Back to Journals » Lung Cancer: Targets and Therapy » Volume 16
Integrated Multi-Omics Approaches for Predicting Immune Checkpoint Inhibitor Response in NSCLC – Insights From Genomics, Proteomics, and Metabolomics
Authors Elayeh E ![]() , Aleidi SM, Aboud O
, Aleidi SM, Aboud O ![]() , Semreen MH
, Semreen MH ![]() , Bustanji YK, Dahabiyeh LA
, Bustanji YK, Dahabiyeh LA
Received 21 July 2025
Accepted for publication 2 October 2025
Published 16 December 2025 Volume 2025:16 Pages 167—198
DOI https://doi.org/10.2147/LCTT.S539777
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Sai-Hong Ou
Eman Elayeh,1 Shereen M Aleidi,1,2 Orwa Aboud,3– 5 Mohammad H Semreen,2,6 Yasser K Bustanji,1,7 Lina A Dahabiyeh8
1Department of Biopharmaceutics and Clinical Pharmacy, School of Pharmacy, The University of Jordan, Amman, 11942, Jordan; 2Department of Medicinal Chemistry, College of Pharmacy, University of Sharjah, Sharjah, 27272, United Arab Emirates; 3Department of Neurology, University of California, Davis, Sacramento, CA, 95817, USA; 4Department of Neurological Surgery, University of California, Davis, Sacramento, CA, 95817, USA; 5Comprehensive Cancer Center, University of California, Davis, Sacramento, CA, 95817, USA; 6Research Institute of Medical and Health Sciences, University of Sharjah, Sharjah, United Arab Emirates; 7College of Medicine, University of Sharjah, Sharjah, 27272, United Arab Emirates; 8Department of Pharmaceutical Sciences, School of Pharmacy, the University of Jordan, Amman, 11942, Jordan
Correspondence: Lina A Dahabiyeh, Department of Pharmaceutical Sciences, School of Pharmacy, The University of Jordan, Amman, 11942, Jordan, Email [email protected]
Background and Purpose: Immune checkpoint inhibitors (ICIs) have improved outcomes in non-small cell lung cancer (NSCLC), yet durable benefit is limited to a subset of patients. Reliable predictive biomarkers are therefore essential. We reviewed genomic, proteomic, and metabolomic studies to evaluate how multi-omics integration advances prediction of ICI efficacy in NSCLC.
Methods: A systematic search of PubMed, ClinicalTrials.gov, and Google Scholar was conducted on April 11, 2024, covering studies published from 2016 through January 2025, to identify omics-based biomarkers of ICI response in NSCLC. In total, 33 genomic, 9 proteomic, and 9 metabolomic studies met inclusion criteria. Each was evaluated using a standardized evidence rubric (0– 14) assessing effect robustness, validation, cohort size, and clinical endpoint relevance.
Results: Genomic predictors of poor response included EGFR and ALK/RET/ROS1 fusions, as well as KRAS co-mutations with STK11, KEAP1, or SMARCA4, all linked to immune-cold phenotypes with low tumor mutational burden (TMB) and poor T-cell infiltration. In contrast, KRAS/TP53 co-mutations, NOTCH family alterations, and BRAF V600E aligned with immune-hot signatures characterized by interferon signaling, PD-L1 upregulation, and cytotoxic T-cell infiltration. Proteomic studies consistently identified chemokines CXCL9 and CXCL10, apoptotic regulators (CASP8, FASLG), and checkpoint proteins (soluble PD-1, PD-L1, LAG-3) as predictive, while acute-phase proteins (SAA1/2, S100A8/9) correlated with resistance. Multi-analyte platforms such as PROphet demonstrated promising risk-stratification potential. Metabolomic profiling linked ICI benefit to higher baseline tryptophan, histidine, and short-chain fatty acids, while resistance was associated with increased 3-hydroxyanthranilic acid, pyruvate, and lipid metabolites indicating immunosuppressive IDO pathway activity.
Conclusion: Multi-omics approaches converge on pathways governing antigenicity, interferon signaling, and immune-metabolic crosstalk. Although promising, most biomarkers require prospective validation in large, uniformly treated cohorts. Integrative strategies—particularly when combined with AI-driven analytics—hold potential to refine patient stratification and guide clinical use of ICIs in NSCLC.
Plain Language Summary: Immune checkpoint inhibitors (ICIs) are a type of cancer drugs that help the body’s immune system find and attack cancer cells. They have improved treatment for non-small cell lung cancer (NSCLC), but they only work well for some patients. Doctors still struggle to predict who will benefit from these treatments. This review looks at how combining different scientific approaches—studying genes (genomics), proteins (proteomics), and small molecules in the body (metabolomics)—can help find better ways to predict treatment success.
Researchers found that certain gene mutations in tumors can affect how well patients respond to ICIs. For example, some mutations are linked to poor outcomes, while others are associated with better survival. Protein studies have also discovered markers in blood and tissue that may help predict who will respond well. In addition, changes in specific body chemicals (metabolites) are linked to treatment results and can give clues about how a patient’s body is reacting to therapy.
Overall, combining these “multi-omics” approaches can help doctors better understand which patients are likely to benefit from immunotherapy and could lead to more personalized and effective treatments in the future.
Keywords: non-small cell lung cancer, immune checkpoint inhibitors, prediction of response, multi-omics, biomarkers
Introduction
Background
Lung cancer is the leading cause of cancer-related mortality worldwide, responsible for nearly one in five cancer deaths.1 Non-small cell lung cancer (NSCLC) accounts for ~80% of cases, with adenocarcinoma, squamous cell carcinoma, and large cell carcinoma as the major subtypes.2,3 Tobacco smoking remains the primary risk factor, while radon exposure is the second leading cause, particularly among non-smokers.3 At diagnosis, nearly half of cases present with metastatic disease, and five-year survival rates remain low, especially for advanced stages.4,5 These figures emphasize the urgent need for improved therapies and reliable biomarkers to guide treatment selection.
Treatment for early-stage NSCLC (I–II) usually combines surgery, radiation, and platinum-based chemotherapy.6 Stage III patients without disease progression may receive immunotherapy, and stage IV patients benefit from molecularly targeted and immunotherapies, including cytokine therapy, adaptive cell transfer, and tumor vaccines.6
Immune checkpoint inhibitors (ICIs) are a type of immunotherapy that block inhibitory pathways used by cancer cells to escape the immune system, thereby helping the body’s immune cells recognize and attack tumors. ICIs have emerged as a promising option due to the limitations of earlier treatments.7 FDA-approved ICIs and their indications for treatment of NSCLC are summarized in Table 1.
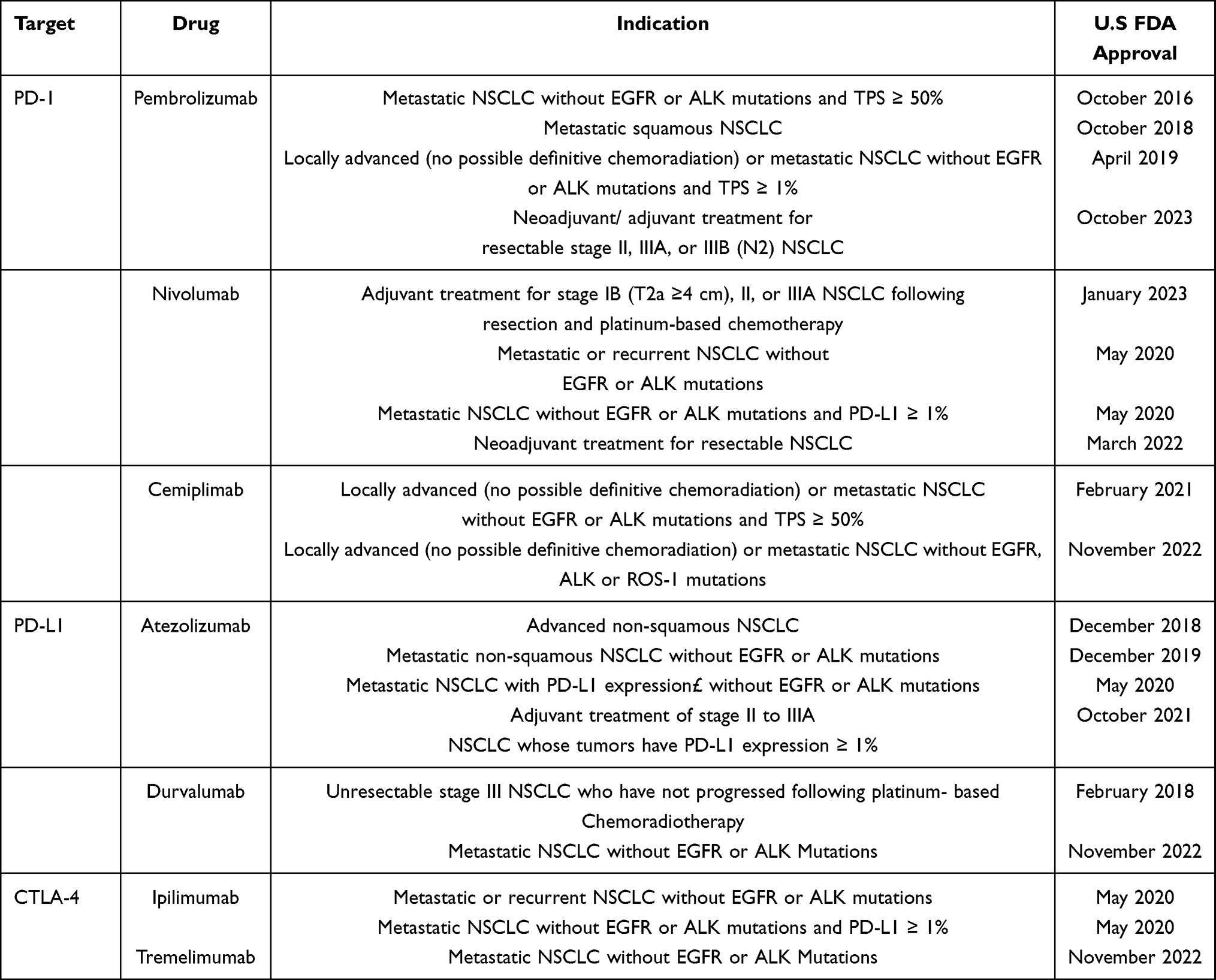 |
Table 1 FDA-Approved Immune Checkpoint Inhibitors and Their Clinical Indications |
Immune checkpoints regulate immune responses, preventing attacks on healthy cells, but their dysfunction contributes to NSCLC progression.7 Programmed Death-1 (PD-1), found on T cells, B cells, and natural killer cells, binds to Programmed Death-Ligand 1 (PD-L1) and PD-L2 on tumor and immune cells, sending inhibitory signals that suppress T cell activity and enable immune evasion.8,9 PD-L1 is expressed in 53–62% of NSCLC cases10 and is often upregulated, contributing to immune evasion by allowing tumors to escape immune detection, particularly in advanced stages.9,11 Another checkpoint, Cytotoxic T-Lymphocyte Antigen-4 (CTLA-4), competes with the co-stimulatory receptor CD28 for binding to B7 molecules on antigen-presenting cells. While CD28 promotes T cell activation, CTLA-4 inhibits it, dampening the immune response.9,11,12 The mechanisms of immune checkpoint blockade are illustrated in Figure 1. Tumors exploit both PD-1/PD-L1 and CTLA-4 pathways to suppress immunity and promote growth.9 These biological insights have directly informed the therapeutic targeting of PD-1/PD-L1 and CTLA-4 in NSCLC.
 |
Figure 1 Mechanism of immune checkpoint blockade. Professional antigen-presenting cells activate naïve T cells via co-stimulatory interactions between B7 (CD80/86)/CD28 and the MHC-II/TCR complex. CTLA-4 inhibitors enhance activation by preventing CTLA-4 from binding to B7 ligands. Activated effector T cells secrete cytokines such as IFNγ, amplifying the immune response against tumors. Tumor-expressed PD-L1 binds to PD-1 on T cells, suppressing activity even in the presence of tumor antigens. Regulatory T cells (Tregs) further inhibit T cell function, promoting an “exhausted” phenotype.PD-1/PD-L1 inhibitors restore anti-tumor immunity by blocking this suppressive interaction. Abbreviations: CTLA-4, cytotoxic T-lymphocyte-associated protein 4; MHC, major histocompatibility complex; PD-1, programmed cell death protein 1; PD-L1, programmed death ligand 1; TCR, T cell receptor. Note: Figure created with BioRender. |
Building on this knowledge, the PD-1/PD-L1 and CTLA-4 pathways have become critical therapeutic targets, with ICIs significantly advancing the treatment of NSCLC by restoring T-cell-mediated anti-tumor immunity. ICIs, including anti-PD-1/PD-L1 and CTLA-4 inhibitors (eg, ipilimumab), are FDA-approved for neoadjuvant, adjuvant, and first-line use—either as monotherapy or in combination with platinum-based chemotherapy.6,10,13–15 In advanced NSCLC, combining ICIs with chemotherapy has improved overall and progression-free survival.16,17 Furthermore, early-stage trials support the integration of ICIs into neoadjuvant and adjuvant settings, demonstrating increased major and complete pathological response rates.18–21 Yet only 20–30% of patients respond,22–27 resistance is common, and immune-related toxicities occur.28 This highlights the need for reliable biomarkers.
Among the biomarkers investigated, PD-L1 was the first to receive FDA approval.29 High tumor proportion score (TPS ≥50%) is generally associated with better outcomes,30–32 yet inconsistent findings across trials,33–35 together with variability in immunohistochemistry assays and intratumoral heterogeneity,36,37 limit its reliability. Microsatellite instability and mismatch repair deficiency (MSI/dMMR) were subsequently approved as predictive biomarkers for pembrolizumab in metastatic or unresectable tumors.38 Although clinical trials demonstrated meaningful response rates,39–41 their extremely low prevalence in NSCLC and methodological challenges constrain clinical applicability.42 More recently, tumor mutational burden (TMB) received tumor-agnostic approval in 2020.29 Elevated TMB is linked to increased neoantigen load and better ICI response,42–46 however, controversies persist regarding appropriate thresholds—≥10 mut/Mb in trials such as CheckMate 227 and 56844,47,48 versus higher cut-offs of ≥16–20 mut/Mb or ≥200 nonsynonymous mutations by whole-exome sequencing.49,50 Added to this are differences in sequencing platforms and bioinformatics pipelines that further undermine reproducibility.51,52 Taken together, PD-L1, MSI/dMMR, and TMB represent the first generation of biomarkers that established proof of principle for biomarker-guided immunotherapy in NSCLC. Yet their inconsistent predictive performance, technical variability, and limited generalizability underscore the need for more robust and integrative strategies.
Several landmark clinical trials and large-scale consortia have driven biomarker development. The KEYNOTE trials validated PD-L1,53,54 while CheckMate studies highlighted mutational load and combination regimens.16,44 Large initiatives like Blueprint55 and TRACERx56 advanced assay harmonization and multi-omic profiling (Supplementary Table S1). Together, these efforts define the foundation of biomarker research in NSCLC, illustrating both the validation of PD-L1 and TMB and the collaborative infrastructure that enables cross-platform standardization.
Nevertheless, translating biomarkers into practice remains fraught with challenges. Clinically, immune-related adverse events, atypical responses, and comorbidities complicate care.57 Technically, assay variability undermines reproducibility.50,55,58,59 Economically, disparities in access and affordability remain pressing, with cost-effectiveness differing across therapy lines and health systems.60 Debates also persist regarding the predictive reliability of PD-L1 and TMB. Some trials show strong associations between high PD-L1 expression and outcomes, while others demonstrate inconsistent value due to assay variability and heterogeneity.55,59 For TMB, thresholds of ≥10 mut/Mb have been widely applied, yet other studies suggest higher cut-offs, further complicating clinical implementation.44,47–50 MSI/dMMR, though highly predictive in colorectal cancer, remains exceedingly rare in NSCLC (<1%), raising questions about clinical utility.61,62 Collectively, these discrepancies highlight the urgent need for more integrative biomarker strategies.
Emerging approaches aim to address these limitations by combining multiple modalities. Multi-omics analyses integrated with artificial intelligence (AI) are generating clinically actionable predictive models.63,64 Blood-based proteomic platforms such as PROphet (Proteomic Prediction of immunotherapy benefit) have shown value alongside PD-L1 testing for treatment planning in metastatic NSCLC.65 Advances in tumor microenvironment (TME) profiling, including spatial proteomics, add predictive value beyond bulk assays.66,67 The gut microbiome has also emerged as a modulator of ICI efficacy, with dysbiosis and antibiotic exposure linked to reduced responses.68
While tumor-intrinsic features such as PD-L1, TMB, and MSI/dMMR remain central to biomarker development, accumulating evidence shows the TME exerts decisive influence. High CD8⁺ T-cell infiltration, tertiary lymphoid structures, and favorable effector-to-suppressor ratios improve outcomes, whereas stromal exclusion, myeloid-derived suppressor cells, and regulatory T-cell dominance promote resistance.69–71 Stromal remodeling and cytokine signaling dynamically modulate immune accessibility and checkpoint pathways. Multi-omics technologies provide powerful means to interrogate these interactions: proteomics captures cytokine and checkpoint profiles; metabolomics reveals nutrient competition and immunosuppressive metabolites; spatial proteomics and transcriptomics map immune–stromal crosstalk.66,67 Integrating these TME-derived signals with tumor-intrinsic biomarkers is therefore essential to overcome the limitations of single-parameter approaches and establish robust composite predictors.
Several reviews have addressed NSCLC biomarkers, but with limitations. Bourbonne et al72 emphasized genomics and transcriptomics with little focus on proteomics and no discussion of metabolomics. Mei et al64 integrated multi-omics with AI but underrepresented proteomics and excluded metabolomics. Yoon et al73 presented a broad overview of metabolomics and advanced omics but lacked NSCLC-specific clinical focus. Other reviews emphasized single-cell or spatial omics74 or circulating biomarkers75 without systematic integration. In contrast, the present review provides a balanced synthesis of genomics, proteomics, and metabolomics, anchored in the TME context. By addressing both strengths and limitations of existing biomarkers, it highlights the need for integrated multi-omics approaches that combine tumor-intrinsic, TME, and host-related factors into clinically actionable models for NSCLC immunotherapy.
Objective
This review explores the predictive landscape of ICI response in NSCLC, with a focus on the emerging role of genomics, proteomics, and metabolomics in identifying novel biomarkers. By highlighting recent advancements and current challenges, the review aims to provide comprehensive insights into how integrative multi-omic approaches can enhance prediction accuracy, inform personalized immunotherapy, and ultimately improve patient outcomes.
Methods
A comprehensive literature search was performed to identify studies reporting on genomic, proteomic, or metabolomic biomarkers predictive of response to ICIs in NSCLC. The search was conducted on April 11, 2024 across three databases: PubMed, ClinicalTrials.gov, and Google Scholar, covering the period from 2016 to January 2025. The search strategy combined controlled vocabulary and free-text terms, using Boolean operators. For example, the PubMed search string was: (“immune checkpoint inhibitors” OR “ICIs”) AND (“NSCLC” OR “non-small cell lung cancer”) AND (“multi-omics” OR “genomics” OR “proteomics” OR “metabolomics”) AND (“response prediction” OR “predictive biomarkers” OR “tumor microenvironment”). The search strategy is summarized in Table 2, and the detailed PubMed Boolean string is provided in Supplementary Table S2.
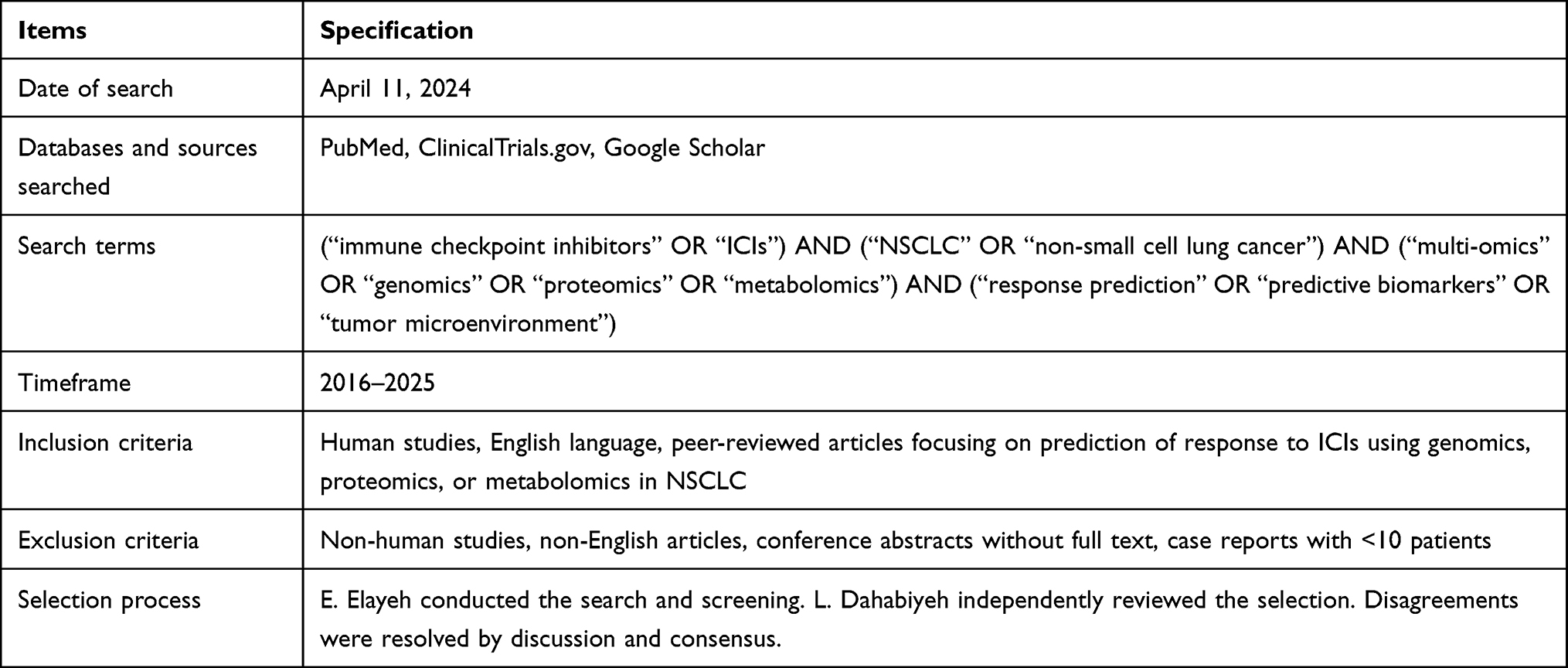 |
Table 2 Summary of Search Strategy for Literature Retrieval |
Eligibility criteria included: (1) human studies, (2) English language, (3) peer-reviewed articles, reviews, clinical trials, or meta-analyses, and (4) studies evaluating genomics, proteomics, or metabolomics in relation to ICI response in NSCLC. Exclusion criteria were: non-human studies, non-English articles, conference abstracts without full text, and case reports with fewer than 10 patients.
Study selection was conducted in two stages: (1) title and abstract screening, and (2) full-text review. The initial screening was performed by E. Elayeh, and independently reviewed by L. Dahabiyeh. Any disagreements were resolved by discussion until consensus was achieved. Duplicate records were removed prior to screening. To ensure methodological rigor, the review followed PRISMA 2020 guidelines for reporting. Risk of bias and study quality were assessed qualitatively according to study design, sample size, and biomarker validation stage.
To complement this qualitative assessment with a standardized approach, we further applied a structured evidence rubric across all three omics domains. Each genomic, proteomic, and metabolomic study was scored on a 0–14 scale derived from seven domains, with each domain contributing 0–2 points: (1) Effect robustness (0 = exploratory, 1 = association without replication, 2 = replicated or consistent across analyses); (2) Statistical validity (0 = descriptive only, 1 = p-value without correction, 2 = multivariate analysis or correction for multiple testing); (3) Clinical endpoint relevance (0 = surrogate only, 1 = intermediate endpoint such as ORR, 2 = hard endpoint such as PFS or OS with HR/CI); (4) Adjustment for confounders (0 = none, 1 = limited adjustment, 2 = comprehensive multivariate adjustment including PD-L1, TMB, stage, etc).; (5) Validation (0 = discovery only, 1 = internal/orthogonal validation, 2 = external cohort validation); (6) Cohort size (0 = <30, 1 = 30–49, 2 = ≥50 patients or multi-center); and (7) Data availability (0 = none, 1 = partial availability, 2 = fully deposited in repositories such as GEO, PRIDE, or MetaboLights). Scores were summed across domains to generate a total between 0 and 14.
For example, a proteomic study reporting differential plasma cytokines in 43 patients with multivariate Cox regression (adjusting for stage and performance status) and orthogonal validation by ELISA scored 13/14. In contrast, a small exploratory metabolomics study with fewer than 20 patients, univariate statistics, no adjustment for PD-L1 or TMB, and no validation scored 6/14.
Based on these standardized scores, all studies were retained in the main text to provide a comprehensive overview of the available evidence. To ensure clarity, we explicitly highlighted high-evidence studies (≥10/14) as robust predictors, while studies scoring <10/14 were clearly designated as exploratory or low-evidence. This approach preserved completeness while also guiding readers toward the most translationally relevant findings. In total, the screening process resulted in the inclusion of 51 eligible studies, comprising 33 genomic, 9 proteomic, and 9 metabolomic investigations.
Results: Omics Approaches Used to Predict Response to ICI Therapy in NSCLC
Omics studies including genomics, transcriptomics, proteomics and metabolomics, are advanced bioanalytical approaches that focus on the identification, quantification and characterization of the entire molecules “omes” within a specific system.76 A genome is defined as the complete genetic material of an organism, while transcriptome is the complete set of RNA transcripts produced by the genome. A proteome refers to the complete set of proteins expressed by an organism, cell, or tissue and metabolome is the entire set of small-molecule chemicals (metabolites with molecular weight <1500 Da) found within a biological sample.77
In the context of this review, these omics approaches were systematically evaluated for their ability to predict response to ICIs in NSCLC, and the results are presented below according to genomic, proteomic, and metabolomic domains.
Genomics
The use of biomarkers in the clinical care of cancer patients has grown in significance with the advent of genomic profiling technology and selective molecular targeted medicines. As predictive biomarkers for treatment decision-making, single-gene/protein or multi-gene “signature”-based assays have been developed to quantify particular molecular pathway dysregulations. It is also possible to include genome-based prognostic biomarkers for various cancer types into clinical prognostic staging systems or practice guidelines.78
Several techniques have been used in identifying genomic alterations for prediction of response to ICI in NSCLC patients.
Next Generation Sequencing in genomics Analysis
Next-generation sequencing (NGS) is among the most frequently reported genomic techniques for predicting immune checkpoint inhibitor (ICI) response in NSCLC.79–82 Characterized by high-throughput, multiplexed, and clonal sequencing, NGS offers significant advantages over traditional Sanger sequencing.79 NGS technologies are broadly categorized into short-read (second-generation) and long-read (third-generation) sequencing.79 Second-generation sequencing enables parallel sequencing of short (250–800 bp) clonally amplified DNA molecules, and involves a pipeline of library preparation, sequencing, and data analysis.83 DNA sequencing approaches include Whole Genome Sequencing (WGS), Whole Exome Sequencing (WES), Epigenome Sequencing, and Targeted Sequencing (TS), depending on the template.84 Long-read technologies, in contrast, can sequence native DNA fragments larger than 10 kb. Although early versions had high error rates, recent improvements have significantly enhanced their accuracy, making them useful for identifying genetic disorders.85 The two primary long-read platforms are Oxford Nanopore Technology (ONT) and Pacific Biosciences (PacBio).
Potential Genomic Mutations for Prediction of Response to ICI in NSCLC
Cancer classification has shifted from tissue-based to genetics-based, driven by initiatives like The Cancer Genome Atlas (TCGA) and the International Cancer Genome Consortium (ICGC), which have deepened understanding of genetic mechanisms, molecular subtypes, and tumor heterogeneity. Platforms such as cBioPortal (https://www.cbioportal.org/) support this shift by allowing exploration of mutation data across cancers. A key success of genomics-guided therapy is the use of vemurafenib in BRAF V600-mutant metastatic melanoma, present in around 50% of screened patients.86 This paradigm demonstrates how genetic alterations can guide therapy, and a similar principle applies in NSCLC, where genomic context modulates ICI response.
To evaluate the robustness of available evidence, we systematically scored studies according to predefined criteria mentioned earlier. Most genomic studies (34/39) achieved high evidence scores (≥10), while four scored below this threshold.87–90 High-scoring studies form the primary evidence base, and lower-scoring studies are presented as exploratory. To improve clarity, we summarized genomic predictors of ICI outcomes across two complementary main tables. Table 3 presents alterations associated with poor response and Table 4 highlights markers linked to favorable response. These tables provide a concise overview of candidate biomarkers, reported outcomes, validation status, and strength of evidence. Full quantitative details, including effect estimates (hazard ratios, odds ratios, confidence intervals, and p-values), are provided in Supplementary Table S3a (Excel file), which contain separate sheet for genomics studies. While the tables summarize the evidence base, Figure 2 provides a mechanistic overview that situates these alterations within broader genomic pathways.
 |
Table 3 Genomic Predictors of Poor Response to ICIs in NSCLC |
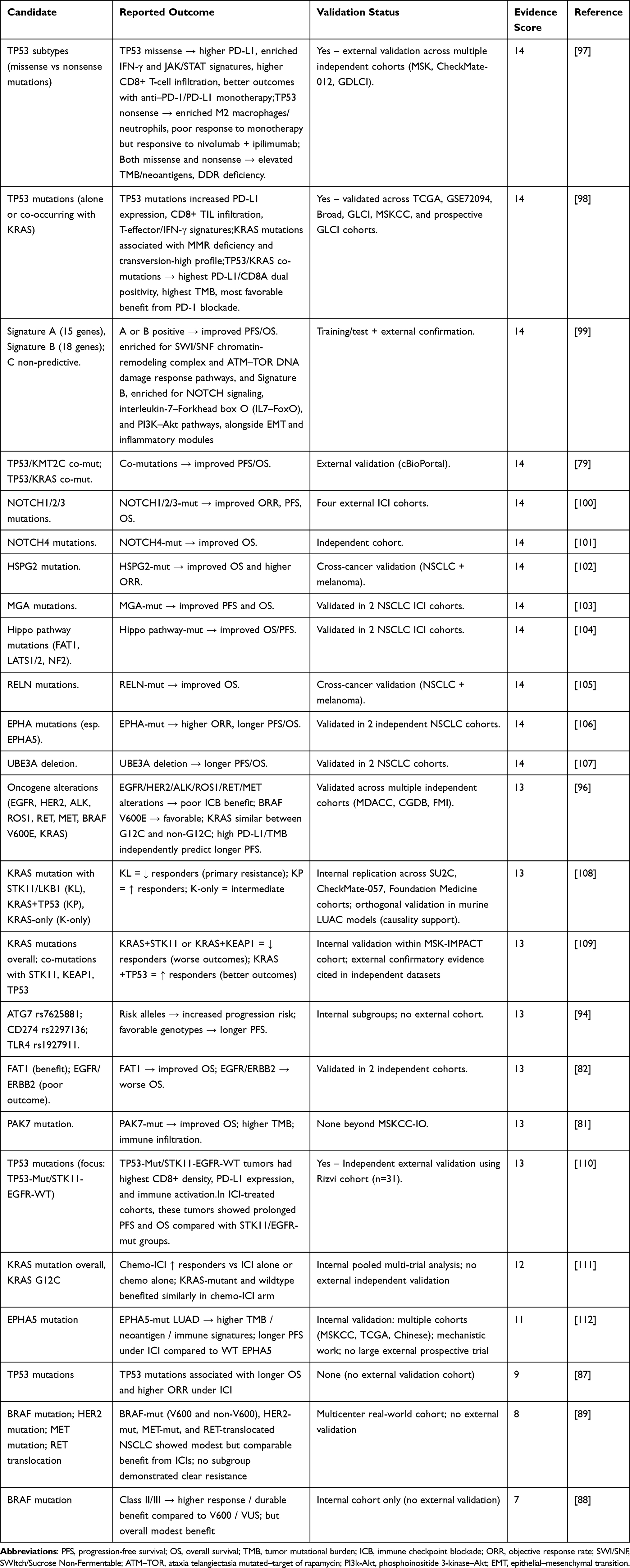 |
Table 4 Genomic Predictors of Favorable Response to ICIs in NSCLC |
 |
Figure 2 NGS workflow, pathways, and genetic mutations implicated in response to immune checkpoint inhibitor therapy in NSCLC. Abbreviations: ALK, anaplastic lymphoma kinase; EGFR, epidermal growth factor receptor; BRAF, v-raf murine sarcoma viral oncogene homolog B1; DLX2, distal-less homeobox 2; EPHA, Ephrin type-A receptor; HER2, human epidermal growth factor receptor 2; HSPG2, heparan sulfate proteoglycan; KRAS, Kirsten rat sarcoma viral oncogene homolog; MAPK, mitogen-activated protein kinase; MGA, MAX gene-associated; PI3K, phosphoinositide 3-kinase; TGFβ, transforming growth factor beta; TP53, tumor protein 53; UBE3A, ubiquitin protein ligase E3A. Note: Figure created with BioRender. |
Predictors of Poor Response
KRAS Co-Mutations with STK11/LKB1
In KRAS (Kirsten rat sarcoma viral oncogene homolog)-mutant NSCLC, concurrent alterations in STK11 (serine/threonine kinase 11, also called LKB1) were consistently associated with primary resistance to PD-1/PD-L1 inhibitors.108,109 These tumors displayed reduced PD-L1 expression, diminished CD8+ T-cell infiltration, and enrichment of immunosuppressive myeloid cells, consistent with an immune-cold phenotype that mediates checkpoint resistance.
KRAS Co-Mutations with KEAP1
Alessi 2021 reported that KEAP1 (Kelch-like ECH-associated protein 1) alterations, particularly in the context of KRAS-mutant NSCLC, were associated with inferior outcomes under checkpoint blockade. KEAP1 loss activates the NRF2 (nuclear factor erythroid 2–related factor 2) pathway, leading to altered oxidative metabolism and immune evasion, which together diminish immunotherapy benefit.95
KRAS Subtype G12D and Other Non-G12C Variants
The G12D variant of KRAS has been reported as a marker of poor response to PD-(L)1 therapy. Stronger evidence indicates that G12D tumors show reduced tumor mutational burden (TMB), lower PD-L1 expression, and poor CD8+ T-cell infiltration, consistent with weaker immunogenicity.91 An exploratory cohort with limited evidence further suggested a similar association (score <10).90 More broadly, non-G12C KRAS variants were less responsive to checkpoint blockade than KRAS G12C, highlighting the relevance of subtype-specific biology.96
EGFR and ERBB2
Two large analyses reached similar conclusions regarding the limited benefit of immunotherapy in EGFR-mutant NSCLC. Lee et al (2017) demonstrated that EGFR-mutant patients derived no overall survival improvement with checkpoint inhibitors compared to docetaxel, while benefit was observed in the EGFR-wild-type subgroup.92 Lee et al (2018) confirmed these findings in a pooled cohort, further supporting EGFR mutation as a predictor of poor outcome under PD-1/PD-L1 blockade. Fang 2019 extended these results by showing that activating alterations in EGFR and ERBB2 (human epidermal growth factor receptor 2) were associated with shorter progression-free survival under immunotherapy.93
TP53 Nonsense Mutations
Sun et al (2020) demonstrated that the nature of TP53 (tumor protein p53) alterations influences outcome. While some TP53 mutations aligned with inflamed phenotypes, nonsense or truncating variants were associated with impaired interferon signaling, lower PD-L1 expression, and reduced clinical benefit from checkpoint inhibition.97
SWI/SNF Complex Mutations
Alessi et al (2021) also investigated the SWI/SNF (SWItch/Sucrose Non-Fermentable) chromatin remodeling complex, including ARID1A, ARID1B, ARID2, PBRM1, SMARCA4, and SMARCB1. While outcomes in unselected NSCLC were not significantly different between SWI/SNF-mutant and wild-type tumors, in the KRAS-mutant subset concurrent SWI/SNF alterations, particularly SMARCA4, were associated with significantly shorter progression-free and overall survival. These findings indicate that SWI/SNF disruption amplifies resistance to immune checkpoint inhibitors in specific molecular contexts.95
Other Oncogenic Drivers: ALK, ROS1, RET, MET
Negrao et al (2021) reported that alterations in ALK (anaplastic lymphoma kinase), ROS1, RET, and MET were linked to poor outcomes under PD-1/PD-L1 blockade. These oncogene-driven tumors typically occur in never-smokers, are characterized by low TMB, and harbor immune-excluded microenvironments, explaining the lack of immunotherapy benefit.96
Germline Immunogenetic Variants
Xin et al (2023) highlighted the role of host germline variants in shaping ICI outcomes. Mutant alleles in ATG7 (autophagy related 7), TLR4 (toll-like receptor 4), and CD274 (encoding PD-L1) were associated with early progression under PD-1/PD-L1 therapy. These variants likely impair autophagy-mediated immune activation, alter innate sensing, or disrupt PD-L1 regulation, collectively contributing to checkpoint resistance.94
Predictors of Favorable Response
KRAS with TP53 (KP Subtype)
Several recurrent genomic alterations in NSCLC have been associated with improved outcomes under ICIs, reflecting their ability to shape an immune-active tumor microenvironment. Among the best studied is the co-occurrence of KRAS and TP53 mutations, often termed the KP subtype. KRAS mutations alone represent a heterogeneous group, but when accompanied by TP53 alterations they consistently align with favorable outcomes across multiple cohorts.98,108,109 Mechanistically, the combined effect of KRAS-driven oncogenic signaling and partial loss of TP53 function appears to generate genomic instability, increased neoantigen load, and activation of interferon pathways, which together promote higher TMB, upregulation of PD-L1, and robust infiltration by cytotoxic CD8-positive T cells. These features collectively define an inflamed or “hot” tumor immune microenvironment that is permissive for checkpoint blockade.
TP53 Mutation (Context Beyond KRAS)
Beyond the KRAS context, TP53 mutations in general have shown favorable associations, although the subtype of TP53 alteration is critical. Missense mutations, which often produce a dysfunctional but still expressed protein, correlate with enhanced immune activation, interferon gamma signaling, and greater T-cell infiltration. These tumors frequently express PD-L1 at higher levels, consistent with an immune escape mechanism that is effectively targeted by PD-1/PD-L1 blockade.12,110 An exploratory cohort with limited evidence further supported this association (score <10).87 In contrast, nonsense or truncating TP53 mutations abolish protein function and have been associated with poor outcomes,97 underscoring the need to differentiate TP53 subtypes when evaluating predictive value.
TP53 Mutation (Context Beyond KRAS)
KRAS mutations more broadly can be associated with benefit, particularly outside the unfavorable backgrounds of STK11/LKB1 or KEAP1. While meta-analyses have shown that EGFR-mutant tumors do not benefit, KRAS-mutant cases often align more closely with the genomic and immunologic features that support ICI efficacy.93,111 Subtype analyses have highlighted that KRAS G12C can be a favorable predictor in specific clinical contexts. For example, in patients with high PD-L1 expression treated in the first-line setting, KRAS G12C has been associated with longer progression-free survival, suggesting that its unique mutational landscape confers greater immunogenicity.80 The mechanism likely relates to the smoking-associated mutational background common in KRAS G12C tumors, which contributes to higher TMB and the presence of diverse neoantigens, thereby supporting immune recognition.
BRAF
BRAF mutations, particularly the V600E variant, have emerged as another favorable context. Although these cases remain relatively rare, stronger evidence suggests that BRAF V600E-mutant NSCLC responds more favorably to checkpoint blockade compared to other oncogenic drivers such as EGFR or ALK.96 Additional exploratory studies with lower evidence levels also reported benefit (scores <10).88,89 The biologic explanation may involve elevated TMB and activation of the MAPK pathway, which interacts with immune signaling pathways to promote tumor antigenicity.
The NOTCH Family
The NOTCH signaling family has also been implicated in favorable ICI response. Mutations in NOTCH1, NOTCH2, and NOTCH3 have been correlated with improved response rates and survival, while NOTCH4 mutations have been independently associated with longer survival in treated cohorts.100,101 The NOTCH pathway regulates T-cell development and activation, and alterations within this pathway may enhance interferon responses and antigen presentation, explaining the observed clinical benefit.
EPHA
Favorable associations have also been identified with alterations in ephrin receptors, particularly EPHA5. Studies demonstrated that EPHA mutations were linked to higher response rates, longer progression-free survival, and improved overall survival.106,112 Mechanistically, EPHA5-mutant tumors displayed increased TMB and enhanced immune infiltration, suggesting that loss of EPHA5 function creates a more immunogenic state.
The Hippo Signaling Pathway
The Hippo signaling pathway, which regulates organ size and tissue homeostasis through control of YAP and TAZ activity, has also been implicated. Mutations in Hippo pathway regulators such as FAT1, LATS1, LATS2, and NF2 were associated with favorable ICI outcomes.104 Disruption of Hippo signaling may enhance tumor immunogenicity by increasing antigen presentation and interferon signaling, thereby facilitating immune-mediated tumor control.
RELN, HSPG2, PAK7, MGA
Other favorable mutations identified across multiple datasets include RELN, which encodes the extracellular matrix protein reelin; HSPG2, encoding the basement membrane proteoglycan perlecan; and PAK7, a serine/threonine kinase involved in cytoskeletal signaling. Each of these genes, when mutated, was associated with longer survival or higher response under PD-1/PD-L1 blockade.81,102,105 The precise mechanisms vary but generally converge on immune activation, either through altered extracellular matrix signaling, improved T-cell access, or modulation of immune checkpoints. MGA (MAX gene associated) further supports this convergence by linking transcriptional deregulation to immune activation. MGA encodes a transcriptional repressor that interacts with the MAX protein, antagonizing the MYC oncogene. Loss of MGA function disrupts this balance, allowing unchecked MYC signaling, which promotes tumor progression. However, MGA-mutant NSCLCs have been reported to exhibit higher TMB, enriched interferon signaling, and increased infiltration by CD8+ T cells.103 This inflamed phenotype provides a mechanistic basis for the improved outcomes observed in ICI-treated patients. In other words, while MGA loss deregulates growth control through MYC, it simultaneously generates a tumor environment more visible to the immune system, which makes ICIs more effective. UBE3A deletion has also been reported as a favorable marker, associated with longer progression-free and overall survival.107 UBE3A encodes a ubiquitin ligase, and its loss may deregulate protein degradation pathways in ways that enhance immune recognition.
Multi-gene signatures integrating DNA damage response and immune signalling
Cucchiara et al99 integrated MSK-IMPACT sequencing with network clustering to define composite predictive signatures in NSCLC patients treated with ICI. Two signatures emerged as favorable: Signature A, enriched for SWItch/Sucrose Non-Fermentable (SWI/SNF) chromatin-remodeling complex and ataxia telangiectasia mutated–target of rapamycin (ATM–TOR) DNA damage response pathways, and Signature B, enriched for NOTCH signaling, interleukin-7–Forkhead box O (IL7–FoxO), and phosphoinositide 3-kinase–Akt (PI3K–Akt) pathways, alongside epithelial–mesenchymal transition (EMT) and inflammatory modules. The presence of either signature was consistently associated with improved PFS and OS across discovery, internal testing, and external validation cohorts, highlighting both genomic instability–linked and immune signaling–linked mechanisms as potential drivers of immunotherapy sensitivity.
Taken together, favorable genomic predictors share several convergent mechanistic themes. The first theme is the inflamed tumor microenvironment. Alterations such as KRAS/TP53 co-mutations, MGA mutations, NOTCH family mutations, and RELN or EPHA5 alterations consistently align with higher levels of cytotoxic CD8-positive T-cell infiltration and activation of interferon signaling pathways. These changes create an immune-hot environment that supports recognition and elimination of tumor cells once the PD-1/PD-L1 axis is blocked. A second theme is enhanced antigenicity. TP53 missense mutations, BRAF V600E, Hippo pathway alterations involving FAT1, LATS1, LATS2, and NF2, as well as ephrin receptor mutations, are associated with higher TMB and a broader neoantigen repertoire. This increased antigenicity improves the likelihood of effective immune surveillance and augments response to checkpoint inhibition. A third theme is checkpoint pathway engagement. Many favorable genomic contexts, including KRAS/TP53, MGA, and NOTCH alterations, are correlated with elevated PD-L1 expression. This suggests that these tumors rely on the PD-1/PD-L1 axis for immune escape, making them particularly sensitive to therapeutic blockade of this pathway. By contrast, poor predictors demonstrate the opposite pattern. KRAS with STK11 or KEAP1 co-mutations, EGFR-driven tumors, and SWI/SNF or SMARCA4 alterations were all associated with immune-cold phenotypes characterized by reduced interferon signaling, lower PD-L1 expression, and exclusion of effector T cells from the tumor bed. Finally, the predictive impact of many alterations is context and treatment dependent. For instance, KRAS G12C was associated with longer progression-free survival in patients with high PD-L1 tumors treated with first-line immunotherapy, but no differential benefit in unselected settings. Similarly, TP53 mutations can predict improved outcomes when they are missense variants, but nonsense or truncating mutations confer resistance. These observations demonstrate that the predictive value of genomic alterations is not absolute, but rather interacts with co-mutation status, PD-L1 expression, TMB, and treatment regimen. Together, these mechanistic insights underscore that genomics stratifies NSCLC into immune-hot and immune-cold archetypes that diverge markedly in their sensitivity to immune checkpoint inhibition.
Heterogeneity and Limitations of Genomics
Genomic predictors of immunotherapy response in NSCLC demonstrate significant heterogeneity, which arises from multiple levels of variability across published studies. Differences in study design are a key driver: some investigations were retrospective analyses of real-world cohorts, while others were prospective clinical trials or exploratory biomarker studies nested within larger treatment protocols. The sequencing platforms employed also vary considerably, ranging from targeted next-generation sequencing panels of a few hundred genes to whole-exome sequencing, RNA sequencing, or broader multi-omics approaches. These methodological differences influence which mutations are captured, how TMB is quantified, and how immune-related gene expression is interpreted. Patient populations also differ substantially across studies, with variability in geography, ethnicity, smoking exposure, histologic subtypes, and prior lines of therapy.
Treatment regimens represent another source of heterogeneity. Some cohorts received anti–PD-1 or anti–PD-L1 monotherapy, whereas others were treated with chemo-immunotherapy combinations, or less commonly CTLA-4–based regimens. Clinical selection factors such as baseline PD-L1 expression and smoking history strongly influence both clinical outcomes and biomarker associations, yet these variables are not uniformly controlled or adjusted across studies. Cohort size further amplifies these discrepancies: small exploratory studies often report striking associations that do not reproduce in larger multicenter datasets, while very large cohorts may dilute signals by pooling heterogeneous patient subsets.
Technical variation also plays a role. Differences in bioinformatic pipelines, variant calling thresholds, and gene-level annotation can lead to discordant classifications of the same genomic alteration across datasets. Endpoint definitions are not always consistent, with some studies reporting objective response rate (ORR), others focusing on progression-free survival (PFS) or overall survival (OS), and some using composite or surrogate endpoints. This lack of uniformity complicates direct comparisons and meta-analyses.
Taken together, these sources of heterogeneity limit the generalizability of individual findings and underscore the importance of validation. Candidate genomic biomarkers require confirmation in large, uniformly treated cohorts with standardized assays, harmonized clinical annotation, and consistent endpoint definitions. Such efforts are essential to distinguish true predictive markers from context-dependent associations driven by study design, patient selection, or technical artifacts. Encouragingly, ongoing large-scale initiatives and prospective trials, such as MSK-IMPACT113 CheckMate 227,44 and the POPLAR and OAK studies10,114 as well as pan-cancer sequencing consortia, are beginning to address these gaps by providing standardized platforms and robust validation cohorts.
Proteomics
Proteomics focuses on the comprehensive examination of proteins, including their structures and activities.115 Analyzing an organism’s dynamic system of continually shifting protein sets, which vary not only across different developmental stages but also between tissues, cell types, and intracellular compartments, is a difficult challenge for proteomics. These dysregulated proteins might act as potential disease biomarkers.115,116 Proteome analysis is now rapidly becoming more successful due to the significant advancements in mass spectrometry (MS) and the coupling of MS with separation methods as two-dimensional gel electrophoresis (2DGE) and Ultrahigh performance liquid chromatography (UHPLC).117,118 Other techniques include protein microarrays, usually called protein chips, which are available in various formats. The four main types of protein microarrays used in cancer research are proteome, antibody, reverse-phase protein array (RPPA), and lectin microarrays. Table 5 presents proteomic predictors of ICI response in NSCLC, detailing the candidate proteins, reported clinical outcomes, validation status, and evidence scores. Full quantitative details, including effect estimates (hazard ratios, odds ratios, confidence intervals, and p-values), are provided in Supplementary Table S3b (Excel file), which contains separate sheet for proteomic studies.
 |
Table 5 Proteomic Predictors of ICI Response in NSCLC |
Techniques Utilized in Proteomics Analysis
The two primary techniques in proteomics analysis for predicting responses to ICIs are MS and protein microarrays. Below is a summary of each method, detailing their principles, subtypes, and respective advantages and disadvantages.
Mass Spectrometry
Mass spectrometry (MS) is a critical tool in proteomics, particularly when integrated with machine learning to identify predictive biomarkers for immunotherapy outcomes.127 Two main quantification strategies are employed: untargeted (broad profiling) and targeted (focused quantification), using either bottom-up (peptide-based) or top-down (intact protein) approaches.128 Common ionization methods include electrospray ionization (ESI) and matrix-assisted laser desorption/ionization (MALDI), enabling the analysis of large polar molecules, with ESI compatible with HPLC and MALDI reducing spectral complexity.129 Key mass analyzers include Orbitrap, TOF, and quadrupole, often used in tandem (MS/MS) for detailed fragmentation and ion analysis.130 In bottom-up proteomics, proteins are digested into peptides for MS analysis, offering high throughput but limited post-translational modifications (PTM) detail.131,132 Top-down proteomics analyzes intact proteins, preserving PTMs and modifications, though it is best suited for small proteins and moderate throughput studies.133 The typical workflow used in proteomics research is illustrated in Figure 3, which outlines key steps from sample preparation and protein digestion to mass spectrometry analysis and data interpretation.
 |
Figure 3 Typical proteomics workflow using MS-based techniques. Abbreviations: SDS-PAGE, sodium dodecyl sulfate–polyacrylamide gel electrophoresis; RP-HPLC, reversed-phase high-performance liquid chromatography; LC/MS, liquid chromatography/mass spectrometry. Note: Figure created with BioRender. |
Protein Microarrays
Protein microarrays (or chips) detect target proteins by immobilizing biomolecules on solid supports for biochemical analysis.134 In cancer research and ICI response prediction, four main types are used: proteome, antibody, reverse-phase (RPPA), and lectin microarrays, offering high-throughput, system-level profiling.135 Proteome arrays immobilize proteins from most ORFs and are made via expression/purification or in vitro transcription and translation (IVTT); while expression-based arrays are comprehensive but costly, IVTT offers a simpler, more economical option despite early-stage limitations.135 Antibody microarrays use antigen-antibody specificity in either planar or bead-based formats. Planar arrays use slides or membranes with high sensitivity and throughput, while bead-based formats capture antibodies on microbeads.136,137 These arrays enable proteome comparisons between cancer and healthy individuals, aiding biomarker discovery. Lectin microarrays target abnormal glycosylation, improving detection of tumor-associated glycoproteins and aiding early cancer diagnosis.138 RPPA uses fixed lysates probed with specific antibodies to assess protein expression and PTMs across many samples with high sensitivity and multiplexing capabilities.139–141
Proteomics Study for the Prediction of Response to ICI in NSCLC
The field of using protein (or peptide) biomarkers in clinical investigations is active and constantly expanding. In cancer research, protein biomarkers are used to guide treatment decisions by stratifying patients according to their likelihood of responding to specific medication142 and to aid in the early detection and prognosis of carcinoma.143–150 One such example of proteomic use for prediction of response to ICI is the VeriStrat test, which is a blood-based proteomic test or host immune classifier (HIC) first developed in 2007 by Biodesix.151 It is used to predict and prognosticate outcomes for patients with advanced NSCLC. By evaluating the host’s response to the tumor using MALDI-TOF analysis, the test determines the degree of disease aggression and assigns patients to one of two groups: VeriStrat-Good (VS-G) or VeriStrat-Poor (VS-P). It has been demonstrated that the test is predictive for EGFR-targeting drugs in second-line treatment, following progression with or following platinum-based chemotherapy.151 According to a recent analysis of the INSIGHT clinical trial (NCT03289780), NSCLC patients can be accurately predicted to respond differently to ICI therapy based on their VS labels, VS Good and VS Poor, regardless of their PD-L1 expression.152 Moreover, individual studies and meta-analyses have demonstrated that patients classified as VS-G before treatment experience significantly better outcomes compared to those classified as VS-P across various treatment regimens and even without active therapy in advanced NSCLC.153–156 Recently, the VS test has demonstrated promise as a prognostic marker for advanced NSCLC treated with immunotherapy.152,157,158 It has also been found to be able to identify which patients would most benefit from the combination of ICI and chemotherapy.152
Potential Protein Biomarkers for Prediction of Response to ICI in NSCLC
All nine proteomic studies included in this review achieved high evidence scores (≥10/14) according to our standardized rubric. This indicates robust statistical design, clinically relevant endpoints, and sufficient cohort sizes, allowing the following results to be interpreted with relatively high confidence.
Predictors of Favorable Response
Chemokines and Cytokines
Increased plasma levels of CXCL9, CXCL10, and interleukin-15 (IL-15) were associated with response to anti–PD-(L)1 therapy. These proteins recruit and sustain CD8+ T cells and natural killer (NK) cells, supporting a T-cell inflamed microenvironment. The same study also demonstrated that higher tumor necrosis factor superfamily ligand 14 (TNFSF14) and Fas ligand (FASLG) were associated with improved overall survival, and that high tumor PD-L1 expression (50–100%) predicted better progression-free and overall survival.119
Checkpoint-Related Proteins
Circulating soluble PD-1 (sPD-1), a decoy receptor form of PD-1, was enriched in responders and linked to improved overall survival in the monotherapy setting.120
Proteomic Classifiers
A machine learning–based proteomic test termed PROphet (Proteomic Prediction of immunotherapy benefit), derived from 388 proteins, stratified patients into groups with differential benefit. The contributing proteins spanned immune-regulatory and stromal pathways, reflecting systemic influences on immunotherapy efficacy.65 Mass spectrometry–based classifiers further identified “sensitive” patients with proteomic patterns enriched for antigen presentation and immune activation functions.121
Adhesion Molecules
High tumor-cell expression of CD44 predicted longer survival, consistent with its role in mediating immune–tumor interactions. In contrast, stromal expression of CD44 carried no predictive value.123
Innate Immune Peptides: Neutrophil Defensins
Berghmans et al126 applied MALDI-MSI on tumor biopsies from NSCLC patients treated with PD-1/PD-L1 blockade. They identified neutrophil defensins 1–3, antimicrobial peptides of the innate immune system, as being enriched in responders. Defensin signal distinguished tumor and immune compartments with area under the curve values around 0.74, and orthogonal immunohistochemistry confirmed defensin staining on the same biopsies. Although based on a small single-center cohort without external validation, these findings suggest that innate immune activation reflected by defensin expression may promote immunotherapy sensitivity.
Predictors of Poor Response
Chemokines and Cytokines
In a larger cohort, CXCL8 (interleukin-8) and CXCL10 were enriched in non-responders both at baseline and during treatment, correlating with poor survival. These findings directly contrast with the Swedish study, in which CXCL10 appeared favorable. This inconsistency likely reflects heterogeneity in cohort size, clinical characteristics, treatment regimens, and assay platforms, and highlights that CXCL10 may act as either a marker of effective T-cell trafficking or of chronic, non-productive inflammation depending on context.119,122 The Swedish study further identified caspase-8 (CASP8), adenosine deaminase (ADA), mucin-16 (MUC16), CD244, interleukin-18 (IL-18), and angiopoietin-2 (ANGPT2) as enriched in non-responders, each associated with shorter survival outcomes.119
Intracellular and Alveolar Proteins
Non-responders also demonstrated enrichment of proteins such as yes-associated protein 1 (YAP1), decapping scavenger (DCPS), and stratifin (SFN), which reflect oncogenic stress and were linked to poor outcomes.120
Inflammatory Mediators
Systemic inflammation was reflected by higher levels of serum amyloid A1 (SAA1), SAA2, and calcium-binding proteins S100A8 and S100A9, all of which amplify myeloid recruitment and immune escape.125 Elevated interleukin-6 (IL-6), a driver of STAT3 signaling and immunosuppressive myeloid differentiation, further supported the link between inflammatory signaling and resistance.122
Mixed or Context-Dependent Predictors
Autoantibodies
Autoantibodies against MYC-associated factor X (MAX) were enriched in responders, whereas those targeting TAP-binding protein (TAPBP) and DEAH-box helicase 29 (DHX29) showed inconsistent associations, underscoring the heterogeneity of humoral immune influences.124
Soluble Checkpoints
Plasma levels of soluble immune checkpoints including PD-1, PD-L1, PD-L2, cytotoxic T-lymphocyte-associated protein 4 (CTLA-4), T-cell immunoglobulin and mucin domain-containing protein 3 (TIM-3), B- and T-lymphocyte attenuator (BTLA), herpesvirus entry mediator (HVEM), CD137 (4–1BB), and lymphocyte-activation gene 3 (LAG-3) were also studied. No consistent association with outcomes was observed.159
Heterogeneity and Limitations in Proteomics
Proteomic predictors of immunotherapy response in NSCLC demonstrated substantial heterogeneity across studies. Sample sizes varied widely, ranging from fewer than 50 patients in some discovery cohorts119,123 to over 140 patients in larger validation efforts.122 Clinical characteristics such as treatment line, PD-L1 expression, smoking status, and histology were not uniformly controlled, complicating cross-cohort comparisons. Different assay platforms were used, including proximity extension assay panels, multiplex ELISAs, and mass spectrometry–based discovery, each with distinct sensitivity and coverage. Endpoint definitions also differed, with some studies focusing on short-term response, others on PFS or OS, and statistical handling ranging from simple logistic regression to multivariate Cox models with internal cross-validation.
An illustrative example of heterogeneity is CXCL10, which was linked to favorable outcomes in the Swedish cohort but unfavorable in the Israeli cohort, highlighting how cohort size, assay design, and inflammatory context can yield opposing results. Similarly, circulating soluble checkpoint proteins showed inconsistent associations, likely reflecting differences in detection sensitivity and biological interpretation as either decoy receptors or tumor burden surrogates. Collectively, these discrepancies limit direct generalization of single markers. Larger multi-institutional studies, harmonized platforms, and standardized endpoints are required to validate proteomic predictors and establish their translational readiness.
Metabolomics
Metabolomics represents the functional readouts of a cellular state that can reveal changes in biological states.160 In contrast to genes and proteins, which are influenced by epigenetic regulation and PTM, respectively, metabolites act as direct indicators of biological activity, making it simpler to associate them with phenotype.161 Because of their drastically changed metabolism, cancer cells create chemicals that are unique to and characteristic of non-physiological environments.160 As metabolites can interact with and target various therapies, metabolomics is an important area for advancing the evaluation of immunotherapy and chemotherapy effectiveness.162 Techniques utilized in metabolomics analysis.
The primary analytical techniques used in metabolomics are MS and nuclear magnetic resonance spectroscopy (NMR). In most of the studies, MS is coupled to a separation technique such as chromatography (eg LC-MS or gas chromatography (GC-MS)) or capillary electrophoresis (eg CE-MS).163
NMR
This technique has a number of advantages, including as high throughput, significant repeatability, simple metabolite identification, and non-destructive sample preparation.164 The fact that NMR is not limited to the study of tissue or bio fluid extracts alone is a significant benefit.165 Since NMR is not inherently destructive it can be used for metabolite imaging in living samples using magnetic resonance spectroscopy (MRS) and magnetic resonance imaging (MRI), unlike LC-MS and GC-MS based approaches. Nevertheless, NMR is 10–100 times less sensitive than LC-MS and GC-MS methods and provide less metabolome coverage (e.g LC-MS can provide details on 1000+ metabolites with concentrations >10 to 100 nM, while an NMR shows details of 50–200 metabolites with concentrations >1 µM).166
LC-MS and GC-MS Analyses
Combining chromatographic separation with mass spectrometry improves metabolite detection by reducing sample complexity and ionization interference, enhancing sensitivity, signal consistency, and data reliability.167 Advances in liquid chromatography, such as capillary monolithic chromatography and Ultra-Performance Liquid Chromatography, have enhanced analysis speed and resolution.168 While gas chromatography offers higher resolution, liquid chromatography–mass spectrometry is preferred in metabolomics due to its ability to analyze a broader range of metabolites, including polar and thermally unstable molecules like amino acids and organic acids that may degrade during gas chromatography.169,170 Liquid chromatography–mass spectrometry also simplifies sample preparation and enables higher sensitivity and specificity in complex biological matrices such as plasma, urine, and tissue extracts. The technique can be tailored to specific metabolite types by adjusting chromatographic settings, mobile phases, and ionization modes.169,170
Several mass spectrometry analyzers are used in metabolomics: quadrupole mass spectrometers are suitable for targeted analysis; time-of-flight mass (TOF) spectrometers are ideal for untargeted studies; ion trap mass spectrometers allow detailed fragmentation; Orbitrap and Fourier transform ion cyclotron resonance mass spectrometers FT-ICR offer high mass accuracy and resolution for complex mixtures; and triple quadrupole mass spectrometers enable highly sensitive targeted quantification via multiple reaction monitoring.171
A typical metabolomics workflow begins with experimental design to select targeted or untargeted approaches. Metabolites are extracted using biphasic liquid methods, depending on the sample and study goals.172,173 Profiling is done using nuclear magnetic resonance or liquid chromatography–mass spectrometry, followed by data processing with software like XCMS, MAVEN, or MZmine3.174 Metabolite identification is based on standards, mass data, and tandem mass spectrometry, using databases such as the Human Metabolome Database and METLIN.175 Statistical analyses identify biomarkers, followed by pathway analysis using resources like the Kyoto Encyclopedia of Genes and Genomes.174,176 The overall steps involved in metabolomics research are summarized in Figure 4, which depicts the typical workflow from experimental design and metabolite extraction through profiling, data processing, compound identification, and pathway analysis.
 |
Figure 4 Typical metabolomics workflow using MS and NMR-based techniques. Abbreviation: CSF, cerebrospinal fluid; NMR, nuclear magnetic resonance; LC/MS, liquid chromatography/mass spectrometry. Note: Figure created with BioRender. |
Metabolomics Study for the Prediction of Response to ICI in NSCLC
Metabolomics studies have been widely employed in cancer research to investigate the underlying disturbed pathways177–180 and identify metabolic biomarkers or patterns associated with cancer using different biological samples including serum,181–185 plasma,186 saliva,187 urine,188,189 and breath.190,191
In contrast to proteomics, the metabolomics literature is more heterogeneous in evidence strength. Of the nine included studies, only four scored ≥10, while the remainder were exploratory and lower-evidence (<10). We therefore highlight high-scoring studies as primary findings and frame the others as preliminary signals requiring further validation. Table 6 presents metabolomic predictors of ICI response in NSCLC, detailing the candidate metabolites, reported clinical outcomes, validation status, and evidence scores. Full quantitative details, including effect estimates (hazard ratios, odds ratios, confidence intervals, and p-values), are provided in Supplementary Table S3c (Excel file), which contains separate sheet for metabolomic studies.
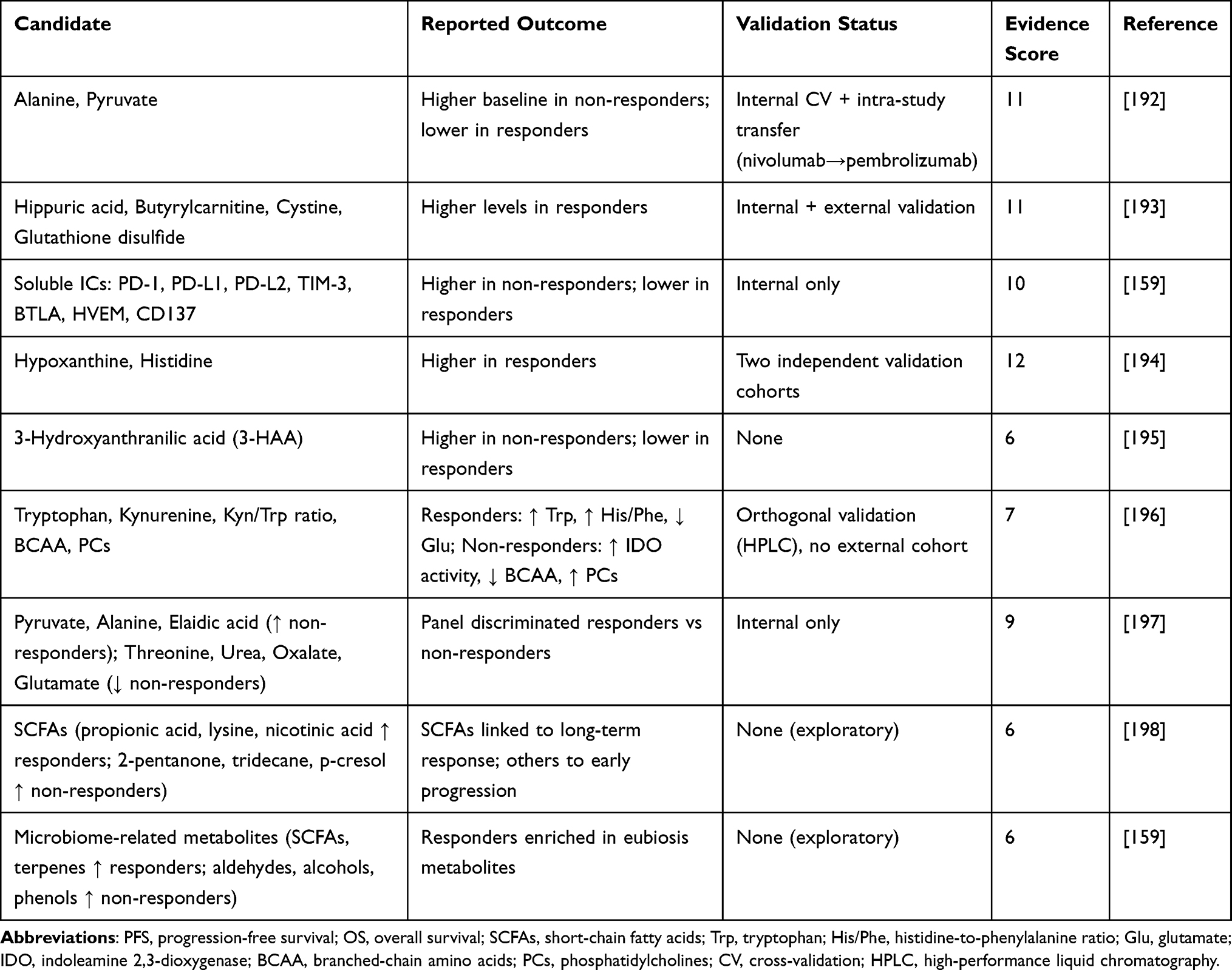 |
Table 6 Metabolomic Predictors of ICI Response in NSCLC |
Potential Metabolite Biomarkers for Prediction of Response to ICI in NSCLC
Predictors of Favorable Response
Amino Acid Metabolism
Lower baseline alanine and pyruvate characterized responders, suggesting reduced glycolytic burden and diminished metabolic competition with lymphocytes (score ≥10).192 Other favorable metabolites included hippuric acid, butyrylcarnitine, cystine, and glutamic acid, reflecting host–microbial co-metabolism and antioxidant balance (scores ≥10).193,194 Exploratory cohorts suggested that responders also showed higher baseline tryptophan and a higher histidine/phenylalanine ratio, consistent with preserved amino acid availability for effector T-cell proliferation (score <10).196
Energy and Cofactor Metabolism
Nicotinic acid (vitamin B3) was enriched in responders, supporting NAD+ biosynthesis, redox balance, and T-cell metabolic fitness; this signal remains exploratory (score <10).198
Microbiome-Derived Metabolites
High-evidence studies identified additional favorable metabolites noted above (hippuric acid, etc). (scores ≥10).193,194 Exploratory cohorts reported short-chain fatty acids (SCFAs) such as propionic acid enriched in responders, consistent with enhanced dendritic cell activation and CD8+ effector function (scores <10).159,198
Predictors of Poor Response
Tryptophan–kynurenine pathway Non-responders demonstrated lower baseline tryptophan and increased indoleamine 2,3-dioxygenase (IDO) activity with accumulation of downstream metabolites such as 3-hydroxyanthranilic acid (3-HAA), which can engage the aryl hydrocarbon receptor and promote T-cell exhaustion; these findings are exploratory (scores <10).195,196
Energy and Cofactor Metabolism
Higher baseline and on-treatment alanine and pyruvate were linked to progression (score ≥10).192 Progressive disease was also associated with reduced threonine, urea, and oxalate, indicating disrupted nitrogen metabolism; this extends the pattern but is exploratory (score <10).197
Lipid and Membrane Remodeling
Non-responders showed increased elaidic acid (a trans-monounsaturated fatty acid) and elevated phosphatidylcholines such as PC C38:0, suggesting intensified membrane synthesis, ER stress, and myeloid activation; these remain exploratory (scores <10).196,197
Microbiome-Derived Metabolites
Exploratory cohorts reported enrichment of 2-pentanone, tridecane, and p-cresol in non-responders, metabolites linked to dysbiosis, oxidative stress, and impaired dendritic cell function (score <10).198
Amino Acid Insufficiency Signatures
Reduced branched-chain amino acids (BCAA), which are needed for mTOR-dependent effector T-cell expansion, were observed in non-responders; these signals are exploratory (scores <10).196,197
Mixed or Context-Dependent Predictors
Amino Acid and Tryptophan Metabolism
While reduced tryptophan and increased IDO activity were associated with poor outcomes in exploratory cohorts, some studies reported preserved tryptophan in responders without uniform changes in the kynurenine/tryptophan ratio, indicating that absolute availability and catabolic flux may both carry predictive value depending on matrix and timing (scores <10).196,197
Energy Metabolism
Alanine and pyruvate trends were largely unfavorable, with context dependence between plasma versus serum and pre- versus on-treatment sampling; the dominant signal (higher alanine/pyruvate → non-response) is supported by higher-evidence work (score ≥10),192 with variability in exploratory cohorts (score <10).197
Heterogeneity and Limitations in Metabolomics
Metabolomic studies also revealed marked heterogeneity in methodology and outcomes. Cohorts were generally small, ranging from fewer than 30 to just over 100 patients,195,197 and biospecimens varied across plasma, serum, and fecal samples, which may capture distinct metabolic environments. Analytical platforms differed, including NMR, LC-MS, and GC-MS, with variability in compound detection and quantification. Reported endpoints spanned short-term response, PFS, and OS, and statistical strategies ranged from univariate comparisons to multivariate machine-learning models.
Contradictory findings emerged particularly in tryptophan metabolism. While lower tryptophan and increased IDO activity with accumulation of 3-hydroxyanthranilic acid were consistently poor predictors in several cohorts, other studies found that preserved tryptophan without consistent kynurenine elevation marked benefit. Similarly, alanine and pyruvate were unfavorable in most contexts but showed variability depending on biofluid source and sampling timepoint. Microbiome-derived metabolites also displayed cohort-specific patterns, with SCFAs generally favorable but other microbial products such as aldehydes, ketones, and phenols enriched in non-responders.
These inconsistencies reflect the influence of geographic and dietary backgrounds, microbiome diversity, and technical variability in metabolite detection. Limited external validation and small sample sizes further restrict generalizability. Standardization of metabolomics pipelines, integration with microbiome sequencing, and prospective validation in larger, uniformly treated NSCLC cohorts are needed to clarify the true predictive value of metabolic signatures.
Cross-Omics Integration of Genomic, Proteomic, and Metabolomic Predictors
In the following section, we integrate genomic, proteomic, and metabolomic evidence to highlight convergent pathways that shape ICI response in NSCLC. Cross-omics synthesis reveals recurring themes—such as interferon signaling, metabolic rewiring, and immune–stromal interactions—that transcend individual omics layers.
Interferon Signaling and T-Cell Inflammation
Multi-omics evidence converges on interferon signaling and T-cell inflammation as favorable predictors of ICI response. At the genomic level, KRAS/TP53 co-mutations have been associated with increased neoantigen load, interferon activation, and CD8⁺ infiltration98,104 Proteomic studies identified higher levels of CXCL9, CXCL10, and IL-15 in responders, reflecting chemokine-driven T-cell recruitment and survival.119 Metabolomic evidence showed enrichment of short-chain fatty acids in responders, which promote dendritic cell activation and interferon-stimulated CD8⁺ effector function.194,198
Adenosine Metabolism and Immune Suppression
Disruption of adenosine metabolism is consistently linked to poor outcomes across omics layers. Genomically, STK11/LKB1 co-mutations (the KL subtype) foster immune-cold phenotypes through adenosine accumulation and myeloid suppressor cell recruitment.104,108 At the proteomic level, elevated adenosine deaminase (ADA) and CASP8 were observed in non-responders, consistent with disrupted purine metabolism and T-cell dysfunction.119 Metabolomic studies showed reduced branched-chain amino acids and accumulation of glycolytic intermediates such as alanine and pyruvate in non-responders, consistent with metabolic exhaustion that synergizes with adenosine-mediated suppression.192,197
Tryptophan–Kynurenine Catabolism
Alterations in tryptophan metabolism emerged as another recurrent mechanism of ICI resistance. At the genomic level, alterations in IDO1 and IDO2, as well as aryl hydrocarbon receptor (AhR) activation signatures, have been reported in NSCLC and associated with immune evasion.100,104 Proteomic studies showed elevated soluble immune checkpoints including PD-1, PD-L1, TIM-3, LAG-3, BTLA, and HVEM in non-responders, reflecting T-cell exhaustion linked to tryptophan depletion and kynurenine accumulation.120,159 Metabolomic profiling further demonstrated lower baseline tryptophan and increased downstream products such as 3-hydroxyanthranilic acid (3-HAA) in non-responders.195,196 Collectively, these cross-omics findings implicate the IDO–kynurenine axis as a central immunosuppressive pathway.
Angiogenesis and Stromal Remodeling
Angiogenesis and stromal remodeling are also recurrently associated with poor ICI outcomes. Genomic loss-of-function mutations in KEAP1 activate NRF2 signaling, impairing antigen presentation and promoting an immune-excluded microenvironment.98,108 Proteomic studies reported elevated angiopoietin-2 (ANGPT2) and IL-6 in non-responders, linking angiogenesis and inflammatory signaling with poor prognosis.119,122 Metabolomic profiles enriched for phosphatidylcholines and trans-fatty acids such as elaidic acid in non-responders further reflect endothelial activation and lipid remodeling that support stromal exclusion.196,197
Although few individual markers have been validated consistently across multiple NSCLC cohorts, integration across omics layers highlights convergent biological pathways—interferon signaling, adenosine metabolism, tryptophan catabolism, and angiogenesis—that collectively distinguish immune-hot from immune-cold tumor states.
To consolidate these findings, Table 7 summarizes cross-omics evidence by integrating genomic, proteomic, and metabolomic predictors of ICI response in NSCLC, highlighting convergent pathways, reported outcomes, validation status, and evidence scores.
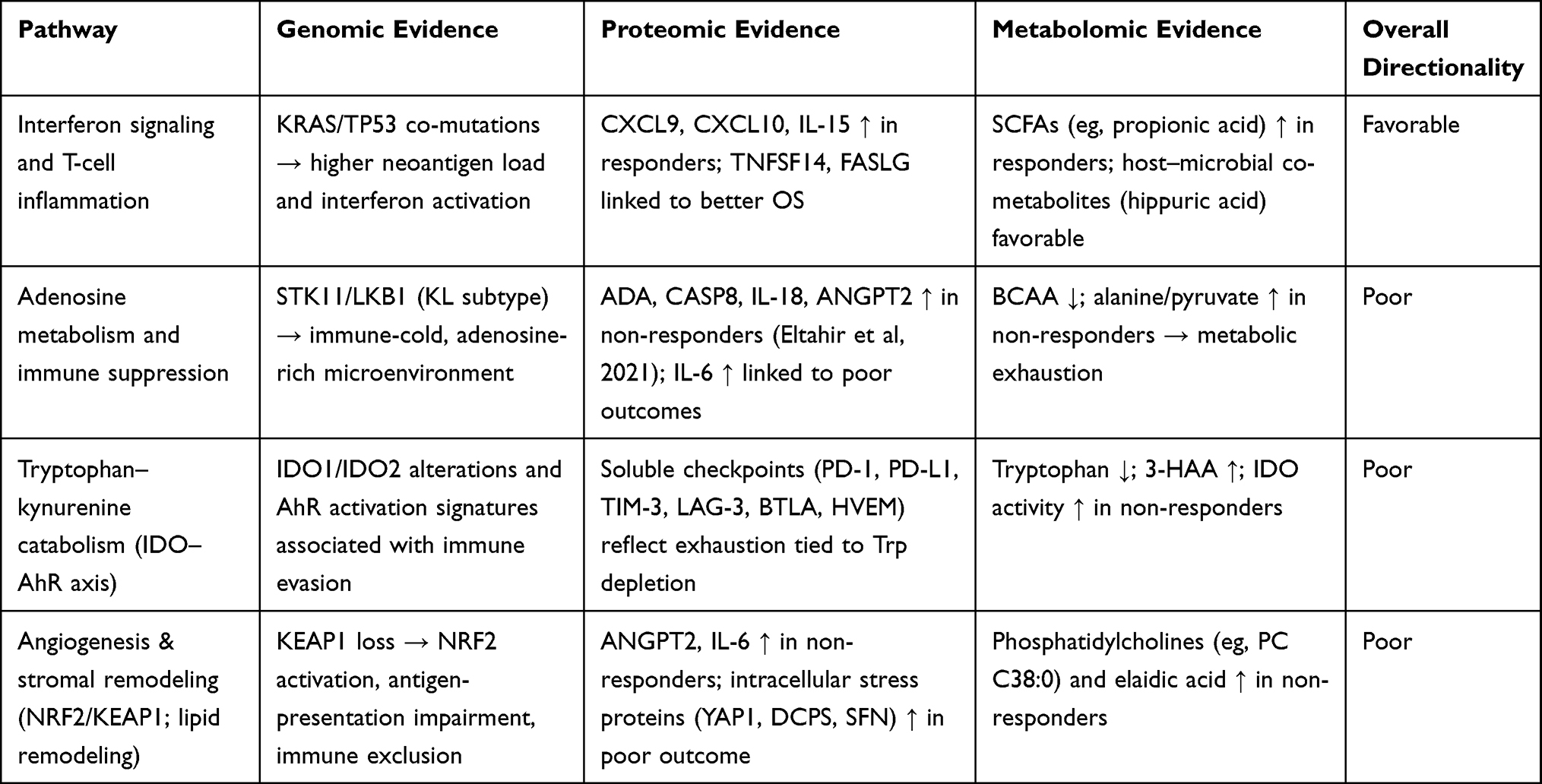 |
Table 7 Cross-Omics Integration of Genomic, Proteomic, and Metabolomic Predictors of Immune Checkpoint Inhibitor Response in NSCLC |
Conclusion
In conclusion, ICIs have reshaped the therapeutic landscape of non-small cell lung cancer (NSCLC), yet accurately predicting benefit remains a major challenge. PD-L1 expression alone has limited precision, underscoring the need for broader predictive strategies. Evidence from 33 genomic, 9 proteomic, and 9 metabolomic studies demonstrates that multi-omics approaches can identify convergent biomarkers that stratify tumors into immune-hot versus immune-cold phenotypes.
Among genomic alterations, KRAS/TP53 co-mutations, NOTCH family variants, and BRAF V600E consistently associate with favorable outcomes, while EGFR, ALK/RET/ROS1 fusions, and KRAS with STK11 or KEAP1 predict resistance. In proteomics, chemokines such as CXCL9 and CXCL10 and soluble checkpoint proteins (sPD-1, sPD-L1, LAG-3) emerged as promising predictors, whereas acute-phase proteins (SAA1/2, S100A8/9) align with poor response. Metabolomic profiling implicated tryptophan and short-chain fatty acids as favorable, while 3-hydroxyanthranilic acid, pyruvate, and lipid metabolites marked resistance. These markers collectively point to pathways of antigenicity, interferon signaling, and metabolic immune regulation.
Although integrative multi-omics shows clear promise, only a subset of biomarkers have reached consistent validation across independent cohorts, and none yet meet the threshold for routine clinical use. Future work should prioritize prospective trials with pre-specified biomarker endpoints, harmonization of assay protocols and data pipelines, and incremental implementation in clinical laboratories. Ultimately, the integration of multi-omics with artificial intelligence may enable robust, individualized prediction of ICI outcomes and more precise treatment decisions for patients with NSCLC.
Future Directions
To advance biomarker development for NSCLC immunotherapy, several priorities emerge. First, prospective trials with pre-specified biomarker endpoints are essential to validate candidate genomic, proteomic, and metabolomic predictors in large, uniformly treated cohorts. Second, standardized assay protocols—including harmonized sequencing pipelines, proteomic panels, and metabolomics workflows—are needed to reduce variability and improve reproducibility across studies. Third, AI-driven integration pipelines that combine multi-omics data with clinical and radiomic features offer a promising approach to capture the complexity of tumor–immune interactions and generate clinically deployable prediction models. Beyond technical harmonization, cost–benefit assessments are required to ensure that proposed biomarkers can be implemented feasibly in routine practice. Finally, incremental implementation pathways should be pursued in clinical laboratories, beginning with targeted, low-cost assays and progressing to comprehensive multi-omics platforms as evidence and infrastructure mature.
A practical roadmap is also required to bridge discovery and clinical implementation. We propose a phased approach: initial analytical validation of candidate biomarkers using standardized protocols; subsequent development of targeted multi-omics panels in CLIA-certified laboratories to ensure regulatory compliance and reproducibility; and finally, prospective clinical trials incorporating these panels to establish predictive utility and cost-effectiveness. Early adoption may focus on single-analyte assays with the strongest evidence (eg, KRAS/TP53 co-mutations, CXCL9/10, tryptophan metabolites), which can be gradually expanded into multiplexed or integrative platforms. This stepwise strategy allows incremental integration into clinical workflows, while reducing barriers to implementation and ensuring that validated biomarkers can meaningfully guide patient management.
Abbreviations
The following abbreviations are used in this manuscript:
ALK, Anaplastic lymphoma kinase; 2DGE, Two-dimensional gel electrophoresis; 3-HAA, 3-hydroxyanthranilic acid; ACT, Adaptive cell transfer; APCs, Antigen-presenting cells; BCAAs, Branched-chain amino acids; BRAF, V-raf murine sarcoma viral oncogene homolog B1;. CTLA-4, Cytotoxic T-lymphocyte–associated antigen 4;. DCB, Durable clinical benefit; DLX2, Distal-less homeobox 2; dMMR, Mismatch repair deficiency; EGFR, Epidermal growth factor receptor; EPHA, Ephrin type-A receptors; FFPE, Formalin-fixed paraffin-embedded; GC-MS, Gas chromatography-mass spectrometry; HER2, Human epidermal growth factor receptor 2; HMDB, Human metabolome database; HSPG2, Heparan sulfate proteoglycan; ICGC, International cancer genome consortium;. ICI, Immune checkpoint inhibitors; IDO, Indoleamine-2,3-dioxygenase; IFN-γ, Interferon-γ; IHC, Immunohistochemistry; IVTT, In vitro transcription and translation; KEGG, Kyoto Encyclopedia of Genes and Genomes; KMT2C, Lysine (K)-specific methyltransferase 2C; KRAS, Kirsten Rat Sarcoma virus oncogene homolog; LC-MS, Liquid chromatography-mass spectrometry; LKB1, Liver kinase B1; LUAD, Lung adenocarcinoma; LUSC, Lung squamous cell carcinoma; MALDI, Matrix-assisted laser desorption/ionization; MAPK, Mitogen-activated protein kinase; METLIN, Metabolite and Tandem MS Database; MGA, MAX gene-associated; MS, Mass spectrometry; MSI, Microsatellite instabilityMSKCC-IO, Memorial Sloan-Kettering Cancer Center-immunotherapy; NAL, Neoantigen load; NGS, Next generation sequencing; NMD, Nonsense-mediated decay; NMR, Nuclear magnetic resonance; NSCLC, Non-small cell lung cancer; ONT, Oxford nanopore technology; ORF, Open reading frame; ORR, Objective response rate; OS, Overall survival; PAK7, P21-activated kinase 7;. PCR, Polymerase chain reaction; PD-1, Programmed death-1;. PD-L1, Programmed cell death-ligand 1; PEA, Proximity extension assay; PFS, Progression free survival; PI3K, Phosphoinositide 3-kinase; PTMs, Post-translational modifications; RELN, Reelin; RP-HPLC, Reversed-phase high-performance liquid chromatography; SCFAs, Short-chain fatty acids; SCLC, Small cell lung cancer; SDS-PAGE, Sodium dodecyl sulfate–polyacrylamide gel electrophoresis; STK11, And serine-threonine kinase 11; TCGA, The cancer genome atlas; TGFβ, Transforming growth factor beta; TILs, Tumor-infiltrating lymphocytes; TIP, Tumor immune profile; TKI, Tyrosine kinase inhibitor; TKRs, Tyrosine kinase receptors; TMB, Tumor mutational burden; TOF, Time of flight; TP53, Tumor protein 53; TPS, Tumor proportion score;. TS, Targeted sequencing; UBE3A, Ubiquitin protein ligase e3a; UHPLC, Ultrahigh performance liquid chromatography; WES, Whole exome sequencing; WGS, Whole genome sequencing.
Ethical Statement
The authors are accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This research received no external funding.
Disclosure
The authors state that there are no conflict of interest regarding the publication of this article.
References
1. Siegel RL, Miller KD, Wagle NS, Jemal A. Cancer statistics, 2023. CA Cancer J Clin. 2023;73(1):17–48. doi:10.3322/caac.21763
2. American Cancer Society. Cancer facts & figures; 2024. Available from: www.cancer.org/content/dam/cancer-org/research/cancer-facts-and-statistics/annual-cancer-facts-and-figures/2024/2024-cancer-facts-and-figures-acs.pdf.
3. Duma N, Santana-Davila R, Molina JR. Non–Small Cell Lung Cancer: Epidemiology, Screening, Diagnosis, and Treatment. Elsevier. 2019:1623–1640.
4. Niu F-Y, Zhou Q, Yang -J-J, et al. Distribution and prognosis of uncommon metastases from non-small Cell Lung Cancer. BMC Cancer. 2016;16:1–6. doi:10.1186/s12885-016-2169-5
5. American cancer society. Lung Cancer survival rates. Available from: https://www.cancer.org/cancer/types/lung-cancer/detection-diagnosis-staging/survival-rates.html.
6. Kim J, Lee H, Huang BW. Lung cancer: diagnosis, treatment principles, and screening. Am Fam Phys. 2022;105(5):487–494.
7. Shiravand Y, Khodadadi F, Kashani SMA, et al. Immune checkpoint inhibitors in cancer therapy. Curr Oncol. 2022;29(5):3044–3060. doi:10.3390/curroncol29050247
8. Alemohammad H, Najafzadeh B, Asadzadeh Z, et al. The importance of immune checkpoints in immune monitoring: a future paradigm shift in the treatment of cancer. Biomed Pharmacother. 2022;146:112516. doi:10.1016/j.biopha.2021.112516
9. Karachaliou N, Cao MG, Teixidó C, et al. Understanding the function and dysfunction of the immune system in Lung Cancer: the role of immune checkpoints. Cancer Biol Med. 2015;12(2):79. doi:10.7497/j.issn.2095-3941.2015.0029
10. Rittmeyer A, Barlesi F, Waterkamp D, et al. Atezolizumab versus docetaxel in patients with previously treated non-small-cell lung cancer (OAK): a Phase 3, open-label, multicentre randomised controlled trial. Lancet. 2017;389(10066):255–265. doi:10.1016/S0140-6736(16)32517-X
11. Freeman GJ, Long AJ, Iwai Y, et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med. 2000;192(7):1027–1034. doi:10.1084/jem.192.7.1027
12. Zhang Y, Zheng J. Functions of immune checkpoint molecules beyond immune evasion. Regulation Cancer Immune Checkpoints. 2020;2020:201–226.
13. Wei SC, Duffy CR, Allison JP. Fundamental mechanisms of immune checkpoint blockade therapy. Cancer Discov. 2018;8(9):1069–1086. doi:10.1158/2159-8290.CD-18-0367
14. Singh N, Temin S, Baker JS, et al. Therapy for stage IV non–small-cell lung cancer without driver alterations: ASCO living guideline. J Clin Oncol. 2022;40(28):3323–3343. doi:10.1200/JCO.22.00825
15. Owen DH, Singh N, Ismaila N, et al. Therapy for stage IV non–small-cell lung cancer with driver alterations: ASCO living guideline, version 2022.2. J Clin Oncol. 2023;41(5):e10–e20. doi:10.1200/JCO.22.02124
16. Borghaei H, Paz-Ares L, Horn L, et al. Nivolumab versus docetaxel in advanced nonsquamous non–small-cell lung cancer. N Engl J Med. 2015;373(17):1627–1639. doi:10.1056/NEJMoa1507643
17. Brahmer J, Reckamp KL, Baas P, et al. Nivolumab versus docetaxel in advanced squamous-cell non–small-cell lung cancer. N Engl J Med. 2015;373(2):123–135. doi:10.1056/NEJMoa1504627
18. Forde PM, Spicer J, Lu S, et al. Abstract CT003: nivolumab (NIVO)+ platinum-doublet chemotherapy (chemo) vs chemo as neoadjuvant treatment (tx) for resectable (IB-IIIA) non-small cell lung cancer (NSCLC) in the phase 3 CheckMate 816 trial. Cancer Res. 2021;81(13_Supplement):CT003–CT003. doi:10.1158/1538-7445.AM2021-CT003
19. X-h J, L-y G, Jiao M, W-j W, L-l J, Guo H. Efficacy and safety of neoadjuvant immunotherapy in resectable nonsmall cell lung cancer: a meta-analysis. Lung Cancer. 2020;147:143–153. doi:10.1016/j.lungcan.2020.07.001
20. Lazzari C, Spagnolo CC, Ciappina G, et al. Immunotherapy in early-stage non-small cell lung cancer (NSCLC): current evidence and perspectives. Curr Oncol. 2023;30(4):3684–3696. doi:10.3390/curroncol30040280
21. Paucek RD, Baltimore D, Li G. The cellular immunotherapy revolution: arming the immune system for precision therapy. Trends Immunol. 2019;40(4):292–309. doi:10.1016/j.it.2019.02.002
22. Garon EB, Rizvi NA, Hui R, et al. Pembrolizumab for the treatment of non–small-cell lung cancer. N Engl J Med. 2015;372(21):2018–2028. doi:10.1056/NEJMoa1501824
23. Herbst RS, Baas P, Kim D-W, et al. Pembrolizumab versus docetaxel for previously treated, PD-L1-positive, advanced non-small-cell lung cancer (KEYNOTE-010): a randomised controlled trial. Lancet. 2016;387(10027):1540–1550. doi:10.1016/S0140-6736(15)01281-7
24. Sharma P, Hu-Lieskovan S, Wargo JA, Ribas A. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell. 2017;168(4):707–723. doi:10.1016/j.cell.2017.01.017
25. Fares CM, Van Allen EM, Drake CG, Allison JP, Hu-Lieskovan S. Mechanisms of resistance to immune checkpoint blockade: why does checkpoint inhibitor immunotherapy not work for all patients? Am Soc Clin Oncol Educ Book. 2019;39:147–164. doi:10.1200/EDBK_240837
26. Powell SF, Rodríguez-Abreu D, Langer CJ, et al. Outcomes with pembrolizumab plus platinum-based chemotherapy for patients with NSCLC and stable brain metastases: pooled analysis of KEYNOTE-021,-189, and-407. J Thorac Oncol. 2021;16(11):1883–1892. doi:10.1016/j.jtho.2021.06.020
27. Gandhi L, Rodríguez-Abreu D, Gadgeel S, et al. Pembrolizumab plus chemotherapy in metastatic non–small-cell lung cancer. N Engl J Med. 2018;378(22):2078–2092. doi:10.1056/NEJMoa1801005
28. Goh KY, Cheng TYD, Tham SC, Lim DW-T. Circulating biomarkers for prediction of immunotherapy response in NSCLC. Biomedicines. 2023;11(2):508. doi:10.3390/biomedicines11020508
29. Wang Y, Tong Z, Zhang W, et al. FDA-approved and emerging next generation predictive biomarkers for immune checkpoint inhibitors in cancer patients. Front Oncol. 2021;11:683419. doi:10.3389/fonc.2021.683419
30. Reck M, Rodríguez–Abreu D, Robinson AG, et al. Updated analysis of KEYNOTE-024: pembrolizumab versus platinum-based chemotherapy for advanced non–small-Cell Lung Cancer with PD-L1 tumor proportion score of 50% or greater. J Clin Oncol. 2019;37(7):537–546. doi:10.1200/JCO.18.00149
31. Hanna N, Johnson D, Temin S, et al. Systemic therapy for stage IV non–small-cell lung cancer: American Society of Clinical Oncology clinical practice guideline update. J Clin Oncol. 2017;35(30):3484–3515. doi:10.1200/JCO.2017.74.6065
32. Oitabén A, Fonseca P, Villanueva MJ, et al. Emerging blood-based biomarkers for predicting immunotherapy response in NSCLC. Cancers. 2022;14(11):2626. doi:10.3390/cancers14112626
33. Kang Y-K, Boku N, Satoh T, et al. Nivolumab in patients with advanced gastric or gastro-oesophageal junction cancer refractory to, or intolerant of, at least two previous chemotherapy regimens (ONO-4538-12, ATTRACTION-2): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2017;390(10111):2461–2471. doi:10.1016/S0140-6736(17)31827-5
34. Janjigian YY, Bendell J, Calvo E, et al. CheckMate-032 study: efficacy and safety of nivolumab and nivolumab plus ipilimumab in patients with metastatic esophagogastric cancer. J Clin Oncol. 2018;36(28):2836–2844. doi:10.1200/JCO.2017.76.6212
35. Davis AA, Patel VG. The role of PD-L1 expression as a predictive biomarker: an analysis of all US food and drug administration (FDA) approvals of immune checkpoint inhibitors. J Immunother Cancer. 2019;7(1):278. doi:10.1186/s40425-019-0768-9
36. Hong L, Negrao MV, Dibaj SS, et al. Programmed death-ligand 1 heterogeneity and its impact on benefit from immune checkpoint inhibitors in NSCLC. J Thorac Oncol. 2020;15(9):1449–1459. doi:10.1016/j.jtho.2020.04.026
37. Schoenfeld AJ, Rizvi H, Bandlamudi C, et al. Clinical and molecular correlates of PD-L1 expression in patients with lung adenocarcinomas. Ann Oncol. 2020;31(5):599–608. doi:10.1016/j.annonc.2020.01.065
38. Marcus L, Lemery SJ, Keegan P, Pazdur R. FDA approval summary: pembrolizumab for the treatment of microsatellite instability-high solid tumors. Clin Cancer Res. 2019;25(13):3753–3758. doi:10.1158/1078-0432.CCR-18-4070
39. Engel KB, Moore HM. Effects of preanalytical variables on the detection of proteins by immunohistochemistry in formalin-fixed, paraffin-embedded tissue. Arch Pathol Lab Med. 2011;135(5):537–543. doi:10.5858/2010-0702-RAIR.1
40. Waalkes A, Smith N, Penewit K, et al. Accurate pan-cancer molecular diagnosis of microsatellite instability by single-molecule molecular inversion probe capture and high-throughput sequencing. Clin Chem. 2018;64(6):950–958. doi:10.1373/clinchem.2017.285981
41. Duffy MJ, Crown J. Biomarkers for predicting response to immunotherapy with immune checkpoint inhibitors in cancer patients. Clin Chem. 2019;65(10):1228–1238. doi:10.1373/clinchem.2019.303644
42. Cristescu R, Mogg R, Ayers M, et al. Pan-tumor genomic biomarkers for PD-1 checkpoint blockade–based immunotherapy. Science. 2018;362(6411):eaar3593. doi:10.1126/science.aar3593
43. Davoli T, Uno H, Wooten EC, Elledge SJ. Tumor aneuploidy correlates with markers of immune evasion and with reduced response to immunotherapy. Science. 2017;355(6322):eaaf8399.
44. Hellmann MD, Ciuleanu T-E, Pluzanski A, et al. Nivolumab plus ipilimumab in lung cancer with a high tumor mutational burden. N Engl J Med. 2018;378(22):2093–2104. doi:10.1056/NEJMoa1801946
45. Hofman P, Heeke S, Alix-Panabières C, Pantel K. Liquid biopsy in the era of immuno-oncology: is it ready for prime-time use for cancer patients? Ann Oncol. 2019;30(9):1448–1459. doi:10.1093/annonc/mdz196
46. Kowanetz M, Zou W, Shames D, et al. OA20. 01 tumor mutation burden (TMB) is associated with improved efficacy of atezolizumab in 1L and 2L+ NSCLC patients. J Thorac Oncol. 2017;12(1):S321–S322. doi:10.1016/j.jtho.2016.11.343
47. Ready N, Hellmann MD, Awad MM, et al. First-line nivolumab plus ipilimumab in advanced non–small-cell lung cancer (CheckMate 568): outcomes by programmed death ligand 1 and tumor mutational burden as biomarkers. J Clin Oncol. 2019;37(12):992–1000. doi:10.1200/JCO.18.01042
48. Marabelle A, Fakih M, Lopez J, et al. Association of tumour mutational burden with outcomes in patients with advanced solid tumours treated with pembrolizumab: prospective biomarker analysis of the multicohort, open-label, Phase 2 KEYNOTE-158 study. Lancet Oncol. 2020;21(10):1353–1365. doi:10.1016/S1470-2045(20)30445-9
49. Klempner SJ, Fabrizio D, Bane S, et al. Tumor mutational burden as a predictive biomarker for response to immune checkpoint inhibitors: a review of current evidence. Oncologist. 2020;25(1):e147–e159. doi:10.1634/theoncologist.2019-0244
50. Merino DM, McShane LM, Fabrizio D, et al. Establishing guidelines to harmonize tumor mutational burden (TMB): in silico assessment of variation in TMB quantification across diagnostic platforms: Phase I of the friends of cancer Res TMB harmonization project. J Immunother Cancer. 2020;8(1):e000147. doi:10.1136/jitc-2019-000147
51. Alexandrov LB, Nik-Zainal S, Wedge DC, et al. Signatures of mutational processes in human cancer. Nature. 2013;500(7463):415–421. doi:10.1038/nature12477
52. Stenzinger A, Allen JD, Maas J, et al. Tumor mutational burden standardization initiatives: recommendations for consistent tumor mutational burden assessment in clinical samples to guide immunotherapy treatment decisions. Genes Chromosomes Cancer. 2019;58(8):578–588. doi:10.1002/gcc.22733
53. Reck M, Rodríguez-Abreu D, Robinson AG, et al. Pembrolizumab versus chemotherapy for PD-L1–positive non–small-cell lung cancer. N Engl J Med. 2016;375(19):1823–1833. doi:10.1056/NEJMoa1606774
54. Mok TS, Wu Y-L, Kudaba I, et al. Pembrolizumab versus chemotherapy for previously untreated, PD-L1-expressing, locally advanced or metastatic non-small-cell lung cancer (KEYNOTE-042): a randomised, open-label, controlled, phase 3 trial. Lancet. 2019;393(10183):1819–1830. doi:10.1016/S0140-6736(18)32409-7
55. Hirsch FR, McElhinny A, Stanforth D, et al. PD-L1 immunohistochemistry assays for lung cancer: results from Phase 1 of the blueprint PD-L1 IHC assay comparison project. J Thorac Oncol. 2017;12(2):208–222. doi:10.1016/j.jtho.2016.11.2228
56. Jamal-Hanjani M, Wilson GA, McGranahan N, et al. Tracking the evolution of non–small-cell lung cancer. N Engl J Med. 2017;376(22):2109–2121. doi:10.1056/NEJMoa1616288
57. Schneider BJ, Naidoo J, Santomasso BD, et al. Management of immune-related adverse events in patients treated with immune checkpoint inhibitor therapy: ASCO guideline update. J Clin Oncol. 2021;39(36):4073–4126. doi:10.1200/JCO.21.01440
58. Kim SY, Kim TE, Park CK, et al. Comprehensive comparison of 22C3 and SP263 PD-L1 expression in non-small-Cell Lung Cancer using routine clinical and conditioned archives. Cancers. 2022;14(13):3138. doi:10.3390/cancers14133138
59. Vega D, Yee L, McShane L, et al. Aligning tumor mutational burden (TMB) quantification across diagnostic platforms: Phase II of the friends of cancer Res TMB harmonization project. Ann Oncol. 2021;32(12):1626–1636. doi:10.1016/j.annonc.2021.09.016
60. Zhang C, Zhang J, Tan J, Tian P, Li W. Cost-effectiveness of pembrolizumab for the treatment of non–small-cell lung cancer: a systematic review. Front Oncol. 2022;12:815587. doi:10.3389/fonc.2022.815587
61. Chang L, Chang M, Chang HM, Chang F. Microsatellite instability: a predictive biomarker for cancer immunotherapy. Appl Immunohistochem Mol Morphol. 2018;26(2):e15–e21. doi:10.1097/PAI.0000000000000575
62. Vanderwalde A, Spetzler D, Xiao N, Gatalica Z, Marshall J. Microsatellite instability status determined by next-generation sequencing and compared with PD-L1 and tumor mutational burden in 11,348 patients. Cancer Med. 2018;7(3):746–756. doi:10.1002/cam4.1372
63. Li Y, Wu X, Fang D, Luo Y. Informing immunotherapy with multi-omics driven machine learning. NPJ Digit Med. 2024;7(1):67. doi:10.1038/s41746-024-01043-6
64. Mei T, Wang T, Zhou Q, Wang T. Multi-omics and artificial intelligence predict clinical outcomes of immunotherapy in non-small cell lung cancer patients. Clin Exp Med. 2024;24(1):60. doi:10.1007/s10238-024-01324-0
65. Christopoulos P, Harel M, McGregor K, et al. Plasma proteome–based test for first-line treatment selection in metastatic non–small cell lung cancer. JCO Precis Oncol. 2024; 8:e2300555.
66. Yaghoubi Naei V, Monkman J, Sadeghirad H, et al. Spatial proteomic profiling of tumor and stromal compartments in non‐small‐Cell Lung Cancer identifies signatures associated with overall survival. Clin Transl Immunol. 2024;13(7):e1522. doi:10.1002/cti2.1522
67. Williams HL, Frei AL, Koessler T, et al. The current landscape of spatial biomarkers for prediction of response to immune checkpoint inhibition. NPJ Precis Oncol. 2024;8(1):178. doi:10.1038/s41698-024-00671-1
68. Routy B, Le Chatelier E, Derosa L, et al. Gut microbiome influences efficacy of PD-1–based immunotherapy against epithelial tumors. Science. 2018;359(6371):91–97. doi:10.1126/science.aan3706
69. Fridman WH, Zitvogel L, Sautès–Fridman C, Kroemer G. The immune contexture in cancer prognosis and treatment. Nat Rev Clin Oncol. 2017;14(12):717–734. doi:10.1038/nrclinonc.2017.101
70. Helmink BA, Reddy SM, Gao J, et al. B cells and tertiary lymphoid structures promote immunotherapy response. Nature. 2020;577(7791):549–555. doi:10.1038/s41586-019-1922-8
71. Petitprez F, Meylan M, de Reyniès A, Sautès-Fridman C, Fridman WH, de Reyniès A. The tumor microenvironment in the response to immune checkpoint blockade therapies. Front Immunol. 2020;11:784. doi:10.3389/fimmu.2020.00784
72. Bourbonne V, Geier M, Schick U, Lucia F. Multi-omics approaches for the prediction of clinical endpoints after immunotherapy in non-small cell lung cancer: a comprehensive review. Biomedicines. 2022;10(6):1237. doi:10.3390/biomedicines10061237
73. Yoon SJ, Lee CB, Chae SU, Jo SJ, Bae SK. The comprehensive “Omics” approach from metabolomics to advanced omics for development of immune checkpoint inhibitors: potential strategies for next generation of cancer immunotherapy. Int J Mol Sci. 2021;22(13):6932. doi:10.3390/ijms22136932
74. Wlosik J, Fattori S, Rochigneux P, Goncalves A, Olive D, Chretien A. Immune Biology of NSCLC Revealed by Single-Cell Technologies: Implications for the Development of Biomarkers in Patients Treated with Immunotherapy. Springer; 2023:29–41.
75. Hita-Millan J, Carracedo A, Fernandez-Rozadilla C. Liquid biopsy biomarkers for immunotherapy in non-small cell lung carcinoma: lessons learned and the road ahead. J Pers Med. 2021;11(10):971. doi:10.3390/jpm11100971
76. Gubb E, Matthiesen R. Introduction to Omics. Bioinformatics Methods in Clinical Research. Springer; 2009:1–23.
77. Valkova N, Kültz D. Constitutive and inducible stress proteins dominate the proteome of the murine inner medullary collecting duct-3 (mIMCD3) Cell line. Biochimica et Biophysica Acta. 2006;1764(6):1007–1020. doi:10.1016/j.bbapap.2006.03.007
78. Goossens N, Nakagawa S, Sun X, Hoshida Y. Cancer biomarker discovery and validation. Transl Cancer Res. 2015;4(3):256. doi:10.3978/j.issn.2218-676X.2015.06.04
79. Shi Y, Lei Y, Liu L, et al. Integration of comprehensive genomic profiling, tumor mutational burden, and PD‐L1 expression to identify novel biomarkers of immunotherapy in non‐small cell lung cancer. Cancer Med. 2021;10(7):2216–2231. doi:10.1002/cam4.3649
80. Cefalì M, Epistolio S, Ramelli G, et al. Correlation of KRAS G12C mutation and high PD-L1 expression with clinical outcome in NSCLC patients treated with anti-PD1 immunotherapy. J Clin Med. 2022;11(6):1627. doi:10.3390/jcm11061627
81. Zeng H, Tong F, Bin Y, et al. The predictive value of PAK7 mutation for immune checkpoint inhibitors therapy in non-small cell cancer. Front Immunol. 2022;13:834142. doi:10.3389/fimmu.2022.834142
82. Fang W, Ma Y, Yin JC, et al. Comprehensive genomic profiling identifies novel genetic predictors of response to anti–PD-(L) 1 therapies in non–small cell lung cancer. Clin Cancer Res. 2019;25(16):5015–5026. doi:10.1158/1078-0432.CCR-19-0585
83. Tucker T, Marra M, Friedman JM. Massively parallel sequencing: the next big thing in genetic medicine. Am J Hum Genet. 2009;85(2):142–154. doi:10.1016/j.ajhg.2009.06.022
84. Podnar J, Deiderick H, Hunicke‐Smith S. Next‐generation sequencing fragment library construction. Curr Protoc Mol Biol. 2014;107(1):7.17.1–7.17.16. doi:10.1002/0471142727.mb0717s107
85. Logsdon GA, Vollger MR, Eichler EE. Long-read human genome sequencing and its applications. Nat Rev Genet. 2020;21(10):597–614. doi:10.1038/s41576-020-0236-x
86. Chapman PB, Hauschild A, Robert C, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364(26):2507–2516. doi:10.1056/NEJMoa1103782
87. Assoun S, Theou-Anton N, Nguenang M, et al. Association of TP53 mutations with response and longer survival under immune checkpoint inhibitors in advanced non-small-cell lung cancer. Lung Cancer. 2019;132:65–71. doi:10.1016/j.lungcan.2019.04.005
88. Murciano-Goroff YR, Pak T, Mondaca S, et al. Immune biomarkers and response to checkpoint inhibition of BRAF V600 and BRAF non-V600 altered lung cancers. Br J Cancer. 2022;126(6):889–898. doi:10.1038/s41416-021-01679-1
89. Guisier F, Dubos-Arvis C, Viñas F, et al. Efficacy and safety of anti–PD-1 immunotherapy in patients with advanced NSCLC with BRAF, HER2, or MET mutations or RET translocation: GFPC 01-2018. J Thorac Oncol. 2020;15(4):628–636. doi:10.1016/j.jtho.2019.12.129
90. Gao G, Liao W, Ma Q, Zhang B, Chen Y, Wang Y. KRAS G12D mutation predicts lower TMB and drives immune suppression in lung adenocarcinoma. Lung Cancer. 2020;149:41–45. doi:10.1016/j.lungcan.2020.09.004
91. Ricciuti B, Alessi J, Elkrief A, et al. Dissecting the clinicopathologic, genomic, and immunophenotypic correlates of KRASG12D-mutated non-small-cell lung cancer. Ann Oncol. 2022;33(10):1029–1040. doi:10.1016/j.annonc.2022.07.005
92. Lee CK, Man J, Lord S, et al. Checkpoint inhibitors in metastatic EGFR-mutated non–small cell lung cancer—A meta-analysis. J Thorac Oncol. 2017;12(2):403–407. doi:10.1016/j.jtho.2016.10.007
93. Lee CK, Man J, Lord S, et al. Clinical and molecular characteristics associated with survival among patients treated with checkpoint inhibitors for advanced non–small cell lung carcinoma: a systematic review and meta-analysis. JAMA Oncol. 2018;4(2):210–216. doi:10.1001/jamaoncol.2017.4427
94. Xin Z, You L, Li J, et al. Immunogenetic polymorphisms predict therapeutic efficacy and survival outcomes in tumor patients receiving PD-1/PD-L1 blockade. Int Immunopharmacol. 2023;121:110469. doi:10.1016/j.intimp.2023.110469
95. Alessi JV, Ricciuti B, Spurr LF, et al. SMARCA4 and other SWItch/sucrose nonfermentable family genomic alterations in NSCLC: clinicopathologic characteristics and outcomes to immune checkpoint inhibition. J Thorac Oncol. 2021;16(7):1176–1187. doi:10.1016/j.jtho.2021.03.024
96. Negrao MV, Skoulidis F, Montesion M, et al. Oncogene-specific differences in tumor mutational burden, PD-L1 expression, and outcomes from immunotherapy in non-small cell lung cancer. J Immunother Cancer. 2021;9(8):e002891. doi:10.1136/jitc-2021-002891
97. Sun H, Liu S, Zhou J, et al. Specific TP53 subtype as biomarker for immune checkpoint inhibitors in lung adenocarcinoma. EBioMedicine. 2020;60:102990. doi:10.1016/j.ebiom.2020.102990
98. Dong Z-Y, Zhong W-Z, Zhang X-C, et al. Potential predictive value of TP53 and KRAS mutation status for response to PD-1 blockade immunotherapy in lung adenocarcinoma. Clin Cancer Res. 2017;23(12):3012–3024. doi:10.1158/1078-0432.CCR-16-2554
99. Cucchiara F, Crucitta S, Petrini I, et al. Gene-network analysis predicts clinical response to immunotherapy in patients affected by NSCLC. Lung Cancer. 2023;183:107308. doi:10.1016/j.lungcan.2023.107308
100. Zhang K, Hong X, Song Z, et al. Identification of deleterious NOTCH mutation as novel predictor to efficacious immunotherapy in NSCLC. Clin Cancer Res. 2020;26(14):3649–3661. doi:10.1158/1078-0432.CCR-19-3976
101. Long J, Wang D, Yang X, et al. Identification of NOTCH4 mutation as a response biomarker for immune checkpoint inhibitor therapy. BMC Med. 2021;19:1–14. doi:10.1186/s12916-021-02031-3
102. Zhang W, Lin Z, Shi F, et al. HSPG2 mutation association with immune checkpoint inhibitor outcome in melanoma and non-small cell lung cancer. Cancers. 2022;14(14):3495. doi:10.3390/cancers14143495
103. Sun L, Li M, Deng L, et al. MGA mutation as a novel biomarker for immune checkpoint therapies in non-squamous non-small cell lung cancer. Front Pharmacol. 2021;12:625593. doi:10.3389/fphar.2021.625593
104. Liu F, Zhang X, Lu M, et al. The association of genomic alterations with PD‐L1 expression in Chinese patients with EGFR/ALK wild‐type lung adenocarcinoma and potential predictive value of Hippo pathway mutations to immunotherapy. Cancer Med. 2024;13(3):e7038. doi:10.1002/cam4.7038
105. Li Z, Wang X, Yang Y, et al. Identification and validation of RELN mutation as a response indicator for immune checkpoint inhibitor therapy in melanoma and non-small cell lung cancer. Cells. 2022;11(23):3841. doi:10.3390/cells11233841
106. Bai H, Duan J, Li C, et al. EPHA mutation as a predictor of immunotherapeutic efficacy in lung adenocarcinoma. J Immunother Cancer. 2020;8(2):e001315. doi:10.1136/jitc-2020-001315
107. Zhang N, Shen J, Gou L, et al. UBE3A deletion enhances the efficiency of immunotherapy in non-small-cell lung cancer. Bioengineered. 2022;13(5):11577–11592. doi:10.1080/21655979.2022.2069328
108. Skoulidis F, Goldberg ME, Greenawalt DM, et al. STK11/LKB1 mutations and PD-1 inhibitor resistance in KRAS-mutant lung adenocarcinoma. Cancer Discov. 2018;8(7):822–835. doi:10.1158/2159-8290.CD-18-0099
109. Chen X, Su C, Ren S, Zhou C, Jiang T. Pan-cancer analysis of KEAP1 mutations as biomarkers for immunotherapy outcomes. Ann Transl Med. 2020;8(4):141.
110. Biton J, Mansuet-Lupo A, Pécuchet N, et al. TP53, STK11, and EGFR mutations predict tumor immune profile and the response to anti–PD-1 in lung adenocarcinoma. Clin Cancer Res. 2018;24(22):5710–5723. doi:10.1158/1078-0432.CCR-18-0163
111. Nakajima EC, Ren Y, Vallejo JJ, et al. Outcomes of first-line immune checkpoint inhibitors with or without chemotherapy according to KRAS mutational status and PD-L1 expression in patients with advanced NSCLC: FDA pooled analysis. Am Soc Clin Oncol. 2022;2022;1.
112. Huang W, Lin A, Luo P, et al. EPHA5 mutation predicts the durable clinical benefit of immune checkpoint inhibitors in patients with lung adenocarcinoma. Cancer Gene Ther. 2021;28(7):864–874. doi:10.1038/s41417-020-0207-6
113. Zehir A, Benayed R, Shah RH, et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat Med. 2017;23(6):703–713. doi:10.1038/nm.4333
114. Fehrenbacher L, Spira A, Ballinger M, et al. Atezolizumab versus docetaxel for patients with previously treated non-small-Cell Lung Cancer (POPLAR): a multicentre, open-label, phase 2 randomised controlled trial. Lancet. 2016;387(10030):1837–1846. doi:10.1016/S0140-6736(16)00587-0
115. Fermin D, Allen BB, Blackwell TW, et al. Novel gene and gene model detection using a whole genome open reading frame analysis in proteomics. Genome Biol. 2006;7:1–13. doi:10.1186/gb-2006-7-4-r35
116. Sabbioni G, Sepai O, Norppa H, et al. Comparison of biomarkers in workers exposed to 24, 6-trinitrotoluene. Biomarkers. 2007;12(1):21–37. doi:10.1080/13547500600807012
117. Görg A, Obermaier C, Boguth G, et al. The current state of two‐dimensional electrophoresis with immobilized pH gradients. ELECTROPHORESIS. 2000;21(6):1037–1053. doi:10.1002/(SICI)1522-2683(20000401)21:6<1037::AID-ELPS1037>3.0.CO;2-V
118. Pelzing M, Neusüß C. Separation techniques hyphenated to electrospray‐tandem mass spectrometry in proteomics: capillary electrophoresis versus nanoliquid chromatography. Electrophoresis. 2005;26(14):2717–2728. doi:10.1002/elps.200410424
119. Eltahir M, Isaksson J, Mattsson JSM, et al. Plasma proteomic analysis in non-small cell lung cancer patients treated with PD-1/PD-L1 blockade. Cancers. 2021;13(13):3116. doi:10.3390/cancers13133116
120. Bar J, Leibowitz R, Reinmuth N, et al. Biological insights from plasma proteomics of non-small cell lung cancer patients treated with immunotherapy. Front Immunol. 2024;15:1364473. doi:10.3389/fimmu.2024.1364473
121. Muller M, Hummelink K, Hurkmans DP, et al. A serum protein classifier identifying patients with advanced non–small cell lung cancer who derive clinical benefit from treatment with immune checkpoint inhibitors. Clin Cancer Res. 2020;26(19):5188–5197. doi:10.1158/1078-0432.CCR-20-0538
122. Harel M, Lahav C, Jacob E, et al. Longitudinal plasma proteomic profiling of patients with non-small Cell Lung Cancer undergoing immune checkpoint blockade. J Immunother Cancer. 2022;10(6):e004582. doi:10.1136/jitc-2022-004582
123. Moutafi MK, Molero M, Morilla SM, et al. Spatially resolved proteomic profiling identifies tumor Cell CD44 as a biomarker associated with sensitivity to PD-1 axis blockade in advanced non-small-Cell Lung Cancer. J Immunother Cancer. 2022;10(8):e004757. doi:10.1136/jitc-2022-004757
124. Dai L, Tan Q, Li L, et al. High-throughput antigen microarray identifies longitudinal prognostic autoantibody for chemoimmunotherapy in advanced non-small cell lung cancer. Mol Cell Proteomics. 2024;23(5):100749. doi:10.1016/j.mcpro.2024.100749
125. Chao Y, Jiang W, Wang X, et al. Discovery of efficacy biomarkers for non-small cell lung cancer with first-line anti-PD-1 immunotherapy by data-independent acquisition mass spectrometry. Clin Exp Immunol. 2022;208(1):60–71. doi:10.1093/cei/uxac021
126. Berghmans E, Jacobs J, Deben C. Mass spectrometry imaging reveals neutrophil defensins as additional biomarkers for anti-PD-(L) 1 immunotherapy response in NSCLC patients. Cancers. 2020;12:863. doi:10.3390/cancers12040863
127. Hortin GL. The MALDI-TOF mass spectrometric view of the plasma proteome and peptidome. Clin Chem. 2006;52(7):1223–1237. doi:10.1373/clinchem.2006.069252
128. Rozanova S, Barkovits K, Nikolov M, Schmidt C, Urlaub H, Marcus K. Quantitative Mass Spectrometry-Based Proteomics: an Overview. In: Marcus K, Eisenacher M, Sitek B, editors. Quantitative Methods in Proteomics. Springer US; 2021:85–116.
129. El-Aneed A, Cohen A, Banoub J. Mass spectrometry, review of the basics: electrospray, MALDI, and commonly used mass analyzers. Appl Spectrosc Rev. 2009;44(3):210–230. doi:10.1080/05704920902717872
130. Niessen WM, Falck D. Introduction to mass spectrometry, a tutorial. Analyzing biomolecular interactions by mass spectrometry. 2015;1–54.
131. Pandey A, Mann M. Proteomics to study genes and genomes. Nature. 2000;405(6788):837–846. doi:10.1038/35015709
132. Ortea I, O’Connor G, Maquet A. Review on proteomics for food authentication. J Proteomics. 2016;147:212–225. doi:10.1016/j.jprot.2016.06.033
133. Chait BT. Mass spectrometry: bottom-up or top-down? Science. 2006;314(5796):65–66. doi:10.1126/science.1133987
134. Templin MF, Stoll D, Schrenk M, Traub PC, Vöhringer CF, Joos TO. Protein microarray technology. Trends Biotechnol. 2002;20(4):160–166. doi:10.1016/S0167-7799(01)01910-2
135. Chen G, Yang L, Liu G, et al. Research progress in protein microarrays: focussing on cancer res. PROTEOMICS–Clin Appl. 2023;17(1):2200036. doi:10.1002/prca.202200036
136. Hakomori S. Glycosylation defining cancer malignancy: new wine in an old bottle. Proc Natl Acad Sci U S A. 2002;99(16):10231–10233. doi:10.1073/pnas.172380699
137. Huang W-L, Li Y-G, Lv Y-C, Guan X-H, Ji H-F, Chi B-R. Use of lectin microarray to differentiate gastric cancer from gastric ulcer. World J Gastroenterol. 2014;20(18):5474. doi:10.3748/wjg.v20.i18.5474
138. Syed P, Gidwani K, Kekki H, Leivo J, Pettersson K, Lamminmäki U. Role of lectin microarrays in cancer diagnosis. Proteomics. 2016;16(8):1257–1265. doi:10.1002/pmic.201500404
139. Paweletz CP, Charboneau L, Bichsel VE, et al. Reverse phase protein microarrays which capture disease progression show activation of pro-survival pathways at the cancer invasion front. Oncogene. 2001;20(16):1981–1989. doi:10.1038/sj.onc.1204265
140. Ramaswamy A, Lin E, Chen I, et al. Application of protein lysate microarrays to molecular marker verification and quantification. Proteome Sci. 2005;3:1–16. doi:10.1186/1477-5956-3-9
141. VanMeter A, Signore M, Pierobon M, Espina V, Liotta LA, Petricoin Iii EF. Reverse-phase protein microarrays: application to biomarker discovery and translational medicine. Expert Rev Mol Diagn. 2007;7(5):625–633. doi:10.1586/14737159.7.5.625
142. Lacombe J, Azria D, Mange A, Solassol J. Proteomic approaches to identify biomarkers predictive of radiotherapy outcomes. Expert Rev Proteomics. 2013;10(1):33–42. doi:10.1586/epr.12.68
143. Turtoi A, Musmeci D, Wang Y, et al. Identification of novel accessible proteins bearing diagnostic and therapeutic potential in human pancreatic ductal adenocarcinoma. J Proteome Res. 2011;10(9):4302–4313. doi:10.1021/pr200527z
144. Abou Gabal AM. Validation of assessment of Bmi-1 on protein and molecular levels in oral dysplasia and squamous cell carcinoma: a diagnostic study. 2017.
145. Choi JY, Jung SW, Kim HY, et al. Diagnostic value of AFP-L3 and PIVKA-II in hepatocellular carcinoma according to total-AFP. World J Gastroenterol. 2013;19(3):339. doi:10.3748/wjg.v19.i3.339
146. Oikonomopoulou K, Li L, Zheng Y, et al. Prediction of ovarian cancer prognosis and response to chemotherapy by a serum-based multiparametric biomarker panel. Br J Cancer. 2008;99(7):1103–1113. doi:10.1038/sj.bjc.6604630
147. Nunes JJ, Pandey SK, Yadav A, Goel S, Ateeq B. Targeting NF-kappa B signaling by artesunate restores sensitivity of castrate-resistant prostate cancer cells to antiandrogens. Neoplasia. 2017;19(4):333–345. doi:10.1016/j.neo.2017.02.002
148. Xu C, Zeng X-H, Wang L, et al. sFRP-4, a potential novel serum marker for chronic hepatitis B-related hepatocellular carcinoma. Hepatobiliary Pancreat Dis Int. 2015;14(2):164–170. doi:10.1016/S1499-3872(15)60352-6
149. Edfeldt K, Daskalakis K, Bäcklin C, et al. DcR3, TFF3, and midkine are novel serum biomarkers in small intestinal neuroendocrine tumors. Neuroendocrinology. 2017;105(2):170–181. doi:10.1159/000452891
150. Lee SH, Lee JS, Lee EJ, et al. Serum reactive oxygen species modulator 1 (Romo1) as a potential diagnostic biomarker for non-small cell lung cancer. Lung Cancer. 2014;85(2):175–181. doi:10.1016/j.lungcan.2014.05.023
151. Goss G, Lee KH, Felip E, et al. Evaluation of VeriStrat, a serum proteomic test, in the randomized, open-label, Phase 3 LUX-Lung 8 trial of Afatinib versus erlotinib for the second-line treatment of advanced squamous cell carcinoma of the lung. Ann Oncol. 2016;27:vi428. doi:10.1093/annonc/mdw383.38
152. Rich P, Mitchell RB, Schaefer E, et al. Real-world performance of blood-based proteomic profiling in first-line immunotherapy treatment in advanced stage non-small Cell Lung Cancer. J Immunother Cancer. 2021;9(10):e002989. doi:10.1136/jitc-2021-002989
153. Prognostic and predictive role of the VeriStrat plasma test in patients with advanced non–small-Cell Lung Cancer treated with erlotinib or placebo in the NCIC Clinical Trials Group BR, Carbone DP, Ding K, Roder H, et al. 21 trial. J Thorac Oncol. 2012;7(11):1653–1660. doi:10.1097/JTO.0b013e31826c1155.
154. Leal TA, Argento AC, Bhadra K, et al. Prognostic performance of proteomic testing in advanced non-small Cell Lung Cancer: a systematic literature review and meta-analysis. Curr Med Res Opin. 2020;36(9):1497–1505. doi:10.1080/03007995.2020.1790346
155. Lee SM, Upadhyay S, Lewanski C, et al. The clinical role of VeriStrat testing in patients with advanced non–small Cell Lung Cancer considered unfit for first-line platinum-based chemotherapy. Eur J Cancer. 2019;120:86–96. doi:10.1016/j.ejca.2019.07.025
156. Molina-Pinelo S, Pastor MD, Paz-Ares L. VeriStrat: A Prognostic and/or Predictive Biomarker for Advanced Lung Cancer Patients? Taylor & Francis; 2014:1–4.
157. Chae YK, Kim WB, Davis AA, et al. Mass spectrometry-based serum proteomic signature as a potential biomarker for survival in patients with non-small cell lung cancer receiving immunotherapy. Transl Lung Cancer Res. 2020;9(4):1015. doi:10.21037/tlcr-20-148
158. Grossi F, Rijavec E, Biello F, et al. P3. 02c-074 Evaluation of a Pretreatment Serum Tests for Nivolumab Benefit in Patients with Non-Small Cell Lung Cancer: topic: IT Biomarkers. J Thorac Oncol. 2017;12(1):S1322. doi:10.1016/j.jtho.2016.11.1870
159. Zizzari IG, Di Filippo A, Scirocchi F, et al. Soluble immune checkpoints, gut metabolites and performance status as parameters of response to nivolumab treatment in NSCLC patients. J Pers Med. 2020;10(4):208. doi:10.3390/jpm10040208
160. Valenti F, Falcone I, Ungania S, et al. Precision medicine and melanoma: multi-omics approaches to monitoring the immunotherapy response. Int J Mol Sci. 2021;22(8):3837. doi:10.3390/ijms22083837
161. Patti GJ, Yanes O, Siuzdak G. Metabolomics: the apogee of the omics trilogy. Nat Rev Mol Cell Biol. 2012;13(4):263–269. doi:10.1038/nrm3314
162. Loo JM, Scherl A, Nguyen A, et al. Extracellular metabolic energetics can promote cancer progression. Cell. 2015;160(3):393–406. doi:10.1016/j.cell.2014.12.018
163. Gika HG, Theodoridis GA, Wilson ID. Metabolic profiling: status, challenges, and perspective. Metabolic profiling: methods and protocols. Meth Mol Biol. 2018;1738:3–13. doi:10.1007/978-1-4939-7643-0_1
164. Markley JL, Brüschweiler R, Edison AS, et al. The future of NMR-based metabolomics. Curr Opin Biotechnol. 2017;43:34–40. doi:10.1016/j.copbio.2016.08.001
165. Blondel C, Khelalfa F, Reynaud S, Fauvelle F, Raveton M. Effect of organochlorine pesticides exposure on the maize root metabolome assessed using high-resolution magic-angle spinning 1H NMR spectroscopy. Environ Pollut. 2016;214:539–548. doi:10.1016/j.envpol.2016.04.057
166. Jeong S, Eskandari R, Park SM, et al. Real-time quantitative analysis of metabolic flux in live cells using a hyperpolarized micromagnetic resonance spectrometer. Sci Adv. 2017;3(6):e1700341. doi:10.1126/sciadv.1700341
167. Zhou B, Xiao JF, Tuli L, Ressom HW. LC-MS-based metabolomics. Mol BioSys. 2012;8(2):470–481. doi:10.1039/C1MB05350G
168. Bowen BP, Northen TR. Dealing with the unknown: metabolomics and metabolite atlases. J Am Soc Mass Spectrom. 2010;21:1471–1476. doi:10.1016/j.jasms.2010.04.003
169. Lee SY, Park N-H, Jeong E-K, et al. Comparison of GC/MS and LC/MS methods for the analysis of propofol and its Metabolites in urine. J Chromatograph B. 2012;900:1–10. doi:10.1016/j.jchromb.2012.05.011
170. Zeki ÖC, Eylem CC, Reçber T, Kır S, Nemutlu E. Integration of GC–MS and LC–MS for untargeted metabolomics profiling. J Pharm Biomed Anal. 2020;190:113509. doi:10.1016/j.jpba.2020.113509
171. Rahman AMA. Clinical Metabolomics Applications in Genetic Diseases. Springer; 2023.
172. Dahabiyeh LA, Hudaib F, Hourani W, et al. Mass spectrometry-based metabolomics approach and in vitro assays revealed promising role of 2, 3-dihydroquinazolin-4 (1H)-one derivatives against colorectal cancer Cell lines. Eur J Pharm Sci. 2023;182:106378. doi:10.1016/j.ejps.2023.106378
173. Dahabiyeh LA, Nimer RM, Rashed M, Wells JD, Fiehn O. Serum-based lipid panels for diagnosis of idiopathic Parkinson’s disease. Metabolites. 2023;13(9):990. doi:10.3390/metabo13090990
174. Chen Y, Li E-M, Xu L-Y. Guide to metabolomics analysis: a bioinformatics workflow. Metabolites. 2022;12(4):357. doi:10.3390/metabo12040357
175. Misra BB. New software tools, databases, and resources in metabolomics: updates from 2020. Metabolomics. 2021;17(5):49. doi:10.1007/s11306-021-01796-1
176. Marco-Ramell A, Palau-Rodriguez M, Alay A, et al. Evaluation and comparison of bioinformatic tools for the enrichment analysis of metabolomics data. BMC Bioinformatics. 2018;19:1–11. doi:10.1186/s12859-017-2006-0
177. Huang Z, Lin L, Gao Y, et al. Bladder cancer determination via two urinary metabolites: a biomarker pattern approach. Mol Cell Proteomics. 2011;10(10):
178. Yu C, Guoxiang X, Tianlu C, et al. Distinct urinary metabolic profile of human colorectal cancer. 2012.
179. Chen T, Xie G, Wang X, et al. Serum and urine metabolite profiling reveals potential biomarkers of human hepatocellular carcinoma. Mol Cell Proteomics. 2011;10(7):1.
180. Ladep NG, Dona AC, Lewis MR, et al. Discovery and validation of urinary metabotypes for the diagnosis of hepatocellular carcinoma in West Africans. Hepatology. 2014;60(4):1291–1301. doi:10.1002/hep.27264
181. Ikeda A, Nishiumi S, Shinohara M, et al. Serum metabolomics as a novel diagnostic approach for gastrointestinal cancer. Biomed Chromatogr. 2012;26(5):548–558. doi:10.1002/bmc.1671
182. Kobayashi T, Nishiumi S, Ikeda A, et al. A novel serum metabolomics-based diagnostic approach to pancreatic cancer. Cancer Epidemiol Biomarkers Prev. 2013;22(4):571–579. doi:10.1158/1055-9965.EPI-12-1033
183. Odunsi K, Wollman RM, Ambrosone CB, et al. Detection of epithelial ovarian cancer using 1H‐NMR‐based metabonomics. Int J Cancer. 2005;113(5):782–788. doi:10.1002/ijc.20651
184. Osl M, Dreiseitl S, Pfeifer B, et al. A new rule-based algorithm for identifying metabolic markers in prostate cancer using tandem mass spectrometry. Bioinformatics. 2008;24(24):2908–2914. doi:10.1093/bioinformatics/btn506
185. Wang J, Yu L-F, Shen P, Wang S-F. Analysis of serum metabonome of patients with breast cancer by gas chromatography-mass spectrometry. Zhejiang Da Xue Xue Bao Yi Xue Ban= J Zhejiang Univ Med Sci. 2009;38(5):478–484.
186. Beger RD, Schnackenberg LK, Holland RD, Li D, Dragan Y. Metabonomic models of human pancreatic cancer using 1D proton NMR spectra of lipids in plasma. Metabolomics. 2006;2:125–134. doi:10.1007/s11306-006-0026-2
187. Yan S-K, Wei B-J, Lin Z-Y, Yang Y, Zhou Z-T, Zhang W-D. A metabonomic approach to the diagnosis of oral squamous Cell carcinoma, oral lichen planus and oral leukoplakia. Oral Oncol. 2008;44(5):477–483. doi:10.1016/j.oraloncology.2007.06.007
188. Ganti S, Weiss RH. Urine metabolomics for kidney cancer detection and biomarker discovery. Elsevier. 2011:551–557.
189. Nam H, Chung BC, Kim Y, Lee K, Lee D. Combining tissue transcriptomics and urine metabolomics for breast cancer biomarker identification. Bioinformatics. 2009;25(23):3151–3157. doi:10.1093/bioinformatics/btp558
190. Poli D, Carbognani P, Corradi M, et al. Exhaled volatile organic compounds in patients with non-small Cell Lung Cancer: cross sectional and nested short-term follow-up study. Respir Res. 2005;6:1–10. doi:10.1186/1465-9921-6-71
191. Phillips M, Cataneo RN, Ditkoff BA, et al. Prediction of breast cancer using volatile biomarkers in the breath. Breast Cancer Res Treatment. 2006;99:19–21. doi:10.1007/s10549-006-9176-1
192. Ghini V, Laera L, Fantechi B, et al. Metabolomics to assess response to immune checkpoint inhibitors in patients with non-small-cell lung cancer. Cancers. 2020;12(12):3574. doi:10.3390/cancers12123574
193. Hatae R, Chamoto K, Kim YH, et al. Combination of host immune metabolic biomarkers for the PD-1 blockade cancer immunotherapy. JCI Insight. 2020;5(2). doi:10.1172/jci.insight.133501.
194. Nie X, Xia L, Gao F, et al. Serum metabolite biomarkers predictive of response to PD-1 blockade therapy in non-small Cell Lung Cancer. Front Mol Biosci. 2021;8:472. doi:10.3389/fmolb.2021.678753
195. Karayama M, Masuda J, Mori K, et al. Comprehensive assessment of multiple tryptophan metabolites as potential biomarkers for immune checkpoint inhibitors in patients with non-small cell lung cancer. Clin Transl Oncol. 2021;23:418–423. doi:10.1007/s12094-020-02421-8
196. Kocher F, Amann A, Zimmer K, et al. High indoleamine-2, 3-dioxygenase 1 (IDO) activity is linked to primary resistance to immunotherapy in non-small cell lung cancer (NSCLC). Transl Lung Cancer Res. 2021;10(1):304. doi:10.21037/tlcr-20-380
197. Mei L, Zhang Z, Li X, Yang Y, Qi R. Metabolomics profiling in prediction of chemo-immunotherapy efficiency in advanced non-small cell lung cancer. Front Oncol. 2023;12:1025046. doi:10.3389/fonc.2022.1025046
198. Botticelli A, Vernocchi P, Marini F, et al. Gut metabolomics profiling of non-small cell lung cancer (NSCLC) patients under immunotherapy treatment. J Transl Med. 2020;18:1–10. doi:10.1186/s12967-020-02231-0
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.