Back to Journals » The Application of Clinical Genetics » Volume 13
Integrative and Analytical Review of the 5-Alpha-Reductase Type 2 Deficiency Worldwide
Authors Batista RL, Mendonca BB ![]()
Received 3 December 2019
Accepted for publication 20 February 2020
Published 14 April 2020 Volume 2020:13 Pages 83—96
DOI https://doi.org/10.2147/TACG.S198178
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Martin Maurer
Video abstract presented by Rafael Loch Batista.
Views: 2111
Rafael Loch Batista, Berenice Bilharinho Mendonca
Unidade de Endocrinologia do Desenvolvimento, Laboratório de Hormônios e Genética Molecular/LIM42, Hospital das Clínicas, Disciplina de Endocrinologia, do Departamento de Clínica Médica, Faculdade de Medicina da Universidade de São Paulo, São Paulo, Brazil
Correspondence: Berenice Bilharinho Mendonca Eneas Carvalho de Aguiar, 255, São Paulo, SP 05403-000, Brazil
Tel +55 1126617512
Email [email protected]
Introduction: The conversion of testosterone into dihydrotestosterone is catalyzed by the 5α-reductase type 2 enzyme which plays a crucial role in the external genitalia virilization. It is encoded by the SRD5A2 gene. Allelic variants in this gene cause a 46,XY DSD with no genotype–phenotype relationship. It was firstly reported in the early 70s from isolated clusters. Since then, several cases have been reported. Putting together, it will expand the knowledge on the molecular bases of androgen milieu.
Methods: We searched for SRD5A2 allelic variants (AV) in the literature (PubMed, Embase, MEDLINE) and websites (ensembl, HGMD, ClinVar). Only cases with AV in both alleles, either in homozygous or compound heterozygous were included. The included cases were analyzed according to ethnicity, exon, domain, aminoacid (aa) conservation, age at diagnosis, sex assignment, gender reassignment, external genitalia virilization and functional studies. External genitalia virilization was scored using Sinnecker scale. Conservation analysis was carried out using the CONSURF platform. For categorical variables, we used X2 test and Cramer’s V. Continuous variables were analyzed by t test or ANOVA. Concordance was estimated by Kappa.
Results: We identified 434 cases of 5ARD2 deficiencies from 44 countries. Most came from Turkey (23%), China (17%), Italy (9%), and Brazil (7%). Sixty-nine percent were assigned as female. There were 70% of homozygous allelic variants and 30% compound heterozygous. Most were missense variants (76%). However, small indels (11%), splicing (5%) and large deletions (4%) were all reported. They were distributed along with all exons with exon 1 (33%) and exon 4 (25%) predominance. Allelic variants in the exon 4 (NADPH-binding domain) resulted in lower virilization (p< 0.0001). The codons 55, 65, 196, 235 and 246 are hotspots making up 25% of all allelic variants. Most of them (76%) were located at conserved aa. However, allelic variants at non-conserved aa were more frequently indels (28% vs 6%; p< 0.01). The overall rate of gender change from female to male ranged from 16% to 70%. The lowest rate of gender change from female to male occurred in Turkey and the highest in Brazil. External genitalia virilization was similar between those who changed and those who kept their assigned gender. The gender change rate was significantly different across the countries (V=0.44; p< 0.001) even with similar virilization scores.
Conclusion: 5ARD2 deficiency has a worldwide distribution. Allelic variants at the NADPH-ligand region cause lower virilization. Genitalia virilization influenced sex assignment but not gender change which was influenced by cultural aspects across the countries. Molecular diagnosis influenced on sex assignment, favoring male sex assignment in newborns with 5α-reductase type 2 deficiency.
Keywords: SRD5A2, 46XY DSD, differences of sexual development, atypical genitalia, dihydrotestosterone, 5α-reductase type 2 deficiency
Introduction
In 1961, Nowakowski e Lenz named as pseudovaginal perineoescrotal hypospadias an disorder of sex development affecting individuals with 46,XY karyotype.1 The reported phenotype included female-like external genitalia, bilateral testes, and male urogenital tracts in which the ejaculatory ducts terminate in a blind-ending vagina. Subsequently, animal studies showed that male external genitalia virilization resulted from the conversion of testosterone into dihydrotestosterone, a reaction catalyzed by 5α-reductase enzyme.2 The 5α-reductase type 2 deficiency syndrome was biochemically and clinically reported in 24 individuals from Dominican Republic and two siblings from North America, concomitantly.3,4 Typically, affected individuals presented virilization (clinical and psychological) at puberty with no gynecomastia. Both studies characterized this syndrome as an autosomal recessive inheritance pattern condition, resulting from the inability to convert testosterone into dihydrotestosterone presenting with a wide range of genital ambiguity at birth and pronounced virilization at puberty.
Defects in the 5α-reductase type 2 enzyme arise from mutations in the SRD5A2 gene.5,6 This gene is made up of five exons and four introns and allelic variants have been reported in the whole gene. Impairment in the 5α-reductase type 2 enzymatic activity results from either homozygous or compound heterozygous allelic variants. Initially, this disorder was reported in clusters around the world in individuals from specific ethnic groups. There are growing evidence reporting affected individuals with a variety of ethnic backgrounds and coming from several geographical areas, suggesting that 5α-reductase type 2 deficiency has a worldwide distribution.
The 5α-reductase type 2 defects have been reported in individuals with several degrees of undervirilization, ranging from typical female external genitalia to hypospadias or isolated micropenis. The causes of divergent phenotypes are still unclear.
Intriguingly, 5α-reductase type 2 deficiency is reported with an extensive phenotype variability, even in affected individuals carrying the same SRD5A2 mutation.7,8 It suggests that factors others than residual 5α-reductase type 2 activity may play a role on 5α-reductase type 2 deficiency phenotype.
This review is focused on allelic variants and polymorphisms in the SRD5A2 gene, molecular mechanisms of 5α-reductase type 2 deficiency and in the genotype–phenotype correlation of this syndrome.
Methods
We searched for SRD5A2 allelic variants in the literature (PubMed, Embase, MEDLINE) and websites (ensemble, HGMD, ClinVar). Only individuals with variants in both alleles (either homozygous or composed heterozygous) and 5α- reductase type 2 deficiency phenotype were included. The variants were analyzed according to ethnicity, exon, domain, aminoacid conservation, age at diagnosis, sex assignment, gender change, external genitalia virilization and functional studies. External genitalia virilization was scored using Sinnecker, which ranges from 1 to 5.9 As Sinnecker’s scores 1–4 are divided into two points (a and b), the whole scale is made up of 9 points. The score of external genitalia virilization was analyzed as a Likert-scale. Based on a previous retrospective study showing a predominance of male sex assignment after 1999,10 all cases included were divided between those born before 1999 and those born after 1999. They were also divided according to country income based on the 2018 World Bank classification (www.worldbank.org). This classification is based on the gross national income (GIN) being divided into four categories: low-income economies are defined as those with a GNI per capita of $1025 or less; lower middle-income economies are those with a GNI per capita between $1026 and $3995; upper-middle-income economies are those with a GNI per capita between $3996 and $12,375; and high-income economies are those with a GNI per capita of $12,376 or more.
Categorical variables were analyzed using Chi-square test followed by the Cramer’s V. Continuous variables were analyzed either by Student t test or ANOVA one way. The Bonferroni’s test was applied for multiple comparisons. Estimative of concordance was made by Kappa index. A p<0.05 was considered as significant.
Steroids and Male Development
Testosterone (T) is the most abundant androgen in the serum. T is synthesized by the Leydig cells of the testes under control of LH.11,12 In male fetuses, T binds to the androgen receptor (AR) and promote the differentiation of Wolffian duct into male internal genitalia, including epididymis, vas deferens, and seminal vesicles. The complex T plus AR is also essential to induce male psychosexuality which starts early during embryo development.13–15 Intracellularly, T is converted into dihydrotestosterone (DHT), a more potent androgen which has 2–5 times higher affinity for AR and 10-fold higher potency of inducing AR signaling than T. In utero, DHT is crucial for growth of the prostate gland and for differentiation of the external genitalia into male genitalia.16,17 In other words, the external genitalia virilization depends on a functioning AR and DHT which, by it is turn, depends on T as substrate for conversion.
Steroids are a particular type of lipids, that are 5α-reduced into more potent steroids by 5α-reductases.18 Basically, substrates for 5α-reductases are 3-oxo (3-keto), Δ 4,5 C19/C21 steroids which include testosterone, progesterone, cortisol and aldosterone as examples. The reaction involves a stereospecific, irreversible breakage of the double bond between carbons 4 and 5 (delta 4,5) with the aid of NADPH cofactor and the insertion of a hydride anion to the α face at carbon C-5 (5α reduction).18,19 Outside of DHT, much of the physiological role of 5α-reduced steroids are still unknown.
The 5α-Reductases Enzymes and 5α-Reductases Genes
5α-RD1 and 5α-RD2 isoenzymes are membrane-associated (microsomal) enzymes, composed of 259 and 254 aminoacids, respectively. Both enzymes catalyze the same reaction (5α-steroid reduction), but they only share a limited degree of homology in protein sequence, are located on different chromosomes, play distinctive biochemical roles and are expressed in different tissues.20,21 5α-RD1 is expressed in fetal scalp and nongenital skin. The 5α-RD1 expression is from 5 to 50 times higher in adults than in fetus, suggesting that enzyme is not related to male fetal development.21 On the other hand, 5α-RD2 is highly expressed in fetal prostates. After birth, 5α-RD1 is expressed in more locations, including the liver, skin, scalp and prostate while 5α-RD2 is expressed in prostate, seminal vesicles, epididymis, liver.18
Their role in mammalian male physiology comes from developmental studies of mammalian embryos showing that 5α-reduction was highest in the prostate and external genitalia prior to their virilization, but very low in Wolffian duct structures.22,23 Thereafter, a generalized defect in the conversion of T to DHT was demonstrated in individuals with a rare disorder of male sex differentiation,3,4 subsequently referred to as 5α-reductase deficiency. Later, a cDNA from rat liver was used to isolate a human 5α-RD cDNA by cross-hybridization with a prostate cDNA library. Further genetic studies in individuals with 5α-reductase deficiency identified a second cDNA from the human prostatic 5α-R.20,24 The first cDNA (from rat liver) was named 5α-RD1 and the second cDNA (from human prostate) was named 5α-RD2.
5α-RD1 and 5α-RD2 isoenzymes are encoded by SRD5A1 and SRD5A2 genes, respectively. Both genes have similar structures, with five coding exons separated by four introns.5,25 The position of the introns is essentially identical. They share approximately 60% of sequence identity, indicating the possibility of a common precursor gene during evolution. The SRD5A1 gene is located on chromosome 5p15 and encodes a 259 amino acid protein (5α-reductase type 1) whereas the SRD5A2 gene is located on 2p23 and encodes a 254 amino acid protein (5α-reductase type 2).2,19 More recently, with the development of genome-wide gene expression profile analysis, a third 5α-RD gene (SRD5A3) was identified, located at 4q12. This gene encodes a 318 aminoacids protein (5α-reductase type 3), which has 19% of homology with 5α-RD1 and 20% of homology with 5α-RD2.
All SRD genes are implicated in human disorders. The main one is the SRD5A2 gene in which several allelic variants have been reported in individuals with 5α-reductase type 2 deficiency, a rare difference of sex differentiation among 46,XY individuals resulting from defective conversion from T to DHT.5,6,8,21,26
The role of the others 5α-reductases in human diseases are still not fully understood. 5α-reductases isoenzymes irreversibly catalyze A-ring reduction of pregnene-based steroids, which includes glucocorticoids and androgens. As 5α-RD1 is highly expressed in liver, 5α-RD1 disruption could impact on steroid metabolism. It was tested in female mice with transgenic disruption of 5α-reductase type 1 (5αRD1-KO) in which 5α-RD1 deficiency resulted in glucocorticoid clearance impairment, predisposes to glucose intolerance and hepatic steatosis upon metabolic challenge.27 The hypothesis that inhibition of 5α-R1 causes metabolic dysfunction in humans was tested in a double-blind randomized controlled parallel study comparing the insulin effects of using finasteride (which inhibits only 5α-RD2), and dutasteride (5α-RD1 and 5α-RD2 inhibitor). Dutasteride was able to modulate insulin sensitivity in human peripheral tissues.28
The role of 5α-RD3 in human diseases is even more recent. SRD5A3 encodes a polyprenol reductase enzyme required for the synthesis of dolichol, a final product of the mevalonate pathway.29 Biallelic mutations in SRD5A3 gene have been reported in individuals with congenital disorders of glycosylation, eventually associated with ophthalmological and neurological features.30,31
Phenotype, Ethnicity and Sex of Rearing
We identified 434 cases of 5α-reductase type 2 deficiency in the literature from 44 different countries (Table 1), which means that this condition has a worldwide distribution. It is noteworthy that many cases have recently been reported in China and Turkey,32–34 besides reports in countries without no previous cases, as Bulgaria.8 Neonatal diagnosis was carried out in 29.7%. Most cases had the 5α-reductase type 2 deficiency diagnosis later in life (mean 12.56 ± 8.41, from 1 to 47 years of age). The diagnosis was done at childhood in 58%, at puberty in 25%, and adulthood in 17%.
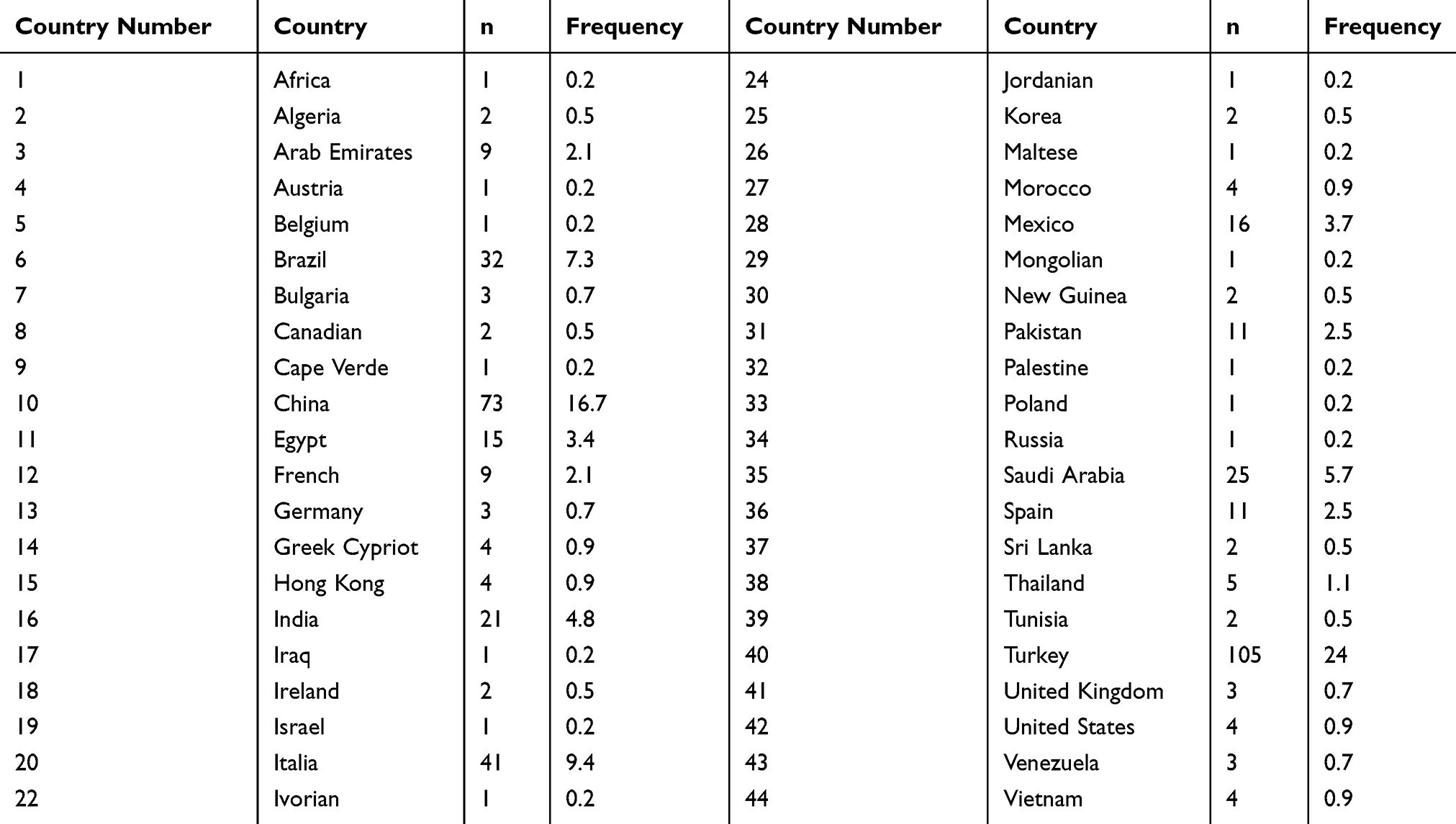 |
Table 1 Country of Birth of the Individuals with 5α-Reductase Type 2 Deficiency Included in This Review |
Most cases were assigned as female (69.4%). The association between the score of external genitalia virilization and sex assignment was significant (p<0.001). This score was higher among those who were assigned as female (6.48 ± 1.82) versus those assigned as male (4.66 ± 1.89), suggesting that external genitalia appearance influenced the choice of assigned sex.
When we divided the cases into those who were diagnosed after and before 1999, the percentage of male sex assignment rise from 26.8% to 42.8% (p<0.0001, X2=17.79). The estimated odds ratio for female sex assignment for individuals with diagnosis after 1999 was 1.7 (1.03–2.83). In the same direction, the rate of sex reassignment from female to male was lower in those who were diagnosed after 1999 (p=0.036, X2=4.39).
In fact, there are a clear temporal trends pointing toward an increased likelihood of affected 46,XY DSD being raised as boys.10 According to our analysis, it is also true for 5α-reductase type 2 deficiency individuals. This probably results of several studies that have shown that many individuals with 46,XY have a male psychosexuality regardless the external genitalia appearance,13,35 especially those with DSD due disorders of androgen synthesis, such as 5α-reductase type 2 deficiency and 17β-Hydroxysteroid dehydrogenase type 3 deficiency.10,13,36,37
To evaluate an eventual impact of income on sex assignment, we divided the patients according to the country’s income. Surprisingly, there was no impact of income on sex designation (p=0.21). However, it is interesting to note that no cases have been reported in countries classified as low-income. The vast majority of cases (86%) were reported by countries with high and upper-middle economies. The absence of reports from low-income countries is maybe due to several reasons, such as barriers for molecular diagnosis, scientific access, and specialized medical assistance.
As molecular diagnosis is valuable for 46,XY DSD management, it is fast becoming the first-line approach for DSD newborns.12,38-40 However, genetic testing is still not available everywhere. As important as molecular advancement is also to make this genetic test easy and feasible for any newborn, anywhere in the world.
Genotype
We found 129 different allelic variants in the SRD5A2 gene in the literature among individuals with 5α-reductase type 2 deficiency. Most are missense mutations (n = 83), but small deletions (n = 12), splicing mutations (n = 6), stop codons (n = 4), small indels (n = 20) and gross deletions (n = 4) have also been described (Human Gene Mutation Database at the Institute of Medical Genetics in Cardiff, Wales, UK: SRD5A2 gene: http://www.hgmd.cf.ac.uk, Clinvar and PubMed) (Figure 1). These variants have been reported in all exons of this gene, but most mutations are mainly located at exons 1 (33%) and at exon 4 (25%), similarly to previously reported.7,25,41 Among the 254 amino acids that make up the 5α-RD2 protein, we found allelic variants in the SRD5A2 gene in 76 of them.
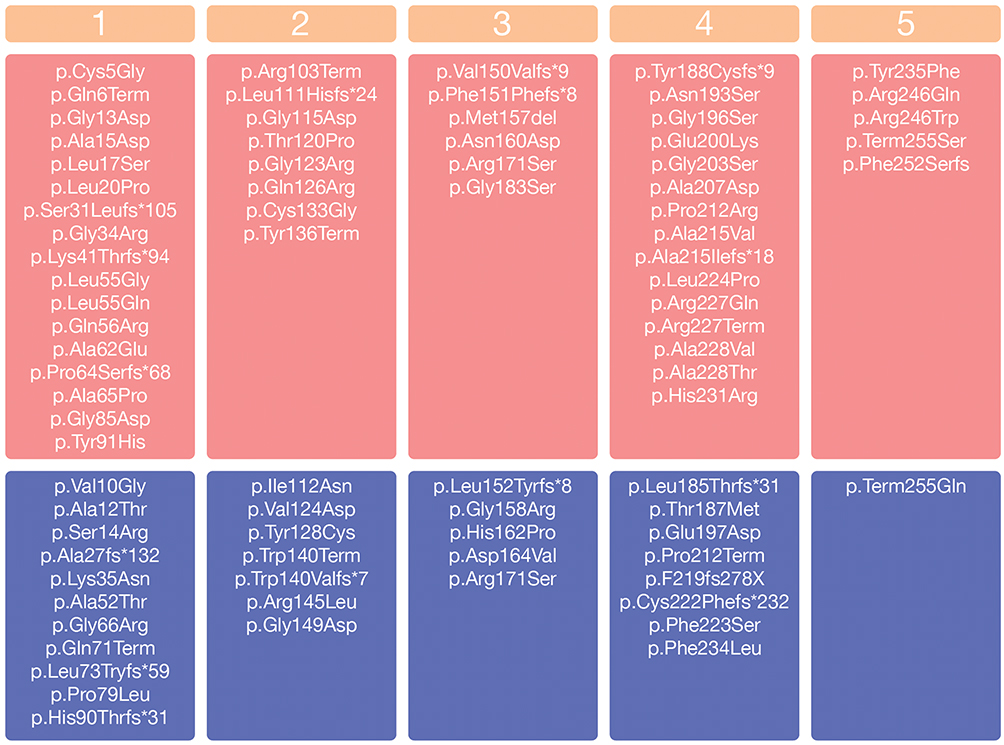 |
Figure 1 Allelic variants in the SRD5A2 related to 5α-reductase type 2 deficiency. The SRD5A2 exons are defined from 1 to 5. Variants in homozygous are in the pinkish-red boxes. Variants reported only as compound heterozygous are in the blue boxes. Note: *It indicates the position of the premature stop codon. |
5α-R2 deficiency is inherited in an autosomal recessive pattern. As in other recessive conditions, the consanguineous population presented a higher frequency of this disease. 5α-R2 deficiency results either from homozygous defects in the SRDA2 gene or compound heterozygosity. Allelic variants in homozygosity are more frequent than compound heterozygous among affect individuals with 5α-R2 deficiency.5,6,42 We identified 70% (305 out 434) of AV in the SRDA2 gene causing 5α-reductase type 2 deficiency in homozygosity and 30% (129 out 434) in composed heterozygosity, reinforcing that most 5α-reductase type 2 deficiency cases are homozygous. This rate is in agreement with a previous study which includes 55 individuals with 5α-R2 deficiency and also with other 5α-RD2 deficiency review.7,43
The positions 196, 227, 235 and 246 are hotspots of the SRD5A2 gene. Collectively, they make up 25% of all AV reported as causative of 5α-reductase type 2 deficiency. These hotspots are suggested based on some SRD5A2 gene mutations that have been reported in individuals from different countries and from different ethnicities. As an example, the p.Arg246Gln mutation was reported in India, Austria, Brazil, Italy, Korea, Pakistan, Dominican Republic, Egypt, China, Saudi Arabia and Mexico6,42,44-48 as well as the p.Gly196Ser in Turkey, China, Bulgaria, Italy, and the United Kingdom.8,33,49-51 On the other hand, many SRD5A2 mutations remain from specific ethnicities, such as p.Pro59Arg from Algeria, p.Asn160Asp from Egypt, c.188_189insTA from India, c.453delC from Italy, p.Gly183Ser from Brazil and p.Ala65Pro from Turkey.7,33,52-54 Therefore, ethnicity does not seem to impact on phenotype–genotype correlation.
Functional Studies on Allelic Variants of the SRD5A2 Gene
Despite a very well-characterized disease, functional studies of the SRD5A2 allelic variants are uncommon. Most reports have been focused in clinical phenotype, laboratory data, and SRD5A2 sequencing whereas the functional consequences of the SRD5A2 variants remain unexplored. As a consequence, only 40 SRD5A2 allelic variants were functionally investigated for their deleterious potential (Table 2). Among the allelic variants that were investigated, most are non-synonymous allelic variants (36 out 40), in all exons (Table 3).
 |
Table 2 Functional Studies of Allelic Variants of the SRD5A2 in Patients with 5α-Reductase Type 2 Deficiency |
 |
Table 3 Sinnecker’s Score* |
The 5α-RD2 is a membrane-bound enzyme that catalyzes the irreversible conversion of testosterone into dihydrotestosterone using NADPH as cofactor.21,55 Based on that, most of the functional studies focused on enzymatic kinetics studies. Enzymatic activity is estimated by Vmax and most non-synonymous 5α-RD2 allelic variants affected the Vmax of 5α-RD2 enzyme (Table 3). Sixteen of these variants result in a protein with no detectable enzyme activity, whereas the remaining give rise to proteins with severely decreased enzymatic activity (Table 3). The variants able to impair the 5α-RD2 activity can be divided into two groups: those that affect the affinity for NADPH and those that affect the ligand-binding (testosterone).6,19,55-57 However, the impact of the residual activity on phenotype is controversial. Allelic variants with 0% of residual function (p.Leu55Gly and p.Arg246Gln) resulted in several degrees of external genitalia virilization,7,51,57 but allowing some degree of external genitalia virilization even without 5α-RD2 residual function.
A 3D protein model for SRD5A2 protein was recently constructed.58 That model suggests that the SRD5A2 protein is comprised of six α-helices and two smaller loops corresponding to transmembrane domains. According to this model, the residues Y98, N102, Y107, L167, R171, H231, and Y235 are in direct contact with NADPH in the binding cavity. Among these residues, three were recurrently reported in individuals with 5α-R2 deficiency (R171, H231, and Y235). Mutations in two of these points (p.R171S and p.H231R) were evaluated in a functional study.59 While the p. H231R primarily affected the ability of the enzyme to bind testosterone, the p.HR171S severely decreased the affinity of the enzyme for NADPH.
Using an informatics tool to analyze the evolutionary relationship between species, the same study was able to select the amino acids of the SRD5A2 gene that are highly conserved. They are: 1,5,10,14,21,33-5, 49, 52, 53, 56, 57, 59, 87, 90, 91, 92, 94, 95, 98, 102, 104, 106, 108, 118, 119, 122, 123, 126, 128, 130, 133, 140, 145, 149, 153, 156, 157, 159-64, 167, 170-3, 175, 178, 180, 181, 183, 184, 186, 188-90, 191-3, 196-8, 200, 201, 203, 20, 210, 212, 216, 217, 219, 220, 224, 227, 228, 231-3, 235, 239, 241, 243, 248, 251, 252 and 254). Thus, we compared all variants in the literature according to amino acid conservation. Not surprisingly, most SRD5A2 variants (76%) were located at conserved amino acids. However, allelic variants at non-conserved amino acids were more frequently indels (28% vs 6%; p<0.01, V=0.38) than those at conserved amino acids and caused lower scores of external genitalia virilization (p = 0.001). It makes sense, once insertions and deletions in protein-coding regions primarily involve amino acids that have a minor impact on the structure and function of the protein.60 On the other hand, frameshifting indels usually are deleterious, leading to mRNA degradation owing to nonsense-mediated mRNA decay or the production of severely truncated proteins, which could explain the lower external genitalia virilization.61 Except by the p.Met157del reported in patients from Turkey,7,33 all the others small indels in the SRD5A2 are frameshifting leading to a premature stop codon (Table 4).
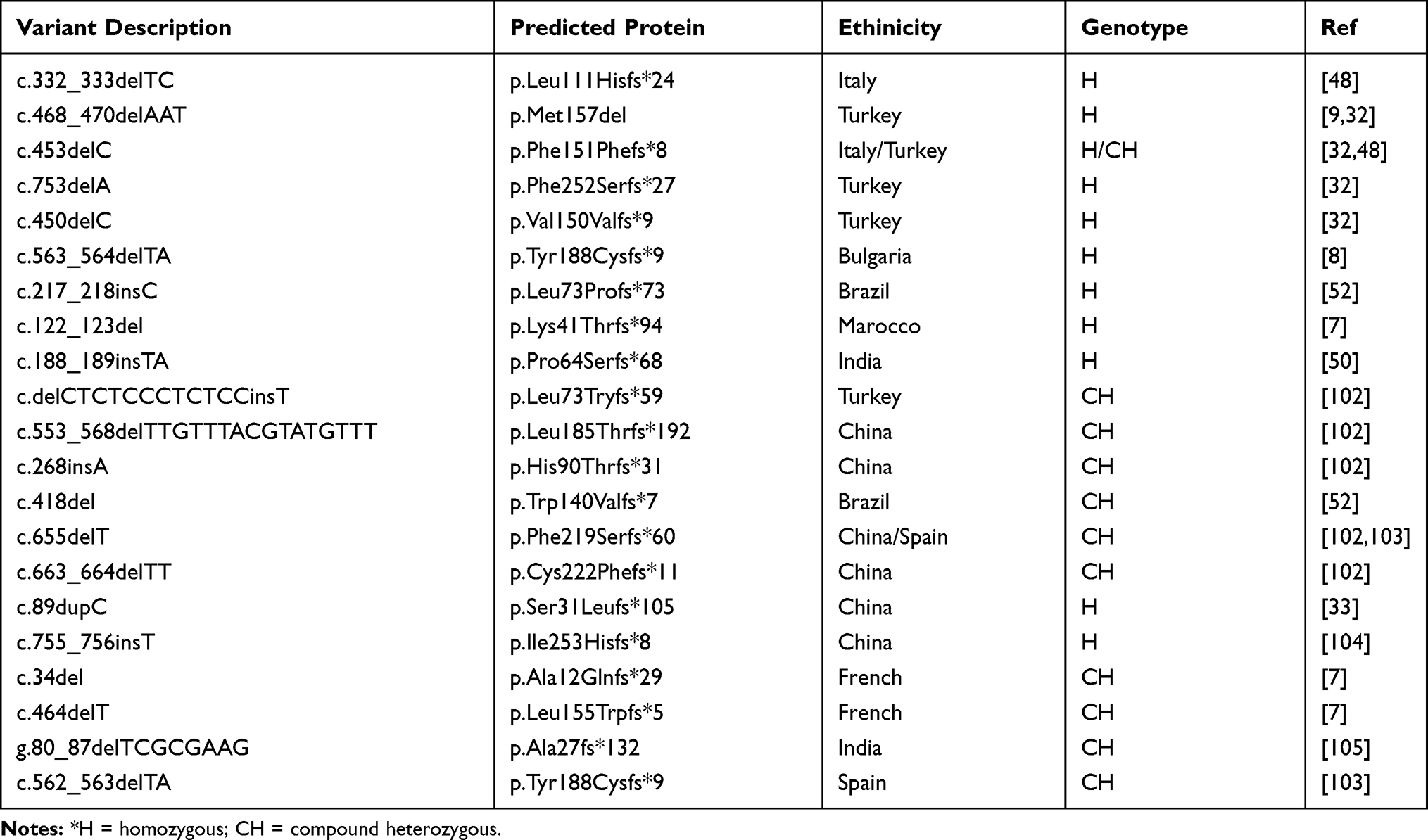 |
Table 4 Indel Variants in the SRD5A2 in Individuals with 5α-Reductase Type 2 Deficiency |
As in other inherited disorders, allelic variants in canonical splicing sites use to be deleterious.62,63 It was also reported in cases of 5α-RD2 deficiency.7,42 These variants usually resulting in exon skipping with a dramatic impact on protein.63 However, splicing is a process that can be disrupted in several ways. Single nucleotide changes (including those in synonymous variants) can disrupt splicing, either abolishing or creating a new splice site, which usually leads to the inclusion of an intron fragment or to an exon skipping.63,64 Although mutations at splicing region are not rare among individuals with 5α-RD2 deficiency only one was functionally investigated (IVS4+2T>C), which resulted on exon 4 skipping leading to a truncated protein of 205 amino acids that lacks 5α-RD2 residual activity.57
Unusual patterns of inheritance have also been reported. In an interesting case of uniparental disomy (UND), a patient was born to nonconsanguineous parents who are carriers of two distinct variants (p.Glu197Asp and p.Pro212Arg). The patient was homozygous for the p.Glu197Asp, indicating an alternative mechanism whereby 5α-R2 deficiency can derive from a single parent.65
In a case series with 14 individuals with 5α-RD2 deficiency from China, two patients carried three mutations in the SRD5A2 gene. The first one with p.Gln6Term, p.Phe234Leu, and p.Lys35Asn and the second one with p.Gly203Ser, p.Arg227Gln, and p.Gly34Arg. The first case had a completely female external genitalia while the second one had only hypospadias.49
Deletion of the entire SRD5A2 gene, as well as exons 1 and 2 deletions, have been reported in the 5α-RD2 deficiency.24,66,67 The understanding of the mechanisms underlying those deletions would be interesting to expand the molecular possibilities of 5α-RD2 deficiency. Genomic deletions give rise from multiple genetic phenomena,68,69 but recombination among repetitive elements in the DNA has grown as a CNV mechanism.70–72 In fact, the chromosomal region of the SRD5A2 gene (chr2:31,522,480-31,580,938) is enriched by mobile DNA elements, especially LINE-1 (long interspersed nuclear elements) sequences as L1Hs (chr2:31575999-31577158), L1PA15 (chr2:31569929-31572454), L1M1(chr2:31540540-31541521), L1MB3(chr2:31540540-31541521), and L1MDa (chr2:31539048-31539981) and also by Alu elements as AluSx1, AluSg4, AluSp, and AluSx3 (verified on genome browser; gene SRD5A2). Due to their ability to mobilize across the genome, both LINE-1 and Alu elements are able to impact on the genome structure and their role in inherited disorders has been increasingly reported.73–75
Well-documented subjects with 5α-RD2 deficiency (including hormonal data) with only a single SRD5A2 variant suggest that other mechanisms beyond SRD5A2 exonic sequences may play a role in this condition.21 Interesting mechanisms of gene disruption have been reported in other 46,XY DSD conditions, especially in subjects with androgen insensitivity syndrome (AIS). As examples, synonymous variants of the Androgen Receptor (AR) gene proved to be causative of both partial and complete phenotype of AIS as well as post-zygotic mosaicism.76,77 Epigenetic alterations compromising AR expression were reported in patients which were negative for AR mutations but presented clinical evidence of AIS.78 Single nucleotide change in the 5ʹUTR region of the AR was able to cause a new open frame region leading to CAIS.79 Finally, a LINE-1 retrotransposon was recently reported as causative of PAIS.73 All of these interesting molecular mechanisms of disease are still underexplored in 5α-RD2 deficiency.
Polymorphisms of the SRD5A2 Gene
Evidence about the role of polymorphism in human diseases have been on debate for a long time. In part because its frequency is relatively high to be causative of diseases and also because some polymorphisms are not located at coding regions. However, techniques to study the whole genome are maturing fast and there are growing evidence that polymorphisms are able to both change phenotypes and cause diseases.80,81 It appears to be true for 5α-R2 deficiency. The most frequent polymorphism of the SRDA2 gene is located at exon 1. A substitution from valine to leucine (p.V89L) proved to decrease the 5α-R2 activity by 30% compared with the wild type.82 This polymorphism was reported as a compound of heterozygous mutations in patients with 5α-R2 deficiency5,7 and it was also reported as causative of isolated hypospadias among Chinese and Indian children.45,83 A recent metanalysis included six studies focused on the relationship between SRD5A2 polymorphism and hypospadias. Using an Egger’s test to rule out the possibility of publication bias, the authors concluded that SRD5A2 polymorphisms might be one of the risk factors of isolated hypospadias.84 Interestingly, this polymorphism is frequent among Chinese and Japanese man and it has been considered as a protector factor to prostate cancer once this cancer is rare among individuals from this ethnicity. On the other hand, the presence of p.V89L polymorphism has been associated with a higher risk of breast cancer.85
Another polymorphism of the SRD5A2 gene is the change from alanine residue to threonine at codon 49 (p.A49T). In vitro studies showed that this polymorphism increases the enzymatic activity of the 5α-R2 enzyme by five folds, which has been suggested as a risk for prostate cancer development, mainly among African-American and Latin-American populations. However, while this risk is suggested,86 it has not been confirmed in other studies.87 On the other hand, a modest association of this polymorphism has been reported in less severe hypospadias.88
The SRD5A2 gene harbors a polymorphic site at the 3ʹunstranslated region (3ʹUTR) where a variable number of dinucleotide TA repeat length exists.89 Variations of these TA repeats occur in tandem, ranging from zero [(TA) 0] or [(TA) 9] to [(TA) 18]. Variations in TA lengths could influence the enzymatic activity of the SRD5A2 gene and it has been reported among Caucasian, African-Americans, Non-Hispanics and South Indian men. A study failed to prove the role of TA repeats favoring breast or ovarian cancer as well as another study failed to prove this polymorphism as causative of isolated hypospadias among North Indian children.89,90 Conversely, another study suggested that TA repeats polymorphism confers a prostate cancer risk among Lebanese man.91
Summarizing, the p.V89L polymorphism is probably associated with hypospadias because it impairs the 5α-R2 activity. The presence of this polymorphism should be taken into account both in cases of isolated hypospadias or as a compound of heterozygous mutations in cases of α-R2 deficiency. The role of others SRD5A2 polymorphisms either as causative of isolated hypospadias or as a contributing factor for breast and prostate cancer remains unclear.
Genotype–Phenotype Relationship in Individuals with 5α-Reductase Type 2 Deficiency
We analyzed the range of external genitalia virilization of recurrent variants in homozygosity reported in the literature in order to estimate the genotype–phenotype relationship. The Sinnecker’s scores of the p.Leu55Gly, reported in individuals from Turkey, Lebanon and Iraq, ranged from 2a to 4b.33,82,92-94 For the p.Ala65Pro variant reported in individuals from Turkey, the Sinnecker’s scores ranged from 2a to 5.33 For the p.pro181Leu variant, which was reported in Saudi Arabia and Turkey, the Sinnecker’s scores ranged from 2a to 4a.33,42,95
Phenotype variability was also observed in mutations at hotspots of the SRD5A2 gene. The p.Thy235Phe, which is located at a very conserved amino acid in direct contact with NADPH, the Sinnecker’s scores ranged from 2a to 5.50,59,95,96 In the p.Gly196Ser variant, that score ranged from 1a to 4a.6,33,50,97 However, some variants were more congruent in their impact on external genitalia virilization. The p.Arg246Gln variant is associated with more virilization (2a – 3a) than both p.Gly183Ser and p.Gln126Arg variants, which consistently caused severe undervirilization (3b – 4b and 4a – 4a, respectively).5,6,33,47,50,54,97
In cases of nonsense variants in homozygosity would be expected more undervirilization. However, individuals from Saudi Arabia carrying nonsense variants in homozygosity (p.Arg227Term and P.Arg103Term), manifested with hypospadias and microphallus,42 showing that phenotypic variability remains even in cases of truncated proteins.
We compared the mean Sinnecker’s score of external genitalia virilization among the types of allelic variants (homozygous, compound heterozygous and indels). The mean score was 5.44 ± 2.2 in those in homozygous, 5.02 ± 2.1 in compound heterozygous, and 6.89 ± 1.1 in indels. There was no difference between homozygous and compound heterozygous (p=0.16). Conversely, when these two groups were compared independently with indels, significant statistical differences were obtained in both instances with p=0.02 and p<0.001, respectively.
We also analyzed the influence of the exon location on external genitalia virilization among 156 individuals with 5α-reductase type 2 deficiency from the literature. There was a significant difference between the mean scores of the external genitalia virilization (F=10.57, p<0.001). The Bonferroni test for multiple comparisons showed that this difference was caused by the lower scores of external genitalia virilization from variants in the exon 4 (p<0.001 vs exon 1; p=0.002 vs exon 2, p<0.001 vs exon 3, and p=0.036 vs exon 5) (Figure 2). The exon 4 is made up of 28 conserved amino acids (28 out 49 = 57%).58 Among the variants in homozygous located at exon 4 in the literature, 66 out of 68 (97%) are in conserved amino acids. Functional analysis of some variants in the exon 4 showed either no residual activity (p.Gly196Ser, p.Glu197Asp, p.Ala2017Asp, p.Pro212Arg, p.Leu224Pro, p.Ala228Val, and p.His230Pro) or alter the affinity of the enzyme for NADPH (p.Asn193Ser and p.Gly196Ser).57,59 Moreover, there are two variants (p.His230Pro and p.His231Arg) located in a stretch of histidine (residues 230–232) which is highly conserved among species, suggesting that these residues are catalytically important.59
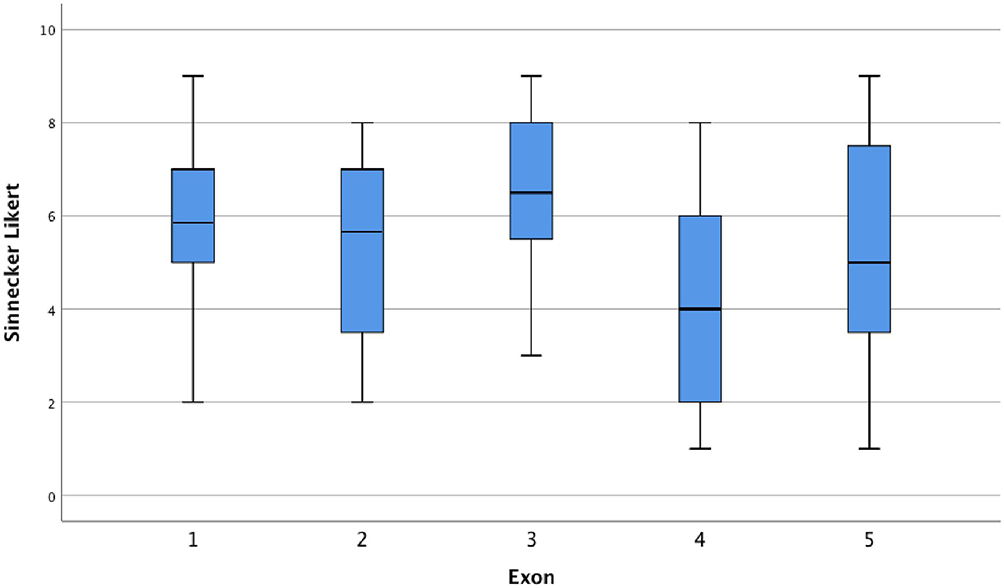 |
Figure 2 Boxplots of the external genitalia virilization accordingly the exonic location of the allelic variants in SRD5A2 gene. |
Collectively, these data show that 5α-reductase type 2 deficiency is a condition without genotype–phenotype correlation. However, the presence of the allelic variant in exon 4 and indel variants influence on phenotype. The fact that relative virilization can occurs even in cases with nonsense variants without any residual 5α-reductase activity suggests that other factors, such as AR-mediated signal transduction activity, environmental factors, and other androgens may play a role in the phenotype of 5α-reductase type 2 deficiency.
Gender Change in 5α-Reductase Type 2 Deficiency
For many reasons, individuals with 46,XY DSD are more likely to present some gender incongruence. However, gender change is more exception than the rule among these individuals.98,99 5α-reductase type 2 deficiency is an important exception. Gender change in individuals with this deficiency is frequently reported, over 50% in some series.13,35
Among all individuals who were included in this review, the percentage rate of gender change was 25% (87 out 349). All but one changed their gender from female to male. We compared the means scores of external genitalia virilization between the individuals who changed and the ones who kept their gender. Both presented similar means of virilization score (5.68 ± 2.07 vs 5.97 ± 1.97; p=0.37, respectively). The rate of gender change was significantly different across the ethnicities (V=0.44; p<0.001). The overall rate of gender change from female to male among countries with more than 10 reported cases ranged from 16% to 70%, suggesting the influence of socio-cultural and environmental factors. The lowest rate of gender change occurred in Turkey and the highest in Brazil.
Human psychosexuality is strongly influenced by androgens.14,100 Briefly, in the presence of androgens, the human psychosexuality is usually direct towards male while female psychosexuality results from the absence of androgens (even in the absence of estrogens).13,15 During certain critical prenatal periods, androgens cat directly on neural regions containing androgen receptors providing organizational changes in the brain is areas related with male sexual behavior.15,101,102 However, if the external virilization depends on dihydrotestosterone, testosterone is enough for the brain virilization. In other words, male psychosexuality develops even in the absence of dihydrotestosterone, which explain the frequency of gender change from female to male in 5α-reductase type 2 deficiency.
Many factors play a role in human psychosexuality development, such as androgen exposure, sex of rearing, and sociocultural and environmental factors. For most 46,XY DSD conditions, the sex of rearing remains as the main predictive factor of gender identity.98,103 Again, 5α-reductase type 2 deficiency is an exception. While some affected individuals maintain the female gender, many change to male gender. This change happens regardless of the external genitalia appearance, most of them at childhood and it occurs even in individuals who underwent gonadectomy. Altogether, these observations evidence the strong role of prenatal androgen exposure in male psychosexuality, reinforcing the importance of the molecular diagnosis in 46,XY DSD.
Conclusion
5α-reductase type 2 deficiency is a rare condition with a worldwide distribution which needs to be considered as a diagnosis in front of all 46,XY newborn with atypical genitalia. It results from allelic variants in the SRD5A2 gene, leading to a broad spectrum of external genitalia phenotypes with no strong genotype–phenotype relationship. However, allelic variants at exon 4 and indels consistently caused more severe phenotypes. The genotype–phenotype incongruence occurs even in individuals carrying the same variant and also in individuals from the same family, suggesting that other factors beyond the 5α-reductase type 2 enzyme play a role in phenotype. These factors are still unclear and constitute a relevant field for future research. Although many allelic variants in the SRD5A2 gene have been reported, other genetic mechanisms that may influence gene expression have not yet been investigated. The role of SRD5A2 polymorphisms in human diseases is debatable. Most cases of 5α-reductase type 2 deficiency were assigned as female at birth due to severe undervirilization of external genitalia. Later, many changed from female to male gender. It clearly favors to base the sex assignment on molecular diagnosis instead on external genitalia appearance. There has been an improvement in the number of 5α-reductase type 2 deficiencies individuals raised as boys. However, the impact of this change on long-term health outcomes requires further research.
Disclosure
The authors declare that they have no conflicts of interest.
References
1. Nowakowski H, Lenz W. Genetic aspects in male hypogonadism. Recent Prog Horm Res. 1961;17:53–95.
2. Imperato-McGinley J, Zhu YS. Androgens and male physiology the syndrome of 5alpha-reductase-2 deficiency. Mol Cell Endocrinol. 2002;198(1–2):51–59. doi:10.1016/S0303-7207(02)00368-4
3. Imperato-McGinley J, Guerrero L, Gautier T, Peterson RE. Steroid 5alpha-reductase deficiency in man: an inherited form of male pseudohermaphroditism. Science. 1974;186(4170):1213–1215. doi:10.1126/science.186.4170.1213
4. Walsh PC, Madden JD, Harrod MJ, Goldstein JL, MacDonald PC, Wilson JD. Familial incomplete male pseudohermaphroditism, type 2. Decreased dihydrotestosterone formation in pseudovaginal perineoscrotal hypospadias. N Engl J Med. 1974;291(18):944–949. doi:10.1056/NEJM197410312911806
5. Mendonca BB, Inacio M, Costa EM, et al. Male pseudohermaphroditism due to steroid 5alpha-reductase 2 deficiency. Diagnosis, psychological evaluation, and management. Medicine (Baltimore). 1996;75(2):64–76. doi:10.1097/00005792-199603000-00003
6. Thigpen AE, Davis DL, Milatovich A, et al. Molecular genetics of steroid 5 alpha-reductase 2 deficiency. J Clin Invest. 1992;90(3):799–809. doi:10.1172/JCI115954
7. Maimoun L, Philibert P, Cammas B, et al. Phenotypical, biological, and molecular heterogeneity of 5α-reductase deficiency: an extensive international experience of 55 patients. J Clin Endocrinol Metab. 2011;96(2):296–307. doi:10.1210/jc.2010-1024
8. Andonova S, Robeva R, Vazharova R, et al. New territory for an old disease: 5-alpha-reductase type 2 deficiency in Bulgaria. Sex Dev. 2017;11(1):21–28. doi:10.1159/000454974
9. Sinnecker GH, Hiort O, Dibbelt L, et al. Phenotypic classification of male pseudohermaphroditism due to steroid 5 alpha-reductase 2 deficiency. Am J Med Genet. 1996;63(1):223–230. doi:10.1002/(SICI)1096-8628(19960503)63:1<223::AID-AJMG39>3.0.CO;2-O
10. Kolesinska Z, Ahmed SF, Niedziela M, et al. Changes over time in sex assignment for disorders of sex development. Pediatrics. 2014;134(3):e710–e715. doi:10.1542/peds.2014-1088
11. Atta I, Ibrahim M, Parkash A, Lone SW, Khan YN, Raza J. Etiological diagnosis of undervirilized male/XY disorder of sex development. J Coll Physicians Surg Pak. 2014;24(10):714–718. doi:10.2014/JCPSP.714718
12. Achermann JC, Domenice S, Bachega TA, Nishi MY, Mendonca BB. Disorders of sex development: effect of molecular diagnostics. Nat Rev Endocrinol. 2015;11(8):478–488. doi:10.1038/nrendo.2015.69
13. Loch Batista R, Inácio M, Prado Arnhold IJ, et al. Psychosexual aspects, effects of prenatal androgen exposure, and gender change in 46,XY disorders of sex development. J Clin Endocrinol Metab. 2019;104(4):1160–1170. doi:10.1210/jc.2018-01866
14. Berenbaum SA, Beltz AM. How early hormones shape gender development. Curr Opin Behav Sci. 2016;7:53–60. doi:10.1016/j.cobeha.2015.11.011
15. Berenbaum SA, Beltz AM. Sexual differentiation of human behavior: effects of prenatal and pubertal organizational hormones. Front Neuroendocrinol. 2011;32(2):183–200. doi:10.1016/j.yfrne.2011.03.001
16. Banerjee PP, Banerjee S, Brown TR, Zirkin BR. Androgen action in prostate function and disease. Am J Clin Exp Urol. 2018;6(2):62–77.
17. Imperato-McGinley J. 5alpha-reductase-2 deficiency and complete androgen insensitivity: lessons from nature. Adv Exp Med Biol. 2002;511:
18. Azzouni F, Godoy A, Li Y, Mohler J. The 5 alpha-reductase isozyme family: a review of basic biology and their role in human diseases. Adv Urol. 2012;2012:530121. doi:10.1155/2012/530121
19. Russell DW, Wilson JD. Steroid 5 alpha-reductase: two genes/two enzymes. Annu Rev Biochem. 1994;63:25–61. doi:10.1146/annurev.bi.63.070194.000325
20. Andersson S, Russell DW. Structural and biochemical properties of cloned and expressed human and rat steroid 5 alpha-reductases. Proc Natl Acad Sci U S A. 1990;87(10):3640–3644. doi:10.1073/pnas.87.10.3640
21. Wilson JD, Griffin JE, Russell DW. Steroid 5 alpha-reductase 2 deficiency. Endocr Rev. 1993;14(5):577–593. doi:10.1210/edrv-14-5-577
22. Wilson JD, Lasnitzki I. Dihydrotestosterone formation in fetal tissues of the rabbit and rat. Endocrinology. 1971;89(3):659–668. doi:10.1210/endo-89-3-659
23. Deykin JD, Balko C, Wilson JD. Recent studies on the mechanism of action of testosterone. N Engl J Med. 1972;287(25):1284–1291. doi:10.1056/NEJM197212212872508
24. Andersson S, Berman DM, Jenkins EP, Russell DW. Deletion of steroid 5 alpha-reductase 2 gene in male pseudohermaphroditism. Nature. 1991;354(6349):159–161. doi:10.1038/354159a0
25. Mendonca BB, Batista RL, Domenice S, et al. Steroid 5α-reductase 2 deficiency. J Steroid Biochem Mol Biol. 2016;163:206–211. doi:10.1016/j.jsbmb.2016.05.020
26. Costa EM, Domenice S, Sircili MH, Inacio M, Mendonca BB. DSD due to 5α-reductase 2 deficiency - from diagnosis to long term outcome. Semin Reprod Med. 2012;30(5):427–431. doi:10.1055/s-00000072
27. Livingstone DE, Di Rollo EM, Mak TC, Sooy K, Walker BR, Andrew R. Metabolic dysfunction in female mice with disruption of 5α-reductase 1. J Endocrinol. 2017;232(1):29–36. doi:10.1530/JOE-16-0125
28. Upreti R, Hughes KA, Livingstone DE, et al. 5α-reductase type 1 modulates insulin sensitivity in men. J Clin Endocrinol Metab. 2014;99(8):E1397–E1406. doi:10.1210/jc.2014-1395
29. Stiles AR, Russell DW. SRD5A3: a surprising role in glycosylation. Cell. 2010;142(2):196–198. doi:10.1016/j.cell.2010.07.003
30. Taylor RL, Arno G, Poulter JA, et al. Association of steroid 5α-reductase type 3 congenital disorder of glycosylation with early-onset retinal dystrophy. JAMA Ophthalmol. 2017;135(4):339–347. doi:10.1001/jamaophthalmol.2017.0046
31. Kara B, Ayhan Ö, Gökçay G, Başboğaoğlu N, Tolun A. Adult phenotype and further phenotypic variability in SRD5A3-CDG. BMC Med Genet. 2014;15:10. doi:10.1186/1471-2350-15-10
32. Cheng J, Lin R, Zhang W, et al. Phenotype and molecular characteristics in 45 Chinese children with 5α-reductase type 2 deficiency from South China. Clin Endocrinol (Oxf). 2015;83(4):518–526. doi:10.1111/cen.12799
33. Abacı A, Çatlı G, Kırbıyık Ö, et al. Genotype-phenotype correlation, gonadal malignancy risk, gender preference, and testosterone/dihydrotestosterone ratio in steroid 5-alpha-reductase type 2 deficiency: a multicenter study from Turkey. J Endocrinol Invest. 2019;42(4):453–470. doi:10.1007/s40618-018-0940-y
34. Gui B, Song Y, Su Z, et al. New insights into 5α-reductase type 2 deficiency based on a multi-centre study: regional distribution and genotype–phenotype profiling of SRD5A2 in 190 Chinese patients. J Med Genet. 2019;56:685–692. doi:10.1136/jmedgenet-2018-105915
35. Cohen-Kettenis PT. Gender change in 46,XY persons with 5alpha-reductase-2 deficiency and 17beta-hydroxysteroid dehydrogenase-3 deficiency. Arch Sex Behav. 2005;34(4):399–410. doi:10.1007/s10508-005-4339-4
36. Mendonca BB, Gomes NL, Costa EM, et al. 46,XY disorder of sex development (DSD) due to 17β-hydroxysteroid dehydrogenase type 3 deficiency. J Steroid Biochem Mol Biol. 2017;165(Pt A):79–85. doi:10.1016/j.jsbmb.2016.05.002
37. Inacio M, Sircili MH, Brito VN, et al. 46,XY DSD due to 17β-HSD3 deficiency and 5α-reductase type 2 deficiency. Adv Exp Med Biol. 2011;707:9–14.
38. Cools M, Nordenström A, Robeva R, et al. Caring for individuals with a difference of sex development (DSD): a Consensus Statement. Nat Rev Endocrinol. 2018;14(7):415–429. doi:10.1038/s41574-018-0010-8
39. Hiort O, Birnbaum W, Marshall L, et al. Management of disorders of sex development. Nat Rev Endocrinol. 2014;10(9):520–529. doi:10.1038/nrendo.2014.108
40. Wisniewski AB, Batista RL, Costa EMF, et al. Management of 46,XY differences/disorders of sex development (DSD) Throughout Life. Endocr Rev. 2019;40:1547–1572. doi:10.1210/er.2019-00049
41. Russell DW, Wilson JD. Chapter 4A - steroid 5α-reductase 2 deficiency. In: Minlptywomd H, editor. Genetic Steroid Disorders. San Diego: Academic Press; 2014:199–214.
42. Alswailem MM, Alzahrani OS, Alghofaili L, et al. Molecular genetics and phenotype/genotype correlation of 5-α reductase deficiency in a highly consanguineous population. Endocrine. 2019;63(2):361–368. doi:10.1007/s12020-018-1767-1
43. Avendaño A, Paradisi I, Cammarata-Scalisi F, Callea M. 5-α-Reductase type 2 deficiency: is there a genotype-phenotype correlation? A review. Hormones (Athens). 2018;17(2):197–204. doi:10.1007/s42000-018-0013-9
44. Ko JM, Cheon CK, Kim GH, Kim SH, Kim KS, Yoo HW. Clinical characterization and analysis of the SRD5A2 gene in six Korean patients with 5alpha-reductase type 2 deficiency. Horm Res Paediatr. 2010;73(1):41–48. doi:10.1159/000271915
45. Yuan S, Meng L, Zhang Y, et al. Genotype-phenotype correlation and identification of two novel SRD5A2 mutations in 33 Chinese patients with hypospadias. Steroids. 2017;125:61–66. doi:10.1016/j.steroids.2017.06.010
46. Sahu R, Boddula R, Sharma P, et al. Genetic analysis of the SRD5A2 gene in Indian patients with 5alpha-reductase deficiency. J Pediatr Endocrinol Metab. 2009;22(3):247–254. doi:10.1515/JPEM.2009.22.3.247
47. Vilchis F, Méndez JP, Canto P, Lieberman E, Chávez B. Identification of missense mutations in the SRD5A2 gene from patients with steroid 5alpha-reductase 2 deficiency. Clin Endocrinol (Oxf). 2000;52(3):383–387. doi:10.1046/j.1365-2265.2000.00941.x
48. Samtani R, Bajpai M, Ghosh PK, Saraswathy KN. SRD5A2 gene mutations–a population-based review. Pediatr Endocrinol Rev. 2010;8(1):34–40.
49. Cheng T, Wang H, Han B, et al. Identification of three novel. Asian J Androl. 2019.
50. Bertelloni S, Baldinotti F, Russo G, et al. 5α-Reductase-2 deficiency: clinical Findings, endocrine pitfalls, and genetic features in a large Italian Cohort. Sex Dev. 2016;10(1):28–36. doi:10.1159/000445090
51. Berra M, Williams EL, Muroni B, et al. Recognition of 5α-reductase-2 deficiency in an adult female 46XY DSD clinic. Eur J Endocrinol. 2011;164(6):1019–1025. doi:10.1530/EJE-10-0930
52. Shabir I, Khurana ML, Marumudi E, Khadgawat R, Ammini AC. Novel nucleotide insertions in two unrelated Indian patients with 5 alpha reductase 2 deficiency leading to premature termination of SRD5A2 enzyme. Steroids. 2013;78(12–13):
53. Di Marco C, Bulotta AL, Varetti C, et al. Ambiguous external genitalia due to defect of 5-α-reductase in seven Iraqi patients: prevalence of a novel mutation. Gene. 2013;526(2):490–493. doi:10.1016/j.gene.2013.04.070
54. Hackel C, Oliveira LE, Ferraz LF, et al. New mutations, hotspots, and founder effects in Brazilian patients with steroid 5alpha-reductase deficiency type 2. J Mol Med (Berl). 2005;83(7):569–576. doi:10.1007/s00109-005-0651-7
55. Makridakis NM, Di Salle E, Reichardt JK. Biochemical and pharmacogenetic dissection of human steroid 5 alpha-reductase type II. Pharmacogenetics. 2000;10(5):407–413. doi:10.1097/00008571-200007000-00004
56. Vilchis F, Valdez E, Ramos L, García R, Gómez R, Chávez B. Novel compound heterozygous mutations in the SRD5A2 gene from 46,XY infants with ambiguous external genitalia. J Hum Genet. 2008;53(5):401–406. doi:10.1007/s10038-008-0274-2
57. Zhu H, Liu W, Han B, et al. Phenotypic and molecular characteristics in eleven Chinese patients with 5α-reductase Type 2 deficiency. Clin Endocrinol (Oxf). 2014;81(5):711–720. doi:10.1111/cen.2014.81.issue-5
58. Katharopoulos E, Sauter K, Pandey AV, Flück CE. In silico and functional studies reveal novel loss-of-function variants of SRD5A2, but no variants explaining excess 5α-reductase activity. J Steroid Biochem Mol Biol. 2019;190:263–272. doi:10.1016/j.jsbmb.2019.01.017
59. Wigley WC, Prihoda JS, Mowszowicz I, et al. Natural mutagenesis study of the human steroid 5 alpha-reductase 2 isozyme. Biochemistry. 1994;33(5):1265–1270. doi:10.1021/bi00171a029
60. de la Chaux N, Messer PW, Arndt PF. DNA indels in coding regions reveal selective constraints on protein evolution in the human lineage. BMC Evol Biol. 2007;7:191. doi:10.1186/1471-2148-7-191
61. Lalonde S, Stone OA, Lessard S, et al. Frameshift indels introduced by genome editing can lead to in-frame exon skipping. PLoS One. 2017;12(6):e0178700. doi:10.1371/journal.pone.0178700
62. Wang GS, Cooper TA. Splicing in disease: disruption of the splicing code and the decoding machinery. Nat Rev Genet. 2007;8(10):749–761. doi:10.1038/nrg2164
63. Anna A, Monika G. Splicing mutations in human genetic disorders: examples, detection, and confirmation. J Appl Genet. 2018;59:253–268. doi:10.1007/s13353-018-0444-7
64. Hunt RC, Simhadri VL, Iandoli M, Sauna ZE, Kimchi-Sarfaty C. Exposing synonymous mutations. Trends Genet. 2014;30(7):308–321. doi:10.1016/j.tig.2014.04.006
65. Chávez B, Valdez E, Vilchis F. Uniparental disomy in steroid 5alpha-reductase 2 deficiency. J Clin Endocrinol Metab. 2000;85(9):3147–3150. doi:10.1210/jcem.85.9.6786
66. Deeb A, Al Suwaidi H, Ibukunoluwa F, Attia S. Phenotype, sex of rearing, gender re-assignment, and response to medical treatment in extended family members with a novel mutation in the SRD5A2 gene. J Clin Res Pediatr Endocrinol. 2016;8(2):236–240. doi:10.4274/jcrpe.2782
67. Fénichel P, Paris F, Philibert P, et al. Molecular diagnosis of 5α-reductase deficiency in 4 elite young female athletes through hormonal screening for hyperandrogenism. J Clin Endocrinol Metab. 2013;98(6):E1055–E1059. doi:10.1210/jc.2012-3893
68. Stankiewicz P, Lupski JR. Structural variation in the human genome and its role in disease. Annu Rev Med. 2010;61:437–455. doi:10.1146/annurev-med-100708-204735
69. Zhang F, Gu W, Hurles ME, Lupski JR. Copy number variation in human health, disease, and evolution. Annu Rev Genomics Hum Genet. 2009;10:451–481. doi:10.1146/annurev.genom.9.081307.164217
70. Lisch D, Burns KH. Editorial overview: genome architecture and expression: mobile elements at work. Curr Opin Genet Dev. 2018;49:iv–v. doi:10.1016/j.gde.2018.05.003
71. Kazazian HH, Moran JV. Mobile DNA in health and disease. N Engl J Med. 2017;377(4):361–370. doi:10.1056/NEJMra1510092
72. Szafranski P, Kośmider E, Liu Q, et al. LINE- and Alu-containing genomic instability hotspot at 16q24.1 associated with recurrent and nonrecurrent CNV deletions causative for ACDMPV. Hum Mutat. 2018;39(12):1916–1925. doi:10.1002/humu.23608
73. Batista RL, Yamaguchi K, Di Santi Rodrigues A, et al. Mobile DNA in endocrinology: LINE-1 retrotransposon causing partial androgen insensitivity syndrome. J Clin Endocrinol Metab. 2019;104:6385–6390. doi:10.1210/jc.2019-00144
74. Hancks DC, Kazazian HH. Roles for retrotransposon insertions in human disease. Mob DNA. 2016;7:9.
75. Ule J. Alu elements: at the crossroads between disease and evolution. Biochem Soc Trans. 2013;41(6):1532–1535. doi:10.1042/BST20130157
76. Batista RL, Di Santi Rodrigues A, Nishi MY, et al. A recurrent synonymous mutation in the human androgen receptor gene causing complete androgen insensitivity syndrome. J Steroid Biochem Mol Biol. 2017;174:14–16. doi:10.1016/j.jsbmb.2017.07.020
77. Batista RL, Rodrigues AS, Machado AZ, et al. Partial androgen insensitivity syndrome due to somatic mosaicism of the androgen receptor. J Pediatr Endocrinol Metab. 2018;31(2):223–228. doi:10.1515/jpem-2017-0095
78. Hornig NC, Ukat M, Schweikert HU, et al. Identification of an AR mutation-negative class of androgen insensitivity by determining endogenous AR activity. J Clin Endocrinol Metab. 2016;101(11):4468–4477. doi:10.1210/jc.2016-1990
79. Hornig NC, de Beaufort C, Denzer F, et al. A recurrent germline mutation in the 5ʹUTR of the androgen receptor causes complete androgen insensitivity by activating aberrant uORF translation. PLoS One. 2016;11(4):e0154158. doi:10.1371/journal.pone.0154158
80. Tirabassi G, Cignarelli A, Perrini S, et al. Influence of CAG repeat polymorphism on the targets of testosterone action. Int J Endocrinol. 2015;2015:298107. doi:10.1155/2015/298107
81. Chen R, Davydov EV, Sirota M, Butte AJ. Non-synonymous and synonymous coding SNPs show similar likelihood and effect size of human disease association. PLoS One. 2010;5(10):e13574. doi:10.1371/journal.pone.0013574
82. Hochberg Z, Chayen R, Reiss N, et al. Clinical, biochemical, and genetic findings in a large pedigree of male and female patients with 5 alpha-reductase 2 deficiency. J Clin Endocrinol Metab. 1996;81(8):2821–2827. doi:10.1210/jcem.81.8.8768837
83. Palmer BW, Reiner W, Kropp BP. Proximal hypospadias repair outcomes in patients with a specific disorder of sexual development diagnosis. Adv Urol. 2012;2012:708301. doi:10.1155/2012/708301
84. Sun L, Zhou M, Liu T. Association between SRD5A2 polymorphism and hypospadias: a meta-analysis. Pharmazie. 2019;74(2):125–128. doi:10.1691/ph.2019.8768
85. Francis A, Sarkar S, Pooja S, et al. SRD5A2 gene polymorphisms affect the risk of breast cancer. Breast. 2014;23(2):137–141. doi:10.1016/j.breast.2013.11.010
86. Li Q, Zhu Y, He J, et al. Steroid 5-alpha-reductase type 2 (SRD5A2) V89L and A49T polymorphisms and sporadic prostate cancer risk: a meta-analysis. Mol Biol Rep. 2013;40(5):3597–3608. doi:10.1007/s11033-012-2434-x
87. Li J, Coates RJ, Gwinn M, Khoury MJ. Steroid 5-{alpha}-reductase Type 2 (SRD5a2) gene polymorphisms and risk of prostate cancer: a HuGE review. Am J Epidemiol. 2010;171(1):1–13. doi:10.1093/aje/kwp318
88. Silver RI, Russell DW. 5alpha-reductase type 2 mutations are present in some boys with isolated hypospadias. J Urol. 1999;162(3 Pt 2):1142–1145. doi:10.1016/S0022-5347(01)68102-3
89. Samtani R, Bajpai M, Ghosh PK, Saraswathy KN. A49T, R227Q and TA repeat polymorphism of steroid 5 alpha-reductase type II gene and hypospadias risk in North Indian children. Meta Gene. 2015;3:1–7. doi:10.1016/j.mgene.2014.11.003
90. Spurdle AB, Hopper JL, Chen X, et al. The steroid 5alpha-reductase type II TA repeat polymorphism is not associated with risk of breast or ovarian cancer in Australian women. Cancer Epidemiol Biomarkers Prev. 2001;10(12):1287–1293.
91. El Ezzi AA, Boyko VG, Baker MT, et al. Association of some polymorphisms in the VDR gene, CYP17 gene and SRD5A2 gene and prostate cancer among lebanese men. Asian Pac J Cancer Prev. 2017;18(1):93–100. doi:10.22034/APJCP.2017.18.1.93
92. Adiyaman PB, Ocal G, Cetinkaya E, et al. 5 alpha steroid reductase deficiency in Turkey. Pediatr Endocrinol Rev. 2006;3(Suppl 3):462–469.
93. Ocal G, Adiyaman P, Berberoğlu M, et al. Mutations of the 5alpha-steroid reductase type 2 gene in six Turkish patients from unrelated families and a large pedigree of an isolated Turkish village. J Pediatr Endocrinol Metab. 2002;15(4):411–421. doi:10.1515/JPEM.2002.15.4.411
94. Walter KN, Kienzle FB, Frankenschmidt A, et al. Difficulties in diagnosis and treatment of 5alpha-reductase type 2 deficiency in a newborn with 46,XY DSD. Horm Res Paediatr. 2010;74(1):67–71. doi:10.1159/000313372
95. Parlak M, Durmaz E, Gursoy S, Bircan I, Akcurin S. Try235Phe homozygous mutation of the steroid 5-a reductase type 2 (SRD5A2) gene in a Turkish patient. Ann Saudi Med. 2014;34(3):254–256. doi:10.5144/0256-4947.2014.254
96. Mazen I, Gad YZ, Hafez M, Sultan C, Lumbroso S. Molecular analysis of 5alpha-reductase type 2 gene in eight unrelated egyptian children with suspected 5alpha-reductase deficiency: prevalence of the G34R mutation. Clin Endocrinol (Oxf). 2003;58(5):627–631. doi:10.1046/j.1365-2265.2003.01763.x
97. Baldinotti F, Majore S, Fogli A, et al. Molecular characterization of 6 unrelated Italian patients with 5alpha-reductase type 2 deficiency. J Androl. 2008;29(1):20–28. doi:10.2164/jandrol.107.002592
98. Callens N, Van Kuyk M, van Kuppenveld JH, et al. Recalled and current gender role behavior, gender identity and sexual orientation in adults with disorders/differences of sex development. Horm Behav. 2016;86:8–20. doi:10.1016/j.yhbeh.2016.08.008
99. Kreukels BPC, Köhler B, Nordenström A, et al. Gender dysphoria and gender change in disorders of sex development/intersex conditions: results from the dsd-LIFE study. J Sex Med. 2018;15(5):777–785. doi:10.1016/j.jsxm.2018.02.021
100. Hines M. Gender development and the human brain. Annu Rev Neurosci. 2011;34:69–88. doi:10.1146/annurev-neuro-061010-113654
101. Hiort O. The differential role of androgens in early human sex development. BMC Med. 2013;11:152. doi:10.1186/1741-7015-11-152
102. Arnold AP. The organizational-activational hypothesis as the foundation for a unified theory of sexual differentiation of all mammalian tissues. Horm Behav. 2009;55(5):570–578. doi:10.1016/j.yhbeh.2009.03.011
103. Bakula DM, Mullins AJ, Sharkey CM, Wolfe-Christensen C, Mullins LL, Wisniewski AB. Gender identity outcomes in children with disorders/differences of sex development: predictive factors. Semin Perinatol. 2017;41:214–217. doi:10.1053/j.semperi.2017.03.010
104. Zhang M, Yang J, Zhang H, Ning G, Li X, Sun S. A novel SRD5A2 mutation with loss of function identified in Chinese patients with hypospadias. Horm Res Paediatr. 2011;76(1):44–49.
105. Nagaraja MR, Rastogi A, Raman R, Gupta DK, Singh SK. Molecular diagnosis of 46,XY DSD and identification of a novel 8 nucleotide deletion in exon 1 of the SRD5A2 gene. J Pediatr Endocrinol Metab. 2010;23(4):379–385.
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.