34. Yoo HC, Yu YC, Sung Y, Han JM. Glutamine reliance in cell metabolism. Exp Mol Med. 2020;52(9):1496–1516. doi:10.1038/s12276-020-00504-8
35. Lu H, Li X, Lu Y, Qiu S, Fan Z. ASCT2 (SLC1A5) is an EGFR-associated protein that can be co-targeted by cetuximab to sensitize cancer cells to ROS-induced apoptosis. Cancer Lett. 2016;381(1):23–30. doi:10.1016/j.canlet.2016.07.020
36. Dell’anna ML, Picardo M. A review and a new hypothesis for non-immunological pathogenetic mechanisms in vitiligo. Pigment Cell Res. 2006;19(5):406–411. doi:10.1111/j.1600-0749.2006.00333.x
37. Vartabedian VF, Savage PB, Teyton L. The processing and presentation of lipids and glycolipids to the immune system. Immunol Rev. 2016;272(1):109–119. doi:10.1111/imr.12431
38. Dasgupta S, Ray SK. Diverse biological functions of sphingolipids in the CNS: ceramide and sphingosine regulate myelination in developing brain but stimulate demyelination during pathogenesis of multiple sclerosis. J Neurol Psychol. 2017;5(1). doi:10.13188/2332-3469.1000035
39. Jana A, Pahan K. Sphingolipids in multiple sclerosis. Neuromolecular Med. 2010;12(4):351–361. doi:10.1007/s12017-010-8128-4
40. Portis IG, de Sousa Lima P, Paes RA, et al. Copper overload in Paracoccidioides lutzii results in the accumulation of ergosterol and melanin. Microbiol Res. 2020;239:126524. doi:10.1016/j.micres.2020.126524
41. Pretzler M, Rompel A. Tyrosinases: a family of copper-containing metalloenzymes. Chemtexts. 2024;10(4):12. doi:10.1007/s40828-024-00195-y
42. Coll RC, Robertson AA, Chae JJ, et al. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat Med. 2015;21(3):248–255. doi:10.1038/nm.3806
43. Van Gorp H, Lamkanfi M. The emerging roles of inflammasome-dependent cytokines in cancer development. EMBO Rep. 2019;20(6). doi:10.15252/embr.201847575
44. Tsvetkov P, Coy S, Petrova B, et al. Copper induces cell death by targeting lipoylated TCA cycle proteins. Science. 2022;375(6586):1254–1261. doi:10.1126/science.abf0529
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Integration of Single Cell and Bulk RNA-Sequencing Reveals Key Genes and Immune Cell Infiltration to Construct a Predictive Model and Identify Drug Targets in Endometriosis
Zhang H, Fang Y, Luo D, Li YH
Journal of Inflammation Research 2025, 18:2783-2804
Published Date: 25 February 2025