Back to Journals » OncoTargets and Therapy » Volume 12
INPP4B promotes colorectal cancer cell proliferation by activating mTORC1 signaling and cap-dependent translation
Authors Ruan X, Liu X, Yang Z, Zhang S ![]() , Li Q, Lin C
, Li Q, Lin C
Received 4 September 2018
Accepted for publication 6 February 2019
Published 23 April 2019 Volume 2019:12 Pages 3109—3117
DOI https://doi.org/10.2147/OTT.S186365
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr XuYu Yang
Xin-Hua Ruan,1,* Xi-Mei Liu,1,* Zhi-Xiang Yang,1,* Shao-Peng Zhang,1,* Quan-Zheng Li,1 Chun-Sheng Lin2
1Department of Cardiac Surgery, TianJin Union Medical Centre, Tianjin, People’s Republic of China; 2Department of Medical Service, TianJin Union Medical Centre, Tianjin, People’s Republic of China
*These authors contributed equally to this work
Background and objective: Inositol polyphosphate 4-phosphatase type II (INPP4B) is overexpressed in CRC tissues, and emerges as an oncogene. However, the mechanism by which INPP4B regulates CRC cell proliferation remains largely unclear. In this study, we aimed to investigate the regulatory mechanisms of INPP4B in CRC.
Materials and methods: The expression levels of mRNA were detected by qRT-PCR. The expression levels of protein were determined by Western blot. Cell Counting Kit-8 (CCK-8) assays and BrdU incorporation assays were performed to evaluate cell proliferation abilities. Bicistronic luciferase assays and the m7GTP pull down assay were performed to measure the cap-dependent translation in cells.
Results: INPP4B promotes CRC cell proliferation by increasing mTORC1 activity. Furthermore, it was shown that the activation of mTORC1 signaling by INPP4B led to increased cap-dependent translation, which is essential for INPP4B-mediated CRC cell proliferation. Finally, it was demonstrated that increased AKT and serum and glucocorticoid-inducible kinase 1 activity contributed to the activation of cap-dependent translation induced by INPP4B.
Conclusion: Collectively, the present study reveals INPP4B promotes colorectal cancer cell proliferation by activating mTORC1 signaling and cap-dependent translation.
Keywords: colorectal cancer, INPP4B, mTORC1, 4E-BP1, cap-dependent translation
Introduction
Colorectal cancer (CRC) is the third most frequently diagnosed tumor type, and ranks as the fourth leading cause of cancer mortality worldwide.1 With the improvement in present living conditions, there has been a marked increase in the incidence of CRC in China. There were ~376,300 new cases of CRC and 191,000 CRC-related deaths in China in 2015.2 Hence, elucidating the molecular basis underlying CRC development is crucial for facilitating the development of novel diagnostic and therapeutic strategies against this disease.
Cap-dependent translation deregulation plays a critical role in the development of many different types of cancers by enhancing the translational efficiency of multiple oncogenic mRNAs involved in cell proliferation, survival, migration, and invasion.3 Furthermore, cap-dependent translation is under the control of the eukaryotic translation initiation factor 4F (eIF4F) complex, which is composed of eukaryotic translation initiation factor 4E (eIF4E), eukaryotic translation initiation factor 4G (eIF4G), and eukaryotic translation initiation factor 4A (eIF4A), and initiates protein translation by recruiting the 40S ribosome subunit to the 5′ cap mRNA.4 In addition, mTOR kinase complex 1 (mTORC1) regulates the assembly of the eIF4F complex by phosphorylating eIF4E-binding protein 1 (4E-BP1). Hypophosphorylated 4E-BP1 represses the eIF4F assembly by competing with eIF4G for a binding site on eIF4E, while the phosphorylation of 4E-BP1 releases the binding to eIF4E, leading to the formation of the eIF4F complex and translation initiation. The abnormal activation of mTORC1 and high phosphorylation of 4E-BP1 have been found in CRC tissues, which can predict the poor prognosis of patients with CRC.5 Furthermore, inhibiting cap-dependent translation can effectively suppress the proliferation and migration of CRC cells,6,7 suggesting that deregulated cap-dependent translation contributes to CRC progression.
Inositol polyphosphate 4-phosphatase type II (INPP4B) is an inositol polyphosphate phosphatase that has the ability to hydrolyze Phosphatidylinositol 3,4-bisphosphate [PtdIns-3,4-P2] to generate PtdIns-3-P. Since PtdIns-3,4-P2 is required for the full activation of Akt, INPP4B negatively regulates Akt activity by reducing the cellular levels of PtdIns-3,4-P2.8 Accordingly, with the inhibitory effect of INPP4B on Akt activity, INPP4B functions as a tumor suppressor in many cancers, such as prostate cancer, lung cancer, bladder cancer, and ovarian cancer.9–12 Interestingly, increasing studies suggest that INPP4B plays an oncogenic role in some types of cancers, including breast cancer and melanoma.13–15 Serum and glucocorticoid-inducible kinase 1 (SGK3), a member of the AGC family of kinases that is highly homologous to Akt, mediates the oncogenic role of INPP4B in these cancers.13–15 In CRC, INPP4B exhibited a significantly elevated expression in tumor tissues, when compared with adjacent noncancerous colon tissues. Furthermore, INPP4B depletion suppressed the proliferation in CRC cells and reduced colon cancer xenograft growth, indicating that INPP4B plays an oncogenic role in CRC. In CRC cells, INPP4B not only activates SGK3, but also positively regulates Akt activity by decreasing the expression of PTEN, a repressor of Akt signaling.9,16 Although the role of SGK3 and Akt in mediating the oncogenic role of INPP4B in CRC has been characterized, it remains unclear how SGK3 and Akt execute their role in CRC cells. Accumulated evidence has demonstrated that SGK3, similar to Akt, can activate mTORC1 to promote cancer progression.17,18 Mechanistically, SGK3 phosphorylates TSC2 and inhibits GTPase activating protein activity of TSC2, leading to the activation of the Rheb GTPase and hence mTORC1.18 Since SGK3 and Akt act downstream of INPP4B in CRC cells, it was presumed that INPP4B may activate mTORC1, which leads to increased cap-dependent translation and CRC cell proliferation.
In the present study, it was found that INPP4B promotes the proliferation of CRC cells by increasing mTORC1 activity. In addition, it was found that INPP4B activates cap-dependent translation, which further verifies the essential role of cap-dependent translation activation in CRC cell proliferation. Finally, it was demonstrated that AKT and SGK3 activation is required for INPP4B-mediated cap-dependent translation and cell proliferation. The present data indicate that INPP4B promotes CRC cell proliferation by activating mTORC1 signaling and cap-dependent translation.
Materials and methods
Antibodies and reagents
Antibodies against P70S6K, pP70S6K (T389), 4EBP-1, p4EBP-1 (S65), eIF4E, eIF4G, cyclinD1, AKT, and SGK3 were obtained from Cell Signaling Technology (Beverly, MA, USA). Anti-INPP4B and anti-tubulin were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). The antibody against HPIP was obtained from Proteintech (Chicago, IL, USA). RAD001 and 4EGI-1 were purchased from Selleck (Houston, TX, USA) and Calbiochem (Darmstadt, Germany), respectively.
Cells and cell culture
SW620 and HCT116 cells (supplied by Shanghai Suer Biotechnology Co., Ltd.) were cultured in DMEM (Thermo Fisher Scientific, Inc., Waltham, MA, USA) containing 10% FBS (Thermo Fisher Scientific) and 100 U/mL penicillin/streptomycin in an incubator with 5% CO2 at 37°C.
In order to generate SW620 cells with a stable expression of INPP4B, SW620 cells were transfected with the pCMV-2B control vector or pCMV-2B-INPP4B vector with Lipofectamine 3000 (Thermo Fisher Scientific), according to the recommendations of the manufacturer. At 48 hours post-transfection, the transfected cells were selected with 0.5 mg/mL of G418 (Sigma-Aldrich Co., St Louis, MO, USA) for 2 weeks.
For establishing stable INPP4B knockdown HCT116 cells, HCT116 cells were infected with lentiviral particles harboring control shRNA or INPP4B shRNA (GeneChem Co., Shanghai, China). After 48 hours of infection, cells were selected in culture medium containing 1 μg/mL of puromycin (Thermo Fisher Scientific) for 10 days.
Western blotting
Cells were washed and lysed in ice-cold RIPA buffer (50 mM of Tris-HCl, pH 7.5, 150 mM of NaCl, 1% NP-40, 1% sodium deoxycholate, 0.1% SDS, 1 mM of ethylenebis (oxyethylenenitrilo) tetraacetic acid (EGTA), and 1 mM of EDTA) with a protease inhibitor cocktail (Roche Applied Sciences, Indianapolis, IN, USA). Then, 50 μg of the total proteins was separated by 10% SDS-PAGE and transferred onto nitrocellulose membranes (Amersham Biosciences, Piscataway, NJ, USA). Afterward, the membranes were blocked with 5% nonfat milk for 1 hour at room temperature, incubated with primary antibodies overnight at 4°C, and incubated with the appropriate horseradish peroxidase–conjugated secondary antibody (Promega Corporation, Madison, WI, USA) for 1 hour. Membrane blot signals were detected using the enhanced chemiluminiscence detection kit (Amersham Biosciences).
SiRNA and transient transfections
For 4E-BP1, AKT, or SGK3 silencing, cells were transfected with siRNAs against 4E-BP1, AKT, or SGK3 using Lipofectamine RNAiMAX (Thermo Fisher Scientific), according to manufacturer’s instructions. Then, cells were subjected to Western blot analysis and bicistronic luciferase assays at 48 hours post-transfection, or subjected to Cell Counting Kit-8 (CCK-8) assay at 24 hours post-transfection.
Bicistronic luciferase assays
Cells were transiently transfected with a dual-Renilla-firefly-luciferase pcDNA3-rLuc-PolioIRES-fLuc reporter, which directs the cap-dependent translation of the Renilla luciferase gene and cap-independent Polio IRES-mediated translation of the firefly luciferase gene. At 48 hours post-transfection, cells were collected for luciferase activity detection using the Dual-Luciferase Reporter Assay System (Promega). The ratio between Renilla and firefly luciferase activity (LucR/LucF) was calculated for cap-dependent translational activity. Three independent experiments were performed.
The m7GTP pull down assay
0.9% NaCl, 1% NP-40, 1% sodium deoxycholate, 0.1% SDS, and 1 mm cells were washed and lysed in ice-cold RIPA buffer (50 mM of Tris-HCl, pH 7.5, 150 mM of EGTA, and 1 mM of EDTA) with a protease inhibitor cocktail (Roche Applied Sciences). Then, the cell lysates were incubated with 30 μL of m7GTP sepharose beads (GE Healthcare, Chalfont St Giles, England). After incubation with rocking for 3 hours at 4°C, the precipitates were washed three times with ice-cold lysis buffer, and used for Western blotting analysis. The original lysate was used as a loading control.
Cell proliferation assays
Cells were seeded at a density of 2×103 cells per well in a 96-well plate, and cell proliferation was measured using CCK-8 (Dojindo Laboratories, Tokyo, Japan) assay, according to manufacturer’s instructions. Absorbance was measured at 450 nm using a microplate reader. Three independent experiments were performed.
BrdU incorporation assays
A FITC BrdU Flow Kit (BD Biosciences, San Jose, CA, USA) was used to detect the cell proliferation. Briefly, bromodeoxyuridine (BrdU, 10 μM) was added to the culture medium and incubated for 4 hours, followed by flow cytometry analysis. Three independent experiments were performed.
Statistical analysis
Data were presented as mean ± SD of samples from three independent experiments. All data were analyzed using the SPSS 17.0 software (SPSS, Chicago, IL, USA). The statistical differences between two groups were analyzed using an unpaired Student’s t-test. One-way ANOVA, followed by Dunnett’s post hoc test, was used to detemrine the statistical significance when comparing more than two groups. A P-value <0.05 was considered statistically significant.
Results
INPP4B promotes the proliferation of colon cancer cells by increasing mTORC1 activity
Given that previous studies have demonstrated that INPP4B promoted AKT and SGK3 activation in CRC cells,9 and the well-characterized role of AKT and SGK3 in activating mTORC1, it was hypothesized that INPP4B might have a positive impact on mTORC1 activity. In order to test this hypothesis, the effect of INPP4B overexpression on mTORC1 activity in SW620 cells was first examined, which expressed relatively low levels of endogenous INPP4B in CRC cells. As expected, the enforced expression of INPP4B significantly enhanced the phosphorylation of P70S6K and 4EBP1, which are two well-known downstream targets of mTORC1 (Figure 1A). In contrast, silencing the expression of INPP4B profoundly decreased the phosphorylation of P70S6K and 4EBP1 in HCT116 cells, expressing relatively high levels of endogenous INPP4B (Figure 1B). Together, these data suggest that INPP4B increases mTORC1 activity in CRC cells.
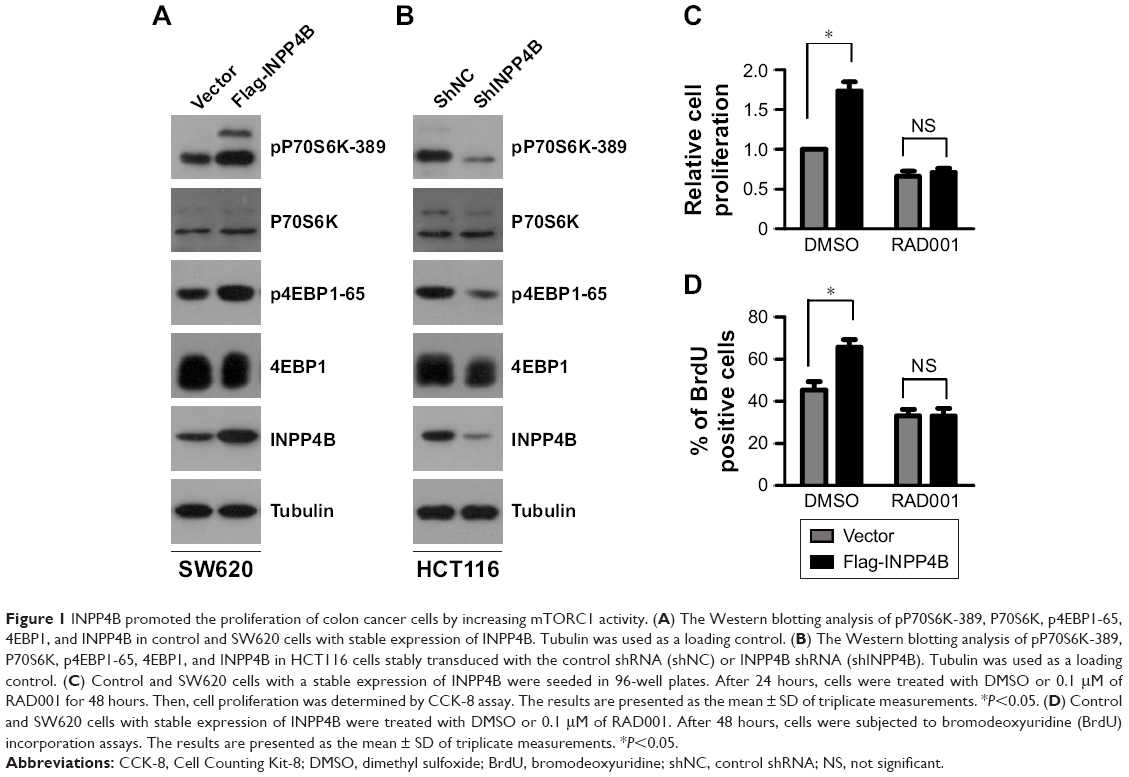 | Figure 1 INPP4B promoted the proliferation of colon cancer cells by increasing mTORC1 activity. (A) The Western blotting analysis of pP70S6K-389, P70S6K, p4EBP1-65, 4EBP1, and INPP4B in control and SW620 cells with stable expression of INPP4B. Tubulin was used as a loading control. (B) The Western blotting analysis of pP70S6K-389, P70S6K, p4EBP1-65, 4EBP1, and INPP4B in HCT116 cells stably transduced with the control shRNA (shNC) or INPP4B shRNA (shINPP4B). Tubulin was used as a loading control. (C) Control and SW620 cells with a stable expression of INPP4B were seeded in 96-well plates. After 24 hours, cells were treated with DMSO or 0.1 μM of RAD001 for 48 hours. Then, cell proliferation was determined by CCK-8 assay. The results are presented as the mean ± SD of triplicate measurements. *P<0.05. (D) Control and SW620 cells with stable expression of INPP4B were treated with DMSO or 0.1 μM of RAD001. After 48 hours, cells were subjected to bromodeoxyuridine (BrdU) incorporation assays. The results are presented as the mean ± SD of triplicate measurements. *P<0.05. |
Subsequently, it was determined whether mTORC1 activity is involved in the regulation of CRC cell proliferation by INPP4B. The CCK-8 assay result revealed that INPP4B overexpression markedly promoted the proliferation of SW620 cells, while the treatment of RAD001, a specific inhibitor of mTORC1, reversed the effect of INPP4B (Figure 1C). BrdU incorporation assay further demonstrated that RAD001 treatment attenuated the increased proliferation of SW620 cells by INPP4B overexpression (Figure 1D). Collectively, these results suggest that the increase in mTORC1 activity contributes to INPP4B-mediated colon cancer cell proliferation.
INPP4B activates cap-dependent translation in colon cancer cells
The above-mentioned results revealed that INPP4B activates mTORC1 in CRC cells. Given that mTORC1 activation positively regulates cap-dependent translation, it was presumed that INPP4B may enhance cap-dependent translation in CRC cells. In order to test this hypothesis, the effect of INPP4B overexpression on cap-dependent translation was first detected by utilizing a dual-luciferase reporter system, which has been widely used for monitoring the cap-dependent translation in cells. As expected, INPP4B overexpression significantly enhanced the cap-dependent translation in SW620 cells (Figure 2A). In contrast, the knockdown of endogenous INPP4B suppressed the cap-dependent translation in HCT116 cells (Figure 2B). Cap-dependent translation is under the control of the eIF4F complex, and its assembly could be accessed through the 7-methyl GTP sepharose bead assay. Next, the effect of INPP4B on the assembly of the eIF4F complex was examined. As shown in Figure 2C, INPP4B overexpression enhanced the formation of the eIF4F complex, as reflected by the increased interaction between eIF4E and eIF4G (Figure 2C). Conversely, INPP4B depletion dramatically impaired the assembly of the eIF4F complex (Figure 2D). In addition, it was explored whether the increase in mTORC1 activity is required for INPP4B-mediated cap-dependent translation. Strikingly, RAD001 treatment reversed the increased cap-dependent translation caused by INPP4B overexpression in SW620 cells (Figure 2E). Collectively, these data suggest that INPP4B enhances cap-dependent translation by activating mTORC1 in colon cancer cells.
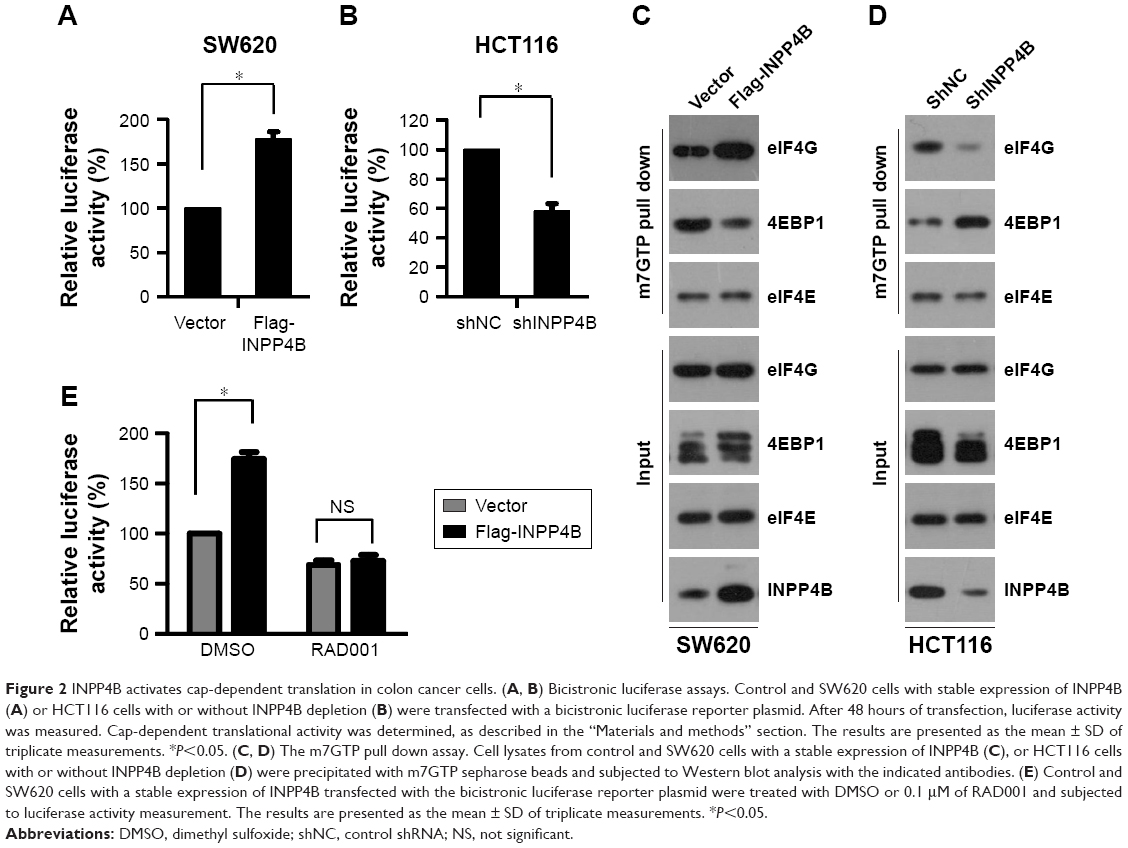 | Figure 2 INPP4B activates cap-dependent translation in colon cancer cells. (A, B) Bicistronic luciferase assays. Control and SW620 cells with stable expression of INPP4B (A) or HCT116 cells with or without INPP4B depletion (B) were transfected with a bicistronic luciferase reporter plasmid. After 48 hours of transfection, luciferase activity was measured. Cap-dependent translational activity was determined, as described in the “Materials and methods” section. The results are presented as the mean ± SD of triplicate measurements. *P<0.05. (C, D) The m7GTP pull down assay. Cell lysates from control and SW620 cells with a stable expression of INPP4B (C), or HCT116 cells with or without INPP4B depletion (D) were precipitated with m7GTP sepharose beads and subjected to Western blot analysis with the indicated antibodies. (E) Control and SW620 cells with a stable expression of INPP4B transfected with the bicistronic luciferase reporter plasmid were treated with DMSO or 0.1 μM of RAD001 and subjected to luciferase activity measurement. The results are presented as the mean ± SD of triplicate measurements. *P<0.05. |
INPP4B promotes colon cancer cell proliferation by activating cap-dependent translation
Next, it was determined whether the enhanced cap-dependent translation contributes to increased cell proliferation by INPP4B. The protein expression levels of cyclinD1, a key effector for regulating cell proliferation, was first detected in SW620 cells with or without INPP4B overexpression. As shown in Figure 3A, INPP4B overexpression markedly enhanced the protein expression levels of cyclinD1 in SW620 cells. However, the RNA expression levels of CCND1 was not significantly altered by INPP4B overexpression (Figure 3B), as assessed by quantitative PCR assay, indicating that INPP4B promotes cyclinD1 expression by increasing its protein translation, but not mRNA transcription. However, the treatment of 4EGI-1, a selective eIF4E/eIF4G interaction inhibitor, almost completely abolished the increase in cyclinD1 expression caused by the INPP4B overexpression (Figure 3A), suggesting that INPP4B promotes cyclinD1 expression by activating cap-dependent translation. Furthermore, the increased proliferation of SW620 cells by INPP4B overexpression was remarkably attenuated by 4EGI-1 treatment, as assessed by the CCK-8 assay (Figure 3C).
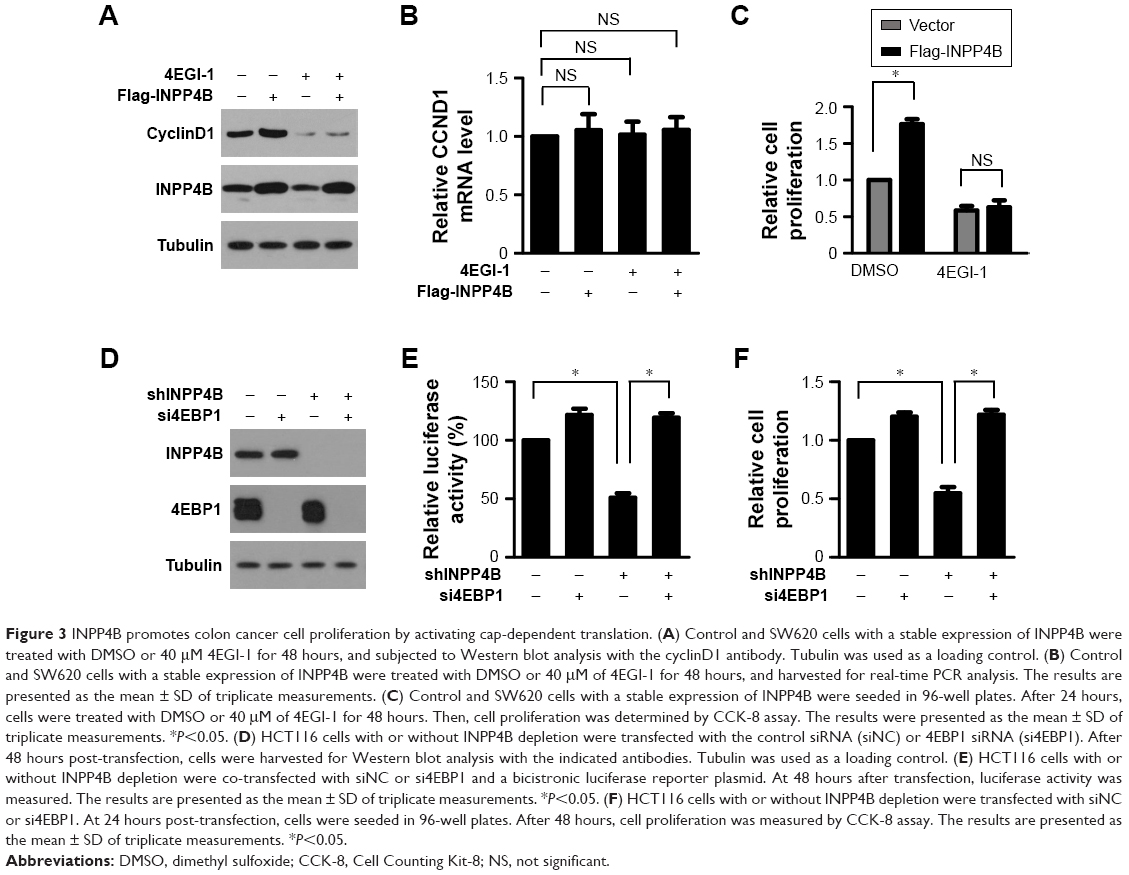 | Figure 3 INPP4B promotes colon cancer cell proliferation by activating cap-dependent translation. (A) Control and SW620 cells with a stable expression of INPP4B were treated with DMSO or 40 μM 4EGI-1 for 48 hours, and subjected to Western blot analysis with the cyclinD1 antibody. Tubulin was used as a loading control. (B) Control and SW620 cells with a stable expression of INPP4B were treated with DMSO or 40 μM of 4EGI-1 for 48 hours, and harvested for real-time PCR analysis. The results are presented as the mean ± SD of triplicate measurements. (C) Control and SW620 cells with a stable expression of INPP4B were seeded in 96-well plates. After 24 hours, cells were treated with DMSO or 40 μM of 4EGI-1 for 48 hours. Then, cell proliferation was determined by CCK-8 assay. The results were presented as the mean ± SD of triplicate measurements. *P<0.05. (D) HCT116 cells with or without INPP4B depletion were transfected with the control siRNA (siNC) or 4EBP1 siRNA (si4EBP1). After 48 hours post-transfection, cells were harvested for Western blot analysis with the indicated antibodies. Tubulin was used as a loading control. (E) HCT116 cells with or without INPP4B depletion were co-transfected with siNC or si4EBP1 and a bicistronic luciferase reporter plasmid. At 48 hours after transfection, luciferase activity was measured. The results are presented as the mean ± SD of triplicate measurements. *P<0.05. (F) HCT116 cells with or without INPP4B depletion were transfected with siNC or si4EBP1. At 24 hours post-transfection, cells were seeded in 96-well plates. After 48 hours, cell proliferation was measured by CCK-8 assay. The results are presented as the mean ± SD of triplicate measurements. *P<0.05. |
Hypo-phosphorylated 4E-BP1 impairs the assembly of the eIF4F complex by competing with eIF4G for binding to eIF4E, leading to the suppression of cap-dependent translation.4 The above-mentioned results revealed that INPP4B depletion suppressed 4E-BP1 phosphorylation and cap-dependent translation in CRC cells. In order to further confirm that INPP4B regulates CRC cell proliferation by controlling cap-dependent translation, the expression of 4EBP1 was silenced in HCT116 cells with or without INPP4B depletion, and it was determined whether 4EBP1 knockdown affects the inhibitory role of INPP4B depletion on the proliferation of HCT116 cells (Figure 3D and E). The CCK-8 assay revealed that 4EBP1 knockdown almost entirely antagonized the suppressive effect of INPP4B depletion on HCT116 cell proliferation (Figure 3F), suggesting that the decrease in cap-dependent translation led to reduced proliferation in INPP4B-depleted HCT116 cells. Taken together, these data indicate that INPP4B promotes colon cancer cell proliferation by activating cap-dependent translation.
Akt and SGK3 regulates cap-dependent translation downstream of INPP4B
The above-mentioned results revealed that INPP4B promotes cap-dependent translation in a mTORC1-dependent manner. Given that previous studies demonstrated that INPP4B activates Akt and SGK3 in CRC cells,9 and that mTORC1 functions as the downstream of both Akt and SGK3, it was speculated that Akt and SGK3 may mediate the action of INPP4B on mTORC1 activity and cap-dependent translation in CRC cells. As expected, the increase in cap-dependent translation and cell proliferation in SW620 cells induced by INPP4B overexpression was abolished by the co-introduction of siRNA against Akt or SGK3 (Figure 4A–C). In contrast, when myr-Akt and myr-SGK3 were co-introduced, the inhibitory effect of INPP4B depletion on cap-dependent translation and cell proliferation in HCT116 cells was eliminated (Figure 4D–F). Together, these results suggest that Akt and SGK3 regulate cap-dependent translation downstream of INPP4B.
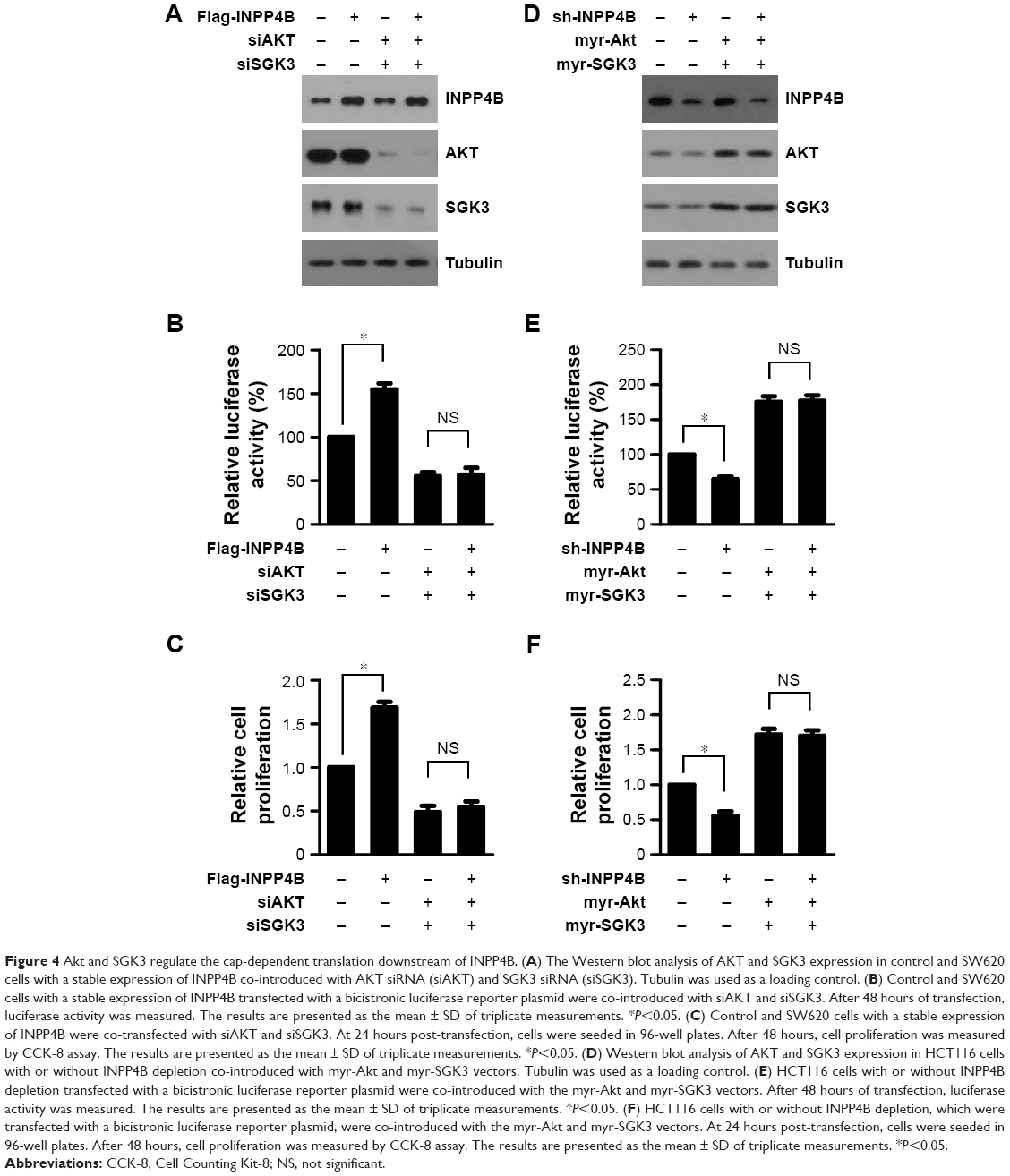 | Figure 4 Akt and SGK3 regulate the cap-dependent translation downstream of INPP4B. (A) The Western blot analysis of AKT and SGK3 expression in control and SW620 cells with a stable expression of INPP4B co-introduced with AKT siRNA (siAKT) and SGK3 siRNA (siSGK3). Tubulin was used as a loading control. (B) Control and SW620 cells with a stable expression of INPP4B transfected with a bicistronic luciferase reporter plasmid were co-introduced with siAKT and siSGK3. After 48 hours of transfection, luciferase activity was measured. The results are presented as the mean ± SD of triplicate measurements. *P<0.05. (C) Control and SW620 cells with a stable expression of INPP4B were co-transfected with siAKT and siSGK3. At 24 hours post-transfection, cells were seeded in 96-well plates. After 48 hours, cell proliferation was measured by CCK-8 assay. The results are presented as the mean ± SD of triplicate measurements. *P<0.05. (D) Western blot analysis of AKT and SGK3 expression in HCT116 cells with or without INPP4B depletion co-introduced with myr-Akt and myr-SGK3 vectors. Tubulin was used as a loading control. (E) HCT116 cells with or without INPP4B depletion transfected with a bicistronic luciferase reporter plasmid were co-introduced with the myr-Akt and myr-SGK3 vectors. After 48 hours of transfection, luciferase activity was measured. The results are presented as the mean ± SD of triplicate measurements. *P<0.05. (F) HCT116 cells with or without INPP4B depletion, which were transfected with a bicistronic luciferase reporter plasmid, were co-introduced with the myr-Akt and myr-SGK3 vectors. At 24 hours post-transfection, cells were seeded in 96-well plates. After 48 hours, cell proliferation was measured by CCK-8 assay. The results are presented as the mean ± SD of triplicate measurements. *P<0.05. |
Discussion
As a negative regulator of PI3K/AKT signaling, INPP4B has been found to exhibit a tumor suppressive role in a variety of cancers, including ovarian, prostate, and breast cancers.9,12,19 However, recent studies have demonstrated that INPP4B also functions as an oncogene in some cancers. In melanoma, INPP4B has been reported to be highly expressed and promote the development of melanoma.14 Furthermore, INPP4B has been found to be aberrantly overexpressed in acute myeloid leukemia patient samples and associated with chemoresistance and poor clinical outcome.20 In some breast cancers harboring oncogenic PIK3CA, INPP4B drives tumorigenesis by mediating SGK3 activation downstream of PI3K.13 A recent study revealed that INPP4B was markedly upregulated in human CRC cells and promoted CRC cell proliferation by activating SGK3 and AKT.9 However, it remains unclear how SGK3 and AKT exert their role in CRC cells. In the present study, it was found that mTORC1 functions downstream of SGK3 and AKT, and contributes to INPP4B-mediated colon cancer cell proliferation (Figure 5).
 | Figure 5 A working model for INPP4B function and mechanism in CRC cell proliferation. |
The aberrant activation of mTORC1 signaling plays a crucial role in the initiation and progression of CRC.21 Activated mTORC1 phosphorylates 4E-BP1, leading to the formation of the eIF4F complex and the initiation of cap-dependent translation.6 Since it was found that INPP4B increased mTORC1 activity, the investigators determined whether INPP4B enhanced cap-dependent translation in CRC cells. It was found that INPP4B promoted 4E-BP1 phosphorylation and cap-dependent translation, while suppressing mTORC1 activity with RAD001 reversed the increase in cap-dependent translation by INPP4B, suggesting that an increase in mTORC1 activity was required for INPP4B-mediated cap-dependent translation. Importantly, inhibiting cap-dependent translation with 4EGI-1 repressed the ability of INPP4B to promote CRC cell proliferation, while 4E-BP1 knockdown abolished the inhibitory effect of INPP4B silencing on cap-dependent translation and CRC cell proliferation, confirming that INPP4B promotes CRC cell proliferation by enhancing cap-dependent translation (Figure 5). Interestingly, we found that INPP4B significantly promoted the expression of cyclinD1 protein, but had little effect on the RNA expression levels of CCND1. These results may be attributed to the fact that CCND1 contains relatively long and structured 5′-untranslated regions, which makes it more dependent on the unwinding activity of eIF4A within the eIF4F complex, and thus highly sensitive to the levels of eIF4E in the cell.22 Since both INPP4B and phosphorylated 4E-BP1 have been reported to be overexpressed in CRC tissues, and it was found that INPP4B enhanced 4E-BP1 phosphorylation in CRC cells, it is interesting to explore the correlation between INPP4B and phosphorylated 4E-BP1 expression in clinical tissues. In addition, cap-dependent translation has also been reported to promote cancer cell migration by enhancing the expression of Snail, a key regulator of cancer metastasis.21 Therefore, further investigation is needed to determine whether INPP4B regulates CRC cell migration.
In contrast to the negative regulation of INPP4B on PI3K/AKT signaling by dephosphorylating PI(3,4)P2 in many types of cells, INPP4B has been reported to enhance this pathway in CRC cells by repressing PTEN expression. Meanwhile, INPP4B also increased SGK3 activity in CRC cells. Both AKT and SGK3 cooperatively regulate CRC cell proliferation driven by INPP4B.9 Consistent with this observation, accumulating evidences suggest the importance of SGK3 in mediating the oncogenic role of INPP4B, such as in breast cancer, melanoma, and NPM1-mutated leukemia.13,14,23 In the present study, it was confirmed that SGK3 activation is required for INPP4B-mediated cap-dependent translation and cell proliferation. Thus, it would be interesting to investigate whether INPP4B promotes cell proliferation by enhancing cap-dependent translation via activating SGK3 in other cancer cells.
Acknowledgments
This work was supported by Dr Quan-Zheng Li and Dr Chun-Sheng Lin, and I extend my thanks for their guidance. At the same time, I would like to thank Xin-Hua Ruan and Xi-Mei Liu for their design and guidance of this experiment. Finally, I would like to thank my colleague Shao-Peng Zhang for his help in experimental materials and modeling.
Disclosure
The authors report no conflicts of interest in this work. SW620 and HCT116 cell lines were obtained from commercial companies.
References
Benson A, Venook AP, Cederquist L, et al. Colon cancer, version 1.2017, NCCN clinical practice guidelines in oncology. J Natl Compr Cancer Network. 2017;15(3):370. doi:10.6004/jnccn.2017.0036 | ||
Chen W, Zheng R, Baade PD, et al. Cancer statistics in China, 2015. CA Cancer J Clin. 2016;66(2):115–132. doi:10.3322/caac.21338 | ||
Pelletier J, Graff J, Ruggero D, Sonenberg N. Targeting the eIF4F translation initiation complex: a critical nexus for cancer development. Cancer Res. 2015;75(2):250–263. doi:10.1158/0008-5472.CAN-14-2789 | ||
Malka-Mahieu H, Newman M, Desaubry L, et al. Molecular pathways: the eIF4F translation initiation complex-new opportunities for cancer treatment. Clin Cancer Res. 2016;23(1):21. doi:10.1158/1078-0432.CCR-14-2362 | ||
Qin X, Jiang B, Zhang Y. 4E-BP1, a multifactor regulated multifunctional protein. Cell Cycle. 2016;15(6):781–786. doi:10.1080/15384101.2016.1151581 | ||
She Q-B, Halilovic E, Ye Q, et al. 4E-BP1 is a key effector of the oncogenic activation of the AKT and ERK signaling pathways that integrates their function in tumors. Cancer Cell. 2010;18(1):39–51. doi:10.1016/j.ccr.2010.05.023 | ||
Ye Q, Cai W, Zheng Y, Evers BM, She Q-B. ERK and AKT signaling cooperate to translationally regulate survivin expression for metastatic progression of colorectal cancer. Oncogene. 2014;33(14):1828–1839. doi:10.1038/onc.2013.122 | ||
Bertucci MC, Mitchell CA. Phosphoinositide 3-kinase and INPP4B in human breast cancer. Ann N Y Acad Sci. 2013;1280(1):1–5. doi:10.1111/nyas.12036 | ||
Hodgson MC, Shao LJ, Frolov A, et al. Decreased expression and androgen regulation of the tumor suppressor gene INPP4B in prostate cancer. Cancer Res. 2011;71(2):572–582. doi:10.1158/0008-5472.CAN-10-2314 | ||
Zhang L, Zeng D, Chen Y, et al. miR-937 contributes to the lung cancer cell proliferation by targeting INPP4B. Life Sci. 2016;155:110–115. doi:10.1016/j.lfs.2016.05.014 | ||
Hsu I, Yeh CR, Slavin S, et al. Estrogen receptor alpha prevents bladder cancer via INPP4B inhibited akt pathway in vitro and in vivo. Oncotarget. 2014;5:7917–7935. doi:10.18632/oncotarget.1421 | ||
Salmena L, Shaw P, Fans I, et al. Prognostic value of INPP4B protein immunohistochemistry in ovarian cancer. Eur J Gynaecol Oncol. 2015;36(3):260–267. | ||
Gasser JA, Inuzuka H, Lau AW, Wei W. Beroukhim R and Toker A: SGK3 mediates INPP4B-dependent PI3K signaling in breast cancer. Mol Cell. 2014;56:595–607. doi:10.1016/j.molcel.2014.09.023 | ||
Chi MN, Guo ST, Wilmott JS, et al. INPP4B is upregulated and functions as an oncogenic driver through SGK3 in a subset of melanomas. Oncotarget. 2015;6:39891–39907. doi:10.18632/oncotarget.5359 | ||
Chen L, Cao Y, Rong D, Wang Y, Cao Y. MicroRNA-605 functions as a tumor suppressor by targeting INPP4B in melanoma. Oncol Rep. 2017;38:1276–1286. doi:10.3892/or.2017.5740 | ||
Ma Q, Wang Y, Zhang H, et al. MiR-1290 contributes to colorectal cancer cell proliferation by targeting INPP4B. Oncol Res. 2017;26:1167–1174. | ||
Bago R, Sommer E, Castel P, et al. The hVps34-SGK3 pathway alleviates sustained PI3K/Akt inhibition by stimulating mTORC1 and tumour growth. Embo J. 2016;35(17):1902–1922. doi:10.15252/embj.201693929 | ||
Wang Y, Zhou D, Chen S. SGK3 is an androgen-inducible kinase promoting prostate cancer cell proliferation through activation of p70 S6 kinase and up-regulation of cyclin D1. Mol Endocrinol. 2014;28(6):935–948. doi:10.1210/me.2013-1339 | ||
Fedele CG, Ooms LM, Ho M, et al. Inositol polyphosphate 4-phosphatase II regulates PI3K/Akt signaling and is lost in human basal-like breast cancers. Proc Natl Acad Sci U S A. 2010;107(51):22231–22236. doi:10.1073/pnas.1015245107 | ||
Dzneladze I, He R, Woolley JF, et al. INPP4B overexpression is associated with poor clinical outcome and therapy resistance in acute myeloid leukemia. Leukemia. 2015;29:1485–1495. doi:10.1038/leu.2015.51 | ||
Wang XW, Zhang YJ. Targeting mTOR network in colorectal cancer therapy. World J Gastroenterol. 2014;20(15):4178. doi:10.3748/wjg.v20.i15.4178 | ||
Nandagopal N, Roux PP. Regulation of global and specific mRNA translation by the mTOR signaling pathway. Translation. 2015;3(1):e983402. doi:10.1080/21690731.2015.1112458 | ||
Jin H, Yang L, Wang L, et al. INPP4B promotes cell survival via SGK3 activation in NPM1-mutated leukemia. J Exp Clin Cancer Res. 2018;37(1):8. doi:10.1186/s13046-018-0675-9 |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.