Back to Archived Journals » Advances in Genomics and Genetics » Volume 5
Insights into the molecular genetics of Kabuki syndrome
Authors Adam M
Received 11 October 2014
Accepted for publication 6 December 2014
Published 23 February 2015 Volume 2015:5 Pages 121—129
DOI https://doi.org/10.2147/AGG.S58588
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr John Martignetti
Margaret P Adam
Department of Pediatrics, Division of Genetic Medicine, University of Washington School of Medicine, Seattle, WA, USA
Abstract: Kabuki syndrome (KS) is a well-recognized multiple congenital anomaly/intellectual disability syndrome characterized by distinctive facial features, congenital heart defects, skeletal anomalies, persistent fingertip pads, postnatal growth retardation, and cognitive impairment to varying degrees. To date, mutations or deletions in two genes (KMT2D and KDM6A) have been identified to cause the majority of cases of KS. Both genes are involved in histone modification and epigenetic regulation of gene expression in early embryogenesis. In this report, we review the clinical features and management of patients with KS, explore the proposed protein interactions and the molecular pathway that may lead to features of KS, and discuss how knowledge of the molecular mechanisms has the potential to inform further disease gene discovery and targeted treatment of the condition.
Keywords: Kabuki syndrome, Kabuki make-up syndrome, KMT2D, KDM6A, histone modification
Introduction
First described as a distinct syndrome by Kuroki et al1 and Niikawa et al2 in 1981, Kabuki syndrome (KS) has become a well-recognized multiple congenital anomaly/intellectual disability syndrome. The original cohort of affected individuals was ascertained based on the following cardinal dysmorphic facial features: long palpebral fissures with eversion of the lateral third of the lower eyelid; arched and broad eyebrows with the lateral third displaying notching or sparseness; short columella with depressed nasal tip; and large, prominent, or cupped ears (Figure 1). Because the facial features resembled those of the actors in the Kabuki theater, the name “Kabuki make-up syndrome” was proposed, although “make-up” was subsequently discarded and the name “Kabuki syndrome” is now used. For a period in the early literature, the term Niikawa–Kuroki syndrome was also used to refer to this condition.
 | Figure 1 Three patients with Kabuki syndrome. |
In addition to the distinctive facial features listed above, the remaining cardinal manifestations of KS as described by Niikawa et al3 are the following: postnatal growth retardation, skeletal anomalies (clinodactyly of the fifth digit; brachydactyly V; brachymesophalangy; and spinal column abnormalities, including sagittal cleft vertebrae, butterfly vertebrae, narrow intervertebral disc space, and/or scoliosis), dermatoglyphic abnormalities (persistence of fingertip pads; absence of digital triradius c and/or d; hypothenar loop patterns), and intellectual disability (typically in the mild to moderate range). Additionally, a large number of associated congenital anomalies and functional differences have been described in individuals with KS, which can aid in clinical diagnosis. Table 1 summarizes the clinical features of more than 500 individuals with KS and the associated management recommendations for individuals who have the condition.3–16
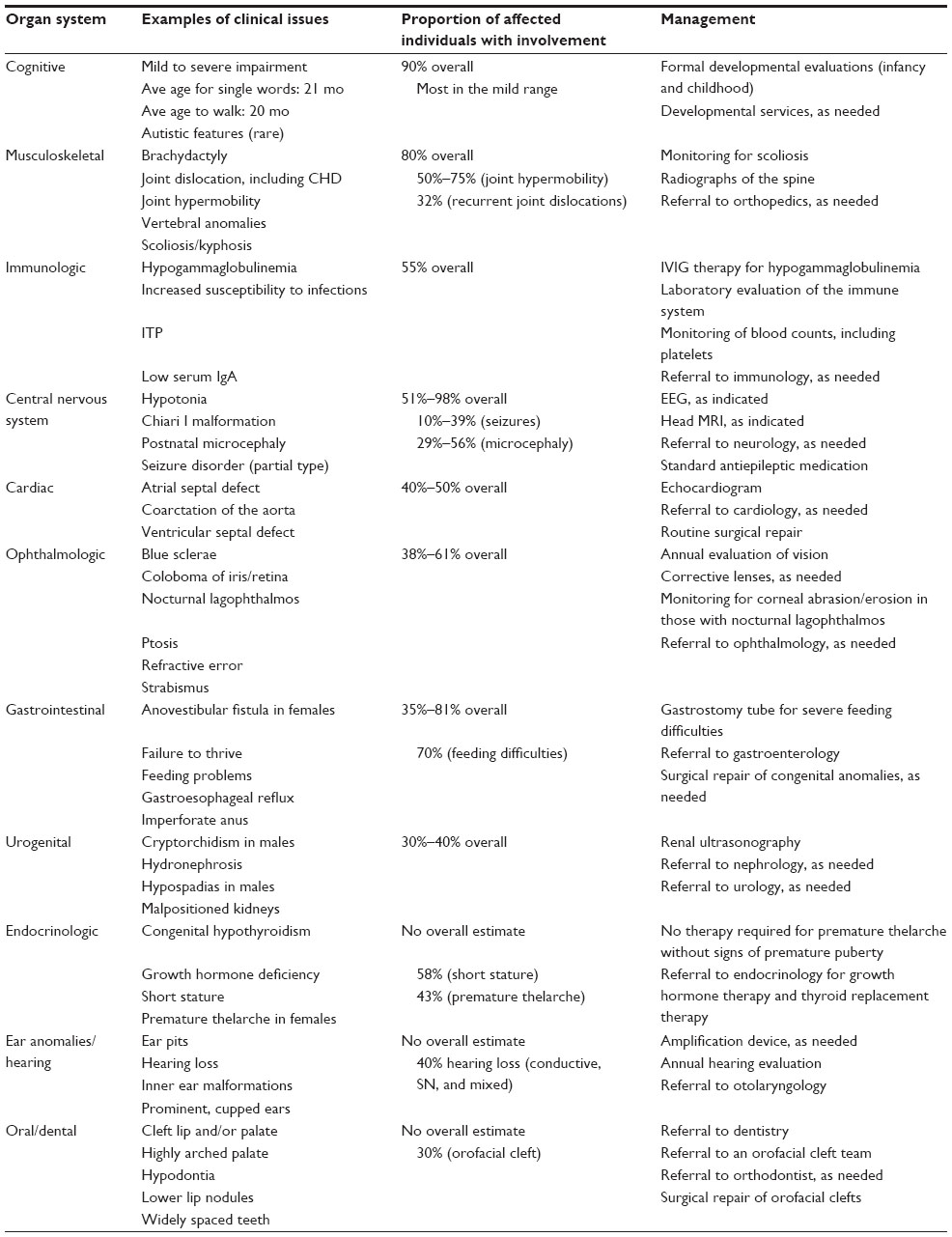 | Table 1 Congenital anomalies, functional differences, and management by organ system in individuals with Kabuki syndrome |
KS has been described in many countries and has been reported in individuals of almost all ethnic groups. Because the first descriptions of this condition originated from Japan, the population prevalence has been studied most extensively in that population, with an estimated birth prevalence of 1:32,000.3 Although the prevalence in other countries and populations presumably approximates that seen in Japan, White et al17 estimated a birth incidence of 1:86,000 in Australia and New Zealand.
Through the use of whole-exome sequencing (WES), heterozygous mutations in KMT2D (NM_003482.3; previously known as MLL2) were first reported in 2010 as a major cause of KS.18 In 2012, Lederer et al19 reported deletions in or involving KDM6A (NM_001291415.1) detected through array comparative genomic hybridization in three individuals with features of KS, confirming the heterogeneous nature of this clinical condition. Once it was determined that more than one gene could lead to features of KS, a phenotypic scoring system was devised by Makrythanasis et al20 to aid clinicians in determining which individuals with features of KS were likely to have a mutation in KMT2D versus those who were not (Table 2). Affected individuals with pathogenic nonsense or missense mutations in KMT2D had a statistically significantly higher mean score using this scoring system (mean score: 6.1) compared to those who were negative for the KMT2D mutation (mean score: 4.5).20 A similar scoring system to determine who may be at highest risk for a pathogenic variant in KDM6A has not yet been proposed.
 | Table 2 Proposed phenotypic scoring system for Kabuki syndrome |
Roles of KMT2D and KDM6A: genomic interactions, mechanisms, and pathways
Precise spatial and temporal regulation of distinct genes and pathways is necessary for normal human embryonic development. Such regulation is in part determined by epigenetic mechanisms, including histone methylation. The three histone lysine methylation states are mono-, di-, and trimethylation. Depending on the location of the lysine residue, these different histone methylation states cause either activation or repression of transcription of a given gene.21
The most common cause of KS is the presence of heterozygous pathogenic variants in KMT2D (formerly MLL2), which codes for a histone 3 lysine 4 (H3K4) N-methyltransferase protein, composed of 5,537 amino acids. KMT2D, which is located on 12q13.12, is 36.3 kb in size and comprises 54 exons. The KMT2D protein contains seven plant homeodomain (PHD) regions, a high-mobility-group-binding motif, a FY(Phe–Tyr)-rich N-terminal motif, a FY(Phe–Tyr)-rich C-terminal domain, a C-terminal Suvar3-9 (SuVar), Enhancer-of-zeste (E[z]), Trithorax (Trx) or SET domain, and a post-SET domain (Figure 2).18 KMT2D specifically modifies the lysine residue at the fourth amino acid position of the histone H3 protein, catalyzing the conversion from nonmethylated to monomethylated and, subsequently, dimethylated H3K4.22,23 The SET domain is responsible for the methyltransferase activity.
 | Figure 2 Protein structure of KMT2D demonstrating the arrangement of the functional domains and the location of the Win motif. |
Recent studies have shed light on the interaction of the MLL family of proteins (MLL1 through MLL4) with other core proteins, whose function is the epigenetic regulation of gene transcription and chromatin structure in early development and in adult organisms.23–25 Di- and trimethylation is only possible when the MLL protein is bound to a minimal core complex that consists of WDR5, RbBP5, Ash2L, and DPY-30, also referred to by the acronym WRAD.26,27 Specifically, Issaeva et al26 demonstrated that all SET1-like complexes, including those that contain KMT2D, share three core proteins (Ash2L, RbBP5, and WDR5) that are essential for methylation of H3K4. While most studies have been performed using MLL1 as a model system, previous studies have confirmed that this interaction also applies to MLL2 (KMT2D).26
The MLL family of proteins has two active sites: a canonical SET domain that monomethylates H3K4 and a dimethylation active site located at the interaction between MLL/WDR5 and the RbBP5/Ash2L heterodimer. Nonsense mutations or deletions may disrupt the canonical SET domain, precluding its enzymatic activity; missense mutations may result in the same inability to methylate H3K4 but not necessarily by disrupting the active site of the canonical SET domain itself. Hannibal et al12 demonstrated that the missense mutations C1430R and C1471Y in the PHD4–6 domains of KMT2D did not affect the ability of KMT2D to bind to or interact with other proteins but did result in decreased DNA-binding ability, which abolished the nucleosome methylation activity of the protein.12,21 The C-terminal portion of the MLL family of genes also contains a WDR5 interaction (Win) motif (Figure 2). Any mutation that disrupts the interaction of MLL and WDR5 or the interaction of the MLL/WDR5 heterodimer with the Ash2L/RbBP5 heterodimer also prohibits the correct assembly of the core complex and abolishes its methylation ability.23,28 Missense mutations in the Win motif eliminate the ability of MLL to bind to WDR5.23,29 Protein modeling has also identified an area of the folded MLL protein that is required for appropriate assembly of the MLL/WDR5 heterodimer to the RbBP5/Ash2L heterodimer. Missense mutations involving the amino acid residues within this so-called Kabuki interacting surface result in the loss of H3K4 methylation activity by weakening the assembly of the MLL/WDR5 heterodimer with the RbBP5/Ash2L heterodimer (Figure 3).23
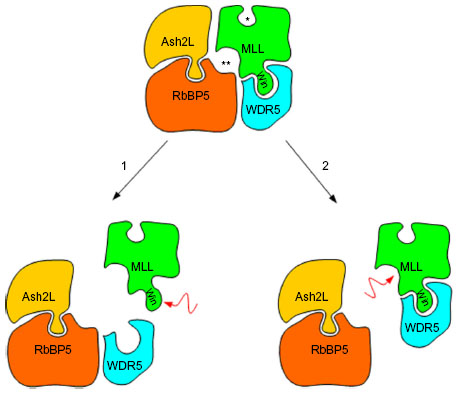 | Figure 3 Proposed model for the interaction of the MLL family of proteins with WDR5 and the RbBP5/Ash2L heterodimer. |
Heterozygous pathogenic variants in KDM6A have been found in a minority of individuals with features of KS.19,30–32 KDM6A is 239 kb in size, comprising 29 exons. The gene is located on Xp11.3 and is thought to escape X-inactivation.33,34 KDM6A is a 1,401-amino acid H3K27 demethylase, meaning that it removes the methyl residue of lysine 27 on histone H3.19,31 The protein contains eight tetratricopeptide repeat elements and one Jumonji C-terminal (JmjC) domain in the C-terminal half.
KMT2D and KDM6A work collaboratively to regulate the epigenetic control of transcriptionally active chromatin.25,31,32 The overall function of the complex is to remove repressive epigenetic marks on chromatin through a histone demethylase and to deposit activating H3K4 methylation marks on chromatin through a histone methyltransferase, which then recruits RNA polymerase II complex. In 2002, Santos-Rosa et al35 demonstrated that H3K4 di- and trimethylation is associated with an active transcriptional state, while H3K27 di- and trimethylation leads to gene silencing.
Most target genes (and their respective functions) of the regulatory pathways in which KMT2D and KDM6A play a role are not yet known, although this is an area of active research. KMT2D and its core complex are able to bind estrogen receptors, which subsequently regulate HOX6A transcription in an estradiol-dependent manner.21,36 Additional proposed target genes have included other HOX gene family members and some of the S100A family of genes, including S100A2, S100A4, S100A5, and S100A6.21,37,38 In a zebrafish model, morpholino knockdown of KDM6A leads to developmental anomalies, including diverse and severe structural anomalies, the proposed mechanism of which is abnormal regulation of the HOX genes.39 In mice, deficiency of KDM6A results in severe congenital heart defects and embryonic lethality,40 possibly by affecting the expression of master regulatory genes that coordinate osteogenesis and adipogenesis in early embryogenesis.41
Overview of studies in large cohorts
Between 55% and 80% of individuals with a clinical diagnosis of KS have an identifiable mutation in KMT2D.12–15,18,20,21,32 The majority of mutations are of the nonsense and frameshift (with similar frequencies) types, followed by (in order of decreasing frequency) missense, splice-site, and in-frame insertion/deletion (indel) mutations.20,21,42,43 Mutations are found more commonly in the 3′ half of the gene (62.7%), and some studies have found an excess of mutations in exons 39 and 48.15,32 Hannibal et al12 noted that several long polyglutamine tracts were encoded in exon 39, which may in part account for an increased frequency of mutations in this exon. Mutations in exons 10, 11, 31, 34, 39, and 48 comprise 62.7% of all discovered mutations; however, as these six exons are the largest and constitute 62% of the entire KMT2D coding sequence, this is not surprising. Micale et al21 demonstrated that messenger (m)RNA derived from individuals with nonsense mutations in KMT2D was degraded through nonsense-mediated decay. Miyake et al43 reported that nontruncating mutations had a tendency to cluster in or around the functional domains of KMT2D, whereas truncating mutations were found to be distributed throughout the entire coding sequence. Micale et al21 found approximately 46 missense mutations in 303 individuals with KS who underwent molecular genetic analysis. Mutations were found in the SET and in the PHD domains but, in general, were also scattered throughout the gene. Presence of mosaic mutations at levels as low as 15% have also been rarely reported.44 Whole-gene or whole-exon deletions or duplications of KMT2D also appear to be rare, accounting for about 5% of cases.44
Mutations in KDM6A are responsible for a minority (possibly up to 12%) of cases of KS.19,21,31,32,45 Mutation types have included the following (listed by decreasing frequency):nonsense or frameshift, larger deletions including KDM6A, splice-site, in-frame nontruncating, and intragenic multiexon deletions.19,21,30–32,45 Taken together, the data pertaining to KMT2D and KDM6A mutation types and locations suggest that the cause of the KS phenotype in affected individuals is haploinsufficiency.
Most mutations in KMT2D are de novo, although a few familial cases have been reported, with autosomal dominant inheritance. Similarly, most deletions and mutations in KDM6A for which parental samples were available have been found to be de novo, although a recent report of a three-generation family, in which a four-base-pair deletion in exon 17 of KDM6A was found in two affected brothers and their less-severely affected mother and maternal grandmother, suggests that X-linked inheritance is possible.31
Genotype–phenotype correlations
Because KMT2D was the first gene identified as causing KS, initial attempts at genotype–phenotype correlations focused on the differentiation of individuals with features of KS who were KMT2D-mutation-positive versus KMT2D-mutation-negative. For example, Makrythanasis et al20 reported that individuals with a KMT2D mutation were more likely to have the following features relative to mutation-negative individuals: arched eyebrows, blue sclerae, broad nasal root, flat nasal tip, large dysplastic ears, thin upper and full lower lips, short stature, joint laxity, intellectual disability, and frequent infections. Although not all mutation-positive individuals have been noted to have typical facial features,13 Bögershausen and Wollnik42 found that individuals with an identified KMT2D mutation had a significantly higher frequency of typical facial features compared to individuals without a KMT2D mutation. Hannibal et al12 and Banka et al15 reported that KMT2D-mutation-positive individuals are more likely to have renal anomalies, feeding problems, premature thelarche in females, joint dislocations, short stature, and palatal malformations in comparison with KMT2D–mutation-negative individuals.
Although most KMT2D-mutation-negative individuals have atypical features, so do up to 30% of mutation-positive individuals.15 It should also be noted that a subset of KMT2D-mutation-negative individuals are likely to have been misdiagnosed and may have a nonclassic presentation of a related condition.46
Only approximately 20 individuals have been identified to date with mutations or deletions in KDM6A.45 Limited genotype–phenotype correlations have focused primarily on the differences between affected males and affected females, as opposed to distinguishing patients with mutations in KDM6A from those who have KMT2D mutations. In females with a KDM6A mutation or deletion, the most consistent finding is short stature, followed by long palpebral fissures.30–32 Reported males have uniformly long palpebral fissures, hyperlaxity, and developmental delay.30,31,43 The degree of cognitive impairment in affected individuals has ranged from mild to severe in affected males and from normal or borderline intelligence quotient to severe intellectual disability in affected females.31 An overall comparison of the features of individuals with KS who have mutations in KMT2D (referred to as KS1) versus those who have mutations or deletions in KDM6A (referred to as KS2) is listed in Table 3.
 | Table 3 Comparison of features between individuals with mutations or deletions in KMT2D (KS1) versus those with mutations or deletions in KDM6A (KS2) |
In general, females with mutations in KDM6A tend to have a milder phenotype than males with mutation in either KDM6A or KMT2D, although at least one female with a mutation in KDM6A had a classic KS phenotype.30,31,43 Because of evidence that KDM6A escapes X-inactivation, this difference cannot necessarily be attributed to favorable X-inactivation in affected females. Miyake et al43 hypothesized that in order for normal males to compensate for being hemizygous for KDM6A, a gene on the Y chromosome (UTY) may serve a functionally similar role as KDM6A. They further hypothesized that in order for an individual to have a normal phenotype, an expression level of the gene product of KDM6A (coupled with the gene product of UTY in males) of 1.0 may be necessary, such that any female with decreased functioning of one KDM6A allele would fall under this threshold and exhibit variable features of the condition depending on the degree of impact that the mutation has on the expression level of KDM6A.
Advances in techniques to elucidate the genetic basis of KS
Approximately 10%–30% of individuals with clinical features of KS will not be found to have mutations or deletions in either KMT2D or KDM6A. Certainly, a subset of these individuals is misdiagnosed.46 Another possibility is that a subset of such individuals has a postzygotic mutation leading to mosaicism for a mutation or deletion in one of these genes. Mosaicism can be difficult to identify without the use of targeted deep sequencing or testing of various tissue types.47 However, this is unlikely to account for the majority of cases that are missed during routine molecular genetic testing of KMT2D and KDM6A.
WES was the technique that identified the most common molecular cause of KS.18 WES has been applied to individuals having a KS phenotype without a known KMT2D or KDM6A mutation and has to date not identified other genes that lead to classic KS. Now that whole-genome sequencing has become available at a lower cost, this may identify mutations in noncoding regions in and around KMT2D and KDM6A, such as promoter or enhancer mutations; however, this is not likely to account for all of the individuals with features of KS who do not have an identifiable mutation within the coding and immediate intron/exon boundary regions of KMT2D and KDM6A.
Similar to other conditions due to mutation or deletion of key components of a developmental pathway, such as the Noonan spectrum of disorders caused by mutations in multiple genes found in the RAS-MAPK pathway,48,49 there are probably other genes within the KMT2D and KDM6A transcriptional complex, its pathway, or a related pathway with similar function that lead to features of KS. For example, a patient with some features of KS was found to have, in addition to a de novo mutation in ANKRD11, which causes KBG syndrome, a de novo missense mutation in KDM1A, which encodes a histone H3 modifier whose function is to remove methyl groups from mono- and dimethylated lysine 4 and lysine 9, similar to the function of KDM6A.50–53 Therefore, targeted evaluation of genes that encode other MLL proteins and their respective interacting protein complexes may lead to the discovery of other genes, mutation or deletion in which leads to a small subset of individuals with features of KS.23
Prospects for management
Knowledge of the types of mutations and the functions of the genes that lead to KS opens the door to the possibility of targeted therapies that can be studied in model systems or in in vitro assays in the laboratory. Recently, Micale et al21 demonstrated that many KMT2D-truncating mutations result in nonsense-mediated mRNA decay, suggesting that readthrough therapy could possibly restore functions of the pathways affected by haploinsufficiency of KMT2D and KDM6A. Their proof-of-principle study involved evaluation of the ability of gentamicin to suppress 14 KMT2D nonsense mutations and two KDM6A nonsense mutations using a previously reported in vitro readthrough reporter system.21,54 Similar to other studies using this compound, the authors21,55 found a statistically significant difference between basal and gentamicin-induced readthrough in eleven KTMT2D and one KDM6A nonsense mutations, suggesting that the sequences surrounding the nonsense mutation had an impact on the success of the treatment. For example, readthrough efficiency was improved if a cytosine was located at a +4 position downstream of the nonsense mutation and/or a uracil was present at a –1 position upstream of the stop codon. This encouraging result suggests that other aminoglycoside compounds (such as amikacin) or nonaminoglycoside readthrough compounds (such as PTC124, NB30, RTC12, and RTC14) could be potential therapies for KS caused by nonsense mutations.21,56 Unfortunately, this therapy would probably not be able to prevent the physical anomalies of KS that are present at birth but could have the potential to affect the neurologic and cognitive issues in affected individuals.
Another experimental type of targeted therapy has focused on modulating gene expression through correction of abnormal epigenetic chromatin states. Studies are currently focused on therapies, such as use of histone deacetylase inhibitors (HDACi), that might promote chromatin opening. In a mouse model of KS in which the mice have a heterozygous mutation in Kmt2d, multiple abnormalities are found in the granule cell layer of the dentate gyrus in the brain, which correlates with hippocampal memory deficits.57 This area of the dentate nucleus has also been associated with adult neurogenesis. Bjornsson et al57 administered oral AR-42, an HDACi, to heterozygous KS mice at two different stages: 1–2 months of age (pups) and 5–6 months of age (adults). They found that such mice demonstrated normalization of the structural and functional deficits in the dentate gyrus, leading to improved neurogenesis and memory, and that this was associated with the return of H3K4 trimethylation. The authors concluded that in the mouse model of KS, HDACi therapy encouraged chromatin opening by influencing histone modification, which subsequently prevented and even reversed the memory deficits. They hypothesized that the intellectual disability in KS is in part due to a deficiency of adult neurogenesis, which can be reversed through pharmacotherapy that encourages an open chromatin state. As ketosis also acts as an endogenous HDACi, the authors further hypothesized that a ketogenic diet may be beneficial as a treatment for this condition, although this has not been systematically studied in humans with KS yet.
Similarly, Freiderich ataxia (FRDA) is a condition that leads to progressive ataxia, cardiomyopathy, scoliosis, muscle weakness, bladder dysfunction, and lower limb spasticity.58 It is caused by abnormal transcriptional repression of FXN, encoding frataxin, with most affected individuals having at least one abnormal allele that has a GAA trinucleotide repeat expansion in the first exon, which leads to transcriptional silencing through repressive chromatin formation and abnormal DNA structures.59 Using knowledge of the mechanism of disease, therapy was trialed using an HDACi both in vitro using patient-derived pluripotent stem cells induced to form neuronal cells and in affected individuals in vivo.60 The drug RG2833 when applied to FRDA neuronal cells led to an epigenetic change to FXN, with subsequent increase in both FXN mRNA levels and frataxin protein. In phase I clinical trials, peripheral blood mononuclear cells taken from patients on study drug demonstrated an increase in FXN mRNA and in H3L9 acetylation, with no safety issues reported.60 This type of proof-of-principle study in humans suggests that drugs that target abnormal epigenetic modifications could be used to treat conditions such as KS in the future.
Conclusion
To date, two genes have been identified that lead to KS; the first gene was discovered using WES, as previous candidate gene approaches failed to discover the underlying cause. The proposed mechanisms that lead to KS involve epigenetic alterations that affect developmental gene expression in early embryogenesis, accounting for the fact that this condition can influence multiple organ systems and developmental processes. However, approximately 10%–30% of individuals with features of KS do not have an identifiable mutation in either KMT2D or KDM6A, suggesting that other genes that encode proteins that couple with KMT2D or KDM6A or in related developmental pathways may account for a small subset of cases. Candidate gene approaches paired with whole-genome technologies could be used to identify such genes. Additionally, advances in the understanding of the molecular mechanisms that lead to KS have the potential to lead to targeted therapies that either bypass nonsense mutations or are able to modify the epigenetic state of chromatin, thus ameliorating disease symptoms.
Acknowledgment
The author thanks Johann Adam for his work on the figures.
Disclosure
The author reports no conflict of interest in this work.
References
Kuroki Y, Suzuki Y, Chyo H, Hata A, Matsui I. A new malformation syndrome of long palpebral fissures, large ears, depressed nasal tip and skeletal anomalies associated with postnatal dwarfism and mental retardation. J Pediatr. 1981;99(4):570–573. | |
Niikawa N, Matsuura N, Fukushima Y, Ohsawa T, Kajii T. Kabuki make-up syndrome: a syndrome of mental retardation, unusual facies, large and protruding ears, and postnatal growth deficiency. J Pediatr. 1981;99(4):565–569. | |
Niikawa N, Kuroki Y, Kajii T, et al. Kabuki make-up (Niikawa-Kuroki) syndrome: a study of 62 patients. Am J Med Genet. 1988;31(3):565–589. | |
Ho HH, Eaves LC. Kabuki make-up (Niikawa-Kuroki) syndrome: cognitive abilities and autistic features. Dev Med Child Neurol. 1997;39(7):487–490. | |
Kawame H, Hannibal MC, Hudgins L, Pagon RA. Phenotypic spectrum and management issues in Kabuki syndrome. J Pediatr. 1999;134(4):480–485. | |
Matsumoto N, Niikawa N. Kabuki make-up syndrome: a review. Am J Med Genet C Semin Med Genet. 2003;117C(1):57–65. | |
Armstrong L, Abd El Moneim A, Aleck K, et al. Further delineation of Kabuki syndrome in 48 well-defined new individuals. Am J Med Genet A. 2005;132A(3):265–272. | |
Ciprero KL, Clayton-Smith J, Donnai D, Zimmerman RA, Zackai EH, Ming JE. Symptomatic Chiari I malformation in Kabuki syndrome. Am J Med Genet A. 2005;132A(3):273–275. | |
Defloor T, van Borsel J, Schrander-Stumpel CT, Curfs LM. Expressive language in children with Kabuki syndrome. Am J Med Genet A. 2005;132A(3):256–259. | |
Schrander-Stumpel CT, Spruyt L, Curfs LM, Defloor T, Schrander JJ. Kabuki syndrome: clinical data in 20 patients, literature review, and further guidelines for preventive management. Am J Med Genet A. 2005;132A(3):234–243. | |
Vaux KK, Jones KL, Jones MC, Schelley S, Hudgins L. Developmental outcome in Kabuki syndrome. Am J Med Genet A. 2005;132A(3):263–264. | |
Hannibal MC, Buckingham KJ, Ng SB, et al. Spectrum of MLL2 (ALR) mutations in 110 cases of Kabuki syndrome. Am J Med Genet A. 2011;155A(7):1511–1516. | |
Li Y, Bögershausen N, Alanay Y, et al. A mutation screen in patients with Kabuki syndrome. Hum Genet. 2011;130(6):715–724. | |
Paulussen AD, Stegmann AP, Blok MJ, et al. MLL2 mutation spectrum in 45 patients with Kabuki syndrome. Hum Mutat. 2011;32(2):E2018–E2025. | |
Banka S, Veeramachaneni R, Reardon W, et al. How genetically heterogeneous is Kabuki syndrome?: MLL2 testing in 116 patients, review and analyses of mutation and phenotypic spectrum. Eur J Hum Genet. 2012;20(4):381–388. | |
Dentici ML, De Pede A, Lepri FR, et al. Kabuki syndrome: clinical and molecular diagnosis in the first year of life. Arch Dis Child. 2014;100(2):158–164. | |
White SM, Thompson EM, Kidd A, et al. Growth, behavior, and clinical findings in 27 patients with Kabuki (Niikawa-Kuroki) syndrome. Am J Med Genet A. 2004;127A(2):118–127. | |
Ng SB, Bigham AW, Buckingham KJ, et al. Exome sequencing identifies MLL2 mutations as a cause of Kabuki syndrome. Nat Genet. 2010;42(9):790–793. | |
Lederer D, Grisart B, Digilio MC, et al. Deletion of KDM6A, a histone demethylase interacting with MLL2, in three patients with Kabuki syndrome. Am J Hum Genet. 2012;90(1):119–124. | |
Makrythanasis P, van Bon BW, Steehouwer M, et al. MLL2 mutation detection in 86 patients with Kabuki syndrome: a genotype-phenotype study. Clin Genet. 2013;84(6):539–545. | |
Micale L, Augello B, Maffeo C, et al. Molecular analysis, pathogenic mechanisms, and readthrough therapy on a large cohort of Kabuki syndrome patients. Hum Mutat. 2014;35(7):841–850. | |
Kim T, Buratowski S. Dimethylation of H3K4 by Set1 recruits the Set3 histone deacetylase complex to 5′ transcribed regions. Cell. 2009; 137(2):259–272. | |
Shinsky SA, Hu M, Vought VE, et al. A non-active-site SET domain surface crucial for the interaction of MLL1 and the RbBP5/Ash2L heterodimer within MLL family core complexes. J Mol Biol. 2014;426(12):2283–2299. | |
Prasad R, Zhadanov AB, Sedkov Y, et al. Structure and expression pattern of human ALR, a novel gene with strong homology to ALL-1 involved in acute leukemia and to Drosophila trithorax. Oncogene. 1997;15(5):549–560. | |
Aziz A, Lui QC, Dilworth FJ. Regulating a master regulator: establishing tissue-specific gene expression in skeletal muscle. Epigenetics. 2010;5(8):691–695. | |
Issaeva I, Zonis Y, Rozovskaia T, et al. Knockdown of ALR (MLL2) reveals ALR target genes and leads to alterations in cell adhesion and growth. Mol Cell Biol. 2007;27(5):1889–1903. | |
Dhar SS, Lee SH, Kan PY, et al. Tans-tail regulation of MLL4-catalyzed H3K4 methylation by H4R3 symmetric dimethylation is mediated by a tandem PHD of MLL4. Genes Dev. 2012;26(24):2749–2762. | |
Zhang P, Lee H, Brunzelle JS, Couture JF. The plasticity of WDR5 peptide-binding cleft enables the binding of the SET1 family of histone methyltransferases. Nucleic Acids Res. 2012;40(9):4237–4246. | |
Dharmarajan V, Lee JH, Patel A, Skalnik DG, Cosgrove MS. Structural basis for WDR5 interaction (Win) motif recognition in human SET1 family histone methyltransferase. J Biol Chem. 2012;287(33):27275–27289. | |
Miyake N, Mizuno S, Okamoto N, et al. KDM6A point mutations cause Kabuki syndrome. Hum Mutat. 2013;34(1):108–110. | |
Lederer D, Shears D, Benoit V, Verellen-Dumoulin C, Maystadt I. A three generation X-linked family with Kabuki syndrome phenotype and a frameshift mutation in KDM6A. Am J Med Genet A. 2014;164A(5):1289–1292. | |
Cheon CK, Sohn YB, Ko JM, et al. Identification of KMT2D and KDM6A mutations by exome sequencing in Korean patients with Kabuki syndrome. J Hum Genet. 2014;59(6):321–325. | |
Greenfield A, Carrel L, Pennisi D, et al. The UTX gene excapes X inactivation in mice and humans. Hum Mol Genet. 1998;7(4):737–742. | |
Xu J, Deng X, Watkins R, Disteche CM. Sex-specific differences in expression of histone demethylase Utx and Uty in mouse brain and neurons. J Neurosci. 2008;28(17):4521–4527. | |
Santos-Rosa H, Schneider R, Bannister AJ, et al. Active genes are tri-methylated at K4 of histone H3. Nature. 2002;419(6905):407–411. | |
Ansari KI, Hussain I, Shrestha B, Kasiri S, Mandal SS. HOXC6 is transcriptionally regulated via coordination of MLL histone methylase and estrogen receptor in an estrogen environment. J Mol Biol. 2011;411(2):334–349. | |
Ansari KI, Mandal SS. Mixed lineage leukemia: roles in gene expression, hormone signaling and mRNA processing. FEBS J. 2010;277(8):1790–1804. | |
Eissenberg JC, Shilatifard A. Histone H3 lysine 4 (H3K4) methylation in development and differentiation. Dev Biol. 2010;339(2):240–249. | |
Lindgren AM, Hoyos T, Talkowski ME, et al. Haploinsufficiency of KDM6A is associated with severe psychomotor retardation, global growth restriction, seizures, and cleft palate. Hum Genet. 2013;132(5):537–552. | |
Lee S, Lee JW, Lee SK. UTX, a histone H3-lysine 27 demethlase, acts as a critical switch to activate the cardiac developmental program. Dev Cell. 2012;22(1):25–37. | |
Hemming S, Cakouros D, Isenmann S, et al. EZH2 and KDM6A act as an epigenetic switch to regulate mesenchymal stem cell lineage specification. Stem Cells. 2014;32(3):802–815. | |
Bögershausen N, Wollnik B. Unmasking Kabuki syndrome. Clin Genet. 2013;83(3):201–211. | |
Miyake N, Koshimizu E, Okamoto N, et al. MLL2 and KDM6A mutations in patients with Kabuki syndrome. Am J Med Genet A. 2013;161A(9):2234–2243. | |
Banka S, Howard E, Bunstone S, et al. MLL2 mosaic mutations and intragenic deletion-duplications in patients with Kabuki syndrome. Clin Genet. 2013;83(5):467–471. | |
Banka S, Lederer D, Benoit V, et al. Novel KDM6A (UTX) mutations and a clinical and molecular review of the X-linked Kabuki syndrome (KS2). Clin Genet. Epub 2014 Feb 14. | |
Schulz Y, Freese L, Mänz J, et al. CHARGE and Kabuki syndromes: a phenotypic and molecular link. Hum Mol Genet. 2014;23(16):4396–4405. | |
Rivière JB, Mirzaa GM, O’Roak BJ, et al; Finding of Rare Disease Genes (FORGE) Canada Consortium. De novo germline and postzygotic mutations in AKT3, PIK3R2 and PIK3CA cause a spectrum of related megalencephaly syndromes. Nat Genet. 2012;44(8):934–940. | |
Sarkozy A, Carta C, Moretti S, et al. Germline BRAF mutations in Noonan, LEOPARD, and cardiofaciocutaneous syndromes: molecular diversity and associated phenotypic spectrum. Hum Mutat. 2009;30(4):695–702. | |
Bezniadow N, Gos M, Obersztyn E. The RASopathies as an example of RAS/MAPK pathway disturbances-clinical presentation and molecular pathogenesis of selected syndromes. Dev Period Med. 2014;18(3):285–296. | |
Shi Y, Lan F, Matson C, et al. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell. 2004;119(7):941–953. | |
Lin T, Ponn A, Hu X, Law BK, Lu J. Requirement of the histone demethylase LSD1 in Snai1-mediated transcriptional repression during epithelial-mesenchymal transition. Oncogene. 2010;29(35):4896–4904. | |
Shao GB, Huang XJ, Gong AH, Zhang ZJ, Lu RZ, Sang JR. Histone demethylase LSD1 and its biological functions. Yi Chuan. 2010;32(4):331–338. | |
Tunovic S, Barkovich J, Sherr EH, Slavotinek AM. De novo ANKRD11 and KDM1A gene mutations in a male with features of KBG syndrome and Kabuki syndrome. Am J Med Genet A. 2014;164A(7):1744–1749. | |
Floquet C, Deforges J, Rousset JP, Bidou L. Rescue of non-sense mutated p53 tumor suppressor gene by aminoglycosides. Nucleic Acids Res. 2011;39(8):3350–3362. | |
Floquet C, Hatin I, Rousset JP, Bidou L. Statistical analysis of readthrough levels for nonsense mutations in mammalian cells reveals a major determinant of response to gentamicin. PLoS Genet. 2012;8(3):e1002608. | |
Bordeira-Carrico R, Pego AP, Santos M, Oliveira C. Cancer syndromes and therapy to stop-codon readthrough. Trends Mol Med. 2012;18(11):667–678. | |
Bjornsson HT, Benjamin JS, Zhang L, et al. Histone deacetylase inhibition rescues structural and functional brain deficits in a mouse model of Kabuki syndrome. Sci Transl Med. 2014;6(256):256ra135. | |
Filla A, De Michele G, Coppola G, et al. Accuracy of clinical diagnostic criteria for Friedreich’s ataxia. Mov Disord. 2000;15(6):1255–1258. | |
Chutake YK, Costello WN, Lam C, Bidichandani SI. Altered nucleosome positioning at the transcription start site and deficient transcriptional initiation in Friedreich ataxia. J Biol Chem. 2014;289(22):15194–15202. | |
Soragni E, Miao W, Iudicello M, et al. Epigenetic therapy for Friedreich ataxia. Ann Neurol. 2014;76(4):489–508. | |
Adam MP, Hudgins L. Kabuki syndrome: a review. Clin Genet. 2005;67(3):209–219. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.