Back to Journals » International Journal of General Medicine » Volume 15
Integrated Management Strategies for Epidermolysis Bullosa: Current Insights
Authors Sait H, Srivastava S, Saxena D
Received 10 March 2022
Accepted for publication 5 May 2022
Published 24 May 2022 Volume 2022:15 Pages 5133—5144
DOI https://doi.org/10.2147/IJGM.S342740
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Scott Fraser
Haseena Sait, Somya Srivastava, Deepti Saxena
Department of Medical Genetics, Sanjay Gandhi Postgraduate Institute of Medical Sciences, Lucknow, Uttar Pradesh, India
Correspondence: Deepti Saxena, Department of Medical Genetics, Sanjay Gandhi Postgraduate Institute of Medical Sciences, Lucknow, Uttar Pradesh, India, Email [email protected]
Abstract: Epidermolysis bullosa (EB) is a group of rare genodermatoses that is characterized by skin fragility resulting from minor trauma. There are four major subtypes, namely, EB simplex, junctional EB, dystrophic EB and Kindler EB, depending upon the localization of defective protein and resulting plane of blister formation. The phenotype is heterogeneous in terms of severity and majority of them present at birth or neonatal period. Currently, the treatment is mainly supportive and requires multidisciplinary care. The complex molecular pathology creates difficulty in discovering a unified curative treatment approach. But with arduous efforts, significant progress has been made in the development of treatment strategies in the last decade. The management strategies range from targeting the underlying causative factor to symptom-relieving approaches, and include gene, mRNA, protein, cell and combination therapies. In this review, we enumerate the promising approaches that are currently under various stages of investigation to provide effective treatment for patients with EB.
Keywords: epidermolysis bullosa, blistering skin disorder, gene replacement, gene editing, antisense oligonucleotides, siRNA therapeutics, spliceosome-mediated RNA trans-splicing, revertant mosaicism, readthrough therapies, squamous cell carcinoma
Corrigendum for this paper has been published
Introduction
Epidermolysis bullosa (EB) is the prototype of genetic disorders of skin fragility where blister formation, skin peeling, erosions and ulceration develop on various body parts such as nail, hair, teeth, oral, ocular, esophageal, tracheal, and genito-urinary system. Majority of these disorders usually present at birth or neonatal period. The severe ones that present at birth can succumb to illness as early as 6 months of age due to fever, sepsis, and failure to thrive. Some types which present in childhood may resolve spontaneously with age and others may present in adulthood with mild symptoms. The symptoms may be localized to hand and foot or generalized to the whole body with significant involvement of other epithelial lined structures leading to multisystem involvement. Significant morbidity may be seen due to recurrent infections, stricture formation in esophageal tract, contractures, acral mutilation, microstomia, anemia, osteoporosis and cutaneous malignancies like squamous cell carcinoma and basal cell carcinoma.
These disorders have profound clinical and genetic heterogeneity. Genetics of EB is complex with the same gene leading to both autosomal dominant (AD) and autosomal recessive (AR) inheritance with different phenotypes. Different genes may lead to same phenotype in different subclasses as well. Many genes are still being discovered and the complete molecular pathogenesis is yet to be deciphered. Figure 1 depicts different proteins and their localization that are involved in pathogenesis of EB.
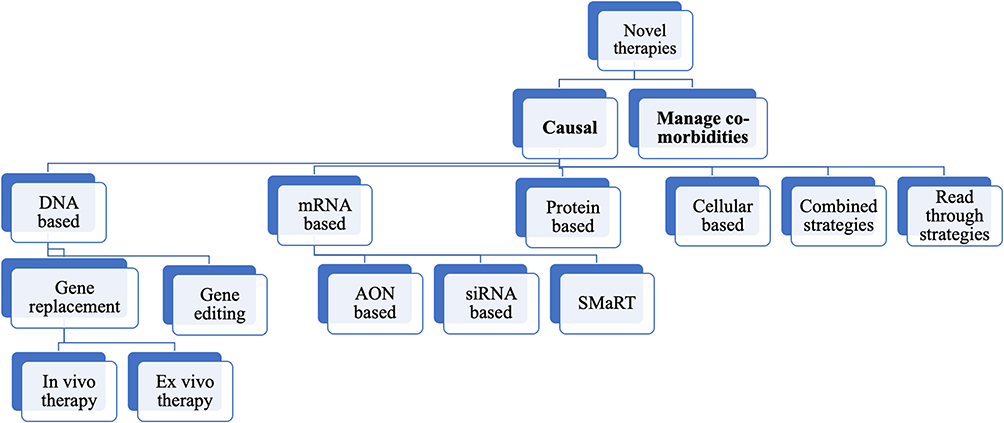 |
Figure 1 Various novel therapeutic strategies that are being explored in management of epidermolysis bullosa. |
Classification: There are 4 major subtypes depending upon the layer of the skin where the defective protein is localized and leads to blister formation in that layer:1
- EB simplex – fragility involving epidermis of skin
- EB junctional – fragility involving lamina lucida of the basement membrane of skin
- EB dystrophic – fragility involving lamina densa of the basement membrane of skin
- Kindler EB – fragility involving skin epidermis or basement membrane or underlying connective tissue
These 4 types are further classified into multiple types based on mode of inheritance, phenotypic severity and isolated or syndromic. According to the latest classification of an international consensus group on inherited EB disorders, there are 14 subtypes of EB simplex, 9 subtypes of EB junctional and 11 subtypes of EB dystrophic.1 Incidence of epidermolysis bullosa varies with type, with EB simplex being the most common and Kindler EB being the rarest with only 250 reported individuals to date.2
Overview of Different Types of EB
- EB simplex (EBS): This type is characterized by non-scarring blisters and erosions triggered by minor mechanical trauma. Seven genes have been implicated and 75% involve keratin 5 and 14, which are produced by basal keratinocytes. These 2 keratins form a heterodimer to form a network and provide strength. The common subtypes are enumerated in Table 1.
- EB junctional (JEB): This type is characterized by mild or severe blisters with little or no trauma and significant oral and mucosal involvement. Genes maintaining lamina lucida are involved.3 Table 2 shows a few common subtypes of junctional EB.
- EB dystrophic (DEB): Due to blistering in deep level of skin, fibrosis, milia and scarring is common. COL7A1 mutation leads to defective type VII collagen, which is the anchoring fibril in the basement membrane.3 Dystrophic EB subtypes are shown in Table 3.
- Kindler EB: Rare form of EB where blisters occur in multiple layers and hence it may mimic any of the 3 other types. Characteristically, they present with blistering, atrophy, poikiloderma, telangiectasia, gum, ocular, esophageal and genito-urinary involvement. Kindler EB has autosomal recessive inheritance and is caused by variation in FERMT1 gene, which codes for kindlin-1. This protein is localized next to basal keratinocytes and has a role in adhesion. Defective kindlin leads to disorganization of keratinocytes.4
 |
Table 1 Most Common Subtypes of EB Simplex |
 |
Table 2 Common Subtypes of Junctional EB |
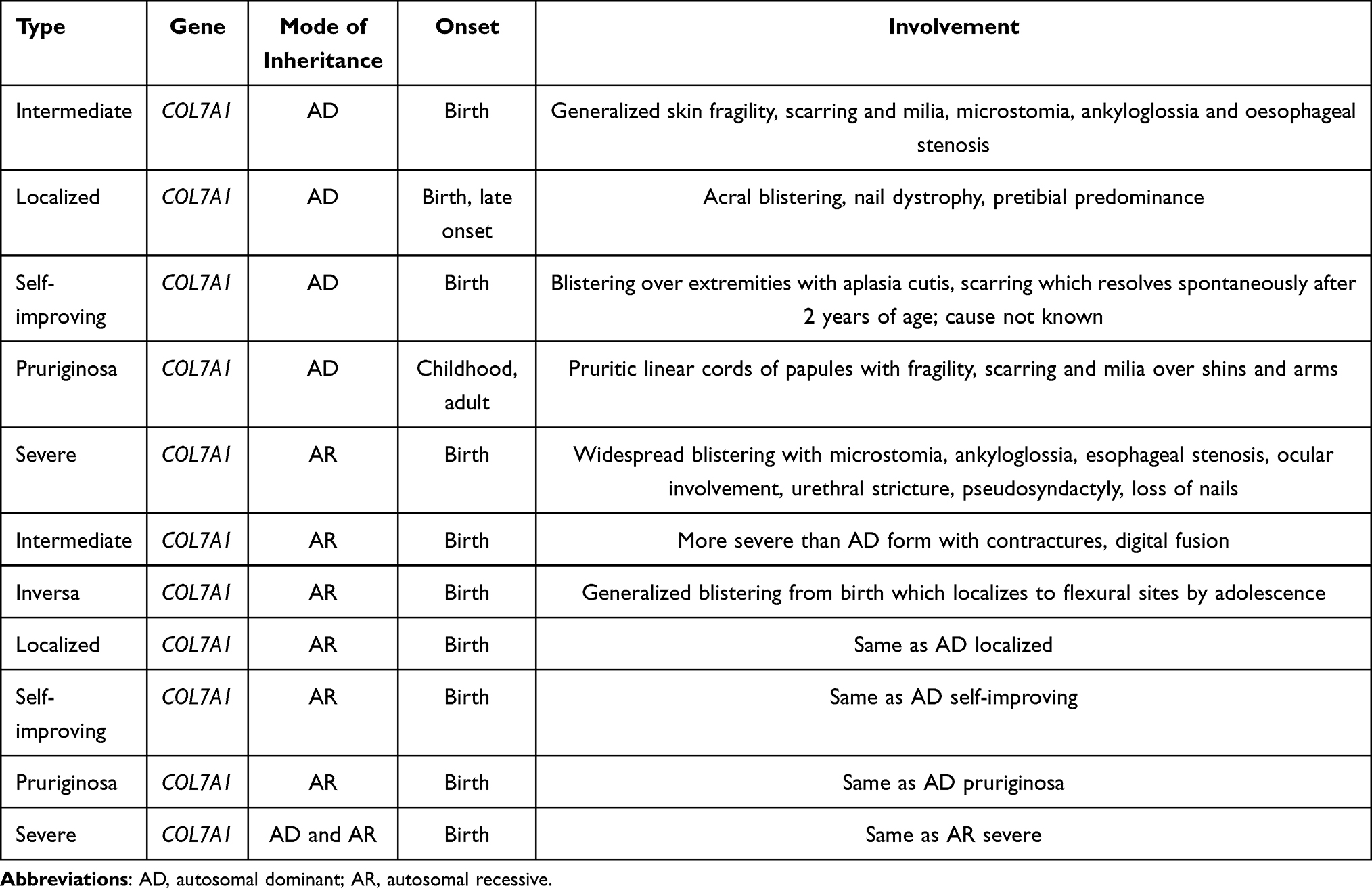 |
Table 3 Most Common Subtypes of Dystrophic EB |
Laboratory Diagnosis
Due to significant clinical overlap between the various subtypes, diagnosis based on clinical features is not entirely possible. Routine histological examination does not have good resolution to identify the planes of cleavage and is therefore not used. Other methods of diagnosis include immunofluorescence mapping (IFM) and transmission electron microscopy (TEM).
Immunofluorescence mapping is a rapid technique where skin biopsy taken from perilesional area is subjected to fluorescent labelled antibodies against the protein. This helps to visualize the layer of skin where cleavage occurs.3 Transmission electron microscopy identifies the ultrastructure aberration in the layer of the skin and delineates additional features such as keratin filaments, desmosomes etc., which may help in subtyping.3 However, both these methods require expertise on specimen transport, processing, and result interpretation. They are labor- and cost-intensive and are therefore not available at all laboratories. Moreover, artefacts may be produced due to use of local anesthetic or tissue handling. Some cases may produce no result when no cleavage or immunofluorescence is seen. Also, these methods do not give any information about the underlying genetic defect. Nevertheless, these techniques have demonstrated high sensitivity (IFM, 97%; TEM, 71%) and specificity (IFM, 100%; TEM, 81%).5 These techniques are complementary to genetic testing and help in confirming the diagnosis in cases where variant of uncertain significance is obtained in next generation sequencing.
At present, next generation sequencing is the gold standard for diagnosis of EB. The concordance of IFM and TEM with next generation sequencing (NGS) is 76% and 78.5%, respectively.6 NGS based on massive parallel sequencing is the most effective approach to identify the candidate gene. Molecular diagnosis not only confirms the subtype, but also identifies those subtle types of EB which do not have ultrastructural abnormalities. Novel genes may also be identified which may help in expanding the classification of EB and provide knowledge on the etiology of the disease. Confirmed molecular diagnosis helps in predicting the disease outcome as well. It provides information on mode of inheritance, which helps in counselling of the family and is also useful for the purpose of prenatal diagnosis.
Next generation sequencing may be offered in the form of targeted panels or whole exome. Targeted panels comprising the testing for common genes have high efficiency (94.3%)7 and sensitivity ranges from 75–98%8 with low cost and rapid turnaround time. Whole exome sequencing has the potential to identify novel disease-causing genes, further improving diagnostic sensitivity. Large in-frame deletion in COL7A1 and KRT5 has also been reported and this can be diagnosed by multiplex ligation-dependent probe amplification (MLPA) or quantitative polymerase chain reaction (PCR).9,10
The clinical practice guidelines for laboratory diagnosis of EB that has been previously published by internationally recognized experts in this field emphasizes in detail the importance of early and accurate diagnosis.11
Management
Currently, there is no definitive treatment and cure remains elusive for epidermolysis bullosa (EB). A patient-centric multidisciplinary approach therefore is required for the effective management of these individuals. One should adopt strategies to ameliorate symptoms that are critically needed to increase patients’ quality of life. Good skin care to prevent infections, encouraging wound healing, pain and pruritus control and minimizing complications by avoiding trauma, management of extracutaneous complications, nutritional support, and occupational therapy are required for effective management. Comprehensive guidelines regarding the interdisciplinary management of EB have been previously published.12
The following section will provide an overview of novel therapies (Figure 2) that are in various stages of experimentation.
 |
Figure 2 The 4 layers of skin with dermo-epidermal junctions and corresponding genes involved in the maintenance of their integrity. Basal keratinocytes secrete KRT5 and KRT14, which form the intermediate filaments and interact with other cytoskeletal proteins in epidermis and lamina lucida to provide integrity and adhesion. Gain of function mutations in KLHL24 gene leads to breakdown of intermediate filaments. AON: Anti sense oligonucleotide, SMaRT: Spliceosome-mediated RNA trans-splicing. |
Novel Experimental Therapies
Supportive approaches do not sufficiently meet the medical needs of those suffering from severe EB subtypes. Hence, strategies to correct or modulate the underlying disease mechanism are essential. This is reflected by the increasing number of trials that are being conducted for this condition in the last decade. The goal of these trials is the prolonged or permanent restoration of functional protein expression through addition, replacement, modification, disruption or correction of the defect at the DNA, RNA, protein, or cellular level. The diversity of clinical pathology presents a major challenge in optimizing patient management.
We divide these strategies into those that target the underlying cause and those that alleviate the associated comorbidities.
Causal Strategies
DNA-Based Strategies
Epidermolysis bullosa (EB) is inherited in both autosomal dominant and recessive forms. Gene therapy strategies mainly focus on replacing genes in recessive forms and silencing genes in dominant forms.
Gene Replacement Therapies
Ex vivo Gene Therapy
This therapy uses viral vectors to replace a missing gene product by isolating patients’ cells and inserting a normal gene in vitro followed by expansion of corrected cells into epidermal sheets and grafting these back onto wounds in patients. This method has been successfully utilized in three patients with junctional EB (JEB) who were carrying mutations in the LAMB3 gene, with a major challenge of identifying the keratinocyte stem cell for the gene therapy. Targeting the holoclone stem cells, which has the greatest capability of self-renewal and proliferation, led to satisfactory results with no adverse effects on long-term follow up.13–15 However, a similar therapeutic effect was not obtained in individuals with recessive dystrophic EB (DEB),16,17 partly owing to large size of the transgene. This explains the varying therapeutic effects amongst different subtypes owing to differences in biology of affected protein. General limitations of ex vivo gene therapy include requirement of multiple biopsies to ensure successful stem cell isolation, and extensive debridement for wound bed preparation to increase engraftment success.18
In vivo Gene Therapy
This is performed via topical delivery of genetically corrected cells using vectors to chronic skin lesions. In preclinical studies, highly branched poly(β-amino ester)/minicircle COL7A1 polymeric nanoparticles for gene delivery in recessive DEB keratinocytes showed positive results.19 Currently, in vivo gene therapy using viral vectors is being investigated for patients with DEB20 along with other clinical trials (Phase III trial, NCT04491604). Potential advantages include low risk of immunological reactions, minimum toxicity, a more stable delivery, low costs, easy manufacturing and reduced interventional burden.
The overall limitation of gene replacement therapy is patient selection. Only those patients who have partial but positive expression of the mutated protein can be included in order to minimize the risk of autoreactivity to the newly-formed wild-type proteins.21
Gene Editing Therapies
Gene editing by designer nucleases such as zinc-finger nucleases (ZNF), transcription activator-like effector nucleases (TALEN) and clustered regularly interspaced short palindromic repeats (CRISPR) /CRISPR-associated protein 9 (Cas9) have revolutionized the field of genetics. These tools can permanently correct the genetic defect at DNA level by utilizing an exogenous donor template and generating double-stranded breaks at the loci of interest and activating endogenous DNA repair mechanisms (non-homologous end joining (NHEJ) or homology direct repair (HDR). While NHEJ can be used to induce disruption of a dominant mutant allele, reframing of a frameshift mutation or skipping of a mutant-bearing exon, HDR can achieve a precise repair and complete restoration of the wild-type genetic sequence.22
In preclinical studies, TALENs were first used to edit primary recessive DEB fibroblasts through homology-directed repair.23 Ex vivo gene disruption using these tools was achieved for dominant negative mutations in COL7A124 and KRT5 genes.25 Another strategy like base editing, where precise point mutations can be generated instead of double-stranded breaks, has also been employed successfully in recessive DEB.26 Most recently, the expansion and refinement of base editing in the form of “prime editing” represents a further advancement that can potentially edit the vast majority of all pathogenic EB mutations.27 The dilemma regarding the efficacy and safety of these techniques, especially the unpredictable off-target effect, precludes its entry into clinical applicability at present.
mRNA-Based Strategies
a) Antisense oligonucleotide (AON): AONs are short fragments of single-stranded DNA or RNA that specifically bind to a complementary sequence in the target pre-mRNA and mask the mutated exon from the splicing machinery, thereby excluding it from the mature mRNA. Thus, a truncated but functional protein can be obtained and is useful where in-frame exons encode non-essential domains and their deletion from the protein is unlikely to result in major structural or functional changes. This method has been successfully employed in patients with DEB due to mutations in exons 13, 70, 73, 80 and 105 of COL7A1 gene.28–31 A double-blind, randomized, intra-subject, placebo-controlled clinical trial of an AON targeting exon 73 of COL7A1 gene in DEB wounds (NCT03605069) is currently being assessed.32 Overall limitations of AON-based therapy include transient effect and its beneficial effect on a limited subset of patients with desired mutations.
b) Small interfering RNA (siRNA): siRNA has gained attention as a potential therapeutic reagent due to its ability to inhibit expression of a mutant mRNA without silencing the wild-type allele and is most suitable for dominant negative mutations. This method has been employed in dominant DEB and EBS.33–35 The long-term safety of siRNA is still not clear due to the off-target effect and intact delivery of siRNA into skin in vivo is a major challenge.
c) Spliceosome-mediated RNA trans-splicing (SMaRT): The endogenous splicing machinery is exploited to replace mutated sequences of an endogenous pre-mRNA transcript with wild-type sequences provided by an exogenously provided RNA trans-splicing molecule.36 The same template can be used to correct a pool of mutations. This type of RNA editing was first achieved for PLEC gene37 followed by KRT1438 and COL7A139,40 genes by in vitro and in vivo methods in preclinical studies.
Protein-Based Strategies
Replacement of a missing or faulty protein with wild type form is currently being evaluated only for recessive DEB and is under Phase II trial (NCT04599881). A pre-clinical study has shown that intravenously injected Recombinant Human Type VII Collagen led to its adherence to skin wounds and restored skin integrity of DEB.41 Poor uptake of protein by skin cells owing to its size, poor accessibility to other extracutaneous tissues, unknown duration of efficacy and immune responses are its downsides.
Cell-Based Strategies
Revertant mosaicism (RM) is a process by which inherited mutation is rescued by a second somatic or postzygotic mutation that results in a functional protein. Because of this nature, it is often considered as a ‘natural gene therapy’. This phenomenon has been reported in all types of EB, particularly in intermediate JEB.42 The mechanisms for revertant mosaicism include formation of back mutations, second-site mutations, intragenic recombination, gene conversion and errors during DNA repair.43 Revertant mosaicism in the skin can be readily detected using sequence analysis of genomic DNA isolated using laser capture microdissection (LCM) of biopsies from suspected revertant areas, as well as immunohistochemistry to detect changes in protein expression.44 This mechanism was exploited and the resultant patchy healthy skin due to revertant mosaicism can be expanded in vitro and can be used to promote healing of affected skin areas.45 Cultured epidermal autografts containing revertant cells have also been used in the management of chronic wounds in patients with recessive DEB.46
Mesenchymal stem/stromal cells (MSCs), which have stem-cell capabilities, immunomodulatory and anti-inflammatory effects, were delivered through various routes (intravenous/ intradermally) and their effects are being assessed in patients with EB. In recessive DEB, intravenous infusion of MSCs resulted in improved wound healing and reduced pain and itching in children.47 Usefulness of MSC subpopulation including bone marrow-derived muse cells were also demonstrated in DEB patients.48 The unresolved doubts with this mode of therapy exist in terms of the type of source, route of delivery, optimal dosage and treatment intervals and potential role of haploidentical donors.
Combined Strategies
A Phase I, open-label, single-centre clinical trial evaluated the efficacy of combined ex vivo gene and cell therapy using patients’ autologous fibroblasts in four recessive DEB patients. Patient fibroblasts were first modified ex vivo using a self-inactivating lentiviral vector that carries COL7A1 cDNA. The gene-modified autologous fibroblasts were then injected intradermally back into the patients. This treatment had no serious adverse effects and was well tolerated by the patients.49
Readthrough Strategies
Nonsense mutations leading to premature stop codon (PTC) formation are present in 15–20% of JEB and recessive DEB patients. In EB, nonsense mutations tend to result in severe phenotype due to complete or near complete absence of functional protein. In premature stop codon readthrough strategies, a random amino acid is incorporated at the PTC position in the mRNA. Depending on the impact of the introduced amino acid on protein folding, stability, and post-translational processing, PTC readthrough therapies can result in the synthesis of a functional full-length protein by inducing a conformational change at the decoding site, causing reduced translational fidelity and incorporation of near-cognate tRNAs at the stop site.50 The common readthrough agents employed in various trials in EB include aminoglycosides geneticin, gentamicin and paromomycin and anti-inflammatory drug amlexanox. This strategy has been utilized in recessive DEB-derived cells51,52 and JEB keratinocytes carrying nonsense mutations within the LAMB3 gene.53 Two-week daily intravenous gentamicin administration in four recessive DEB children not only improved wound healing for at least 3 months, but also markedly increased C7 expression and anchoring fibrils.51 Three-weekly intravenous gentamicin infusions (7.5 mg/kg/day) in five severe JEB neonates led to improved skin lesions in four subjects.54 Optimal dosing, treatment intervals, and the cumulative toxicity profile still need better definition.
Novel Strategies to Modify Disease and Alleviate Comorbidities
Blister Management
Topical application of a small molecule drug, diacerein, was found to significantly reduce the number of blisters and their recurrence in RCTs of EBS.55 Upregulation of IL-1ß due to accumulation of mutated keratins is a characteristic feature of few subtypes of EB. This rhein prodrug has been shown to reduce expression of K14 and inhibit IL-1ß converting enzyme and found to be safe and effective as a 1% topical formulation.
Apremilast, a phosphodiesterase 4 inhibitor (PDE-4) that suppresses Th1/ Th17 activation has already been approved for treatment of psoriatic arthritis and oral ulcerations in Behcet's disease. Since EBS fluid was found to have high levels of Th17 cytokines, this drug was tried and a dramatic reduction in blisters were found in three patients and two of them had sustained clinical remission.56
High Mobility Group Box‐1 (HMGB1) is a peptide responsible for mobilizing MSCs from bone marrow and recruiting them to damaged skin for repair. Based on preliminary data, a Phase II, single-arm, non-randomized, uncontrolled clinical trial (UMIN000029962) to assess its role in recessive DEB showed satisfactory results in blister/erosion reduction.
Pruritus Management
The role of neurokinin-1 receptor (NK1R) antagonist serlopitant is currently being evaluated in patients with any EB subtype.57
A double-blind RCT is currently in place to evaluate the role of low-dose topical calcipotriol ointment on improving wound healing in DEB.58 This is based on the antiproliferative role of calcipotriol on keratinocytes.
A monoclonal antibody drug, dupilumab, has already been approved for moderate to severe atopic dermatitis and is currently being evaluated for EB-pruriginosa.59 It is an anti-interleukin-4 receptor alpha (IL-4Rα) monoclonal antibody that inhibits both IL-4 and IL-13 signalling and modulates Th2-mediated immune mechanisms.
Wound Healing
Thymosin β4 is a naturally-produced polypeptide and has several wound healing properties, including anti-inflammation, anti-fibrosis, pro-angiogenesis, stem cell recruitment and keratinocyte migration promotion.60 Currently, a single-blind Phase II clinical trial (NCT03578029) evaluating the efficacy of a topical thymosin β4 dermal gel on paired wounds in 15 JEB/DEB patients is underway.
A triterpene extract in sunflower oil, oleogel- S10 has been shown to promote wound healing through inflammation modulation, stimulation of keratinocyte migration and altered epidermal differentiation. It acts irrespective of the underlying molecular pathology in EB.61 Currently, preliminary results from a Phase III double-blind, randomized, placebo-controlled “EASE” trial (NCT03068780) are showing sustained wound closure benefits in recessive DEB patients.
Deformities Correction
Chronic wounds lead to repeated cycles of inflammation eventually culminating in progressive fibrosis followed by tissue destruction. This in turn results in mitten hand and foot causing severe disability. Transforming growth factor-beta (TGF-ß), a pro-inflammatory cytokine, plays a key role in EB-associated fibrosis.62 Thus, modulating the expression of TGF-ß1 can help in reduction of fibrosis and the role of angiotensin II antagonist with anti-fibrotic effects, losartan, is currently being evaluated in children and adolescents with recessive DEB.63
Risk of Skin Malignancy
One of the deadly complications of EB that significantly reduces the life span of these individuals is the risk of developing aggressive squamous cell carcinoma (SCC) due to repeated wounds, infections and inflammation of skin. In comparison to the general population, these individuals are at 70-fold higher risk of developing SCC.64 Conventional treatments in the form of local excision, radiotherapy or chemotherapy can be detrimental owing to their adverse effects. Hence, it is important to strike a balance between tumour suppression and its adverse effects on wound healing.
Few groups have evaluated the role of cetuximab, a monoclonal antibody targeting epidermal growth factor receptor (EGFR) on EB patients with advanced cutaneous SCC as EB-associated SCC often express EGFR. However, they found limited beneficial effects of the drug on survival but a better response is seen when administered early in the course of illness.65,66
Currently, a multicentre Phase II clinical trial in recessive DEB patients with late stage, metastatic or unresectable SCC is in place to evaluate the role of rigosertib, a PLK1 (polo-like kinase-1) inhibitor that has a strong and selective apoptotic effect in recessive DEB-SCC cells (NCT03786237).
Anti-PD1 (programmed death-1) monoclonal antibodies are also being evaluated on EB patients with metastatic SCC (Eudra CT‐No. 2016‐002811‐16). PD1 is predominantly expressed on T cells, and, by binding to its ligands PD-L1 and PD-L2 expressed on tumor cells, induces a negative signal that leads to effector T cell suppression. Specific antibodies that block these interactions can thus lead to reactivation of the immune system and improvement of anti-tumour immune responses.
To conclude, there has been tremendous progress in understanding the molecular genetics and underlying pathological mechanisms of EB over the past few decades. Many preclinical and clinical attempts to develop new treatments for EB are currently in place. Gene replacement therapy is an exciting approach and few studies have reached Phase III trials for both ex vivo and in vivo approaches and this can benefit a subset of people with EB. mRNA-based therapies have better safety standards but their effects are transient in nature and their use is restricted to a subset of people bearing specific mutations. Despite rapid advancements in genome editing tools and their great potential, their entry at the clinical level in EB is still precluded by their unpredictable off-target effects. Readthrough therapies, though restricted to nonsense mutations, showed promising results in Phase I and II trials. Other regenerative cell-based therapies and strategies to mitigate the effect of comorbidities are currently in Phase I, II and III trials and can significantly improve the quality of life of individuals with EB. But one should also weigh the risks of some of these technologies against their potential benefits before their clinical application. With advancing technologies, refinement of current innovative strategies with improved safety profiles for successful treatment of this group of incurable diseases is not far from reach. The search for new methods of treatment will keep expanding and provides hope for a definitive cure.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Has C, Bauer JW, Bodemer C, et al. Consensus reclassification of inherited epidermolysis bullosa and other disorders with skin fragility. Br J Dermatol. 2020;183(4):614–627. doi:10.1111/bjd.18921
2. Fine JD. Epidemiology of inherited epidermolysis bullosa based on incidence and prevalence estimates from the National Epidermolysis Bullosa Registry. JAMA Dermatol. 2016;152(11):1231–1238. doi:10.1001/jamadermatol.2016.2473
3. Mariath LM, Santin JT, Schuler-Faccini L, Kiszewski AE. Inherited epidermolysis bullosa: update on the clinical and genetic aspects. An Bras Dermatol. 2020;95(5):551–569. doi:10.1016/j.abd.2020.05.001
4. Bardhan A, Bruckner-Tuderman L, Chapple IL, et al. Epidermolysis bullosa. Nat Rev Dis Primers. 2020;6(1):1–27. doi:10.1038/s41572-020-0210-0
5. Yiasemides E, Walton J, Marr P, Villanueva EV, Murrell DF. A comparative study between transmission electron microscopy and immunofluorescence mapping in the diagnosis of epidermolysis bullosa. Am J Dermat Opathol. 2006;28(5):387–394. doi:10.1097/01.dad.0000211510.44865.6d
6. Mahajan R, Manjunath S, Madakshira MG, et al. Correlating clinical and laboratory diagnostic modalities in the diagnosis of epidermolysis bullosa in a resource-poor setting. J Cutan Pathol. 2022;49(5):454–459. doi:10.1111/cup.14208
7. Rossi S, Castiglia D, Pisaneschi E, et al. Immunofluorescence mapping, electron microscopy and genetics in the diagnosis and sub-classification of inherited epidermolysis bullosa: a single-centre retrospective comparative study of 87 cases with long-term follow-up. J Eur Acad Dermatol Venereol. 2021;35(4):1007–1016. doi:10.1111/jdv.17060
8. Lucky AW, Dagaonkar N, Lammers K, Husami A, Kissell D, Zhang K. A comprehensive next-generation sequencing assay for the diagnosis of epidermolysis bullosa. Pediatr Dermatol. 2018;35(2):188–197. doi:10.1111/pde.13392
9. Has C, Schumann H, Leppert J, et al. Monoallelic large intragenic KRT5 deletions account for genetically unsolved cases of epidermolysis bullosa simplex. J Invest Dermatol. 2017;137(10):2231–2234. doi:10.1016/j.jid.2017.05.016
10. Chmel N, Bornert O, Hausser I, et al. Large deletions targeting the triple-helical domain of collagen VII lead to mild acral dominant dystrophic epidermolysis bullosa. J Invest Dermatol. 2018;138(4):987–991. doi:10.1016/j.jid.2017.11.014
11. Has C, Liu L, Bolling MC, et al. Clinical practice guidelines for laboratory diagnosis of epidermolysis bullosa. Br J Dermatol. 2020;182(3):574–592. doi:10.1111/bjd.18128
12. EB-CLINET. Completed EB guidelines [Internet]; 2022 [cited April 22, 2022]. Available from: https://www.eb-clinet.org/clinical-guidelines/completed-eb-guidelines/.
13. Mavilio F, Pellegrini G, Ferrari S, et al. Correction of junctional epidermolysis bullosa by transplantation of genetically modified epidermal stem cells. Nat Med. 2006;12(12):1397–1402. doi:10.1038/nm1504
14. Bauer JW, Koller J, Murauer EM, et al. Closure of a large chronic wound through transplantation of gene-corrected epidermal stem cells. J Investig Dermatol. 2017;137(3):778–781. doi:10.1016/j.jid.2016.10.038
15. Hirsch T, Rothoeft T, Teig N, et al. Regeneration of the entire human epidermis using transgenic stem cells. Nature. 2017;551(7680):327–332. doi:10.1038/nature24487
16. Siprashvili Z, Nguyen NT, Gorell ES, et al. Safety and wound outcomes following genetically corrected autologous epidermal grafts in patients with recessive dystrophic epidermolysis bullosa. JAMA. 2016;316(17):1808–1817. doi:10.1001/jama.2016.15588
17. Eichstadt S, Barriga M, Ponakala A, et al. Phase 1/2a clinical trial of gene- corrected autologous cell therapy for recessive dystrophic epidermolysis bullosa. JCI Insight. 2019;4(19):e130554. doi:10.1172/jci.insight.130554
18. Marinkovich MP, Tang JY. Gene therapy for epidermolysis bullosa. J Invest Dermatol. 2019;139(6):1221–1226. doi:10.1016/j.jid.2018.11.036
19. Zeng M, Alshehri F, Zhou D, et al. Efficient and robust highly branched poly(β-amino ester)/minicircle COL7A1Polymeric nanoparticles for gene delivery to recessive dystrophic epidermolysis bullosa keratinocytes. ACS Appl Mater Interfaces. 2019;11(34):30661–30672. doi:10.1021/acsami.9b13135
20. Marinkovich MP, Vinzant S, Karkala V, et al. 305 In vivo correction of recessive dystrophic epidermolysis bullosa (RDEB) by direct cutaneous COL7A1 gene replacement: results of a phase 1–2 trial. In: Society for Investigative Dermatology (SID) 2020 meeting abstract supplement. J Investig Dermatol. 2020;140(7):S37.
21. Gaucher S, Lwin SM, Titeux M, et al. EB Gene trial: patient preselection outcomes for the European GENEGRAFT ex vivo phase I/II gene therapy trial for recessive dystrophic epidermolysis bullosa. Br J Dermatol. 2020;182(3):794–797. doi:10.1111/bjd.18559
22. March OP, Kocher T, Koller U. Context-dependent strategies for enhanced genome editing of genodermatoses. Cells. 2020;9(1):112. doi:10.3390/cells9010112
23. Osborn MJ, Starker CG, McElroy AN, et al. TALEN-based gene correction for epidermolysis bullosa. Mol Ther. 2013;21(6):1151–1159. doi:10.1038/mt.2013.56
24. Shinkuma S, Guo Z, Christiano AM. Site-specific genome editing for correction of induced pluripotent stem cells derived from dominant dystrophic epidermolysis bullosa. Proc Natl Acad Sci U S A. 2016;113(20):5676–5681. doi:10.1073/pnas.1512028113
25. Aushev M, Koller U, Mussolino C, Cathomen T, Reichelt J. Traceless targeting and isolation of gene-edited immortalized keratinocytes from epidermolysis bullosa simplex patients. Mol Ther Methods Clin Dev. 2017;6:112–123. doi:10.1016/j.omtm.2017.06.008
26. Osborn MJ, Newby GA, McElroy AN, et al. Base editor correction of COL7A1 in recessive dystrophic epidermolysis bullosa patient-derived fibroblasts and iPSCs. J Invest Dermatol. 2020;140(2):338–347.e5. doi:10.1016/j.jid.2019.07.701
27. Anzalone AV, Randolph PB, Davis JR, et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature. 2019;576(7785):149–157. doi:10.1038/s41586-019-1711-4
28. Bremer J, Bornert O, Nyström A, et al. Antisense oligonucleotide-mediated exon skipping as a systemic therapeutic approach for recessive dystrophic epidermolysis bullosa. Mol Ther Nucleic Acids. 2016;5(10):e379. doi:10.1038/mtna.2016.87
29. Goto M, Sawamura D, Nishie W, et al. Targeted skipping of a single exon harboring a premature termination codon mutation: implications and potential for gene correction therapy for selective dystrophic epidermolysis bullosa patients. J Invest Dermatol. 2006;126(12):2614–2620. doi:10.1038/sj.jid.5700435
30. Turczynski S, Titeux M, Tonasso L, Décha A, Ishida-Yamamoto A, Hovnanian A. Targeted exon skipping restores type VII collagen expression and anchoring fibril formation in an in vivo RDEB model. J Invest Dermatol. 2016;136(12):2387–2395. doi:10.1016/j.jid.2016.07.029
31. Bornert O, Kühl T, Bremer J, van den Akker PC, Pasmooij AM, Nyström A. Analysis of the functional consequences of targeted exon deletion in COL7A1 reveals prospects for dystrophic epidermolysis bullosa therapy. Mol Ther. 2016;24(7):1302–1311. doi:10.1038/mt.2016.92
32. Marinkovich MP, Sridhar K, Karkala V, et al. 306 Topical QR-313, an antisense oligo- nucleotide, in the treatment of dystrophic epidermolysis bullosa. J Investig Dermatol. 2020;140:S37.
33. Morgan CP, Allen DSI, Millington-Ward S, O’Dwyer GE, Palfi A, Jane Farrar G. A mutation-independent therapeutic strategy for dominant dystrophic epidermolysis bullosa. J Invest Dermatol. 2013;133(12):2793–2796. doi:10.1038/jid.2013.241
34. Pendaries V, Gasc G, Titeux M, Tonasso L, Mejía JE, Hovnanian A. siRNA-mediated allele-specific inhibition of mutant type VII collagen in dominant dystrophic epidermolysis bullosa. J Invest Dermatol. 2012;132(6):1741–1743. doi:10.1038/jid.2012.11
35. Atkinson SD, McGilligan VE, Liao H, et al. Development of allele-specific therapeutic siRNA for keratin 5 mutations in epidermolysis bullosa simplex. J Invest Dermatol. 2011;131(10):2079–2086. doi:10.1038/jid.2011.169
36. Koller U, Wally V, Bauer JW, Murauer EM. Considerations for a successful RNA trans-splicing repair of genetic disorders. Mol Ther Nucleic Acids. 2014;3(4):e157. doi:10.1038/mtna.2014.10
37. Wally V, Klausegger A, Koller U, et al. 5’ trans-splicing repair of the PLEC1 gene. J Invest Dermatol. 2008;128(3):568–574. doi:10.1038/sj.jid.5701152
38. Peking P, Breitenbach JS, Ablinger M, et al. An ex vivo RNA trans-splicing strategy to correct human generalized severe epidermolysis bullosa simplex. Br J Dermatol. 2019;180(1):141–148. doi:10.1111/bjd.17075
39. Tockner B, Kocher T, Hainzl S, et al. Construction and validation of an RNA trans-splicing molecule suitable to repair a large number of COL7A1 mutations. Gene Ther. 2016;23(11):775–784. doi:10.1038/gt.2016.57
40. Peking P, Koller U, Duarte B, et al. An RNA-targeted therapy for dystrophic epidermolysis bullosa. Nucleic Acids Res. 2017;45(17):10259–10269. doi:10.1093/nar/gkx669
41. Woodley DT, Wang X, Amir M, et al. Intravenously injected recombinant human type VII collagen homes to skin wounds and restores skin integrity of dystrophic epidermolysis bullosa. J Invest Dermatol. 2013;133(7):1910–1913. doi:10.1038/jid.2013.10
42. Pasmooij AM, Nijenhuis M, Brander R, Jonkman MF. Natural gene therapy may occur in all patients with generalized non-Herlitz junctional epidermolysis bullosa with COL17A1 mutations. J Invest Dermatol. 2012;132(5):1374–1383. doi:10.1038/jid.2011.477
43. Kiritsi D, Garcia M, Brander R, et al. Mechanisms of natural gene therapy in dystrophic epidermolysis bullosa. J Invest Dermatol. 2014;134(8):2097–2104. doi:10.1038/jid.2014.118
44. Lim YH, Fisher JM, Choate KA. Revertant mosaicism in genodermatoses. Cell Mol Life Sci. 2017;74(12):2229–2238. doi:10.1007/s00018-017-2468-2
45. Gostyński A, Pasmooij AM, Jonkman MF. Successful therapeutic transplantation of revertant skin in epidermolysis bullosa. J Am Acad Dermatol. 2014;70(1):98–101. doi:10.1016/j.jaad.2013.08.052
46. Matsumura W, Fujita Y, Shinkuma S, et al. Cultured epidermal autografts from clinically revertant skin as a potential wound treatment for recessive dystrophic epidermolysis bullosa. J Invest Dermatol. 2019;139(10):2115–2124.e11. doi:10.1016/j.jid.2019.03.1155
47. Petrof G, Lwin SM, Martinez-Queipo M, et al. Potential of systemic allogeneic mesenchymal stromal cell therapy for children with recessive dystrophic epidermolysis bullosa. J Invest Dermatol. 2015;135(9):2319–2321. doi:10.1038/jid.2015.158
48. Fujita Y, Nohara T, Takashima S, et al. Intravenous allogeneic multilineage-differentiating stress-enduring cells in adults with dystrophic epidermolysis bullosa: a Phase 1/2 open-label study. J Eur Acad Dermatol Venereol. 2021;35(8):e528–e531. doi:10.1111/jdv.17201
49. Lwin SM, Syed F, Di WL, et al. Safety and early efficacy outcomes for lentiviral fibroblast gene therapy in recessive dystrophic epidermolysis bullosa. JCI Insight. 2019;4(11):e126243. doi:10.1172/jci.insight.126243
50. Cogan J, Weinstein J, Wang X, et al. Aminoglycosides restore full-length type VII collagen by overcoming premature termination codons: therapeutic implications for dystrophic epidermolysis bullosa. Mol Ther. 2014;22(10):1741–1752. doi:10.1038/mt.2014.140
51. Woodley DT, Cogan J, Hou Y, et al. Gentamicin induces functional type VII collagen in recessive dystrophic epidermolysis bullosa patients. J Clin Invest. 2017;127(8):3028–3038. doi:10.1172/JCI92707
52. Atanasova VS, Jiang Q, Prisco M, et al. Amlexanox enhances premature termination codon read-through in COL7A1 and expression of full length type VII collagen: potential therapy for recessive dystrophic epidermolysis bullosa. J Invest Dermatol. 2017;137(9):1842–1849. doi:10.1016/j.jid.2017.05.011
53. Hammersen J, Neuner A, Wild F, Schneider H. Attenuation of severe generalized junctional epidermolysis bullosa by systemic treatment with gentamicin. Dermatology. 2019;235(4):315–322. doi:10.1159/000499906
54. Lincoln V, Cogan J, Hou Y, et al. Gentamicin induces LAMB3 nonsense mutation readthrough and restores functional laminin 332 in junctional epidermolysis bullosa. Proc Natl Acad Sci U S A. 2018;115(28):E6536–E6545. doi:10.1073/pnas.1803154115
55. Wally V, Hovnanian A, Ly J, et al. Diacerein orphan drug development for epidermolysis bullosa simplex: a Phase 2/3 randomized, placebo-controlled, double-blind clinical trial. J Am Acad Dermatol. 2018;78(5):892–901.e7. doi:10.1016/j.jaad.2018.01.019
56. Castela E, Tulic MK, Rozières A, et al. Epidermolysis bullosa simplex generalized severe induces a T helper 17 response and is improved by apremilast treatment. Br J Dermatol. 2019;180(2):357–364. doi:10.1111/bjd.16897
57. Chiou AS, Choi S, Barriga M, et al. Phase 2 trial of a neurokinin-1 receptor antagonist for the treatment of chronic itch in patients with epidermolysis bullosa: a randomized clinical trial. J Am Acad Dermatol. 2020;82(6):1415–1421. doi:10.1016/j.jaad.2019.09.014
58. Guttmann-Gruber C, Piñón Hofbauer J, Tockner B, et al. Impact of low-dose calcipotriol ointment on wound healing, pruritus and pain in patients with dystrophic epidermolysis bullosa: a randomized, double-blind, placebo-controlled trial. Orphanet J Rare Dis. 2021;16(1):473. doi:10.1186/s13023-021-02062-2
59. Shehadeh W, Sarig O, Bar J, Sprecher E, Samuelov L. Treatment of epidermolysis bullosa pruriginosa-associated pruritus with dupilumab. Br J Dermatol. 2020;182(6):1495–1497. doi:10.1111/bjd.18855
60. Yang WS, Kang S, Sung J, Kleinman HK. Thymosin β4: potential to treat epidermolysis bullosa and other severe dermal injuries. Eur J Dermatol. 2019;29(5):459–467. doi:10.1684/ejd.2019.3642
61. Schwieger-Briel A, Ott H, Kiritsi D, Laszczyk-Lauer M, Bodemer C. Mechanism of Oleogel-S10: a triterpene preparation for the treatment of epidermolysis bullosa. Dermatol Ther. 2019;32(4):e12983. doi:10.1111/dth.12983
62. Fritsch A, Loeckermann S, Kern JS, et al. A hypomorphic mouse model of dystrophic epidermolysis bullosa reveals mechanisms of disease and response to fibroblast therapy. J Clin Invest. 2008;118(5):1669–1679. doi:10.1172/JCI34292
63. Nyström A, Thriene K, Mittapalli V, et al. Losartan ameliorates dystrophic epidermolysis bullosa and uncovers new disease mechanisms. EMBO Mol Med. 2015;7(9):1211–1228. doi:10.15252/emmm.201505061
64. Mallipeddi R. Epidermolysis bullosa and cancer. Clin Exp Dermatol. 2002;27(8):616–623. doi:10.1046/j.1365-2230.2002.01130.x
65. Diociaiuti A, Steinke H, Nyström A, et al. EGFR inhibition for metastasized cutaneous squamous cell carcinoma in dystrophic epidermolysis bullosa. Orphanet J Rare Dis. 2019;14(1):278. doi:10.1186/s13023-019-1262-7
66. Medek K, Koelblinger P, Koller J, et al. Wound healing deficits in severe generalized recessive dystrophic epidermolysis bullosa along anticancer treatment with cetuximab. J Dtsch Dermatol Ges. 2019;17(4):448–450. doi:10.1111/ddg.13802
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.