Back to Journals » Journal of Inflammation Research » Volume 18
Integration of Network Pharmacology, Molecular Docking, and Experimental Validation to Identify the Effect of EGCG on Alleviating Chondrocyte Inflammatory Damage by Targeting ER Stress-STAT3 Crosstalk
Authors Zhao MJ ![]() , Yin JL, Luo JH, Ge YS, Huang CM, Meng TT, Wang XZ, Huang XH, Chen LL, Zhai YQ, Wu XB, Ding DF
, Yin JL, Luo JH, Ge YS, Huang CM, Meng TT, Wang XZ, Huang XH, Chen LL, Zhai YQ, Wu XB, Ding DF
Received 3 September 2025
Accepted for publication 15 October 2025
Published 30 October 2025 Volume 2025:18 Pages 15165—15185
DOI https://doi.org/10.2147/JIR.S564356
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Ujjwol Risal
Min-Jun Zhao,1– 3 Jian-Li Yin,1– 3 Jia-Hui Luo,1– 3 Yang-Shuo Ge,2,3 Chun-Meng Huang,2,3 Ting-Ting Meng,2,3 Xue-Zong Wang,4 Xin-Hui Huang,2,3 Liao-Lin Chen,2,3 Yu-Qing Zhai,2,3 Xu-Bo Wu,1,5 Dao-Fang Ding1– 3
1Department of Rehabilitation Therapy, The Second Rehabilitation Hospital of Shanghai, Shanghai, People’s Republic of China; 2Institute of Rehabilitation Medicine, shanghai Academy of Traditional Chinese Medicine, Shanghai, People’s Republic of China; 3School of Rehabilitation Science, Shanghai University of Traditional Chinese Medicine, Shanghai, People’s Republic of China; 4Shi’s Center of Orthopedics and Traumatology, Shuguang Hospital Affiliated to Shanghai University of Traditional Chinese Medicine, Shanghai, People’s Republic of China; 5Department of Rehabilitation Medicine of Shanghai Pudong New Area People’s Hospital, Shanghai, People’s Republic of China
Correspondence: Xu-Bo Wu, Email [email protected] Dao-Fang Ding, Email [email protected]
Background: Osteoarthritis (OA) is the most common joint disorder and a major global health burden. Epigallocatechin-3-gallate (EGCG), a green tea-extracted polyphenol, shows therapeutic potential for OA, but a comprehensive understanding of its mechanisms is essential to enhance clinical utility.
Methods: EGCG-related targets were identified utilizing the TCMSP, BATMAN-TCM, PharmMapper, and SwissTargetPrediction databases. OA-related targets were retrieved from GeneCards, DisGeNET, OMIM, and TTD databases. A protein-protein interaction (PPI) network was constructed using Cytoscape 3.10.1, and the CytoNCA plugin was used for topological analysis to identify the core targets. To clarify EGCG’s therapeutic mechanisms in OA, we performed systematic functional annotation via Gene Ontology (GO) enrichment and interrogated relevant biological pathways using the Kyoto Encyclopedia of Genes and Genomes (KEGG) database. Furthermore, molecular docking was applied to assess the binding affinity between EGCG and key targets. Finally, in vitro experiments using primary chondrocytes stimulated with IL-1β were conducted to validate the predictions from network pharmacology.
Results: 488 EGCG-related targets were identified, with 172 overlapping OA-related genes. Four core genes were identified, including STAT3, TP53, AKT1, and JUN. GO enrichment analysis revealed 2163 biological processes, 92 cellular components, and 223 molecular functions; KEGG analysis identified 175 enriched signaling pathways. Molecular docking showed EGCG’s binding affinity to core target STAT3 was approximately − 8.1 kcal/mol. In vitro experiments showed that EGCG reduced IL-1β-induced catabolic and inflammatory responses in chondrocytes, which is linked with attenuated endoplasmic reticulum (ER) stress. Moreover, the involvement of STAT3 in the effects of EGCG that alleviate ER stress in OA has been established, highlighting its therapeutic potential.
Conclusion: This study reveals that EGCG may ameliorate the metabolic imbalance of extracellular matrix (ECM) and inflammatory responses by modulating the activity of the ER stress-STAT3 crosstalk.
Keywords: OA, EGCG, network pharmacology, ER stress, STAT3
Introduction
OA is a chronic and progressive joint disorder that is particularly prevalent in joints that are subjected to frequent use or substantial mechanical stress. An epidemiological study revealed that the prevalence of OA increases with age, affecting approximately 7.6% of the global population and projected to rise by 60–100% by 2050.1 Notably, OA ranks as the seventh leading cause of disability worldwide among individuals aged ≥70 years, primarily targeting the knee joint.2 The medical costs of OA in various high-income countries have been estimated to account for 1–2.5% of these nations’ gross domestic product,3 with its economic impact far exceeding direct healthcare expenditures.4,5 A major pathological characteristic of OA is the gradual deterioration of articular cartilage, comprising chondrocytes that are situated within a dense ECM.6 In OA, chondrocyte dysfunction impairs ECM synthesis, thereby reducing the tissue’s regenerative capacity.7
ER, as a critical membranous organelle, is essential for the processes of protein synthesis and folding, lipid production, and calcium storage.8 Studies have demonstrated that ER stress was linked with degenerated cartilage.9–12 Various cellular stressors, along with errors in translation or mutations in matrix or chaperone proteins, hinder the production and secretion of ECM proteins, leading to the accumulation of protein aggregates within the ER lumen.13 Chondrocytes undergo ER stress when the sustained accumulation of misfolded or unfolded proteins within the ER exceeds the cell’s ability to properly fold.14,15 This situation subsequently triggers the unfolded protein response (UPR).16,17
The ER is emerging as a new focal site for initiating endogenous cell death pathways. A growing body of evidence indicates that ER stress-induced apoptosis is a significant factor contributing to the progression of OA.18–20 Under severe and prolonged ER stress, the UPR activates unique pathways that lead to cell death through apoptosis. Notably, ER stress has been implicated in the development of OA-related inflammation.21–24 IL-1β appears to trigger ER stress, thereby amplifying OA-associated inflammation and promoting chondrocyte apoptosis. Signal Transducer and Activator of Transcription 3 (STAT3) is a key molecule regulating inflammation in chondrocytes.25 During the progression of OA, the activation of the STAT3 pathway can be modulated by inflammatory cytokines such as interleukin-6 (IL-6) and tumor necrosis factor-α (TNF-α).26 Further studies have revealed a close crosstalk between ER stress and STAT3 signaling. It has been reported that ER stress promotes the phosphorylation of STAT3 via the protein kinase R-like ER kinase (PERK) pathway, thereby amplifying the inflammatory response.27 Another study has confirmed that knockdown of PA28γ in mouse chondrocytes activated STAT3, which in turn reciprocally inhibits the excessive activation of ER stress induced by tert-butyl hydroperoxide (TBHP), thereby forming a bidirectional regulatory loop.28 This suggests that there is a tight association between ER stress and STAT3 signaling in chondrocytes, and targeted regulation of their crosstalk provides a potential avenue for OA treatment.
EGCG is the most abundant polyphenol found in green tea, which exhibits various biological activities such as antiviral, antioxidant, anti-aging, anti-inflammatory, and neuroprotective properties.29–31 EGCG suppresses the expression of pro-inflammatory cytokines (IL-1β, TNF-α, IL-6) in primary human chondrocytes.32,33 Furthermore, it counteracts IL-1β-induced downregulation of transforming growth factor-β (TGF-β) synthesis, while simultaneously promoting the expression of type II collagen (COL2A1) and aggrecan (ACAN) in chondrocytes.34 Importantly, EGCG’s regulation of ER stress is widely validated in multi-system cellular models. For instance, EGCG enhances viability in human trabecular meshwork cells by suppressing ER stress.35 Additionally, it attenuates reactive oxygen species (ROS) production in granulosa cells and targets the eIF2α/ATF4 pathway to alleviate ER stress, thereby mitigating thyroid hormone-induced dysfunction.36 Another study demonstrated EGCG downregulates ER stress markers (eg, Glucose Regulated Protein 78, GRP78) in high-glucose-exposed podocytes, thereby inhibiting glucose-induced stress.37 In endothelial cells, EGCG blocks ER stress-linked NOD-like receptor thermal protein domain associated protein 3(NLRP3) inflammasome activation, protecting against inflammation and apoptosis.38 Collectively, these findings suggest that EGCG alleviates ER stress by suppressing inflammation and other stress responses, including ROS production and high-glucose exposure.
Accordingly, we employed network pharmacology and molecular docking to predict EGCG’s molecular targets and elucidate the mechanisms underlying its effects on OA. Potential therapeutic targets of EGCG in OA were first identified through network pharmacology analysis, followed by computational evaluation of binding affinities between EGCG and prioritized targets via molecular docking. EGCG exhibits the strongest binding affinity with STAT3. We further confirmed the interactions and regulatory mechanisms between STAT3 and ER stress in IL-1β-induced chondrocyte damage, as well as whether EGCG exerts protective effects through the STAT3-mediated inflammatory pathway and ER stress. The overall experimental design is illustrated in the graphical abstract.
Materials and Methods
Network Pharmacology-Based Analysis
Retrieval of EGCG Targets
To identify potential drug targets of EGCG, several online databases were utilized, including the Traditional Chinese Medicine Systems Pharmacology Database and Analysis Platform (TCMSP) (https://www.tcmsp-e.com), the Bioinformatics Analysis Tool for Molecular Mechanism Traditional Chinese Medicine (BATMAN-TCM) (http://bionet.ncpsb.org.cn/batman-tcm/index.php), and PharmMapper (https://www.lilab-ecust.cn/pharmmapper/index.html). To obtain a more comprehensive set of targets, the molecular structure of EGCG was obtained from PubChem, and its corresponding simplified molecular input line entry system (SMILES) notation was obtained. The SMILES string was then entered into the Swiss Target Prediction (https://swisstargetprediction.ch) with the species set as “Homo sapiens” and a probability threshold of greater than 0. Subsequently, the UniProt (https://www.uniprot.org/) was utilized to standardize the nomenclature of the gene names.39 After removing duplicates, a list of potential EGCG targets will be established.
Acquisition of OA-Related Targets
Targets associated with OA were collected through the OMIM (https://www.omim.org/), GeneCards (https://www.genecards.org), Therapeutic Target Database (https://db.idrblab.net/ttd/), and DisGeNET (https://disgenet.com/) databases using the keyword “Osteoarthritis”.40 After removing duplicates, the retained targets were then used for subsequent analyses.
Establishment of the PPI Network
The predicted EGCG targets and OA-related targets were uploaded to the online platform “Venny 2.1.0” (https://bioinfogp.cnb.csic.es/tools/venny/index.html) to identify the overlapping targets. A Venn diagram was generated to visualize the overlapping genes. The common targets were then imported into the STRING (https://string-db.org) for protein-protein interaction (PPI) analysis, with the minimum required interaction score set at 0.9. The species was defined as “Homo sapiens”. Isolated nodes were removed. The resulting PPI data were downloaded as a TSV file and imported into Cytoscape 3.10.1 for visualization. The CytoNCA plugin was applied to analyze the network topology of protein nodes, using Degree Centrality (DC, reflecting the node’s direct connection ability), Betweenness Centrality (BC, measuring the node’s mediating role in network paths), and Closeness Centrality (CC, characterizing the node’s proximity to other nodes) as parameters for identifying key targets.41
GO and KEGG Pathway Enrichment Analyses
To explore the potential biological functions of the identified targets, the overlapping targets between EGCG and OA were submitted to the Metascape database (https://metascape.org/), with parameters set to a minimum overlap of 3 and a P-value cutoff of 0.01 for Gene Ontology (GO) analysis. Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway profiling was performed with the DAVID bioinformatics resource (https://davidbioinformatics.nih.gov/), obtaining data related to Cellular Component (CC), Molecular Function (MF), and Biological Process (BP). The top 20 GO terms and the top 30 KEGG pathways based on P-values were selected for further analysis. The results were visualized using the Bioinformatics online platform (http://www.bioinformatics.com.cn/).
Molecular Docking
EGCG’s 3D structure from PubChem was optimized in Chem3D 18.1 and saved as a MOL2 file. Then, the top four targets from the drug-disease PPI network were acquired through the Protein Data Bank (PDB, https://www.rcsb.org/), with the organism set to “Homo sapiens”, the experimental method specified as X-ray diffraction, and a resolution of less than 3 Å. Suitable target proteins for docking were selected from the search results. The PDB files of these proteins were imported into PyMOL to remove solvents and organic molecules, and the cleaned structures were saved again in PDB format. In this study, molecular docking was performed using CB-DOCK2 (https://cadd.labshare.cn/cb-dock2/php/index.php), a server for protein-ligand blind docking developed by Liu et al.42 This tool, known as CB-DOCK2 (CB-DOCK2: enhanced protein-ligand blind docking through the integration of cavity detection, docking, and fitting of homologous templates), employs a cavity detection technique grounded in the curvature of protein surfaces (CurPocket) to direct AutoDock Vina (version 1.1.2) during the molecular docking process. Receptor and ligand files were uploaded into CB-DOCK2 to assess cavity size and Vina docking scores. In molecular docking, the estimated affinity values represent the binding strength and serve as a quantitative indicator of ligand-receptor interaction intensity; numerically lower (more negative) values reflect stronger binding affinity. A binding energy of −5.0 kcal/mol or lower was considered indicative of significant molecular-target interactions. The docking conformations were visualized using PyMOL (https://www.pymol.org/).
Experimental Validation
Reagents and Antibodies
Epigallocatechin-3-gallate (≥95%, ST1011) was purchased from Beyotime Institute of Biotechnology (Shanghai, China). Tunicamycin (TM, 11089-65-9) was obtained from Yuanye Bio-Technology Co., Ltd. (Shanghai, China). Type II collagenase (C6885) and dimethyl sulfoxide (DMSO, 67-68-5) were obtained from Sigma-Aldrich (St. Louis, MO, USA). Dulbecco’s Modified Eagle Medium (DMEM, LM-D1110) and fetal bovine serum (FBS, FB-1058) were purchased from BIOSERA (Cholet, France). The BCA protein assay kit (23227) was purchased from Thermo Fisher Scientific (Waltham, MA, USA). Penicillin-streptomycin solution (C0222) was acquired from the Beyotime Institute of Biotechnology (Shanghai, China). Primary antibodies against β-actin (AF0003), Matrix Metallopeptidase 13 (MMP13) (AF7479), GRP78 (AF0171), and P-IRE-1α (AF8542) were also obtained from Beyotime Institute of Biotechnology. Primary antibodies against inositol-requiring enzyme 1α (IRE-1α) (A17940), eukaryotic initiation factor 2α (eIF-2α) (A21221), and P-eIF-2α (AP0692) were purchased from ABclonal (Wuhan, China). Antibodies against inducible nitric oxide synthase(iNOS) (F0177), cyclooxygenase-2 (COX2) (F0327), STAT3 (F0200), and C188-9 (S8605) were purchased from Selleck Chemicals (Houston, TX, USA). Primary antibodies against COL2A1 (sc-28887), MMP13 (sc-30073), Matrix Metallopeptidase 3 (MMP3) (sc-21732), and SRY-box transcription factor 9(SOX9) (sc-20095) were obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA). The IL-6 antibody (GB11117) was provided by Servicebio (Wuhan, China). The C/EBP homologous protein (CHOP) (cat. #2895), B-cell lymphoma-2 (BCL-2) (cat. #15071), BCL-2 Associated X Protein (BAX) (cat. #2772), P-STAT3 (Ser272) (cat. #9134), anti-mouse IgG, HRP-linked antibody (cat. #7076), and anti-rabbit IgG, HRP-linked antibody (cat. #7074) were obtained from Cell Signaling Technology (Danvers, MA, USA).
Isolation and Culture of Primary Chondrocytes
One-day-old Sprague-Dawley rats (Shanghai Sipur-Bikai Laboratory Animal Co., LTD. [Certificate No. SCXK (Shanghai) 2018-0006]) were euthanized with excess isoflurane, then soaked in 75% ethanol for 10 minutes and washed 3 times with PBS. Under sterile conditions, the articular cartilage tissue was dissected, thoroughly rinsed in PBS, and cut into small pieces approximately 1 mm × 1 mm × 1 mm. These tissue pieces were digested with 0.5% Type II collagenase (C6885, Sigma-Aldrich) and incubated in a shaker at 37°C and 150 rpm for approximately 60 minutes. The supernatant was collected and centrifuged at 1500 rpm for 5 minutes, after which the supernatant was discarded. The cell pellet was resuspended in DMEM (LM-D1110, BIOSERA) containing 10% FBS (FB1058, BIOSERA) and 1% penicillin-streptomycin (C0222, Beyotime) and seeded into a 10 cm culture dish. The cells were cultured in an incubator at 37°C with 5% CO2. All animal experiments comply with the ARRIVE guidelines.
Cell Viability Assay
Chondrocyte viability was assessed using the Cell Counting Kit-8 assay kit (BS350B) from Biosharp. Chondrocytes were seeded at a density of 1 × 104 cells per well in a 96-well plate and allowed to adhere overnight before treatment. Cells were then exposed to various concentrations of EGCG (0, 1, 5, 10, 50, 100, and 200 μM) for 24 and 48 hours. Following treatment, 10 µL of CCK-8 reagent was added to each well, and the microplate was incubated at 37 °C for 120 minutes. The absorbance at 450 nm was then quantified using a spectrophotometric plate reader (BioTek). Cell proliferation activity (%) was calculated using the formula (OD test - OD blank) / (OD control - OD blank) × 100%.
Chondrocyte Grouping and Treatment
After culturing chondrocytes from passages 1 to 2 for 24 hours, the cells were randomly divided into five groups according to the results of the cell viability assays: control group, IL-1β-treated group, low-concentration EGCG group, medium-concentration EGCG group, and high-concentration EGCG group.
The treatments were as follows: the control group received no treatment. For the remaining groups, an in vitro inflammation model was established by stimulating chondrocytes with 10 ng/mL IL-1β, as previously described.43 The low-, medium-, and high-concentration EGCG groups were co-treated with IL-1β and EGCG at final concentrations of 1, 10, and 50 μM, respectively, for 24 hours. To further investigate the role of ER stress in EGCG-mediated effects, an additional group was treated with 2 μg/mL Tunicamycin (TM), a classical inducer of ER stress.
Immunofluorescence Staining
Chondrocytes were fixed using Cell Tissue Fixative (P00989, Beyotime) and subsequently permeabilized with 0.3% Triton X-100. After blocking with 3% bovine serum albumin (BSA) for 1 hour at 4°C, the cells were incubated overnight at 4°C with primary antibodies against MMP13, SOX9, and COL2A1 (all diluted 1:500). The following day, cells were washed three times with PBS and incubated in the dark for 1 hour with secondary antibodies: goat anti-mouse (A32723; Invitrogen) or goat anti-rabbit (A32732; Invitrogen), diluted 1:1000. Nuclei were counterstained with DAPI (Proteintech, CM07245) for 10 minutes in the dark. Fluorescence images were captured using a fluorescence microscope (Olympus, IX73), and fluorescence intensities were quantified using GraphPad Prism 10.0.
Western Blot Analysis
Chondrocytes were seeded into 6-well plates at a density of 3 × 105 cells per well. When the cells reached approximately 80% confluence, they were subjected to the indicated treatment for 24 hours. The cells were collected and lysed on ice with RIPA buffer (G2002, Servicebio) supplemented with a cocktail of protease and phosphatase inhibitors. The concentration of protein was measured using the BCA Protein Assay kit. Equal volumes of protein from each group were separated by SDS-PAGE gel electrophoresis, followed by transfer to PVDF membranes (IPVH00010, Immobilon-P). The membranes were blocked for 1 hour at room temperature with a quick blocking solution (PS108P, Epyzime). Primary antibodies (as shown in 2.2.1. Reagents and Antibodies) were diluted according to the manufacturer’s instructions and incubated with the membranes at 4°C overnight, and subsequently washed three times with Tris-buffered saline with Tween-20 (TBST). The secondary antibodies (as shown in 2.2.1. Reagents and Antibodies) were added and incubated at room temperature for 1 hour. The membranes were then washed three more times with TBST, and the protein bands were visualized using an ECL luminescence kit (BL520A, BioSharp, China).
Statistical Analysis
All experiments were conducted in triplicate. Statistical analysis was performed using GraphPad Prism 10.0 (GraphPad Software, USA). Data were analyzed using one-way analysis of variance (ANOVA), followed by Tukey’s honestly significant difference (HSD) post hoc test for pairwise comparisons between groups. p < 0.05 was statistically significant.
Results
Network Pharmacology-Based Analysis
Target Prediction of EGCG and OA
Based on the predictions from several databases, including TCMSP, BATMAN-TCM, and PharmMapper, 488 potential targets of EGCG were identified (Table S1). In addition, concurrently, 1618 OA-associated targets were identified through the consolidation of four disease-associated databases: OMIM, GeneCards, Therapeutic Target Database (TTD), and DisGeNET, after removing duplicates (Figure 1a) (Table S2). A Venn diagram was used to display the overlap between EGCG-related targets and OA-related targets (Figure 1b).
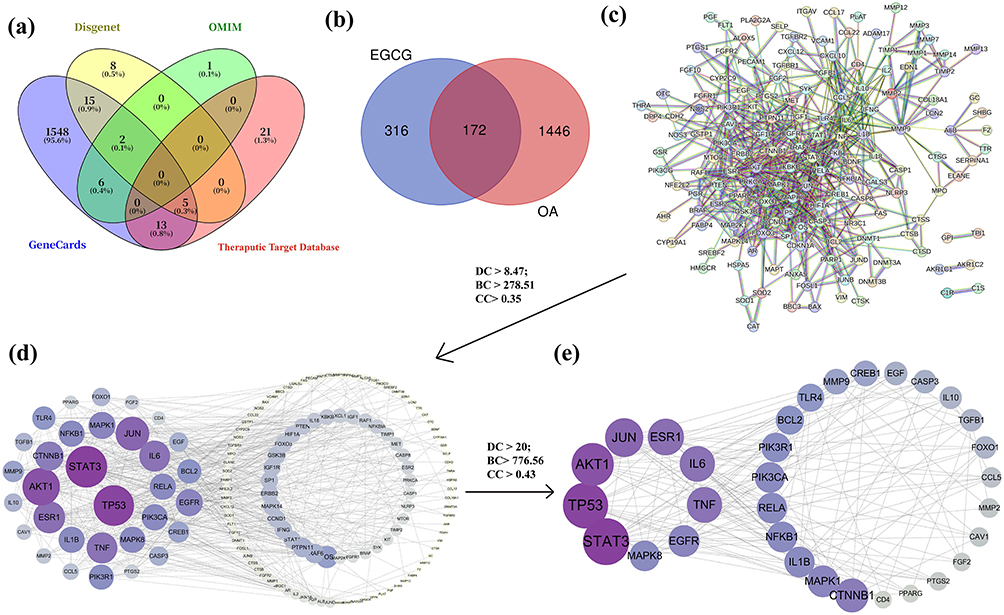 |
Figure 1 Targets of EGCG in OA. (a) Venn diagram of targets associated with OA; (b) Overlapping genes between EGCG targets and OA-related targets; (c) PPI network of shared targets between EGCG and OA; (d) 32 key targets of EGCG in OA, listed on the left; (e) The top nine key targets of EGCG in OA, listed on the left. |
Construction of the PPI Network
A total of 172 shared targets between EGCG and OA were entered into the STRING 11.0 database, with a required score of highest confidence (0.90). The resulting PPI network contained 171 nodes and 621 edges (Figure 1c) (Table S3). To further investigate the interactions among these proteins, the PPI network was visualized using Cytoscape software. After removing isolated nodes, 146 nodes were retained. Subsequently, CytoNCA, a Cytoscape plugin for network centrality analysis, was applied to identify hub genes within the network (Table S4). Genes with values above the average for DC > 8.47, BC > 278.51, and CC > 0.35 were selected to construct a subnetwork, resulting in the identification of 32 core targets (Figure 1d). A second round of screening, using more stringent thresholds (DC > 20; BC > 776.56; CC > 0.43), further refined the network to a smaller set of highly relevant targets (Figure 1e). Among these, the top four key proteins, including STAT3, TP53, AKT1, and JUN, were selected for subsequent molecular docking analyses.
GO and KEGG Enrichment Analysis
Among the 172 genes in the core PPI network, GO analysis identified 2163 BP (Table S5A), 223 MF terms (Table S5B), and 92 CC (Table S5C). The top 20 significantly enriched terms in each category are shown in Figure 2a. KEGG pathway analysis revealed 175 statistically significant pathways (Table S6), with the top 30 (based on P-values) displayed in a bubble chart (Figure 2b). Here, color intensity reflects significance, and dot size represents the number of genes.
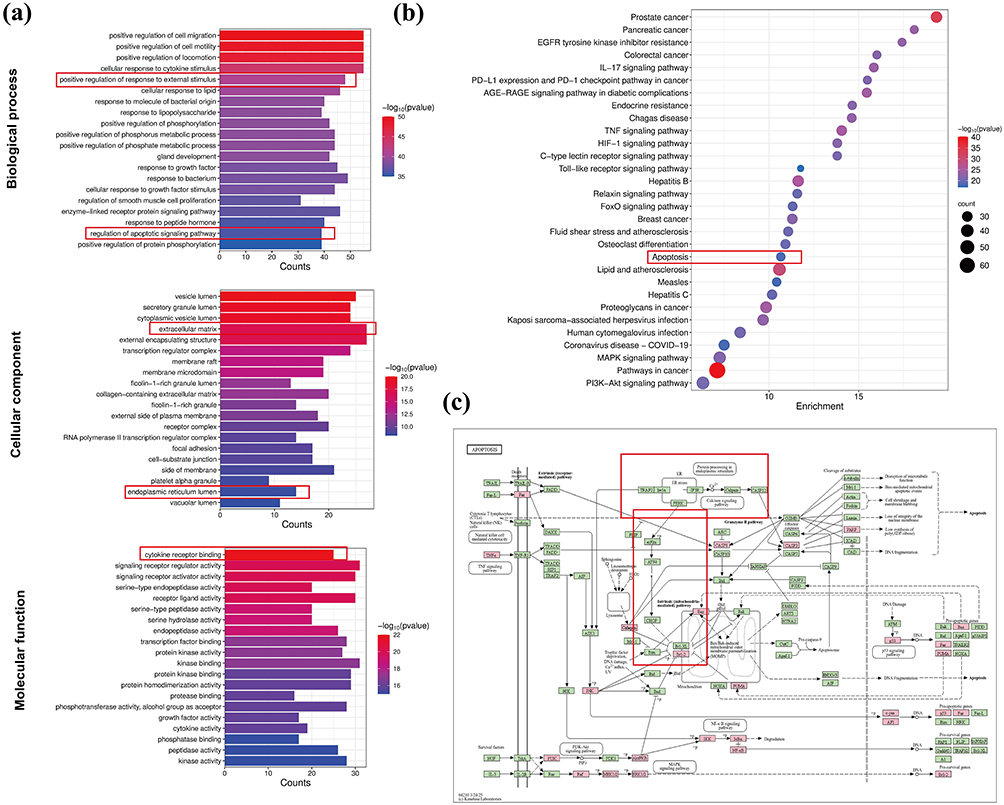 |
Figure 2 Functional Enrichment Analysis of EGCG Targets. (a) Top 20 GO terms for BP, CC, and MF; (b) Top 30 KEGG pathways shown in the bubble plot; (c) Distribution of EGCG target proteins within the apoptosis pathway. Red nodes represent potential targets of EGCG, and green nodes represent related targets within the pathway. |
GO analysis revealed that the main association of BP terms was with cell motility, cytokine response, external stimulus response, and regulation of apoptotic signaling. CC terms were enriched in collagen-containing ECM and ER lumen, while MF terms included cytokine receptor binding, serine-type endopeptidase activity, transcription factor binding, and kinase-related activities. Notably, both BP and KEGG results indicated significant enrichment in apoptosis-related pathways. Further KEGG analysis (https://www.genome.jp/kegg/pathway.html) revealed that 26 target genes were involved in the apoptosis pathway (Figure 2c). Literature evidence also supports a strong link between ER stress and apoptosis, consistent with the CC enrichment of ER lumen-related terms.20,44,45 These findings suggest that EGCG’s therapeutic effects in OA may be closely associated with the regulation of ER stress.
Molecular Docking
We performed molecular docking of EGCG with four core proteins (STAT3, TP53, AKT1, JUN) to assess the binding potential between protein receptors and ligands. As shown in Table 1, the binding affinity was assessed based on docked scores. The results showed that EGCG showed favorable binding affinity with all four targets, with binding energies ranging from −5.9 to −8.1 kcal/mol. The 3D molecular docking conformations illustrating these interactions between EGCG and the core proteins were visualized using PyMOL software (Figure 3). The strong binding affinity between EGCG and STAT3 (−8.1 kcal/mol) suggests a robust interaction. Moreover, STAT3 was identified as one of the most central nodes in the PPI network. Given its pivotal role in mediating cytokine-induced signaling, inhibition of STAT3 activation has been shown to suppress inflammatory cascades and mitigate cartilage degradation in OA.25,46 Therefore, we speculate that the protective effect of EGCG on OA may be associated with its ability to inhibit STAT3 activation.
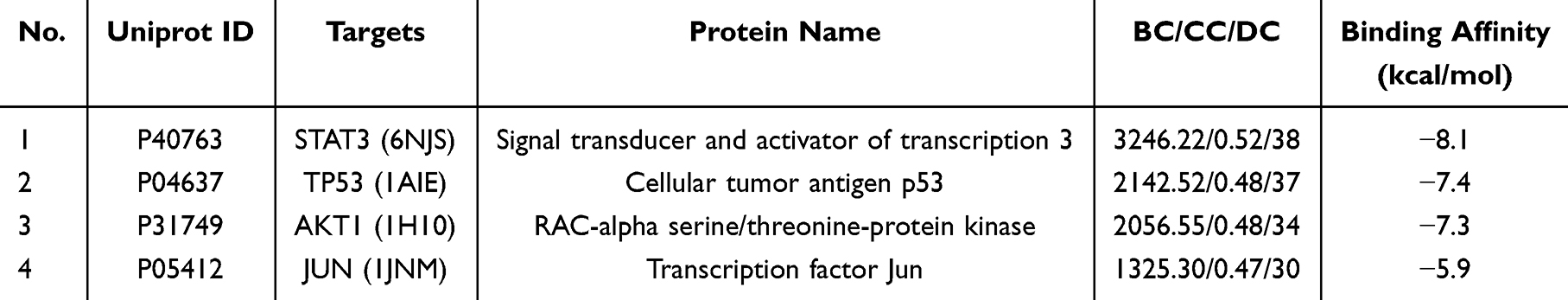 |
Table 1 Key Information for the 4 Core Targets |
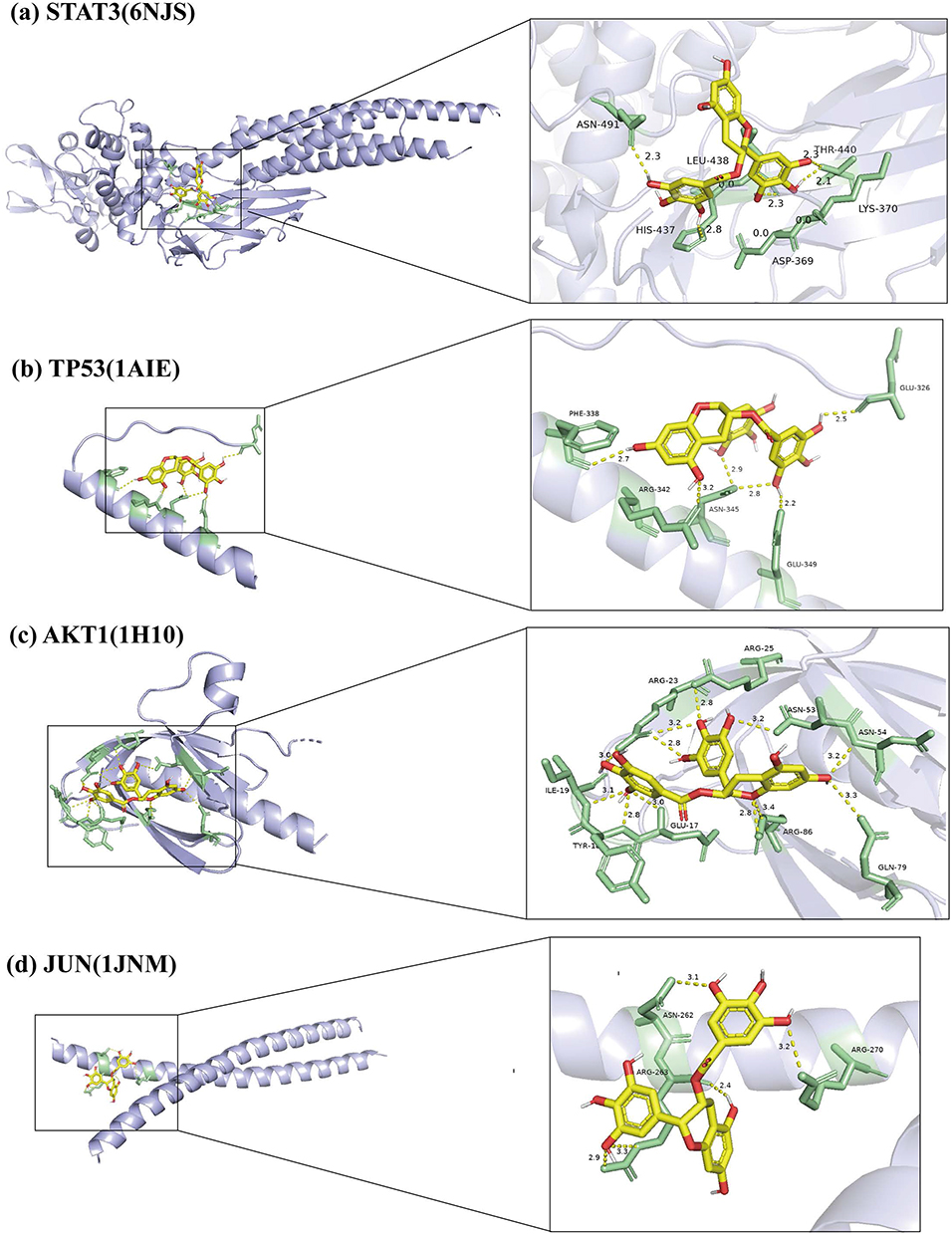 |
Figure 3 Molecular docking model 3D diagram. EGCG binds to STAT3 (a), TP53 (b), AKT1 (c), and JUN (d). |
Experimental Validation in vitro
Effects of EGCG on Chondrocytes Viability
The chemical structure of EGCG is shown in Figure 4a. To investigate the effects of EGCG on chondrocyte viability, cells were exposed to increasing concentrations of EGCG (1, 5, 10, 50, 100, and 200 μM) for 24 and 48 hours. The results demonstrated that EGCG concentrations below 50μΜ maintained cell viability above 80% after 24 hours of treatment (Figure 4b and c). Based on these findings, three concentrations: low (1 μM), medium (10 μM), and high (50 μM) were selected for subsequent 24-hour treatment experiments to further evaluate the effects of EGCG on chondrocytes.
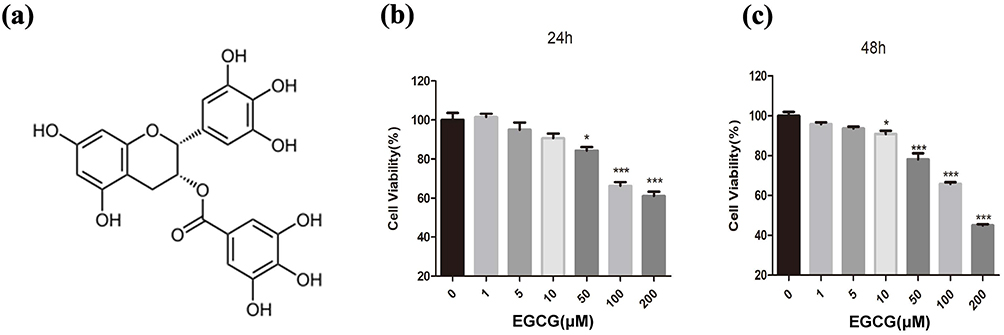 |
Figure 4 The effects of EGCG on chondrocyte viability. (a) Chemical structure of EGCG; (b and c) The cytotoxic effects of EGCG on chondrocytes were assessed across a range of concentrations over 24 and 48 hours using a CCK-8 assay kit. The data in the figures are expressed as means ± S.D. *p < 0.05, ***p < 0.001. n = 3. |
EGCG Inhibits IL-1β-Mediated Inflammatory Response and ECM Catabolism in Chondrocytes
Disruption of the balance between ECM synthesis and degradation plays a pivotal role in the progression of OA, with inflammatory mediators accelerating ECM breakdown.47 To investigate the effects of EGCG on IL-1β-induced inflammation and ECM degradation in chondrocytes, Western blot analyses were performed. As shown in Figure 5a–e, IL-1β stimulation markedly increased the protein expression of MMP13, COX2, iNOS, and IL-6 in chondrocytes.
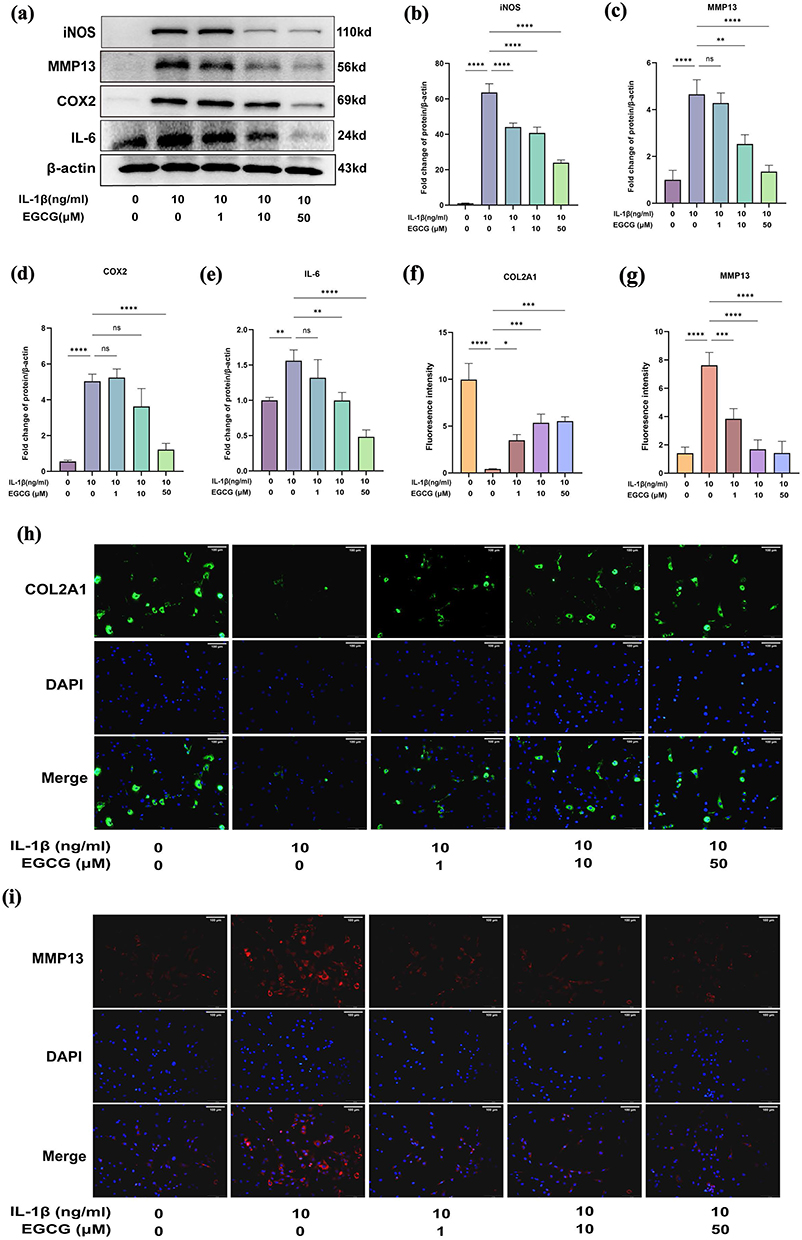 |
Figure 5 EGCG Attenuates IL-1β-Induced Inflammatory Response and Catabolism in Chondrocytes. Chondrocytes were treated with IL-1β (10 ng/mL) in the presence or absence of EGCG (1, 10, 50 μM) for 24 hours. The expression levels of inflammation- and catabolism-related proteins in chondrocytes were subsequently evaluated. (a-e) Western blot analysis of iNOS, MMP13, COX2, and IL-6 expression. (f-i) Immunofluorescence co-staining with DAPI to detect COL2A1 and MMP13 in the nucleus (scale bar: 100 μm). The data in the figures are expressed as means ± S.D. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, ns p > 0.05, n = 3. |
Further immunofluorescence staining of MMP13 and type II collagen confirmed the results of the Western blot in Figure 5a–e, as illustrated in Figure 5f–i. IL-1β-treated chondrocytes exhibited decreased fluorescence intensity of COL2A1 (green, Figure 5h) and increased intensity of MMP13 (red, Figure 5i), indicating ECM degradation. These alterations were partially mitigated by EGCG treatment.
EGCG Reduces the IL-1β-Induced Increase in ER Stress Markers
ER stress plays a significant role in the pathological mechanisms of OA. Under homeostatic conditions, the ER chaperone protein GRP78 (also known as BiP) remains tightly bound to ER stress sensors, preventing their activation. However, under pathological conditions, GRP78 preferentially binds to misfolded proteins, leading to the release and subsequent phosphorylation of ER stress sensors.8 CHOP is a well-characterized transcription factor that induces apoptosis when ER stress remains unresolved.48
To evaluate the effects of EGCG on ER stress in IL-1β-treated chondrocytes, we measured the levels of GRP78 and CHOP. Immunoblotting results (Figure 6a–c) indicated that the expression levels of ER stress markers were markedly up-regulated in the IL-1β-treated group compared to the control group. In contrast, EGCG treatment attenuated the IL-1β-induced upregulation of these markers, indicating a reduction in ER stress.
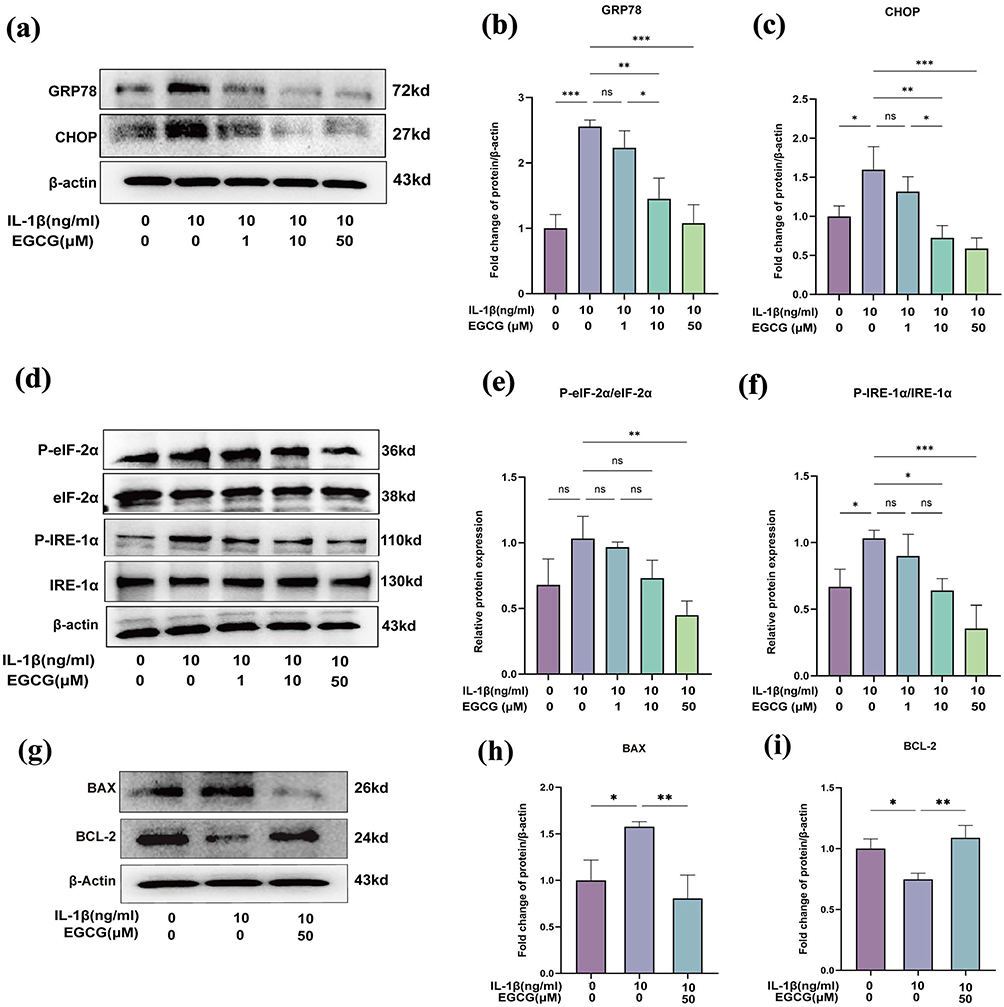 |
Figure 6 EGCG Alleviates IL-1β-Induced ER Stress. (a–c) Western blot analysis of ER stress markers GRP78 and CHOP in chondrocytes treated with or without EGCG (1, 10, 50 μM) under IL-1β stimulation; (d–f) Western blot analysis of key proteins associated with ER stress signaling pathways following EGCG treatment, including eIF-2α, phosphorylated eIF-2α (P-eIF-2α), IRE-1α, and phosphorylated IRE-1α (P-IRE-1α). (g–i) Western blot analysis of BAX and BCL-2 expression in chondrocytes treated with or without 50 μM EGCG under IL-1β stimulation. The data in the figures are expressed as ± S.D. *p < 0.05, **p < 0.01, ***p < 0.001, ns p > 0.05, n = 3. |
EGCG Mitigates ER Stress-Induced UPR and Subsequent Apoptosis in Chondrocytes
We further explored EGCG’s regulatory effects on the major UPR signaling pathways. Signal transduction during ER stress leads to the selective activation of downstream cascades. Specifically, activated PERK inhibits overall protein synthesis by phosphorylating the translation initiation factor eIF-2α.49 EGCG, particularly at a concentration of 50 μM, significantly reduced the phosphorylation of eIF-2α mediated by PERK (Figure 6d and e).
IRE-1α, a type I transmembrane protein located in the ER, undergoes oligomerization and autophosphorylation upon ER stress, thereby activating its ribonuclease activity.50 Western blot analysis revealed that in the IRE-1α pathway, the administration of 50 μM EGCG markedly suppressed IL-1β-mediated IRE-1α phosphorylation (Figure 6d and f).
CHOP is recognized as a key mediator that shifts the UPR toward apoptosis by downregulating BCL-2 levels, thereby diminishing cellular resistance to intrinsic and extrinsic apoptotic signals.51 To demonstrate the anti-apoptotic effects of EGCG, we assessed the expression of BAX and BCL-2 in chondrocytes. The results showed that, compared with the IL-1β-treated group, EGCG treatment significantly decreased BAX expression and increased BCL-2 expression (Figure 6g–i). Overall, these findings suggest that EGCG has a broad impact on the UPR and its downstream apoptotic pathways.
EGCG May Promote ECM Anabolism by Alleviating ER Stress
The increased protein synthesis and cartilage matrix degeneration may result in an excessive protein load within the ER, thereby inducing ER stress.52 To further investigate whether the protective effects of EGCG on ECM synthesis-related factors are mediated by ER stress, TM, an ER stress activator, was employed. Immunofluorescence analysis (Figure 7a–d) revealed that EGCG alone mitigated the IL-1β-induced suppression of COL2A1 expression, the expression levels of COL2A1 and SOX9 were reduced again upon TM treatment. These findings suggest that ER stress can partially counteract or interfere with the protective effects of EGCG on ECM synthesis in chondrocytes. Modulation of ER stress thus represents a pivotal mechanism underlying EGCG activity, indicating that its effects are closely linked to the ER stress signaling pathway.
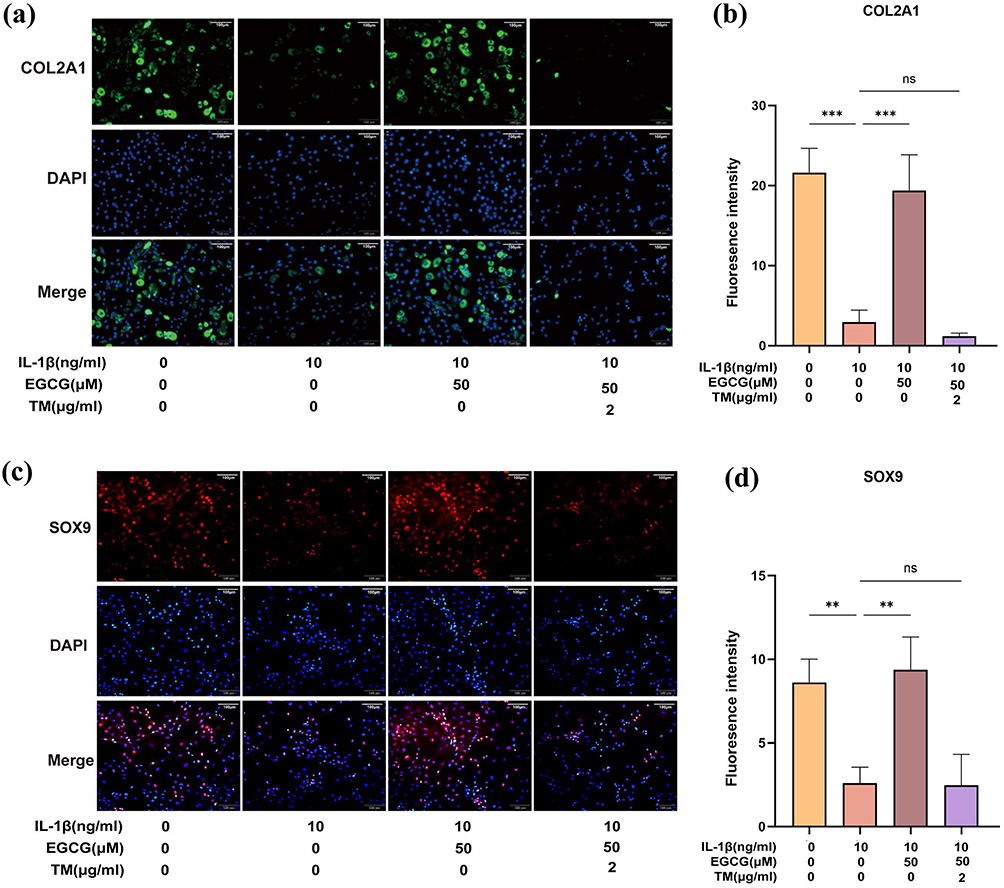 |
Figure 7 EGCG reduces chondrocyte apoptosis; TM-induced ER stress reversed the protective effects of EGCG against IL-1β-induced matrix reduction in chondrocytes. (a and b) COL2A1 immunofluorescence staining. Markedly increased green bright puncta indicated the upregulated expression of COL2A1 (bar: 100 μm). (c and d) SOX9 immunofluorescence staining. Markedly increased red bright puncta indicated the upregulated expression of SOX9 (bar: 100 μm). Fluorescence intensity was quantified using ImageJ software. The data in the figures are expressed as means ± S.D., **p < 0.01, ***p < 0.001, ns p > 0.05; n = 3. |
The Effect of EGCG on ER Stress in IL-1β-Stimulated Chondrocytes Was Blocked by TM
To further elucidate the mechanism by which EGCG modulates ER stress in IL-1β-induced chondrocytes, we examined the expression of ER stress-related genes in response to TM. As illustrated in Figure 8, TM administration markedly attenuated EGCG-mediated regulation of ER-stress-associated proteins, including the canonical markers GRP78 and CHOP (Figure 8a–c). Moreover, TM reversed the EGCG-induced suppression of IRE-1α and eIF-2α phosphorylation (Figure 8d–f). Consistent with these observations, EGCG significantly diminished the overall stress burden in the TM-induced ER-stressed chondrocyte model (Figure 8g–l). Collectively, these findings indicate that the cytoprotective effects of EGCG on chondrocytes are, at least in part, mediated through modulation of the ER-stress signaling pathway.
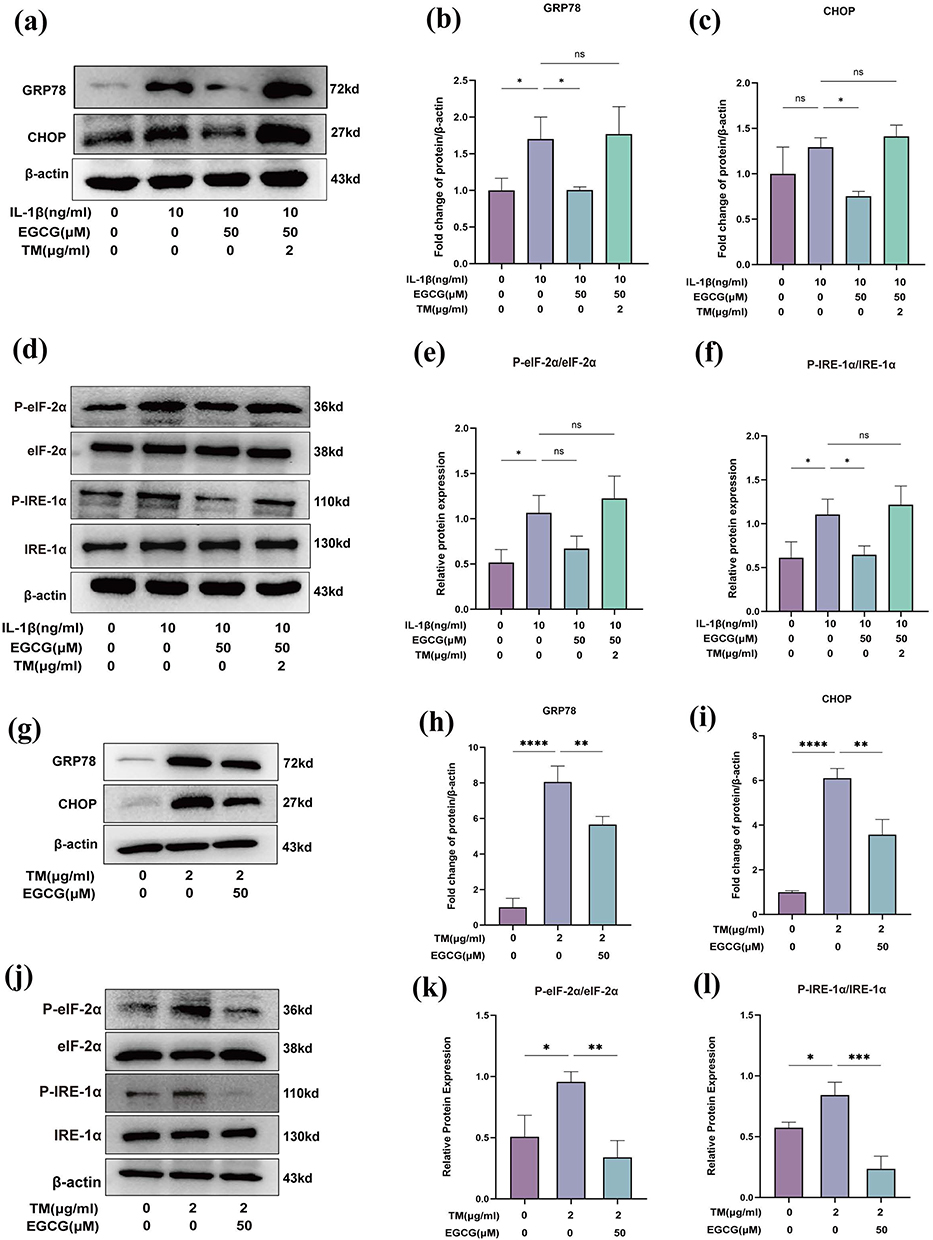 |
Figure 8 TM Reverses the Ameliorative Effects of EGCG on IL-1β-Induced ER Stress in Chondrocytes. (a) Western blot analysis of GRP78, CHOP; (b and c) Quantitative analysis of protein expression levels; (d) Western blot analysis of IRE-1α, phosphorylated IRE-1α (P-IRE-1α), eIF-2α, and phosphorylated eIF-2α (P-eIF-2α); (e and f) The ratio of P-IRE-1α to total IRE-1α and P-eIF-2α to total eIF-2α; (g) Western blot analysis of GRP78 and CHOP in TM-induced chondrocytes; (h and i) Quantitative analysis of protein expression levels; (j) Western blot analysis of IRE-1α, phosphorylated IRE-1α (P-IRE-1α), eIF-2α, and phosphorylated eIF-2α (P-eIF-2α) in TM-induced chondrocytes; (k and l) The ratio of P-eIF-2α to total eIF-2α and P-IRE-1α to total IRE-1α. The data in the figures are expressed as means ± S.D. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, ns p > 0.05; n = 3. |
EGCG Attenuates ER Stress Through STAT3 Signaling
STAT3 is a central target of EGCG in OA therapy, with a notable binding affinity to the EGCG ligand. Western blot analysis revealed that IL‑1β stimulation markedly increased STAT3 phosphorylation at the Ser727 site (P-STAT3^S727). Notably, intervention with EGCG (50 μM) significantly suppressed IL‑1β-induced STAT3 phosphorylation (Figure 9a and b).
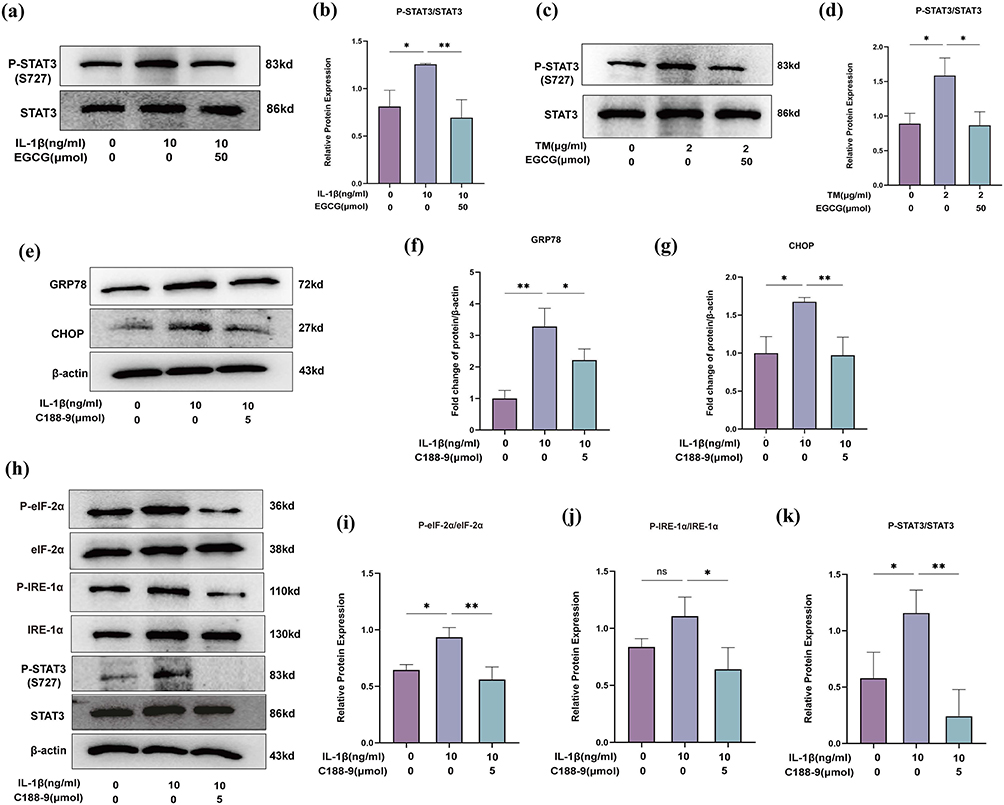 |
Figure 9 EGCG decreases the phosphorylation of STAT3 in IL-1β-induced and TM-induced chondrocytes. (a) Western blot analysis of STAT3 and phosphorylated-STAT3 (P-STAT3) in the IL-1β group. (b) The ratio of P-STAT3 to total STAT3 in the IL-1β group; (c) Western blot analysis of STAT3 and phosphorylated-STAT3 (P-STAT3) in the TM group; (d) The ratio of STAT3 to total STAT3 in the TM group. (e) Western blot analysis of GRP78, CHOP; (f and g) Quantitative analysis of protein expression levels; (h) Western blot analysis of IRE-1α, phosphorylated IRE-1α (P-IRE-1α), eIF-2α, and phosphorylated eIF-2α (P-eIF-2α), STAT3, and phosphorylated STAT3; (i–k) The ratio of P-IRE-1α to total IRE-1α, P-eIF-2α to total eIF-2α, and P-STAT3 to total STAT3. The data in the figures are expressed as means ± S.D. (*p < 0.05, **p < 0.01; n = 3). |
Sustained ER stress has been reported to activate the JAK1/STAT3 axis, thereby driving inflammation.53 To further validate this, chondrocytes were treated with TM (2 µg/mL) to induce ER stress. Under these conditions, P‑STAT3 (S727) was markedly up-regulated, whereas co‑treatment with EGCG (50 μM) effectively attenuated TM‑induced STAT3 hyperphosphorylation (Figure 9c and d). These findings suggest that EGCG modulates the STAT3 signaling pathway, at least in part, by alleviating ER stress.
To further examine the potential connection, STAT3 signaling was pharmacologically silenced with the selective inhibitor C188-9 in IL-1β-stimulated chondrocytes, and the resultant impact on ER stress was assessed. Blockade of STAT3 activation markedly attenuated ER stress (Figure 9e–k), providing compelling evidence for a functional crosstalk between the STAT3 pathway and ER stress.
Discussion
EGCG, a polyphenolic compound, has been reported to exert protective effects in various inflammation-related diseases, including neural injury,54 non-alcoholic fatty liver disease,55 and retinopathy.56 Its therapeutic potential in OA has gained increasing attention, as it not only displays potent anti-inflammatory properties but also alleviates cartilage degeneration by modulating OA-related gene expression.57,58 Building on these findings, we employed an integrated network pharmacology approach to systematically explore the molecular mechanisms underlying the effects of EGCG in OA.
Through this analysis, we identified 172 potential therapeutic targets of EGCG in OA. GO enrichment revealed that these targets are mainly involved in responses to cytokine stimuli, external stressors, and inflammation-related biological processes. KEGG pathway analysis further suggested that inhibition of ER stress-induced apoptosis may be a critical mechanism by which EGCG exerts its therapeutic effects (Figure 2b).
In OA pathogenesis, chondrocyte apoptosis represents a pivotal pathological event that, together with inflammation-driven ECM metabolic dysregulation, accelerates cartilage destruction.59 Among the endogenous apoptotic pathways, ER stress-mediated apoptosis plays a central role.60 ER stress in OA cartilage has been shown to activate apoptotic signaling in chondrocytes.61,62 Under the regulation of pro-inflammatory cytokines, ER stress-related molecules are upregulated, disrupting the collagen framework essential for chondrocyte survival.63 This, in turn, promotes the production of matrix metalloproteinase (MMPs) and pro-inflammatory mediators, shifting the ECM synthesis-degradation balance toward net loss.64–66
Consistent with previous findings,29,67 our study confirmed that EGCG inhibits chondrocyte apoptosis. Importantly, we first demonstrated a previously unreported mechanism whereby EGCG mitigates inflammation-induced apoptosis by alleviating ER stress. Notably, in this process, ER stress interacts with the IL-1β-mediated STAT3 signaling pathway, exerting chondrodestructive effects. We observed that EGCG can inhibit the inflammatory reaction and protect chondrocyte ECM from degradation in the IL-1β-treated model (Figures 4 and 5). Given the crucial role of ER homeostasis in maintaining chondrocyte function and cartilage integrity, we further explored whether EGCG’s chondroprotective effects might also involve modulation of ER stress.
GRP78 and CHOP are pivotal biomarkers of ER stress. GRP78 is upregulated during the early phase of ER stress, facilitating cellular adaptation to the buildup of misfolded proteins and marking the onset of the stress response.68,69 Sustained and unresolved ER stress activates the UPR, culminating in CHOP transcription.68 The upregulation of CHOP expression signifies the transition from pro-survival mechanisms to apoptotic pathways.48 Both GRP78 and CHOP, as critical effector proteins in ER stress, play central roles in inflammation-driven chondrocyte degeneration.70 Our experimental results revealed that EGCG significantly inhibited the expression of ER stress markers GRP78 and CHOP in IL-1β-stimulated chondrocytes.
GRP78 preferentially binds misfolded proteins, thereby releasing stress sensors such as PERK and IRE-1α. Subsequently, PERK phosphorylates eIF-2α, leading to a reduction in global protein synthesis and thereby limiting the influx of newly synthesized proteins into the ER.49 A prior study demonstrated PERK and eIF-2α phosphorylation in IL-1β-exposed human juvenile costal chondrocytes,71 suggesting that elevated IL-1β levels in arthritic conditions may induce ER stress in chondrocytes. We analyzed the main branches of the UPR, specifically IRE-1α and eIF-2α, to evaluate the effects of EGCG on IL-1β-induced ER stress in chondrocytes. Our results showed that IL-1β stimulation markedly increased the phosphorylation of IRE-1α and eIF-2α in chondrocytes, whereas EGCG treatment, especially at 50 μM, significantly reduced their phosphorylation levels. Subsequently, our experiments demonstrated that EGCG markedly downregulated the expression of the BAX (a pro-apoptotic factor) and upregulated the expression of BCL-2 (an anti-apoptotic protein). This result suggested that EGCG may inhibit chondrocyte apoptosis by suppressing ER stress.
To further validate the protective effects of EGCG on chondrocytes by alleviating ER stress, a classical ER stress inducer, TM, which inhibits N-linked glycosylation and disrupts N-glycoprotein folding, was used to treat chondrocytes.72 The results showed that TM reversed the EGCG-induced upregulation of COL2A1 and SOX9 in inflammatory chondrocytes and reactivated ER stress markers and key UPR branches. Additionally, EGCG treatment suppressed the TM-induced upregulation of ER stress markers and major UPR signaling pathways in an in vitro model of ER stress. A study demonstrated that sustained ER stress impairs type II collagen folding and secretion, resulting in its intracellular accumulation within the dilated ER lumen and contributing to matrix loss in cartilage tissue.73 CHOP-deficient mice exhibit significantly reduced cartilage degradation, accompanied by elevated Col2A1 and ACAN expression in chondrocytes.74 In addition, CHOP has been implicated in the regulation of MMP13 expression, possibly through interaction with C/EBPβ, which directly binds to the MMP13 promoter in human chondrocytes.75 As a member of the same transcription factor family, CHOP is known to exert a dominant-negative regulatory effect on C/EBPβ activity, further highlighting the complex interplay between ER stress signaling and cartilage matrix degradation.76 Collectively, these findings underscore that EGCG may maintain chondrocyte homeostasis and cartilage integrity by preventing excessive activation of the UPR pathway.
STAT3 exhibited the strongest binding affinity with EGCG, implying that STAT3 functions as a pivotal node via which EGCG delivers its protective actions. As a central transcription factor in the JAK-STAT signaling pathway, the activation of STAT3 and the UPR pathway may produce synergistic effects that further intensify inflammatory responses and ER stress.77 Avalle et al reported that ER-localized STAT3 acts as a “gatekeeper” of ER-mitochondrial Ca2+ signaling.78,79 Mechanistically, STAT3 can translocate to mitochondria via Ser727 phosphorylation, modulate calcium flux to coordinate mitochondrial and ER function.78,80 Activation of the STAT3 pathway upregulates NLRP3 inflammasome or hypoxia-inducible factor 1α (HIF-1α), which promotes pro-inflammatory cytokine expression, reduces mitochondrial membrane potential, induces ROS production, and ultimately triggers ER stress.81,82 Additionally, recent research has revealed a regulatory interplay between STAT3 and ER stress.83,84 A study conducted by Chou et al provided additional evidence that cadmium (Cd) fosters the interaction between IRE-1α and STAT3, which enhances the phosphorylation of STAT3 at Ser727, thus hindering its signaling function and worsening cell apoptosis.85 This forms a positive feedback loop “ER stress-STAT3 activation-exacerbated ER stress”, which plays a central role in the mutual amplification of inflammation and ER stress.
Our study demonstrated that EGCG significantly suppresses IL-1β-induced STAT3 (Ser727) phosphorylation, indicating that EGCG modulates this pathway in IL-1β-stimulated chondrocytes. Interestingly, we also found that TM increased the phosphorylated of STAT3 (P-STAT3) (Figure 9c and d), whereas inhibition of STAT3 led to a reduction in ER stress (Figure 9e–k). These findings are consistent with previous studies,27,84,86 which further suggest that EGCG may alleviate chondrocyte injury by disrupting the positive feedback loop between ER stress and STAT3 activation. Consistent with the effects observed with EGCG, other polyphenolic compounds, such as resveratrol, curcumin and Gallotannin, have also been reported to modulate ER stress and inflammatory signaling pathways in OA, highlighting a shared potential among polyphenols to target inflammatory and ER-related pathways in chondrocytes.87–89 However, EGCG, as a green tea-derived catechin, exhibits distinct antioxidant and anti-inflammatory properties in OA,90,91 which may confer additional advantages in modulating STAT3 and ER stress simultaneously. Our findings support the potential of EGCG as a natural, dietary-derived compound with therapeutic promise in OA, and further studies are warranted to explore its efficacy in preclinical models or as an adjunctive therapy.
This work still has several limitations. We acknowledge that using chondrocyte cultures may overlook heterogeneity in STAT3 and ER stress responses among different subpopulations. A multicellular coculture system incorporating both synovial cells and chondrocytes might better mimic the in vivo microenvironment and more accurately reflect the complex interplay governing these signaling pathways. While our in vitro model offers mechanistic insights, in vivo validation is necessary. Future work will utilize OA rat models [eg, Destabilization of the Medial Meniscus (DMM) model or Monosodium Iodoacetate (MIA) injection] to assess EGCG’s effectiveness in reducing joint inflammation, cartilage degradation, and ER stress/STAT3 activation and multiple ER stress inducers or gene-editing technologies should be used in to comprehensively validate EGCG’s regulatory mechanisms within the ER stress network. Additionally, the dose-dependent effects of EGCG remain incompletely understood. Evidence shows high concentrations induce cytotoxicity via several mechanisms: disrupting calcium homeostasis/mitochondrial function, or auto-oxidizing to form EGCG auto-oxidation products (EAOPs), which synergize with EGCG to exacerbate cellular damage;92–94 Our CCK-8 data show >75% chondrocyte viability at 50 μM during the intervention, balancing efficacy and safety. Thus, higher doses (>50 μM) likely mask its protective effects due to cytotoxicity. Future studies should also clarify the dose-effect relationship of EGCG within its anti-inflammatory range to define the optimal dosing for clinical translation.
Overall, our findings suggest that EGCG confers chondroprotective effects, at least in part, by modulating the crosstalk between ER stress and STAT3 signaling. Nevertheless, the exact mechanisms by which EGCG affects the relationship between ER stress-related apoptosis and inflammatory responses in OA remain unclear. Although additional research is crucial for a comprehensive understanding of the complex roles of EGCG in treating OA, our current findings emphasize its promising potential as a therapeutic approach for this challenging condition.
Conclusion
The present study demonstrates that EGCG restores ECM homeostasis and attenuates inflammatory responses in IL-1β-induced chondrocytes by modulating the ER-stress-STAT3 interaction. These findings offer novel mechanistic insights into how EGCG affects OA-related cellular pathways, underscoring its potential relevance for therapeutic strategies. Further in vivo studies are warranted to validate these effects and explore their clinical applicability.
Data Sharing Statement
The data sets that were utilized and/or examined throughout the course of this study can be obtained from the corresponding authors if requested for justifiable reasons.
Ethics Approval and Consent to Participate
The study was conducted in compliance with the laboratory animal guideline for ethical review of animal welfare and approved by the Ethics Committee of Shanghai University of Traditional Chinese Medicine (PZSHUTCM2311290001). All animal experiments comply with the ARRIVE guidelines.
Author Contributions
MJZ: Writing-Original Draft; Methodology; Investigation. JLY; JHL: Visualization; Software; Data Curation. YSG; CMH; TTM: Validation; Data Curation; Writing-Review & Editing. XZW; XHH; LLC; YQZ: Visualization; Software; Writing-Review & Editing. XBW; DFD: Writing-Review & Editing; Project administration; Conceptualization; Resources.
All authors took part in drafting, revising, or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This study was supported by the National Natural Science Foundation of China (grant numbers 81902306; 82174406); Shanghai Municipal Health Commission Traditional Chinese Medicine Research Project (grant number 2024QN012); The Investigator-initiated Trial Program of Shanghai Pudong New Area Health Commission (the Medical and Industrial Integration Program) (grant number 2025-PWYC-11).
Disclosure
The authors declare no competing interests.
References
1. Courties A, Kouki I, Soliman N, Mathieu S, Sellam J. Osteoarthritis year in review 2024: epidemiology and therapy. Osteoarthritis Cartilage. 2024;32(11):1397–1404. doi:10.1016/j.joca.2024.07.014
2. Allen KD, Thoma LM, Golightly YM. Epidemiology of osteoarthritis. Osteoarthritis Cartilage. 2022;30(2):184–195. doi:10.1016/j.joca.2021.04.020
3. Hunter DJ, Schofield D, Callander E. The individual and socioeconomic impact of osteoarthritis. Nat Rev Rheumatol. 2014;10(7):437–441. doi:10.1038/nrrheum.2014.44
4. Barnett R. Osteoarthritis. Lancet. 2018;391(10134):1985. doi:10.1016/S0140-6736(18)31064-X
5. Hunter DJ, Bierma-Zeinstra S. Osteoarthritis. Lancet. 2019;393(10182):1745–1759. doi:10.1016/S0140-6736(19)30417-9
6. Lee YT, Mohd Yunus MH, Yazid MD, Ugusman A. Unraveling the path to osteoarthritis management: targeting chondrocyte apoptosis for therapeutic intervention. Front Cell Dev Biol. 2024;12:1347126. doi:10.3389/fcell.2024.1347126
7. Cardoneanu A, Macovei LA, Burlui AM, et al. Temporomandibular joint osteoarthritis: pathogenic mechanisms involving the cartilage and subchondral bone, and potential therapeutic strategies for joint regeneration. Int J Mol Sci. 2022;24(1):171. doi:10.3390/ijms24010171
8. Wen Z, Lin J, Xie W, Shan Y, Zhen G, Li Y. Insights into the underlying pathogenesis and therapeutic potential of endoplasmic reticulum stress in degenerative musculoskeletal diseases. Mil Med Res. 2023;10(1):54. doi:10.1186/s40779-023-00485-5
9. Hughes A, Oxford AE, Tawara K, Jorcyk CL, Oxford JT. Endoplasmic reticulum stress and unfolded protein response in cartilage pathophysiology; Contributing factors to apoptosis and osteoarthritis. Int J Mol Sci. 2017;18(3):665. doi:10.3390/ijms18030665
10. Takada K, Hirose J, Yamabe S, Uehara Y, Mizuta H. Endoplasmic reticulum stress mediates nitric oxide-induced chondrocyte apoptosis. Biomed Rep. 2013;1(2):315–319. doi:10.3892/br.2013.52
11. Lai Z, Li C, Zhang J, et al. Unveiling MiR-3085-3p as a modulator of cartilage degeneration in facet joint osteoarthritis: a novel therapeutic target. J Orthop Translat. 2025;50:235–247. doi:10.1016/j.jot.2024.11.007
12. Gao Y, Wei H, Peng X, Wang C, Zhu H, Yin J. ER stress-induced YAP upregulation leads to chondrocyte phenotype loss in age-related osteoarthritis. Front Pharmacol. 2024;15:1476255. doi:10.3389/fphar.2024.1476255
13. Tian Y, Feng X, Zhou Z, et al. Ginsenoside compound k ameliorates osteoarthritis by inhibiting the chondrocyte endoplasmic reticulum stress-mediated IRE1α-TXNIP-NLRP3 axis and pyroptosis. J Agric Food Chem. 2023;71(3):1499–1509. doi:10.1021/acs.jafc.2c06134
14. Oakes SA, Papa FR. The role of endoplasmic reticulum stress in human pathology. Annu Rev Pathol. 2015;10:173–194. doi:10.1146/annurev-pathol-012513-104649
15. Hetz C, Papa FR. The unfolded protein response and cell fate control. Mol Cell. 2018;69(2):169–181. doi:10.1016/j.molcel.2017.06.017
16. Ahmadi A, Hayes AW, Karimi G. Resveratrol and endoplasmic reticulum stress: a review of the potential protective mechanisms of the polyphenol. Phytother Res. 2021;35(10):5564–5583. doi:10.1002/ptr.7192
17. Wang M, Kaufman RJ. Protein misfolding in the endoplasmic reticulum as a conduit to human disease. Nature. 2016;529(7586):326–335. doi:10.1038/nature17041
18. Yang H, Wen Y, Zhang M, et al. MTORC1 coordinates the autophagy and apoptosis signaling in articular chondrocytes in osteoarthritic temporomandibular joint. Autophagy. 2020;16(2):271–288. doi:10.1080/15548627.2019.1606647
19. Wang Y, Fan A, Lu L, et al. Exosome modification to better alleviates endoplasmic reticulum stress induced chondrocyte apoptosis and osteoarthritis. Biochem Pharmacol. 2022;206:115343. doi:10.1016/j.bcp.2022.115343
20. Lu Y, Zhou J, Wang H, et al. Endoplasmic reticulum stress-mediated apoptosis and autophagy in osteoarthritis: from molecular mechanisms to therapeutic applications. Cell Stress Chaperones. 2024;29(6):805–830. doi:10.1016/j.cstres.2024.11.005
21. Tang Y, Zhou X, Cao T, et al. Endoplasmic reticulum stress and oxidative stress in inflammatory diseases. DNA Cell Biol. 2022;41(11):924–934. doi:10.1089/dna.2022.0353
22. Li Z, Huang Z, Zhang H, et al. IRE1-mTOR-PERK axis coordinates autophagy and ER stress-apoptosis induced by p2x7-mediated ca (2+) influx in osteoarthritis. Front Cell Dev Biol. 2021;9:695041. doi:10.3389/fcell.2021.695041
23. Zhou J, Wang Q. Daphnoretin relieves IL-1β-mediated chondrocytes apoptosis via repressing endoplasmic reticulum stress and NLRP3 inflammasome. J Orthop Surg Res. 2022;17(1):487. doi:10.1186/s13018-022-03316-w
24. Li W, Cao T, Luo C, et al. Crosstalk between ER stress, NLRP3 inflammasome, and inflammation. Appl Microbiol Biotechnol. 2020;104(14):6129–6140. doi:10.1007/s00253-020-10614-y
25. Latourte A, Cherifi C, Maillet J, et al. Systemic inhibition of IL-6/stat3 signalling protects against experimental osteoarthritis. Ann Rheum Dis. 2017;76(4):748–755. doi:10.1136/annrheumdis-2016-209757
26. Cheng C, Shan W, Huang W, et al. ACY-1215 exhibits anti-inflammatory and chondroprotective effects in human osteoarthritis chondrocytes via inhibition of STAT3 and NF-κb signaling pathways. Biomed Pharmacother. 2019;109:2464–2471. doi:10.1016/j.biopha.2018.11.017
27. Meares GP, Liu Y, Rajbhandari R, et al. PERK-dependent activation of JAK1 and STAT3 contributes to endoplasmic reticulum stress-induced inflammation. Mol Cell Biol. 2014;34(20):3911–3925. doi:10.1128/MCB.00980-14
28. Mo H, Sun K, Hou Y, et al. Inhibition of PA28γ expression can alleviate osteoarthritis by inhibiting endoplasmic reticulum stress and promoting STAT3 phosphorylation. Bone Joint Res. 2024;13(11):659–672. doi:10.1302/2046-3758.1311.BJR-2023-0361.R2
29. Zhu W, Tang H, Cao L, et al. Epigallocatechin-3-o-gallate ameliorates oxidative stress-induced chondrocyte dysfunction and exerts chondroprotective effects via the keap1/nrf2/are signaling pathway. Chem Biol Drug Des. 2022;100(1):108–120. doi:10.1111/cbdd.14056
30. Huang H, Cheng T, Yang C, et al. Intra-articular injection of (-)-epigallocatechin 3-gallate (EGCG) ameliorates cartilage degeneration in Guinea pigs with spontaneous osteoarthritis. Antioxidants. 2021;10(2):178. doi:10.3390/antiox10020178
31. Akhtar N, Haqqi TM. Epigallocatechin-3-gallate suppresses the global interleukin-1beta-induced inflammatory response in human chondrocytes. Arthritis Res Ther. 2011;13(3):R93. doi:10.1186/ar3368
32. Rasheed Z, Anbazhagan AN, Akhtar N, Ramamurthy S, Voss FR, Haqqi TM. Green tea polyphenol epigallocatechin-3-gallate inhibits advanced glycation end product-induced expression of tumor necrosis factor-alpha and matrix metalloproteinase-13 in human chondrocytes. Arthritis Res Ther. 2009;11(3):R71. doi:10.1186/ar2700
33. Ansari MY, Ahmad N, Haqqi TM. Oxidative stress and inflammation in osteoarthritis pathogenesis: role of polyphenols. Biomed Pharmacother. 2020;129:110452. doi:10.1016/j.biopha.2020.110452
34. Andriamanalijaona R, Kypriotou M, Baugé C, et al. Comparative effects of 2 antioxidants, selenomethionine and epigallocatechin-gallate, on catabolic and anabolic gene expression of articular chondrocytes. J Rheumatol. 2005;32(10):1958–1967.
35. Zhou L, He JN, Du L, et al. Epigallocatechin-3-gallate protects trabecular meshwork cells from endoplasmic reticulum stress. Oxid Med Cell Longev. 2022;2022:7435754. doi:10.1155/2022/7435754
36. Sun Y, Wu M, Feng H, et al. Epigallocatechin gallate ameliorates granulosa cell developmental via the eukaryotic initiation factor 2 alpha/activating transcription factor 4 pathway in hyperthyroid female rats. Antioxidants. 2025;14(9):1092. doi:10.3390/antiox14091092
37. Xiang C, Xiao X, Jiang B, et al. Epigallocatechin‑3‑gallate protects from high glucose induced podocyte apoptosis via suppressing endoplasmic reticulum stress. Mol Med Rep. 2017;16(5):6142–6147. doi:10.3892/mmr.2017.7388
38. Wu J, Xu X, Li Y, et al. Quercetin, luteolin and epigallocatechin gallate alleviate TXNIP and NLRP3-mediated inflammation and apoptosis with regulation of AMPK in endothelial cells. Eur J Pharmacol. 2014;745:59–68. doi:10.1016/j.ejphar.2014.09.046
39. Xie Y, Qin X, Zhou T, et al. Investigating the protective effect of loganin in ovariectomy‑induced bone loss through network pharmacology and molecular docking. Exp Ther Med. 2024;28(5):417. doi:10.3892/etm.2024.12706
40. Liu X, Cui S, Li W, Xie H, Shi L. Elucidation of the anti-colon cancer mechanism of phellinus baumii polyphenol by an integrative approach of network pharmacology and experimental verification. Int J Biol Macromol. 2023;253(Pt 6):127429. doi:10.1016/j.ijbiomac.2023.127429
41. Tang Y, Li M, Wang J, Pan Y, Wu F. CytoNCA: a cytoscape plugin for centrality analysis and evaluation of protein interaction networks. Biosystems. 2015;127:67–72. doi:10.1016/j.biosystems.2014.11.005
42. Liu Y, Yang X, Gan J, Chen S, Xiao Z, Cao Y. CB-dock2: improved protein-ligand blind docking by integrating cavity detection, docking and homologous template fitting. Nucleic Acids Res. 2022;50(W1):W159–W164. doi:10.1093/nar/gkac394
43. Xu Z, Ke T, Zhang Y, Guo L, Chen F, He W. Danshensu inhibits the IL-1β-induced inflammatory response in chondrocytes and osteoarthritis possibly via suppressing NF-κb signaling pathway. Mol Med. 2021;27(1):80. doi:10.1186/s10020-021-00329-9
44. Sovolyova N, Healy S, Samali A, Logue SE. Stressed to death - mechanisms of ER stress-induced cell death. Biol Chem. 2014;395(1):1–13. doi:10.1515/hsz-2013-0174
45. Li R, Sun K. Regulation of chondrocyte apoptosis in osteoarthritis by endoplasmic reticulum stress. Cell Stress Chaperones. 2024;29(6):750–763. doi:10.1016/j.cstres.2024.11.001
46. Lu X, Xu Y, Li X, et al. Selective STAT3 inhibitor STX-0119 alleviates osteoarthritis progression by modulating the STAT3/PPARγ signaling pathway. Biochem Pharmacol. 2024;227:116420. doi:10.1016/j.bcp.2024.116420
47. Maldonado M, Nam J. The role of changes in extracellular matrix of cartilage in the presence of inflammation on the pathology of osteoarthritis. Biomed Res Int. 2013;2013:284873. doi:10.1155/2013/284873
48. Hu H, Tian M, Ding C, Yu S. The c/EBP homologous protein (CHOP) transcription factor functions in endoplasmic reticulum stress-induced apoptosis and microbial infection. Front Immunol. 2018;9:3083. doi:10.3389/fimmu.2018.03083
49. Saito A, Ochiai K, Kondo S, et al. Endoplasmic reticulum stress response mediated by the PERK-eIF2(alpha)-ATF4 pathway is involved in osteoblast differentiation induced by BMP2. J Biol Chem. 2011;286(6):4809–4818. doi:10.1074/jbc.M110.152900
50. Yu C, Zhang Z, Xiao L, et al. IRE1α pathway: a potential bone metabolism mediator. Cell Prolif. 2024;57(10):e13654. doi:10.1111/cpr.13654
51. Tan L, Register TC, Yammani RR. Age-related decline in expression of molecular chaperones induces endoplasmic reticulum stress and chondrocyte apoptosis in articular cartilage. Aging Dis. 2020;11(5):1091–1102. doi:10.14336/AD.2019.1130
52. Rellmann Y, Eidhof E, Dreier R. Review: ER stress-induced cell death in osteoarthritic cartilage. Cell Signal. 2021;78:109880. doi:10.1016/j.cellsig.2020.109880
53. Gong J, Wang X, Wang T, et al. Molecular signal networks and regulating mechanisms of the unfolded protein response. J Zhejiang Univ Sci B. 2017;18(1):1–14. doi:10.1631/jzus.B1600043
54. Kim S, Seong K, Kim W, Jung J. Epigallocatechin gallate protects against hypoxia-induced inflammation in microglia via NF-κb suppression and nrf-2/HO-1 activation. Int J Mol Sci. 2022;23(7):4004. doi:10.3390/ijms23074004
55. Zuo G, Chen M, Zuo Y, et al. Tea polyphenol epigallocatechin gallate protects against nonalcoholic fatty liver disease and associated endotoxemia in rats via modulating gut microbiota dysbiosis and alleviating intestinal barrier dysfunction and related inflammation. J Agric Food Chem. 2024;72(16):9067–9086. doi:10.1021/acs.jafc.3c04832
56. Perdices L, Fuentes-Broto L, Segura F, et al. Systemic epigallocatechin gallate protects against retinal degeneration and hepatic oxidative stress in the p23h-1 rat. Neural Regen Res. 2022;17(3):625–631. doi:10.4103/1673-5374.320990
57. Rasheed Z, Rasheed N, Al-Shaya O. Epigallocatechin-3-o-gallate modulates global microRNA expression in interleukin-1β-stimulated human osteoarthritis chondrocytes: potential role of EGCG on negative co-regulation of microRNA-140-3p and ADAMTS5. Eur J Nutr. 2018;57(3):917–928. doi:10.1007/s00394-016-1375-x
58. Gelse K, Pöschl E, Aigner T. Collagens--structure, function, and biosynthesis. Adv Drug Deliv Rev. 2003;55(12):1531–1546. doi:10.1016/j.addr.2003.08.002
59. Rahmati M, Nalesso G, Mobasheri A, Mozafari M. Aging and osteoarthritis: central role of the extracellular matrix. Ageing Res Rev. 2017;40:20–30. doi:10.1016/j.arr.2017.07.004
60. Almanza A, Carlesso A, Chintha C, et al. Endoplasmic reticulum stress signalling - from basic mechanisms to clinical applications. FEBS J. 2019;286(2):241–278. doi:10.1111/febs.14608
61. Guan M, Yu Q, Zhou G, et al. Mechanisms of chondrocyte cell death in osteoarthritis: implications for disease progression and treatment. J Orthop Surg Res. 2024;19(1):550. doi:10.1186/s13018-024-05055-6
62. Takada K, Hirose J, Senba K, et al. Enhanced apoptotic and reduced protective response in chondrocytes following endoplasmic reticulum stress in osteoarthritic cartilage. Int J Exp Pathol. 2011;92(4):232–242. doi:10.1111/j.1365-2613.2010.00758.x
63. Sim HJ, Cho C, Kim HE, et al. Augmented ERAD (ER-associated degradation) activity in chondrocytes is necessary for cartilage development and maintenance. Sci Adv. 2022;8(3):eabl4222. doi:10.1126/sciadv.abl4222
64. Mei J, Xiao N, Xi Y, et al. Regulation of apoptosis and interaction with cartilage degeneration in osteoarthritis. Front Cell Dev Biol. 2025;13:1571448. doi:10.3389/fcell.2025.1571448
65. Scheiber AL, Guess AJ, Kaito T, et al. Endoplasmic reticulum stress is induced in growth plate hypertrophic chondrocytes in g610c mouse model of osteogenesis imperfecta. Biochem Biophys Res Commun. 2019;509(1):235–240. doi:10.1016/j.bbrc.2018.12.111
66. Mehana EE, Khafaga AF, El-Blehi SS. The role of matrix metalloproteinases in osteoarthritis pathogenesis: an updated review. Life Sci. 2019;234:116786. doi:10.1016/j.lfs.2019.116786
67. Yang D, Cao G, Ba X, Jiang H. Epigallocatechin-3-o-gallate promotes extracellular matrix and inhibits inflammation in IL-1β stimulated chondrocytes by the PTEN/miRNA-29b pathway. Pharm Biol. 2022;60(1):589–599. doi:10.1080/13880209.2022.2039722
68. Ibrahim IM, Abdelmalek DH, Elfiky AA. GRP78: a cell’s response to stress. Life Sci. 2019;226:156–163. doi:10.1016/j.lfs.2019.04.022
69. Ownby SL, Fortuno LV, Au AY, Grzanna MW, Rashmir-Raven AM, Frondoza CG. Expression of pro-inflammatory mediators is inhibited by an avocado/soybean unsaponifiables and epigallocatechin gallate combination. J Inflamm. 2014;11(1):8. doi:10.1186/1476-9255-11-8
70. Hu S, Wang S, He J, Bian Y. Tetramethylpyrazine alleviates endoplasmic reticulum stress‑activated apoptosis and related inflammation in chondrocytes. Mol Med Rep. 2022;25(1):12. doi:10.3892/mmr.2021.12528
71. Oliver BL, Cronin CG, Zhang-Benoit Y, Goldring MB, Tanzer ML. Divergent stress responses to IL-1beta, nitric oxide, and tunicamycin by chondrocytes. J Cell Physiol. 2005;204(1):45–50. doi:10.1002/jcp.20261
72. Lin JH, Li H, Yasumura D, et al. IRE1 signaling affects cell fate during the unfolded protein response. Science. 2007;318(5852):944–949. doi:10.1126/science.1146361
73. Gualeni B, Rajpar MH, Kellogg A, et al. A novel transgenic mouse model of growth plate dysplasia reveals that decreased chondrocyte proliferation due to chronic ER stress is a key factor in reduced bone growth. Dis Model Mech. 2013;6(6):1414–1425. doi:10.1242/dmm.013342
74. Collins JA, Kim CJ, Coleman A, et al. Cartilage-specific sirt6 deficiency represses IGF-1 and enhances osteoarthritis severity in mice. Ann Rheum Dis. 2023;82(11):1464–1473. doi:10.1136/ard-2023-224385
75. Uehara Y, Hirose J, Yamabe S, et al. Endoplasmic reticulum stress-induced apoptosis contributes to articular cartilage degeneration via c/EBP homologous protein. Osteoarthritis Cartilage. 2014;22(7):1007–1017. doi:10.1016/j.joca.2014.04.025
76. Chiribau C, Gaccioli F, Huang CC, Yuan CL, Hatzoglou M. Molecular symbiosis of CHOP and c/EBP beta isoform LIP contributes to endoplasmic reticulum stress-induced apoptosis. Mol Cell Biol. 2010;30(14):3722–3731. doi:10.1128/MCB.01507-09
77. Duvigneau JC, Luís A, Gorman AM, et al. Crosstalk between inflammatory mediators and endoplasmic reticulum stress in liver diseases. Cytokine. 2019;124:154577. doi:10.1016/j.cyto.2018.10.018
78. Avalle L, Camporeale A, Morciano G, et al. STAT3 localizes to the ER, acting as a gatekeeper for ER-mitochondrion ca(2+) fluxes and apoptotic responses. Cell Death Differ. 2019;26(5):932–942. doi:10.1038/s41418-018-0171-y
79. Gao L, Liu T, Li X. Identification and analysis of the endoplasmic reticulum stress hub genes in sepsis-associated ARDS. Sci Rep. 2025;15(1):31007. doi:10.1038/s41598-025-16644-8
80. Cao T, Zhang W, Wang Q, et al. Cancer SLC6a6-mediated taurine uptake transactivates immune checkpoint genes and induces exhaustion in CD8(+) t cells. Cell. 2024;187(9):2288–2304. doi:10.1016/j.cell.2024.03.011
81. Ji Y, Lin J, Liu R, et al. Celecoxib attenuates hindlimb unloading-induced muscle atrophy via suppressing inflammation, oxidative stress and ER stress by inhibiting STAT3. Inflammopharmacology. 2024;32(2):1633–1646. doi:10.1007/s10787-024-01454-7
82. Xie Q, Wang J, Li R, et al. IL-6 signaling accelerates iron overload by upregulating DMT1 in endothelial cells to promote aortic dissection. Int J Biol Sci. 2024;20(11):4222–4237. doi:10.7150/ijbs.99511
83. Chen C, Zhang X. IRE1α-XBP1 pathway promotes melanoma progression by regulating IL-6/STAT3 signaling. J Transl Med. 2017;15(1):42. doi:10.1186/s12967-017-1147-2
84. Zheng Y, Dai H, Chen R, et al. Endoplasmic reticulum stress promotes sepsis-induced muscle atrophy via activation of STAT3 and smad3. J Cell Physiol. 2023;238(3):582–596. doi:10.1002/jcp.30950
85. Chou X, Ma K, Shen Y, Min Z, Wu Q, Sun D. Dual role of inositol-requiring enzyme 1α (IRE-1α) in cd-induced apoptosis in human renal tubular epithelial cells: endoplasmic reticulum stress and STAT3 signaling activation. Toxicology. 2021;456:152769. doi:10.1016/j.tox.2021.152769
86. An X, Yin M, Shen Y, et al. Dimethyl fumarate ameliorated pyroptosis in contrast-induced acute renal injury by regulating endoplasmic reticulum stress and JAK2-STAT3 pathway. Ren Fail. 2025;47(1):2504633. doi:10.1080/0886022X.2025.2504633
87. Feng K, Ge Y, Chen Z, et al. Curcumin inhibits the PERK-eIF2α-CHOP pathway through promoting SIRT1 expression in oxidative stress-induced rat chondrocytes and ameliorates osteoarthritis progression in a rat model. Oxid Med Cell Longev. 2019;2019:8574386. doi:10.1155/2019/8574386
88. Hecht JT, Veerisetty AC, Wu J, et al. Primary osteoarthritis early joint degeneration induced by endoplasmic reticulum stress is mitigated by resveratrol. Am J Pathol. 2021;191(9):1624–1637. doi:10.1016/j.ajpath.2021.05.016
89. Kim SM, Han Y, Yu S, Kim SJ. Gallotannin attenuates 2‑deoxy‑d‑glucose‑induced dedifferentiation and endoplasmic reticulum stress through inhibition of inositol‑requiring enzyme 1 downstream p38 kinase pathway in chondrocytes. Mol Med Rep. 2019;20(6):5249–5256. doi:10.3892/mmr.2019.10773
90. Suzuki T, Ohishi T, Tanabe H, Miyoshi N, Nakamura Y. Anti-inflammatory effects of dietary polyphenols through inhibitory activity against metalloproteinases. Molecules. 2023;28(14):5426. doi:10.3390/molecules28145426
91. Dai M, Shi J, Wang T, et al. Epigallocatechin-3-gallate protects against osteoarthritis-induced chondrocytes dysfunction by regulating PLa2g2a. Front Pharmacol. 2025;16:1624818. doi:10.3389/fphar.2025.1624818
92. Cai Y, Zhao L, Qin Y, He Y. High dose of epigallocatechin-3-gallate inhibits proliferation and induces apoptosis of h9c2 cardiomyocytes through down-regulation of SIRT1. Pharmazie. 2015;70(1):12–16.
93. Wei Y, Chen P, Ling T, et al. Certain (-)-epigallocatechin-3-gallate (EGCG) auto-oxidation products (EAOPs) retain the cytotoxic activities of EGCG. Food Chem. 2016;204:218–226. doi:10.1016/j.foodchem.2016.02.134
94. Miao Y, Sun X, Gao G, et al. Evaluation of (-)-epigallocatechin-3-gallate (EGCG)-induced cytotoxicity on astrocytes: a potential mechanism of calcium overloading-induced mitochondrial dysfunction. Toxicol In Vitro. 2019;61:104592. doi:10.1016/j.tiv.2019.104592
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Xian Huang Fang Inhibits the PKCδ/MAPK Signaling Pathway to Attenuate Renal Tubular Epithelial Cell Senescence and Thereby Delay Renal Interstitial Fibrosis
Wang Y, Shao M, Li T, Du X, Chen K, Xu L
Drug Design, Development and Therapy 2025, 19:6991-7004
Published Date: 13 August 2025