Back to Journals » International Journal of Nanomedicine » Volume 18
Innovative Strategies of Reprogramming Immune System Cells by Targeting CRISPR/Cas9-Based Genome-Editing Tools: A New Era of Cancer Management
Authors Allemailem KS ![]() , Alsahli MA
, Alsahli MA ![]() , Almatroudi A
, Almatroudi A ![]() , Alrumaihi F
, Alrumaihi F ![]() , Al Abdulmonem W
, Al Abdulmonem W ![]() , Moawad AA, Alwanian WM
, Moawad AA, Alwanian WM ![]() , Almansour NM, Rahmani AH, Khan AA
, Almansour NM, Rahmani AH, Khan AA
Received 24 June 2023
Accepted for publication 16 September 2023
Published 29 September 2023 Volume 2023:18 Pages 5531—5559
DOI https://doi.org/10.2147/IJN.S424872
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Lijie Grace Zhang
Khaled S Allemailem,1 Mohammed A Alsahli,1 Ahmad Almatroudi,1 Faris Alrumaihi,1 Waleed Al Abdulmonem,2 Amira A Moawad,3 Wanian M Alwanian,1 Nahlah Makki Almansour,4 Arshad Husain Rahmani,1 Amjad Ali Khan5
1Department of Medical Laboratories, College of Applied Medical Sciences, Qassim University, Buraydah, Saudi Arabia; 2Department of Pathology, College of Medicine, Qassim University, Buraydah, Saudi Arabia; 3Friedrich-Loeffler-Institut, Institute of Bacterial Infections and Zoonoses, Jena, Germany; 4Department of Biology, College of Science, University of Hafr Al Batin, Hafr Al Batin, Saudi Arabia; 5Department of Basic Health Sciences, College of Applied Medical Sciences, Qassim University, Buraydah, Saudi Arabia
Correspondence: Amjad Ali Khan, Department of Basic Health Sciences, College of Applied Medical Sciences, Qassim University, Buraydah, 51452, Saudi Arabia, Tel +966-16-3800050 Ext. 15445, Fax +966-16-3801628, Email [email protected]
Abstract: The recent developments in the study of clustered regularly interspaced short palindromic repeats/associated protein 9 (CRISPR/Cas9) system have revolutionized the art of genome-editing and its applications for cellular differentiation and immune response behavior. This technology has further helped in understanding the mysteries of cancer progression and possible designing of novel antitumor immunotherapies. CRISPR/Cas9-based genome-editing is now often used to engineer universal T-cells, equipped with recombinant T-cell receptor (TCR) or chimeric antigen receptor (CAR). In addition, this technology is used in cytokine stimulation, antibody designing, natural killer (NK) cell transfer, and to overcome immune checkpoints. The innovative potential of CRISPR/Cas9 in preparing the building blocks of adoptive cell transfer (ACT) immunotherapy has opened a new window of antitumor immunotherapy and some of them have gained FDA approval. The manipulation of immunogenetic regulators has opened a new interface for designing, implementation and interpretation of CRISPR/Cas9-based screening in immuno-oncology. Several cancers like lymphoma, melanoma, lung, and liver malignancies have been treated with this strategy, once thought to be impossible. The safe and efficient delivery of CRISPR/Cas9 system within the immune cells for the genome-editing strategy is a challenging task which needs to be sorted out for efficient immunotherapy. Several targeting approaches like virus-mediated, electroporation, microinjection and nanoformulation-based methods have been used, but each procedure offers some limitations. Here, we elaborate the recent updates of cancer management through immunotherapy in partnership with CRISPR/Cas9 technology. Further, some innovative methods of targeting this genome-editing system within the immune system cells for reprogramming them, as a novel strategy of anticancer immunotherapy is elaborated. In addition, future prospects and clinical trials are also discussed.
Keywords: CRISPR/Cas9, tumor microenvironment, immune response, molecular targeted therapy, cancer immunotherapy, nanotechnology, clinical study
Introduction
Cancer is well characterized by genomic instability resulting in structural alterations that build up with tumor progression.1,2 A subpopulation of undifferentiated cancer cells, known as cancer stem cells (CSCs) are found within the tumor bulk. These cells are responsible for cancer initiation, recurrence and show therapeutic resistance and these cells have a potential role in immune surveillance evasion. Recent literature reveals the deeper interplay between CSCs and immune cells within tumor microenvironment (TME), as surprisingly, some major immune cells have a pivotal role in driving the expansion of CSCs.3 The immune system recognizes the cancer and is poised to eliminate it, but sometimes the immune system is held in check by some inhibitory receptors and ligands. Recent advances in molecular targeted therapy as a novel treatment modality have revolutionized cancer management strategy and taken it to new heights. Different approaches of immunotherapy like immune checkpoint inhibitors (ICIs) and adoptive cell transfer (ACT) have led to satisfying clinical outcomes. However, their efficacies vary and these novel strategies are still very expensive and only a subset of cancer patients have benefited from them.4
The immune surveillance function of different adaptive and innate immune cells within the TME are suppressed by multiple mechanisms.5,6 A continuous selective pressure exerted by the host immune response in context of cancer, often results in tumor variants being skilled in surveillance evasion and enable the tumor cells to survive. Some of the known tumor evasion strategies include (a) secretion of immunosuppressive cytokines like vascular endothelial growth factor (VEGF), transforming growth factor-β (TGF-β), and interleukin-10 (IL-10) into the TME,7,8 (b) cancer cell enclosing down-modulation of antigen processing and presentation pathways,9 (c) suppressive cells’ accumulation within cellular microenvironment such as CD4+CD25+FoxP3+ regulatory T cells (Tregs),10,11 (d) shedding of damage, stress or transformation markers from cancer cells’ surface, including major histocompatibility complex (MHC) class I protein-related sequence A (MICA) and -B (MICB),12 and (e) immune checkpoint ligand upregulation, especially concerning the cytotoxic T lymphocyte antigen-4 (CTLA-4) and programmed cell death-1 (PD-1) pathways.13
The complex biology of immune checkpoint is currently the subject of intense study and challenging mysteries have to be sorted out in near future. Accordingly, advanced research is going on to understand the applications of clustered regularly interspaced short palindromic repeats/associated system 9 (CRISPR/Cas9)-based genetic and epigentic editing as precise therapeutics to counter the immune evasion strategies of cancer cells to get rid of this global menace.
CRISPR/Cas9 has emerged as an advanced multipurpose cutting edge genome-editing technology to thoroughly study different cancers.14,15 In the past few years this system has been used to focus on the mechanism of cancer progression and the advancements in various cell-based therapies. This endonuclease system is also utilized as a programmable strategy to modulate several gene functions by instantly targeting multiple genomic loci designed in a single experiment.16 The genome engineering which uses CRISPR/Cas9-based strategy is quite trustworthy in curing a wide range of diseases like genetic disorders, cardiovascular diseases, neurodegeneration, viral infections, cancer and immunological disorders.17
The targeting of CRISPR/Cas9-based genome editing tools directly to different immune system cells has proved indispensable to engineer these cells to identify complex immune mechanism network thoroughly with emphasis on therapeutic applications. This genome editing tool is not limited to engineering only T cells, as different non-T immune cells such as hematopoietic stem and progenitor cells (HSPC) and B cells have been genetically modified to study disease modeling systems.18 The CRISPR/Cas9-based genome editing in CAR-T cells can be used to block alloreactivity to allogenic receptors to improve the efficacy by the deletion of immune checkpoint and death receptors.19 It also increases the safety by minimizing the potential for cytokine release syndrome. Furthermore, chimeric antigen receptor (CAR) T cells can be engineered with endogenous control over CAR expression profiles.20 CRISPR/Cas9 can be employed to model different pre-disease and disease states such as myelodysplastic syndrome, clonal hematopoiesis of indeterminate potential (CHIP), and acute megakaryoblastic leukemia (AMKL).21,22 In addition, this genome-editing strategy can be used to engineer therapeutic cells for the correlation or disruption of disease-related genes, in addition to the introduction of disease-protection genes such as antiviral antibodies in B cells.23
The effective use of CRISPR/Cas9 methodology involves proper targeting, escaping and minimizing some undesirable off-target mutations for suitable clinical applications.24 To achieve the complete function of this genome-editing technology, it is very crucial to engineer, in parallel, an efficient nanoformulation to deliver this endonuclease system within specific cells or nuclei.
In this review the current updates of crosstalk between TME and immune system cells are described. In addition, CRISPR/Cas9 structure, immunomodulation of different immune system cells against different cancers by reprogramming different protein expression and immune checkpoint blockade pathways are discussed. We describe the different delivery approaches of CRISPR/Cas9 genome-editing system to different immune system cells in parallel, to study the role of engineering these cells as an immunotherapeutic approach to combat cancer progression.
Crosstalk Between TME and Immune System Cells
The tumor microenvironment (TME) and associated immune system cells are multitudinous, mainly including tumor associated macrophages (TAMs), T cells, natural killer (NK) cells, dendritic cells (DCs), mast cells, and myeloid derived suppressor cells (MDSCs) (Figure 1). All these cells perform a significant role in TME, resistance to infection and other diseases.25 The inflammatory microenvironment plays a significant role in TME, and immune system has both positive and negative effects on tumorigenesis. It has been demonstrated by increasing evidence that there is a reciprocal interaction between different immune cells and the CSCs. Within the TME, some major immune cells drive CSC expansion and concomitantly elicit protumorigenic immune cell activities, thus promoting CSC-specific avoidance of immune detection and destruction3 (Figures 2 and 3).
 |
Figure 1 Tumor microenvironment components. It is a complex ecosystem consisting of heterogenous cells such as tumor cells, apoptotic cells, NK cells, stroma cells, Regulatory T cells, exhausted lymphocytes, CD8+ T cells, Dendritic cells, Myeloid cells in a network of dysregulated vasculature and collagen. A pocket of diminished oxygen and higher lactate level with acidic pH medium is produced by densely packed cancer cells. |
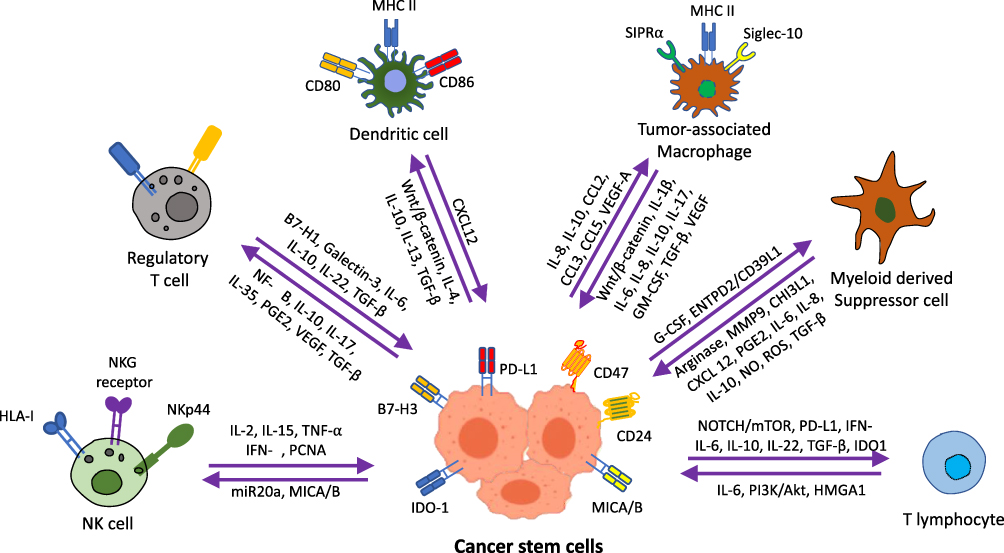 |
Figure 2 A diagram illustrating the crosstalk between cancer stem cells and different immune system cells. |
 |
Figure 3 Outcome of the crosstalk between cancer stem cells and the immune system cells. |
Since CSCs may be derived from reprogramming of oncogenes, the identification of CSC related genes is speculated to generate new cancer treatment targets.26 Currently, the novel applications of CRISPR-based genome editing in CSCs have offered new ways for cancer treatment. The CSC pluripotency and self-renewal capacity is maintained by Nanog gene.27 In addition, Androgen receptors (AR) play a significant role in malignant behavior in different cancers.28 It has been observed that ovarian cancer stem cells (OCSCs) present poor prognosis of ovarian tumor. The novel role of CRISPR/Cas9 has been helpful to report the interaction of Nanog and AR signaling axis, which may contribute to or induce the regulation of OCSC.29 In other CSCs, CRISPR/Cas9 tool has also been used to knockdown the transcription factor YB-1 gene. This knockdown leads to inhibition of melanoma stem cells and breast cancer proliferation, promoting cell cycle arrest and apoptosis.30 In colorectal cancer (CRC), dysfunctional signaling of Wnt gene leads to the development of CSCs.31 CRISPR/Cas9 system has been used to perform Adenomatous Polyposis Coli (APC) truncation mutation. Besides this in CRC models, the inhibitors of MEK enhance Wnt response. Thus, it has been demonstrated that inhibition of MEK increases Wnt activity that enhances the genetic markers related to stemness and tumor recurrence.32 This strategy can provide an efficient way to treat CRC. Furthermore, suppressed CSC properties are employed by CRISPR/Cas9-based REG4 knockdown in CRC with both KRAS and APC mutations inhibiting Wnt/β-linked protein signaling.33 These novel observations have helped to explore the CSC stemness regulatory mechanisms and provide new ways for targeting CSCs as a strategy of cancer treatment.
The TAMs express anti-inflammatory marker genes, such as interleukin-10 (IL-10) and IL-1 receptor-⍺ (IL-1R⍺), and contribute to cancer growth. In addition, monocytes are recruited by TAMs towards TME with the help of secreted chemotactic factors of CCL-2, −5, −7, −8 and −12. These can be polarized to M2-like phenotype with the stimulation of IL-4, −6, −10 −13 and transforming growth factor-β (TGF-β).34,35
DCs are present in almost all tissues and build a communication between innate and adaptive immunity, besides playing a significant role in specific immunity.36,37 DCs are important components in different TMEs, regulated considerably by IL-10, prostaglandin E2 (PGE2), and vascular endothelial growth factor (VEGF).38 Still, the role of DCs at TME is controversial and research is going on to find its significant role in different cancers (Figure 2).
Another type of host immune cells in TME include MDSCs, having two major populations: monocytic and granulocytic MDSCs.39 Different growth factors, matrix metalloproteinases (MMPs) and cytokines proliferate MDSCs and express different markers on plasma membrane as immune suppressive molecules.40 These cells coordinate with other immune system cells and regulate immune response by triggering immune suppression phenotypes.41
T cells are one of the best studied immune system cells that act as perfect killer cells for cancer progression, but still they lack the cancer development task due to some properties such as low activity, exhaustion and aging.42 The T cell immunity is suppressed by chemokines, cytokines and enzymes from TAMs in TME through different signaling pathways (Figure 2).
NK cells play a significant role in TME and perform their killing activity by inflammatory cytokines of interferon-⍺ (IFN-⍺), tumor necrosis factor-⍺ (TNF-⍺), IL-12 and other ILs. The proliferation and intrinsic functions of NK cells are mainly regulated by secreted cytokines of TGF-β and PGE through paracrine and autocrine ways.43
Mast cells are usually involved in angiogenesis, wound healing, and tissue remodeling.44 These cells also take part in tumor cell proliferation and metastasis through the release of mediators involved in remodeling TME.45 The Mast cells promote inflammation by the production of proinflammatory markers,46 and can regulate immune response by cytokine and chemokine hydrolysis.47 In TME, the level of immune-suppressive factors, inflammatory factors and chemokines from Mast cells help in the establishment of immunosuppressive and inflammatory conditions.48 A diagrammatic illustration of crosstalk between cancer stem cells and different immune system cells at the TME is represented in Figure 2.
There is enough evidence that demonstrates how CSCs evade the immune check and surveillance by exerting their effects on NK cells, DCs, TAMs, MDSCs, Tregs, and TILs. The interactions between all these immune system cells and CSCs provide a significant insight into the mechanisms by which immune surveillance is evaded by the cancer cells (Figures 2 and 3). The outcome of crosstalk between CSCs and different immune system cells is summarized in Figure 3.
Different immunotherapeutic modalities have been approved by US Food and Drug Administration (FDA) to treat cancer patients. Some major cancer immunotherapeutic strategies include cytokines and monoclonal antibodies administration and adoptive cell transfer (ACT) of ex vivo “educated” immune system cells. Active immunotherapy includes the use of oncolytic viruses, anti-cancer vaccines, and immune checkpoint inhibitors. All these anticancer strategies could be highly expensive, produce secondary complications, and not be uniform to all types of cancers. Modern approaches to effectively combat the diverse cancers at genetic level include the use of CRISPR/Cas9 genome-editing tools, which can directly reprogram different types of CSCs or immune system cells to eradicate the cancer completely.
CRISPR/Cas9 Biology and Mechanism of Action
The CRISPR/Cas system is a well-known defense mechanism found in most prokaryotes against invading bacteriophages, plasmids and mobile genetic elements.49,50 The prokaryotes acquire short genome segments from such invaders and integrate it within their genetic code, that serves as a molecular memory during any subsequent infection from the same infectious elements.51,52 These acquired sequences are then transcribed as a part of CRISPR array to form CRISPR RNA (crRNA). The crRNA serves as a guide to Cas endonuclease for any infectious genetic material that matches the previous genetic target.53 The endonuclease activity of Cas is also determined by protospacer adjacent motif (PAM), a 2–6 bp nucleotide sequence that serves as a double check to distinguish self from the foreign genetic material before its degradation.
Among different subtypes of CRISPR/Cas systems, the most common and best studied type is CRISPR/Cas9. This endonuclease system has been adopted as a robust, accurate, efficient and programmable method of genome targeting and its editing.54 The Streptococcus pyogenes CRISPR/Cas9 complex (SpCas9) reveals a bilobed architecture with target recognition (REC) and nuclease (NUC) lobes. These lobes possess a positively charged groove at their interface to accommodate single guide RNA (sgRNA): DNA heteroduplex. The NUC lobe contains HNH and RuvC domains, whereas the REC lobe binds with sgRNA and DNA heteroduplex14 (Figure 4).
 |
Figure 4 Three-dimensional structure of Streptococcus pyogenes Cas9-sgRNA-DNA ternary complex shown by (a) Cas9, (b) sgRNA, (c) Cas9-sgRNA complex, (d) target DNA, (e) Cas9-sgRNA-target DNA complex (CRISPR/Cas9) ribbon representation, and (f) CRISPR/Cas9 space-filling model representation, acquired from protein data bank (PDB) https://www.rcsb.org, PDB ID: 4OO8 and edited by UCSF Chimera. |
Cas9 endonuclease forms a complex with either CRISPR RNA (crRNA) and trans-activating crRNA (tracrRNA). However, crRNA and tracrRNA can be engineered to form a single RNA complex called single-guide RNA (sgRNA) that forms a complex with Cas9 enzyme, that targets a complementary genetic sequence for cleavage. A short ∼20-bp nucleotide sequence present in crRNA recognizes the target sequence. In addition, a short sequence (5´-NGG-3´) known as protospacer adjacent motif (PAM) is required very near to downstream of the target sequence for Cas9-mediated cleavage.55 Directly upstream of the PAM, between third nucleotide, the cleavage of phosphodiester bond of target DNA takes place that results in blunt-end double-stranded break (DSB) (Figure 5).54
 |
Figure 5 Cleavage of target DNA by CRISPR/Cas9 followed by multiple genomic modifications. A DNA repair through HDR pathway creates defined insertions, deletions or other specific modifications. However, due to some mistakes in DNA repair by endogenous NHEJ pathway, variable length insertions and/or deletions (indels) can be formed. |
In eukaryotes, DSB repair mechanisms involve either homology directed repair (HDR) or nonhomologous end-joining (NHEJ).56 The HDR is a more precise editing mechanism as compared to NHEJ repair pathway. The HDR repair strategy requires a homologous DNA sequence as a repair template. HDR mechanism can be used to knock-in some exogenous donor sequences within a target DNA. However, NHEJ pathway is error-prone, it requires no template and the ligation of two nascently cleaved DNA strands often occurs with addition of deletion of adjacent nucleotides. This usually results in insertion-deletion (indel) mutations, that result in frame shift mutation, resulting in knock-out of genes (Figure 5).
Cas nucleases have been recently modified in different ways to broaden their action at target locations. Some of the modifications include fusion of adapter proteins, such as transcriptional activators/repressors, deaminases, and reverse transcriptase.56 With these modifications, CRISPR/Cas system has opened a new setup in genome-editing technology, disease management and therapeutic strategies.
Recently new Cas9 variants have been engineered with mutations in RuvC or HNH domains, and one such Cas9 variant exhibits only nickase activity.57 Furthermore, such Cas9 variants can be further fused with reverse transcriptase for prime editing and deaminases and these variants result in adenine/cytidine base editing.56 A completely inactive form of Cas9, dead Cas9 (dCas9) can be engineered by introducing mutations in both nuclease domains. This variant of Cas9 (dCas9) can be fused with other effector proteins such as transcriptional repressors or activators to achieve programmable RNA guided epigenetic regulations.58
Role of CRISPR/Cas9 in Reprogramming Immune System Behavior and Its Novel Targeting Approaches Within Immune System Cells
Precise CRISPR/Cas9-based genetic and epigenetic manipulation of immune system cells for anticancer response is now considered as a revolutionary move in immunotherapeutic approach to combat cancer progression. The CRISPR/Cas9-based immune system reprogramming comprises locus-specific host immunity enhancement, tumor immunogenicity improvement and tumor immunoevasion suppression. The CRISPR/Cas9-based technology is used in adoptive T cells and NK cell transfer, cytokine stimulation, antibody design and overcoming immune checkpoints. In addition, this genome-editing system is used in autologous T cells and NK cells to express different novel antigen designs and chimeric antigen receptors (CARs) targeted at specific antigens for solid and hematological cancers (Figure 6).
 |
Figure 6 CRISPR/Cas9-mediated reprogramming of immune system behavior in different immune system cells. |
A precise and efficient delivery of CRISPR/Cas9 system as ribonucleoproteins (RNPs) or plasmids within target cells or tissues is challenging and needs to be sorted out in the near future. Some of the challenges include fast degradation or denaturation of RNPs during their formulation and delivery steps, large size of Cas9, highly negative charge of sgRNA and toxic effects of plasmids. The existing delivery strategies of CRISPR/Cas9 can be mainly divided into physical, viral and nanomaterial based nanoformulation approaches (Figure 7). Viral vectors are used to perform transduction of cells with different viruses to deliver DNA encoding gRNA and Cas9 protein. The most frequently used viral vectors include lentiviruses, adeno-associated viruses (AAVs) and adenoviruses (ADVs). These viral vector delivery approaches for CRISPR/Cas9 can also have oncogenic potential and immunity complications in humans.59 AAVs have been commonly exploited for CRISPR gene delivery in vivo.60 However, AAV vectors can package up to 4.7 kbp only. Thus only smaller CRISPR/Cas9 systems such as S. aureus Cas9 (SaCas9) (3.16kb)61 or C. jejuni Cas9 (CjCas9) (2.95kb)62 can be used for proper packaging with its sgRNA into a single AAV vector.
 |
Figure 7 Delivery approaches and expression of CRISPR/Cas9 system in vivo. The delivery approaches can be through physical methods, viral vectors and by different nanomaterial based carriers. Abbreviations: PEI, polyethyleneimine; β-CD, β-cyclodextrin; CPP, cell penetrating protein; AuNPs, gold nanoparticles; ZIF, zeolite imidazole; LNPs, lipid NPs; AdV, adenovirus; LV, lentivirus. |
Due to different modification flexibility options which limit the use of viral delivery approaches, non-viral delivery of CRISPR/Cas9 through different nanomaterial/nanoformulations is preferred for clinical applications.63,64 Non-viral delivery approach for Cas9 mRNA has been a significant alternative to bypass many challenges associated with viral delivery.65,66 However, these systems also face some limitations as continuous expression of Cas9 protein and lower transfection efficiency, limit the use of these platforms.67
Recently different nanoformulations have been engineered to deliver this endonuclease system within target sites.14 These nanoformulations include cationic lipid nanoparticles (NPs) and lipoplexes,68 zeolitic imidazole frameworks,69 DNA clews70 and gold NPs71 (Figure 7). However, it is a challenging task to control the size, stability, loading capacity, and uniformity of these resulting nanoformulations.72
Role of CRISPR/Cas9 in Engineering T Cells and Its Delivery Approaches
T cells are significantly involved in fighting against pathogenic microbes and cancer cells. The T cells are highly active in producing effector molecules like proinflammatory cytokines, IL-2, IFN- γ and TNF-⍺. In addition, cancer cells exploit T cell inhibitory receptors like programmed cell death protein-1 (PD-1) by expressing their cognate ligand, ie, programmed cell death protein ligand-1 (PD-L1).73,74 Thus cancer cells apply different tactics and inhibitory mechanisms to diminish T cell effector functions that impairs the tumor elimination.
To overcome this, several immunotherapy approaches have been devised to optimize T cell effector functions with the help of CRISPR/Cas9-based genome-editing strategy (Figure 8). One strategy includes the employment of reprogrammed tumor infiltrating lymphocytes (TILs) for the treatment of solid tumors.75 Another approach makes use of genetically engineered T cells. In this approach, T cell antigen specificity is redirected and targeted to cells that have defined subset of antigen/s.76,77 This can be accomplished by integration of a traditional T cell receptor (TCR),78,79 or a CAR.80 The CAR T cells are furnished with a receptor that comprises a variable region of high affinity monoclonal antibody (mAb) specific for a defined tumor antigen, eg, CD19. It is fused with a signaling domain of CD3ζ and one or more signaling domains from a costimulatory receptor for maximum T cell effector functions.81,82
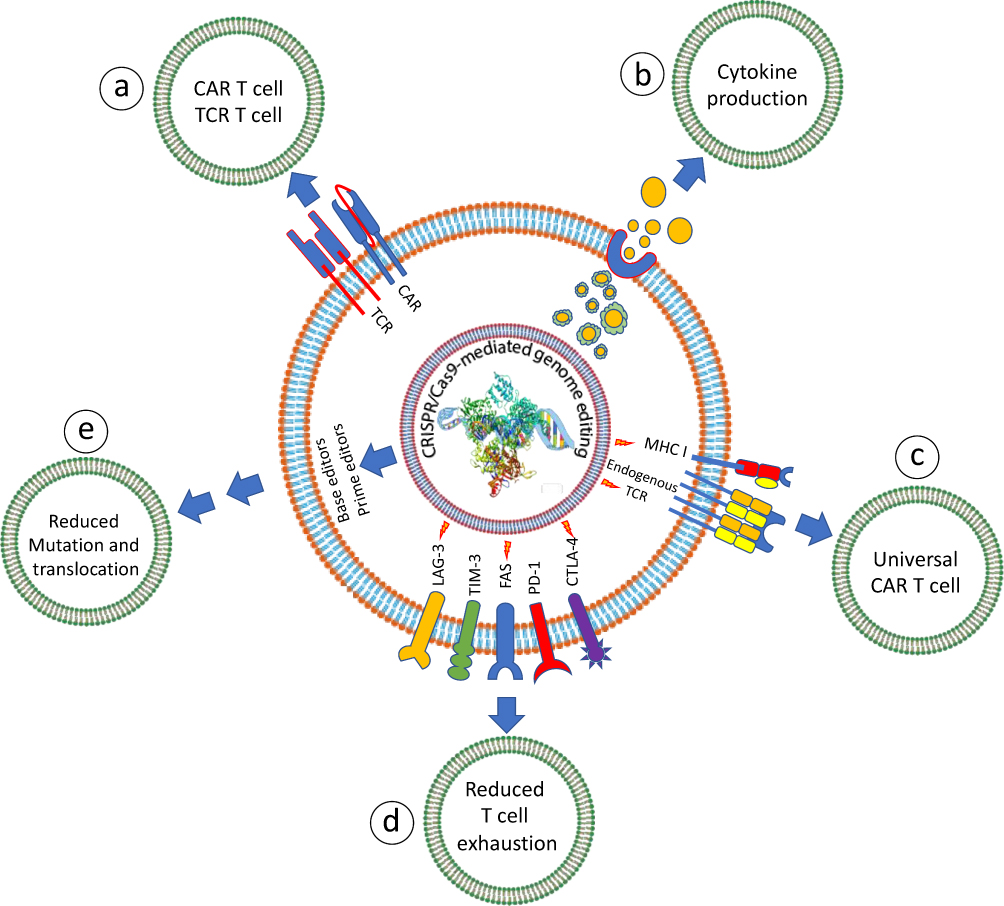 |
Figure 8 CRISPR/Cas9-mediated enhancement of ATC therapy functions. (a) CRISPR/Cas9 mediates safer integration of CAR or TCR into T-cells for a uniform surface expression, (b) genes that drive cytokine suppression or production are manipulated by CRISPR/Cas9 to enhance the cytokine production, (c) the knocking of MHC-I and endogenous TCR in off-the-shelf T cells, host-vs-graft reaction and graft-vs-host diseases are neglected, respectively, (d) more persistent and durable T-cells are produced by knocking out the checkpoint inhibitors either separately or in combination, (e) safer T cells are produced by denying unwanted mutations by the use of prime editors and base editors. |
The recent advances in CRISPR/Cas9 genome-editing technology have been successfully used to modify T cell lines,83 primary murine and human T cells.84,85 The CRISPR/Cas9 system has also been used to employ T cell effector functions,86,87 inactivate inhibitory receptor expression88 or to redirect T cell antigen specificity by targeting TCRs or CARs to the endogenous TCR-⍺ chain (TRAC) locus.78,79
This gene-editing tool box has also been used for knocking in genes,83 silencing genes84 and for induced activation of genes in T cells89 (Figure 8). The CRISPR/Cas9-based genome editing strategy has also been used in T cells to prevent HIV infection,90 stabilization of regulatory T cell phenotype,91 and redirecting T cell antigen specificity.79 In addition, this genome-editing strategy is used for TCR signaling, regulation of T cell effector molecule production and investigating pathogenic cytokine receptor expression by T cells.92
CRISPR/Cas9 system has been used to genetically alter human T cells; as on March 1st, 2020, 21 clinical trials were registered for using this gene-editing technique. Out of these trials, 48% used CRISPR/Cas9 strategy.93–95 Different cancers like non-small cell lung cancer (NSCLC) and esophageal squamous cell carcinoma (ESCC) express immune checkpoint PD-1 ligands (PD-L1) that bind with PD-1 receptors present on host T cells. This circuit inhibits cytokine production and proliferation, thus enabling immunoevasion (Figure 8).96,97 The use of anti-PD-1 and anti-PD-L1 mAbs have been successful in different cancers like liver cancer,98 lung cancer,99 melanoma,100 and Hodgkin’s lymphoma.101 However, some severe toxicity issues have also been reported in this inhibitor therapy.102
The genetic disruption of T cell PD-1 expression is perceived as an alternative and potentially safer immunotherapeutic opportunity. The safety of PD-1 knockout T cell reinfusion was observed in 12 patients suffering from metastatic NSCLC and 17 patients with advanced ESCC, respectively.93,94 In both these trials, adoptive cell transfer (ACT) was performed, whereby peripheral blood mononuclear cells (PBMCs) were collected, followed by CRISPR/Cas9-mediated knockout of PD-1 (PDCD1 gene) ex vivo. After selection and expansion of the edited PMBCs, these engineered cells were reinfused back to the patients. The regimen was well tolerated with no severe events observed. Approximately 18% of NSCLC and 35% of ESCC patients exhibited stable disease. In addition, it was demonstrated that co-transfection of sgRNA plasmid DNA (pDNA) and Cas9 through electroporation in 12 enrolled patients resulted in low median editing efficiency of 5.81%.94
The whole genome sequencing by Cas-OFFinder for 2086 targeted sites, exhibited no off-target events.103 This illustrates the greatest success of CRISPR/Cas9 in clinical applications. From both trials, it was found that only minor adverse effects occurred including fever, fatigue, skin rash, and joint pain during the treatment strategy. This supports the use of CRISPR/Cas9-based ACT for clinical use. However, in both trials, beyond therapeutic efficacy, the interest was more focused on role of CRISPR/Cas9 in editing efficacy, better expansion of tumor-reactive T cells, superior antigen specificity and clear understanding of T cell subtypes undergoing the gene editing process, required for improved patient response. All these observations support that CRISPR/Cas9-mediated immunotherapy has a greater future ahead. The role of CRISPR/Cas9 in mediating gene therapy for the enhancement of different functions of TCR and CAR T-cells is further summarized in Table 1.
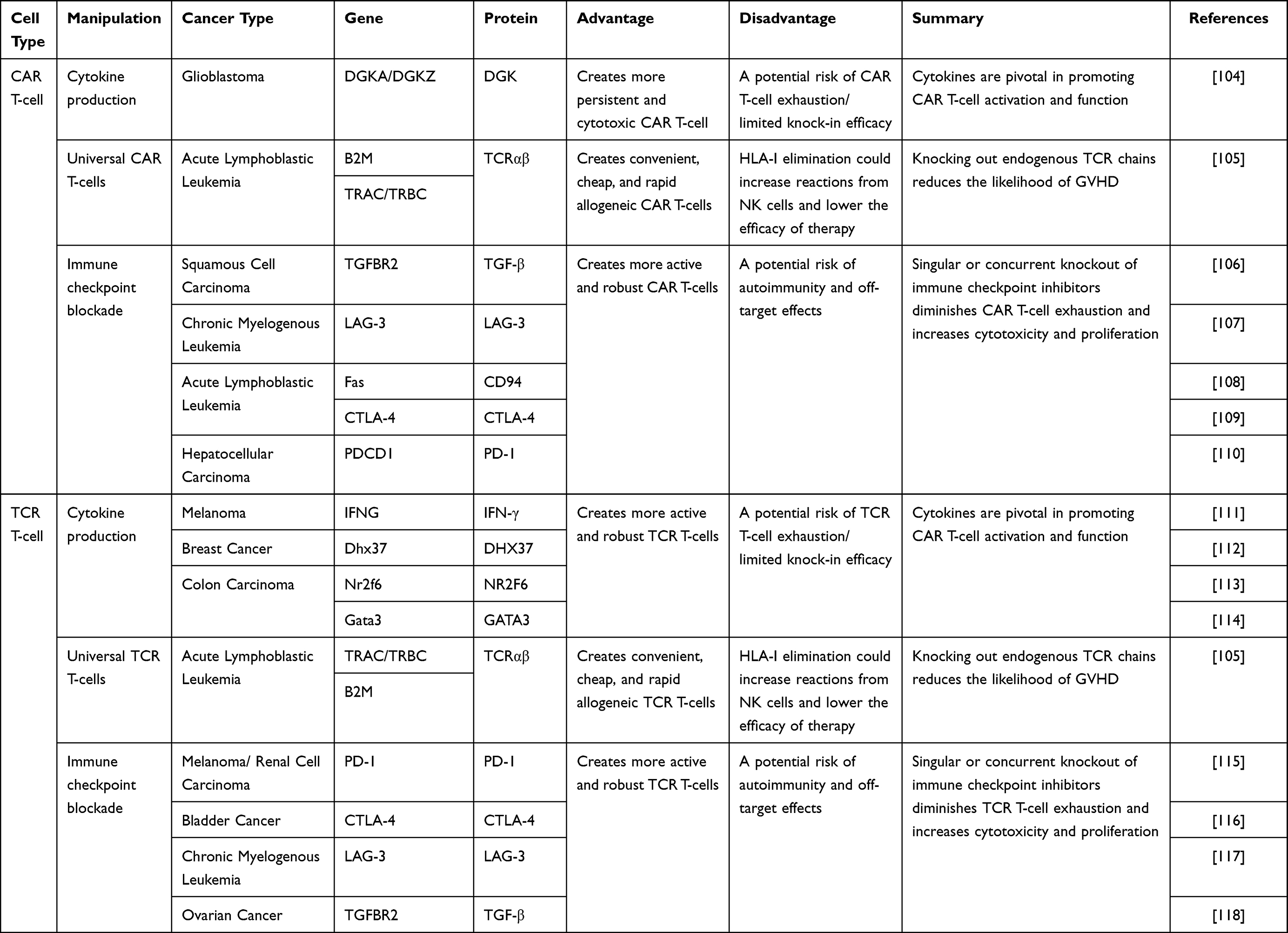 |
Table 1 Role of CRISPR/Cas9-mediated gene therapy to enhance the functions of TCR and CAR T-cells |
A proper selection of delivery protocol and the form of CRISPR/Cas9, either as mRNA or plasmid for targeting within T cells or other immune system cells significantly affects the genome-editing strategy. In some experiments following the gene knockout (KO), Cas9 and gRNA have been delivered as plasmids and mRNA, with separate delivery of gRNAs or as RNPs. The incorporation of Cas9 and gRNA together as plasmids for the delivery approach face some limitation as higher amount of plasmids create some toxicity effects.119 It occurs mostly due to pathogen-associated molecular patterns’ recognition within the plasmid DNA.120,121 Furthermore, delivery of plasmids poses the complication of random DNA integration within the cellular genome.122
To avoid cytotoxicity, mRNA is directly targeted which can be modified by methylation at 5ʹ end and addition of poly A tail on 3ʹ end, to similarize the endogenous mRNA.123 This approach was used in human T cells, although the gRNA delivery was performed by transfection.124,125 Besides gRNA and Cas9, HDR template is also required as CRISPR/Cas9 tool for transportation within the cells. HDR templates can be provided as circular plasmids, ssDNA, dsDNA or AAV-incorporated DNA molecules.126 All three template types have been used effectively to make some changes within T cell genome.
Some transfection techniques which are suitable to other cells have not been proven to be equally applicable for T cells. For example, microinjection, involving a microscopic needle as a transportation strategy directly within the nucleus is highly efficient but challenging and laborious, and is restricted to generation of transgenic animals.127,128
Nucleofection and electroporation allow efficient delivery of different nucleic acids to T cells. Electrical current is used in both of these methods to form pores in plasma membrane to permit the cargo entry by diffusion and movement along the electrical field.129 In contrast to electroporation, nucleofection involves a combination of electrical pulse specific to cell-types and reagents as delivery agents directly within the nucleus.130 Cas9 mRNA and gRNA have been efficiently delivered within T cells by electroporation.80 However, optimized protocol has been established in 2018 by Akiko Seki and Sascha Rutz for the delivery of CRISPR/Cas9 as RNPs by nucleofection to the primary T cells.131 Since then, due to its simplicity and effectiveness, this delivery approach has been most frequently used to deliver CRISPR/Cas9 cargo to T cells.132
Recently, it was suggested that this approach can be further improved by encapsulating Cas9 RNPs into poly-L-glutamic acid (PLGA) NPs.133 Furthermore, the cellular uptake mechanism of micropinocytosis, based on transduction by osmocytosis and propanebetaine (iTOP), was recently demonstrated to deliver RNPs to human cells including primary T cells.134,135 This strategy could also be used to deliver CRISPR/Cas9 genome-editing tools to T cells.
Furthermore, gold nanoparticles (AuNPs) have been used quite efficiently as delivery agents in different types of cells including T cells. AuNPs have been used to deliver CRISPR/Cas9 components within T cells albeit with poor efficiency.136 In contrast, T cells are resistant to lipofection, a method involving delivery of liposomes and other lipid nanoparticles (LNPs) encapsulating genome editing contents to the target cells.137
Role of CRISPR/Cas9 in Engineering NK Cells and Its Delivery Approaches
NK cells are well-known cytotoxic lymphocytes, recognizing and eliminating transformed cells and orchestrating adaptive anticancer immunity.138,139 However, in TME, the NK cells usually get exhausted and some strategies such as checkpoint blockade are under investigation to overcome this exhaustion, to boost their anticancer potential.140 CRISPR/Cas9-mediated genome-editing can significantly modulate different pathways that facilitate NK cell exhaustion and can arm these lymphocytes with novel CARs to specifically target the cancer cells. However, the challenge of CRISPR/Cas9 delivery within NK cells remains a major bottleneck for its therapeutic purpose.139
A recent novel study demonstrated that CRISPR/Cas9 mediates knockout (KO) of key inhibitory signaling molecules [(eg, programmed cell death 1 (PDCD1) and A disintegrin and metalloprotease 17 (ADAM17)], to improve the functionalities of peripheral blood (PB)-NK cells. This approach significantly modified 90% of PB-NK cells with enhanced cytokine production and anticancer cytotoxicity approach in immune suppressive TME.141 Furthermore, the researchers expanded these NK cells without the loss of activity to clinically relevant number. This novel approach led to the clinical translatability of engineered NK cells with CRISPR/Cas9 genome editing tools.141
NK cells play a significant antiviral and antitumor role through numerous mechanisms like direct cytotoxicity, antibody-dependent cell-mediated cytotoxicity (ADCC) and chemokine/cytokine secretion. NK cells lack antigen-specificity recognition capability unlike T cells but perform critical anticancer immune roles.142 The use of NK cell immunotherapy represents a fascinating and dynamic strategy for cancer treatment. After the target recognition, NK cells release perforin and granzyme that eradicate the target cells. The elimination of target cells also occurs through IFN- γ, TNF-⍺, Fas ligand (FasL) and tumor necrosis factor-related apoptosis-inducing ligand (TRAIL), that activate the DNA fragmentation and ultimately the apoptosis of target cancer cells142 (Figure 9). Due to less severe side effects, NK cells have emerged as safer and cost effective as compared to CAR T cells for cancer immunotherapy.143,144
 |
Figure 9 CRISPR/Cas9-based genome-engineering of NK cells to enhance its immunotherapy efficacy against cancer cells. Genome engineering improves the cytotoxicity, reduces sensitivity to TME, increases in vivo proliferation and persistence through autocrine cytokines’ stimulation. |
The anticancer effect of NK cells requires further improvement in some conditions, like their exhaustion during tumor progression. The decreased anticancer effector function of NK cells also occurs due to its differentiation to type 1 innate lymphoid cells (ILC1), which possess decreased anticancer potential.145,146 Some previous strategies to enhance NK cell immunotherapy include the use of cytokines, antibodies, or gene-editing and these approaches were used to overcome immune suppression.147,148
CRISPR/Cas9 genome-editing strategy offers a flexible system in editing the NK cells ex vivo for adoptive therapy. This technique has helped a lot to manipulate the tumors in situ to enhance their susceptibility for NK surveillance.138,139 NK cell tumor immunotherapy has been explored for hematopoietic malignancies recently. Like CAR-T immunotherapy, CAR-engineered NK cells possess tumor target specificity and cytotoxicity.149,150 The ongoing preclinical and clinical applications on engineered CAR-NK cell-based immunotherapy against different cancers have been explored.144,151
CAR-NK-based bispecific T-cell engager (CD19-ENG) has been produced by combining CRISPR/Cas9 with some other gene-editing strategy to produce cells capable of targeting CD22+ B-cells leukemia as well as redirecting T cells to eradicate malignant CD19+ B cells. This strategy improves the anticancer activity and helps to prevent any immune escape by the tumor. This study has shown that CD22 specific engineered CAR-NK cells augmented CD19 T cell targeting of B-cell malignancies.152 This combined cytolytic target killing of cancer cells opens a new window in gene editing with substantial improvement in current B-cell therapy and other cancers.
To improve the anticancer efficacy of NK cells, multiple pathways are implicated. Some pathways are enhanced while at the same time other pathways need to be suppressed. CRISPR/Cas9 gene editing system is very well suited for these requirements. The anticancer potential of NK cells is enhanced in several ways like arming with CARS, enhancing activating pathways, improving NK cell infiltration and targeting inhibitory pathways (Figure 9).
Arming with CARs
NK cells can be armed with CARs via CRISPR/Cas9 genome-editing approach to improve their specific recognition of cancer cell surface markers. CARs consist of single chain variable fragment (ScFv) that is specific to a cancer antigen. This domain is attached to an intracellular signaling domain that transduces activation signal upon the recognition of antigen. Some NK cells have been engineered with CARs which act both by in vitro and in vivo targeting of the cancer antigen.153,154 In parallel, for the improvement of tumor recognition, NK cells can be armed with Pan-specific CAR-like molecules representing an alternative strategy (Figure 10).
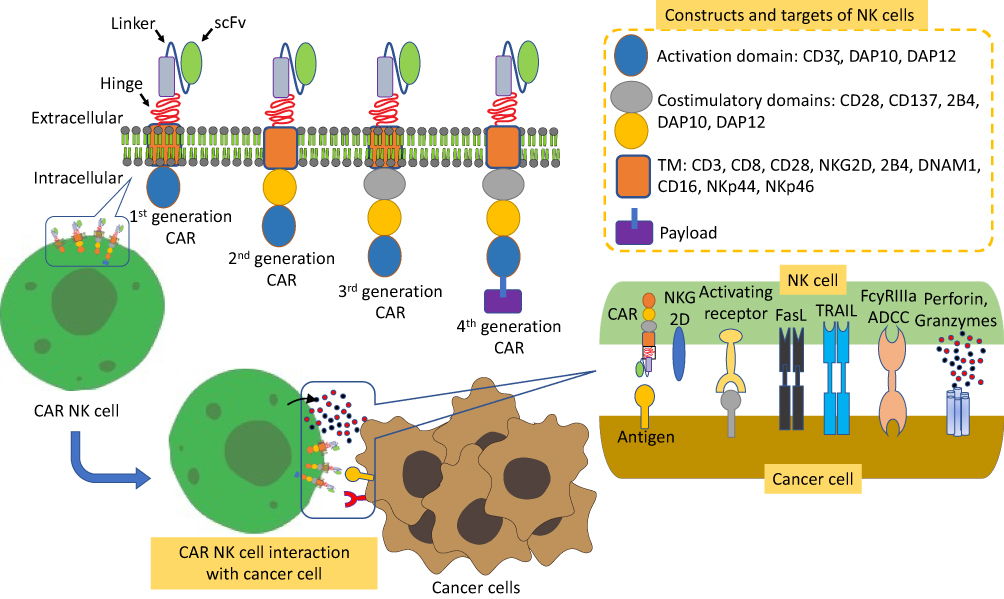 |
Figure 10 Diagrammatic representation of the overview of constructs and targets of CAR-NK cells. |
Tumor evasion usually occurs by losing the original tumor antigens from cancer cells, thus multiple-ligand recognition mode by NK cells seems to be a superior strategy, leading to enhanced antitumor responses. For example, most normal cells express, very poorly or not at all, human NKG2D ligands like MHC class I chain-related proteins A and B (MICA and MICB), and six UL16-binding proteins (ULBPs). In contrast, virus-infected cells and cancer cells express these ligands excessively.155 As compared to conventional single CARs, the tumor recognition is improved by more than a single cancer marker. Thus the fusion of full-length NKG2D polypeptide with CD3f exhibits potent efficacy against NKG2D ligand-positive cancer cells when it is expressed on NK cells or T cells.156,157 In addition, this strategy antagonizes the immunosuppressive TME by targeting NKG2D ligand-positive Tregs and MDSCs in tumors.156 The intracellular domains of NK cell CARs may differ from the intracellular domain of T cells and only DAP12 signaling domain alone shows improved activating effects when compared with CD3f.158,159 However, when used in combination with B4,160 CD3f, DAP10,161 and 4–1BB demonstrated superior triggering capacity compared with CD3f alone. It is fascinating that only DAP12 showed CD3f-independent and CD3f-comparable activation effects. It suggests that DAP12 may be playing a predominant role in activating the NK cell signaling (Figure 10).
Enhancing Activation Pathways
The NK cell response could be initiated and prolonged by some activating receptors and activating cytokines like IL-2, IL-15, IL-18 and IL-21.148 NK cells derived from cord blood have been transduced to express IL-15 and these cells exhibited enhanced survival in xenograft Raji lymphoma murine model.162 In addition, the mutant form of IL-2 called Super-2, binds, with high affinity, to IL-2Rb, reverses the exhaustion state and promotes NK cells’ continuous proliferation without inducing the Treg cells’ expansion.163 In addition to the gene-editing of NK cells for adoptive transfer therapy, boosting the in situ tumor-derived ligand/s for NK cell-stimulating receptors. This exemplifies a novel strategy for enhancing the activating pathways and amplifying anticancer response of these cells. This novel approach is achieved by CRISPR/Cas9-based transcriptional activation. A good example is the expression of NKG2D ligand, MICA, that has been transcriptionally stimulated effectively by CRISPR/Cas9.164
Improving NK Cell Infiltration
Upon adoptive infusion, the success of cell based immunotherapy depends upon efficient NK cell migration and homing to disease sites. CRISPR/Cas9-based gene technology is highly applicable to enhance the expression of tumor specific chemokine receptors on NK cells. Patients with renal cell carcinoma (RCC) have been observed to have enhanced expression of CXCR2 ligands and it correlates with intra-tumoral infiltration of NK cell subpopulation.165 Thus, genetic manipulation of human NK cells to express CXCR2 has been reported with enhanced ability to migrate specifically along a chemokine gradient of recombinant CXCR2 ligands in vitro and in RCC mouse model.165 This demonstrates that for the improved infiltration of NK cells within cancerous mass, CXCR2 symbolizes a significant chemokine receptor that can be overexpressed on these cells. Furthermore, the immune cell infiltration has also been promoted by CCL-19-CCR7 axis.166
A significant migratory ability and improved tumor homing of NK cells were observed by engineering these cells with chemokine receptor CCR7 for its ligand CCL-19 and CCL-21.167,168 However, tumor tissue stroma displays an inhibitory action against such chemokine-driven NK cell infiltration. A forced expression of heparinase in addition to specific CAR has been shown to render CAR T cells with enhanced capability to degrade the extra cellular matrix (ECM).169 This approach of forced heparinase expression is also applicable for NK cell infiltration within primary tumors in addition to recruitment for metastatic sites.170
Targeting Inhibitory Pathways
It has been observed that at cancerous sites, NK cell functional exhaustion occurs via the detrimental modulation of checkpoint receptor signaling.140 The blockade of some checkpoint receptors like CD96, NKG2A or T cell immunoreceptor with Ig and ITIM domains (TIGIT) were reported to boost NK anticancer immunity.171–173 In addition, emerging immunosuppressive pathways contribute to the hypofunctional status of cancer-associated NK cells. Any genetic disruption of such pathways might enhance the effector functions of NK cells for adoptive therapy. The CRISPR/Cas9-based targeting of cytokine inducible SH2-containing protein (Cish) gene has been found to enhance the cytotoxic potential of primary human NK cells towards Daudi B lymphoma cells.174
NK cells are well-known to resist the ordinary transfection, thus making the Cas9 delivery difficult. However, transduction via integrating (retroviruses and lentiviruses) and non-integrating (adenoviruses and AAVs) are applicable strategies.175,176 The ex vivo CRISPR/Cas9-mediated genome editing in lymphocytes has been widely reported. The in vivo genome-editing by this endonuclease system is still a challenging task. In addition to viral delivery, nanoformulation based delivery systems are gaining fast recognition, as such formulations possess good modification flexibility.
Electroporation of human NK cells has been presented as an efficient CRISPR/Cas9-mediated genome-editing system by a Lonza 4D nucleofector system.174,177 The human NK cell electroporation with Cas9 RNPs in P3 Primary Cell Nucleofector solution was performed by using pulse-code “EN-138” which resulted in proportional indel rates up to 30%. This led to 60% reduction in transforming growth factor β receptor 2 (Tgfβr2) mRNA expression level and rendered NK cells more resistant to TGF-β in vitro.177 However, electroporation leads to poor survival of NK cells. This limitation was overcome to some extent by using optimized buffers and pulse codes, as pulse-code “CM-137” with mannitol supplemented phosphate buffer was found optimal with more than 80% NK cell viability uptake of large FITC-dextran molecules that mimic Cas9 complex with 45–80% indel rates in primary human NK cells.174 Interestingly, this approach led to no effect, partial effect or total loss of protein tyrosine phosphatase receptor type c (Ptprc) gene. The Cas9 RNP electroporation of NK cells represents a promising approach for some genetic modifications for adoptive immunotherapy.
Role of CRISPR/Cas9 in Engineering Macrophages and Its Delivery Approaches
Although CRISPR/Cas9-based genome editing has revolutionized the ability to manipulate immune system responses, cell-type specific blockades hamper the genetic approaches to study some cells. Macrophages are significant innate immune cells that act in homeostasis, tissue development, repair as well as during microbial infections. These cells are difficult to genetically manipulate due to sensitive circuits that sense foreign nucleic acids. However, primary bone marrow derived macrophages (BMMs) have been successfully manipulated by CRISPR/Cas9 system.178 However, low transfection and transduction efficacies in these cells result in little editing efficiency. In addition, millions of cells are required for proper biochemical and screening approaches. Furthermore, short life-span of macrophages limits proper selection of individual mutant clones, such as knockout of a second gene or protein over-expression. Due to these limitations, various studies are based on immortalized macrophage cell lines. However these cell lines do not reiterate important metabolic and inflammatory pathways of primary cells.179
It has been reported that some cell surface protein interactions between cancer cells and macrophages prevent phagocytosis. One good example is the interaction between CD47, an overexpressed cancer cell surface protein and macrophage signal regulatory protein-⍺ (SIRP-⍺).180 The interaction between CD47 and SIRP-⍺ is sufficient to bypass phagocytosis. Interaction of CD47 with SIRP-⍺ promotes its phosphorylation on the cytoplasmic tail,181 resulting in binding and activation of Src homology phosphatase-1 (SHP-1) and SHP-2.182 This results in blockade of phagocytosis by blocking the accumulation of myosin-IIA at phagocytic synapse. Thus, blocking the signaling between CD47 and SIRP-α is supposed to be a promising therapeutic target183 (Figure 11).
 |
Figure 11 Different approaches of enhancing macrophage immunotherapy by CRISPR/Cas9-mediated genome editing. Some major enhancements include, (a) SIRP-⍺-CD47 blockade, (b) bispecific T cell engagers, (c) enhanced cytokine secretion, (d) secretion of antibodies, (e) Tie2-driven cytokine secretion, and (f) Tumor mediated antibody-dependent phagocytosis of opsonized tumor cells. |
Recently, anti-CD47 mAbs were designed to block the interaction between CD47 and SIRP-α. This blocking strategy has shown novel efficacy in preclinical studies for different human cancers both in vitro and in animal xenotransplantation models.184,185 In different tumor models, an engineered antibody variant of SIRP-⍺, CV1, has been used as an adjuvant to facilitate macrophage-mediated phagocytosis with low toxicity and enhanced cancer mass penetration. However, a large antigen sink for the employment of Ab-based approach was observed due to overexpression of CD47. This strategy reduces the bioavailability with enhanced toxicity potential to normal cells.186 Targeting SIRP-α with anti-SIRP-α Abs is an alternative approach to block CD47 that significantly enhances the killing of cancer cells by phagocytosis in vitro (Figure 11).
Nevertheless, the penetration of Abs within the cancerous mass remains a major challenge for therapeutic efficacy. Thus SIRP-α-targeting Abs require an easy tumor penetration potential to interact and block the tumor infiltrating macrophages (TIMs) for performing feasible therapeutic contact.187,188 Recently Arg-coated Au NPs have been used for the delivery of CRISPR/Cas9 system within macrophages to knockout SIRP-α expression. This approach demonstrated almost 90% delivery efficiency along with approximately 30% gene editing efficiency, providing a novel approach of cancer immunotherapy.189
Tumor-associated macrophages (TAMs) are indispensable components of TME that play a role in orchestrating extracellular matrix remodeling, angiogenesis, cancer cell proliferation, immunosuppression and metastasis. TAMs also play a role in resisting chemotherapeutic agents and checkpoint blockade immunotherapy.190 Recently, CRISPR/Cas9 system was delivered by engineering Cas9 association with cationic arginine-coated AuNPs (ArgAuNPs) to HeLa cells.189 This strategy was used to transport the endonuclease to generate SIRP-α knockout macrophages. The nanoformulation efficiently co-delivers Cas9 and sgRNA to knock out the “don’t eat me signal” in macrophages, preventing the phagocytosis of cancer cells. The treated macrophages presented 4-fold increased innate phagocytic capability achieved by turning off this signal. The elimination of cancer cells in this way makes this strategy a promising tool to generate weaponized macrophages for cancer immunotherapy191 (Figure 11).
Cationic lipid-assisted NPs (CLAN) made from poly(ethylene glycol)-block-poly(lactide-co-glycolide) (PEG-b-PLGA) were synthesized to encapsulate macrophage-specific promoter driven Cas9 expression plasmids (pM 330 and pM458). The CRISPR/Cas9 plasmids were targeted to macrophages by these NPs. These cells and their precursor monocytes were able to express Cas9 specifically both in vivo and in vitro. After further encoding a gRNA targeting Ntn1 (sgNtn1) into the plasmid, the resultant CLANpM330/sgNtn1 efficiently disrupted the Ntn1 gene in targeted macrophages and their precursor monocytes, and led to reduced netrin-1 expression.192
A macrophage specific direct CRISPR/Cas9 RNP delivery was performed into mice through tail-vein injection. The nanocomposite was fabricated by ArgNPs with Cas9 protein engineered to incorporate 20-glutamate (Cas9E20). The Cas9E20 and ArgNPs along with specified sgRNAs were complexed as a single nanoformulation. The systemic injection of this nanoformulation provided more than 8% phosphatase and tensin homolog (PTEN) gene editing in macrophages of spleen and liver, respectively.193 This approach thus illustrates a potential selective immunotherapeutic strategy for the treatment of macrophage-related diseases.191,194
Role of CRISPR/Cas9 in Engineering Dendritic Cells and Its Delivery Approaches
Dendritic cells (DCs) play a significant role in regulating innate and adaptive immunity by acting as sentinels for invaders and initiating innate immune response as counteract. As APCs, DCs initiate antigen-specific adaptive immune response.195,196 DCs perform pathogen clearance, cancer cell killing, and tolerance to dietary antigens or microbiome bacteria. DCs thus help in shaping host-microbiome and host-pathogen interactions and the etiology of autoimmune disorders. Thus, these cells are thought to be chief targets to develop new techniques of immunotherapies.197
It is a challenging task to efficiently manipulate genetically human DCs, although gene repression approaches by RNAi have been reported.198 Mice models have been generally used to study DC biology, but humans and mice differ in both adoptive and innate immunity.199 To address all such limitations, a CRISPR/Cas9 strategy was used to construct a targeted knockout (KO) directly in human monocyte-derived DCs (moDCs) with a median efficiency of >94% across >300 genes. This method was utilized to perform a genetic screen in moDCs, recognizing the possibilities by which DCs tune their response to human microbiome lipopolysaccharides (LPs). Furthermore, the donor specific response to LPs underlined the significance of evaluating immune phenotypes in donor-derived cells. In addition, the identification of candidate genes that control this specificity to pinpoint the determinants of inter-individual variations in immunity has been revealed.200
The treatment of end-stage organ failure due to some cancerous mass can be performed by tissue transplantation. However, immune rejection is a major challenge and T cells perform a major task in this direction. As professional APCs, the blocking of co-stimulatory signaling molecule CD40 in DCs inhibits the activation of T cells and promotes transplant tolerance. In this scenario, Cas9 mRNA (mCas9) and gRNA targeting CD40 was synthesized and encapsulated into poly(ethylene glycol)-block-poly(lactide-co-glycolide) (PEG-block-PLGA)-based cationic lipid assisted NPs (CLANmCas9/gCD40). This nanoformulation efficiently delivered mCas9/gCD40 within DCs to disrupt CD40 at the genomic level, both in vivo and in vitro. The intravenous injection of this nanoformulation into acute mouse skin transplant model, CLANmCas9/gCD40 significantly mediated the gene disruption of CD40 and inhibited T cell activation. This approach led to prolonged graft survival by reducing its damage. This methodology could be a promising approach to reprogram DCs with CRISPR/Cas9 system to subside transplant rejection.201
Role of CRISPR/Cas9 in Engineering B Cells and Its Delivery Approaches
B cells and their downstream effector cells (plasma cells) are central to humoral immune system as these are the only cells that secrete antibodies.202 These antigen-specific antibodies assist to protect the host from infection through neutralization and opsonization of toxins and pathogens.203 Besides playing a critical role in development and maintenance of immunological memory, B cells also present exogenous antigens to T cells on MHC class II.204
CRISPR/Cas9 genome editing system could be used as a novel approach to modulate B cell functions. CRISPR/Cas9 has been used previously to enhance or modulate some functions of B cells. The survival and differentiation of B cells were demonstrated to be efficiently affected by the CRISPR/Cas9 system.178 This genome-editing system could edit the immunoglobulin genes of hybridoma or B cells under in vitro conditions, thereby altering the kind of immunoglobulin synthesis by these cells.205,206
It is very significant to devise a proper delivery system of this genome editing system to B cells. CRISPR/Cas9 system can be efficiently transported to B cells to study its effects on the expression of specific molecules, survival and cellular differentiation.178 This genome editing system has been used to edit the immunoglobulin genes of B cells or hybridoma cells in vitro, and was used to change the type of antibody produced by these cells.205,206 Several researchers have developed different nanoformulations to deliver CRISPR/Cas9 into cells.207 Still, there is a lack of proper delivery system for CRISPR/Cas9 to B cells in vivo. B cells could take-up almost 30% AuNPs in 24 h post injection.208 This study supported the concept that such nanocarriers could be used as transporters for CRISPR/Cas9 system to immune system organs and may be taken-up by B cells in vivo. However, previous reports demonstrated that surface properties of NPs like size, PEG-ylation and charge, could affect the uptake efficiency of immune system cells.209 Several types of nanoformulations have been used to deliver CRISPR/Cas9 system within B cells under in vivo conditions.207 In this regard, a self-assembled DNA nanoclew carrying Cas9/gRNA ribonucleoprotein complex was delivered under in vivo conditions for tumor gene editing.70
Site-specific engineering of primary human B cells was performed by using CRISPR/Cas9 system through chemically modified sgRNA and purified Cas9 protein. It was demonstrated that Cas9 protein, AAVS1-targeting sgRNA, and AAV6 as a DNA donor for homologous recognition to introduce a DNA sequence encoding splice acceptor-EGFP into the AAVS1 locus of B cells. This system achieved a genetic and protein KO of CD19 at a pace of >70%. This novel approach for genetically engineering B cells opened a window for different applications in basic research, antibody designing and production and cellular therapeutics.210
Clinical Trials Based on CRISPR/Cas9 to Modulate Immune System
In order to portray the potential of genome-editing by CRISPR/Cas9 in different immune system cells, different clinical trials have been carried out over the past decade. The clinical settings involving CRISPR/Cas9 genome-editing platforms were set to assess the maladies such as AIDS, β-thalassemia, hemophilia B, sickle cell anemia, and other different malignancies.211 In 2009, CCR5 gene was knocked out by using zinc finger nucleases (ZFN). During 2016–2017, the first CRISPR was registered for human trials (NCT02793856). Similar trials soon followed this, keeping the significance of CRISPR/Cas9 genome-editing strategy in protocol. This strategy was more time-efficient, cost-effective, and simple as compared to ZFNs or transcription activator-like effector nucleases (TALENs).212
Adoptive T cell therapy (ATC) for cancer management has been a major motivation of recent genome-editing trials. In 2016, the first Phase I clinical trial involving CRISPR was performed in Chinese PLA General Hospital. In this clinical setting, PD-1-KO primary T cells were used on stage IV metastatic NSCLC patients (NCT02793856).94 They opted to use CRISPR/Cas9 to disrupt cytokine-induced SH2 (CISH) in TILs collected from gastrointestinal tract tumor sites registered as (NCT04089891) and (NCT04426669). CISH protein suppresses cytokine signalling which is induced in CD8+ T cells upon TCR stimulation and prevents anticancer function of T cells.213 Almost 250 clinical settings have been registered for CAR T cells, some of which have been dedicated to use genome-editing strategy.214
The safe and effective CRISPR/Cas9-engineered universal CD19/CD22 CAR T cells were used in six patients suffering from relapsed/refractory acute lymphoblastic leukemia. No graft vs host disease (GVHD), chromosomal translocation and genotoxicity were reported.215 In T-cell malignancies, the expression of CD7 can be abrogated by the gene editing strategy. T-cell tumors excessively express this marker and functional UCART7 has been designed to eliminate T-ALL in vivo/in vitro, avoiding the risk of GVHD.216 This tactic has become the foundation of current clinical settings in subjects with lymphoma or T-cell leukemia (NCT03690011). The current clinical settings also include the investigation of immune checkpoint inhibitors (ICI). Disruption of PD-1 by CRISPR/Cas9 is being carried out to generate mesothelin-directed CAR T cells ((NCT03747965) and UCARTs (NCT03545815). Furthermore, a recent trial is examining endogenous hematopoietic progenitor kinase 1 (HPK1) efficacy in CD19 CAR T cells. This protein negatively regulates the activation of TCR signal, thus it can be a novel immunotherapy target.217 There are so many clinical settings for using CAR T cells, however, no registered CRISPR/Cas9 transduced CAR-NK cell clinical trials have been registered. Some more clinical trials using CRISPR/Cas9 genome-editing technology for ACT as a treatment strategy for different cancers are listed in Table 2.
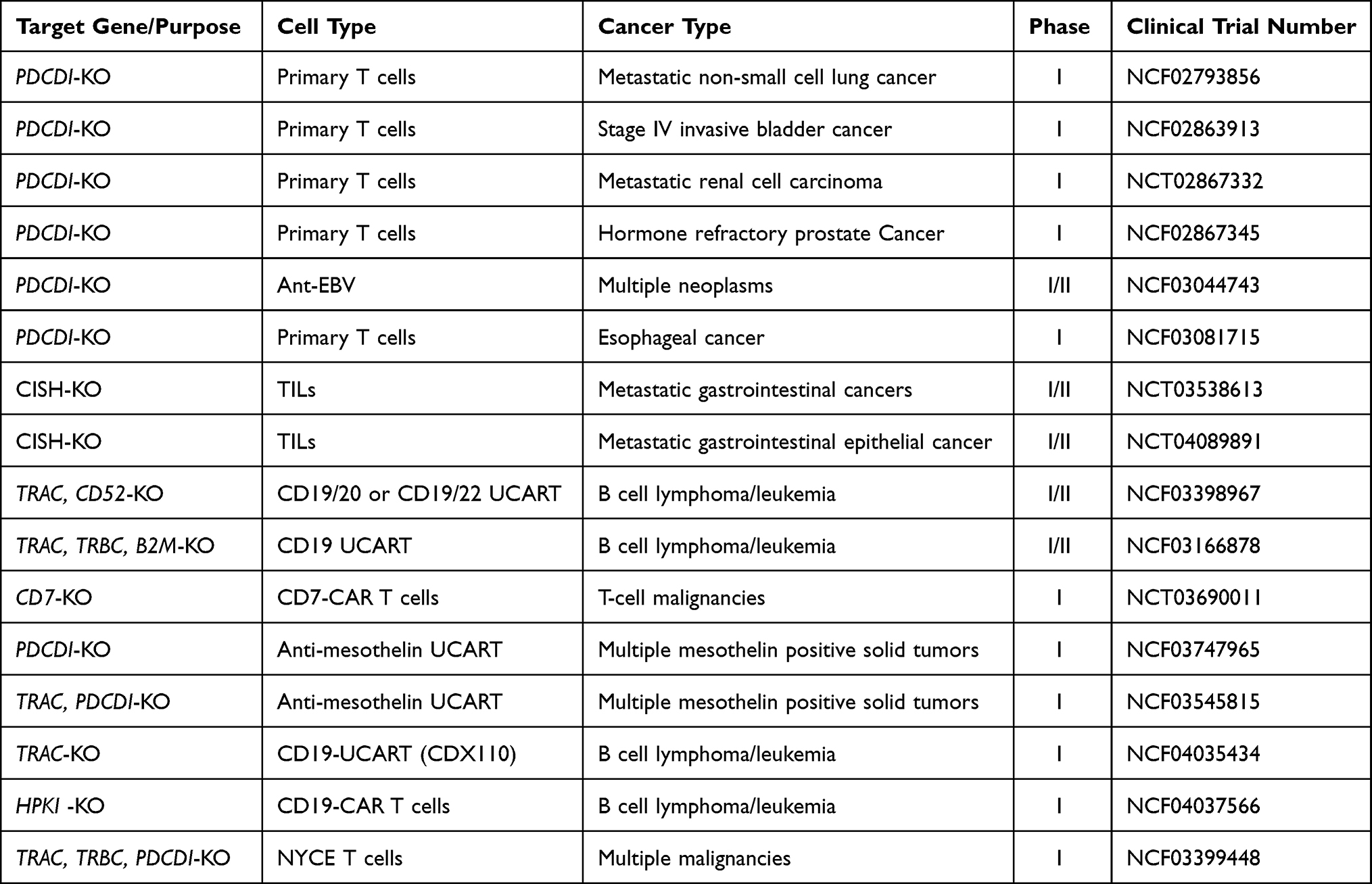 |
Table 2 Clinical trials using CRISPR/Cas9 genome-editing technology for ACT as a treatment strategy for different cancers |
Challenges, Limitations and Future Prospects of CRISPR/Cas9 in Modulating Immune System
Even though encouraging advancement has been achieved in cancer immunotherapy, in depth knowledge of tumor immunology with advanced cancer immunotherapeutics is still highly in demand. With the expansion of CRISPR/Cas9-mediated genome-editing technology, researchers have to exploit this cutting edge strategy to further probe the molecular basis of cancer progression and application in reprogramming different immune system cells. This advanced genome-editing approach may further unravel the mysteries of immune checkpoints and profoundly address the resistance mechanisms involved in cancer immunotherapy in the near future. On the clinical side, CRISPR/Cas9 genome-editing approach will certainly help to engineer further advancement in CAR or TCR T cells. Some new innovative therapeutic immune checkpoints other than PD-1 and CTLA-4 may be disclosed by CRISPR/Cas9-based screening approaches. CRISPR/Cas9-based diamond-cut genome-editing strategy can also make a significant revolution in therapeutic antibodies’ production. CRISPR/Cas9-mediated genetic manipulation can boost immunogenic or antigen-presenting activities within cancerous mass or surrounding immune cells with efficacious cancer vaccines.
In complex eukaryotic cells, the off-target effects of CRISPR/Cas9 genome-editing are a major concern, thus limiting their therapeutic applications.218 The off-target effects can be significantly reduced by using well-optimized and engineered CRISPR/Cas9 system. In addition, CRISPR/Cas9 presents some immunogenic toxicity issues within eukaryotic organisms including humans, as some studies have shown that human subjects possessed pre-existing antibodies against Cas9 protein.219
Currently, CRISPR/Cas9 system is delivered through viral, physical, and nanomaterial-based strategies. Each method presents some advantages besides having other limitations. Thus, choosing an appropriate delivery vehicle targeting the right site with maximum packaging load, and precise delivery are quite challenging and need to be sorted out in the near future.220 Despite the great promise and prospects of applying CRISPR/Cas9 genome-editing technology, special caution needs to be taken before fully addressing the safety protocols and ethical issues in related clinical trials.221 Taken together, CRISPR/Cas9-based genome-editing strategy is believed to transform cancer immunotherapy towards more practical and authorized treatment modalities in cancer clinics.
Conclusion
Recent innovations in CRISPR/Cas9-based genome-editing strategy in different immune system cells have led to the emergence of new promising applications in cancer therapeutics. The stimulation of different immune system cells against varied cancers is totally focussed on destroying them with minimal harmful effects on non-cancerous healthy cells. Recently good progress has been made by precise immunosurveillance and destruction of cancer cells by CRISPR/Cas9-engineered immune cells within the TME, but the task is still challenging and needs to be sorted out in the near future. By using CRISPR/Cas9 system to simultaneously knock out endogenous TCR, MHC-I and different immune checkpoints to construct universal and TCR T cells researchers have been able to advance tumor immunotherapy. This genome-editing system has presented enormous potential to enhance NK cell immunotherapy by arming these cells with CAR constructs, activation pathways, filtration into tumors and by antagonizing inhibitory pathways. Among all immune system cells, NK cells and T cells have shown the most promising results to improve their anticancer activity by using CRISPP/Cas9 genome-editing tools. However, the delivery approaches of CRISPR/Cas9 genome-editing tools to different immune system cells are in parallel challenging as there are many hurdles to be overcome to set the clinical translation of this genome-editing system. Some viral and nanomaterial-based carriers have been designed to introduce CRISPR/Cas9 system within different immune system cells. Besides the targeting approach challenges, the off-target effects of CRISPR/Cas9 are also considered as major limitations of this genome-editing tool, however, some off-target effects have been sorted out by using engineered Cas9 variants. Some other issues related to Cas9 protein include some immunogenic toxicity in eukaryotic organisms. Furthermore, human-health related ethical concerns need to be addressed before accepting the cancer immunotherapy as a routine clinical cancer treatment approach. Nevertheless, with the cooperative employment of biochemistry, biotechnology and nanotechnology, CRISPR/Cas9 will offer an innovative strategy to boost immune surveillance to combat different cancers in the near future.
Abbreviations
AAVs, adeno-associated viruses; ACT, adoptive cell transfer; ADVs, adenoviruses; CAR, chimeric antigen receptor; CLAN, cationic lipid-assisted NPs; CRISPR/Cas9, clustered regularly interspaced short palindromic repeats/associated protein 9; crRNA, CRISPR RNA; CSCs, cancer stem cells; CTLA-4, cytotoxic T lymphocyte antigen-4; DCs, dendritic cells; DSB, double-stranded break; GVHD, graft vs host disease; HDR, homology directed repair; IFN-⍺, interferon-⍺; IL-10, interleukin-10; KO, knockout; mAb, monoclonal antibody; MDSCs, myeloid derived suppressor cells; MHC-I, major histocompatibility complex-I; MICA, MHC class I protein-related sequence A; MMPs, matrix metalloproteinases; NHEJ, nonhomologous end-joining; NK cell, natural killer cell; NPs, nanoparticles; NUC, nuclease; PAM, protospacer adjacent motif; PBMCs, peripheral blood mononuclear cells; PD-1, programmed cell death-1; PGE2, prostaglandin E2; PTEN, phosphatase and tensin homolog; REC, recognition; RNPs, ribonucleoproteins; sgRNA, single guide RNA; TALENs, transcription activator-like effector nucleases; TAMs, tumor associated macrophages; TCR, T-cell receptor; TGF-β, transforming growth factor-β; TILs, tumor infiltrating lymphocytes; TME, tumor microenvironment; TNF-⍺, tumor necrosis factor-⍺; TRAIL, tumor necrosis factor-related apoptosis-inducing ligand; VEGF, vascular endothelial growth factor; ZFN, zinc finger nucleases.
Acknowledgment
Researchers would like to thank the Deanship of Scientific Research, Qassim University for funding publication of this project.
Disclosure
The authors declare no conflicts of interest in this work.
References
1. Stratton MR, Campbell PJ, Futreal PA. The cancer genome. Nature. 2009;458(7239):719–724. doi:10.1038/nature07943
2. Podlaha O, Riester M, De S, Michor F. Evolution of the cancer genome. Trends Genet. 2012;28(4):155–163. doi:10.1016/j.tig.2012.01.003
3. Lei MM, Lee TK. Cancer stem cells: Emerging key players in immune evasion of cancers. Front Cell Dev Biol. 2021;9:692940. doi:10.3389/fcell.2021.692940
4. Robert C. A decade of immune-checkpoint inhibitors in cancer therapy. Nat Commun. 2020;11(1):3801. doi:10.1038/s41467-020-17670-y
5. Thorsson V, Gibbs DL, Brown SD, et al. The immune landscape of cancer. Immunity. 2018;48(4):812–830. doi:10.1016/j.immuni.2018.03.023
6. Binnewies M, Roberts EW, Kersten K, et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat Med. 2018;24(5):541–550. doi:10.1038/s41591-018-0014-x
7. Courau T, Nehar-Belaid D, Florez L, et al. TGF-β and VEGF cooperatively control the immunotolerant tumor environment and the efficacy of cancer immunotherapies. JCI insight. 2016;1(9). doi:10.1172/jci.insight.85974
8. Vahl JM, Friedrich J, Mittler S, et al. Interleukin-10-regulated tumour tolerance in non-small cell lung cancer. Br J Cancer. 2017;117(11):1644–1655. doi:10.1038/bjc.2017.336
9. Burr ML, Sparbier CE, Chan KL, et al. An evolutionarily conserved function of polycomb silences the MHC class I antigen presentation pathway and enables immune evasion in cancer. Cancer cell. 2019;36(4):385–401. doi:10.1016/j.ccell.2019.08.008
10. Curiel TJ, Coukos G, Zou L, et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med. 2004;10(9):942–949. doi:10.1038/nm1093
11. Saito T, Nishikawa H, Wada H, et al. Two FOXP3+ CD4+ T cell subpopulations distinctly control the prognosis of colorectal cancers. Nat Med. 2016;22(6):679–684. doi:10.1038/nm.4086
12. Ferrari de Andrade L, Kumar S, Luoma AM, et al. Inhibition of MICA and MICB Shedding Elicits NK-Cell–Mediated Immunity against Tumors Resistant to Cytotoxic T CellsActivating NK Cells to Target Tumors Resistant to T Cells. Cancer Immunol Res. 2020;8(6):769–780. doi:10.1158/2326-6066.CIR-19-0483
13. Gordon SR, Maute RL, Dulken BW, et al. PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nature. 2017;545(7655):495–499. doi:10.1038/nature22396
14. Allemailem KS, Alsahli MA, Almatroudi A, et al. Current updates of CRISPR/Cas9‐mediated genome editing and targeting within tumor cells: an innovative strategy of cancer management. Cancer Commun. 2022;42:1257–1287. doi:10.1002/cac2.12366
15. Allemailem KS, Almatroodi SA, Almatroudi A, et al. Recent Advances in Genome-Editing Technology with CRISPR/Cas9 Variants and Stimuli-Responsive Targeting Approaches within Tumor Cells: A Future Perspective of Cancer Management. Int J Mol Sci. 2023;24(8):7052. doi:10.3390/ijms24087052
16. Cong L, Ran FA, Cox D, et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339(6121):819–823. doi:10.1126/science.1231143
17. Strong A, Musunuru K. Genome editing in cardiovascular diseases. Nat Rev Cardiol. 2017;14(1):11–20. doi:10.1038/nrcardio.2016.139
18. Kim S, Hupperetz C, Lim S, Kim CH. Genome editing of immune cells using CRISPR/Cas9. BMB Rep. 2021;54(1):59. doi:10.5483/BMBRep.2021.54.1.245
19. Xu Y, Chen C, Guo Y, Hu S, Sun Z. Effect of CRISPR/Cas9-edited PD-1/PD-L1 on tumor immunity and immunotherapy. Front Immunol. 2022;13:848327. doi:10.3389/fimmu.2022.848327
20. Dimitri A, Herbst F, Fraietta JA. Engineering the next-generation of CAR T-cells with CRISPR-Cas9 gene editing. Mol Cancer. 2022;21(1):78. doi:10.1186/s12943-022-01559-z
21. Schieber M, Marinaccio C, Bolanos LC, et al. FBXO11 is a candidate tumor suppressor in the leukemic transformation of myelodysplastic syndrome. Blood Cancer J. 2020;10(10):98. doi:10.1038/s41408-020-00362-7
22. Nguyen AT. CRISPR/Cas9 Screen for Small Molecule Inhibitors of Clonal Hematopoiesis of Indeterminate Potential [Doctoral dissertation]. Harvard-M.I.T; 2020.
23. Nahmad AD, Raviv Y, Horovitz-Fried M, et al. Engineered B cells expressing an anti-HIV antibody enable memory retention, isotype switching and clonal expansion. Nat Commun. 2020;11(1):5851. doi:10.1038/s41467-020-19649-1
24. Shmakov S, Abudayyeh OO, Makarova KS, et al. Discovery and functional characterization of diverse class 2 CRISPR-Cas systems. Mol Cell. 2015;60(3):385–397. doi:10.1016/j.molcel.2015.10.008
25. Luster AD, Alon R, von Andrian UH. Immune cell migration in inflammation: present and future therapeutic targets. Nat Immunol. 2005;6(12):1182–1190. doi:10.1038/ni1275
26. Zomer A, Ellenbroek SI, Ritsma L, Beerling E, Vrisekoop N, Van Rheenen J. Brief report: Intravital imaging of cancer stem cell plasticity in mammary tumors. Stem cells. 2013;31(3):602–606. doi:10.1002/stem.1296
27. Noh KH, Kim BW, Song KH, et al. Nanog signaling in cancer promotes stem-like phenotype and immune evasion. J Clin Invest. 2012;122(11):4077–4093. doi:10.1172/JCI64057
28. Chang C, Lee SO, Yeh S, Chang TM. Androgen receptor (AR) differential roles in hormone-related tumors including prostate, bladder, kidney, lung, breast and liver. Oncogene. 2014;33(25):3225–3234. doi:10.1038/onc.2013.274
29. Ling K, Jiang L, Liang S, et al. Nanog interaction with the androgen receptor signaling axis induce ovarian cancer stem cell regulation: studies based on the CRISPR/Cas9 system. J Ovarian Res. 2018;11(1):1–6. doi:10.1186/s13048-018-0403-2
30. Yang F, Cui P, Lu Y, Zhang X. Requirement of the transcription factor YB-1 for maintaining the stemness of cancer stem cells and reverting differentiated cancer cells into cancer stem cells. Stem Cell Res Ther. 2019;10(1):1–5. doi:10.1186/s13287-019-1360-4
31. Krausova M, Korinek V. Wnt signaling in adult intestinal stem cells and cancer. Cell Signal. 2014;26(3):570–579. doi:10.1016/j.cellsig.2013.11.032
32. Zhan T, Ambrosi G, Wandmacher AM, et al. MEK inhibitors activate Wnt signalling and induce stem cell plasticity in colorectal cancer. Nat Commun. 2019;10(1):2197. doi:10.1038/s41467-019-09898-0
33. Hwang JH, Yoon J, Cho YH, Cha PH, Park JC, Choi KY. A mutant KRAS‐induced factor REG4 promotes cancer stem cell properties via Wnt/β‐catenin signaling. Int J cancer. 2020;146(10):2877–2890. doi:10.1002/ijc.32728
34. Mantovani A, Sica A, Sozzani S, Allavena P, Vecchi A, Locati M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004;25(12):677–686. doi:10.1016/j.it.2004.09.015
35. Balkwill F. Cancer and the chemokine network. Nat Rev Cancer. 2004;4(7):540–550. doi:10.1038/nrc1388
36. Steinman RM. Decisions about dendritic cells: past, present, and future. Annu Rev Immunol. 2012;30:1–22. doi:10.1146/annurev-immunol-100311-102839
37. Xiangrong C, Jingbo L. Dendritic cells play a role in the specific cellular immunity. J Immunol. 2001;17(3):231–234.
38. Youlin K, Weiyang H, Simin L, Xin G. Prostaglandin E2 inhibits prostate cancer progression by countervailing tumor microenvironment-induced impairment of dendritic cell migration through LXRα/CCR7 pathway. J Immunol Res. 2018;2018:1. doi:10.1155/2018/5808962
39. Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. 2009;9(3):162–174. doi:10.1038/nri2506
40. Condamine T, Gabrilovich DI. Molecular mechanisms regulating myeloid-derived suppressor cell differentiation and function. Trends Immunol. 2011;32(1):19–25. doi:10.1016/j.it.2010.10.002
41. Ostrand-Rosenberg S, Sinha P, Beury DW, Clements VK. Cross-talk between myeloid-derived suppressor cells (MDSC), macrophages, and dendritic cells enhances tumor-induced immune suppression. In: Seminars in cancer biology. Academic Press; 2012:275–281.
42. Adeegbe DO, Nishikawa H. Natural and induced T regulatory cells in cancer. Front Immunol. 2013;4:190. doi:10.3389/fimmu.2013.00190
43. De Visser KE, Eichten A, Coussens LM. Paradoxical roles of the immune system during cancer development. Nat Rev Cancer. 2006;6(1):24–37. doi:10.1038/nrc1782
44. Weller K, Foitzik K, Paus R, et al. Mast cells are required for normal healing of skin wounds in mice. FASEB J. 2006;20(13):2366–2368. doi:10.1096/fj.06-5837fje
45. Conti P, Castellani ML, Kempuraj D, et al. Role of mast cells in tumor growth. Ann Clin Lab Sci. 2007;37(4):315–322.
46. Frenzel L, Hermine O. Mast cells and inflammation. Joint Bone Spine. 2013;80(2):141–145. doi:10.1016/j.jbspin.2012.08.013
47. Caughey GH. Mast cell tryptases and chymases in inflammation and host defense. Immunol Rev. 2007;217(1):141–154. doi:10.1111/j.1600-065X.2007.00509.x
48. Parker KH, Beury DW, Ostrand-Rosenberg S. Myeloid-derived suppressor cells: critical cells driving immune suppression in the tumor microenvironment. Adv Cancer Res. 2015;128:95–139.
49. Barrangou R, Fremaux C, Deveau H, et al. CRISPR provides acquired resistance against viruses in prokaryotes. Science. 2007;315(5819):1709–1712. doi:10.1126/science.1138140
50. Brouns SJ, Jore MM, Lundgren M, et al. Small CRISPR RNAs guide antiviral defense in prokaryotes. Science. 2008;321(5891):960–964. doi:10.1126/science.1159689
51. Xu Y, Li Z. CRISPR-Cas systems: overview, innovations and applications in human disease research and gene therapy. Comput Struct Biotechnol J. 2020;18:2401–2415. doi:10.1016/j.csbj.2020.08.031
52. Bolotin A, Quinquis B, Sorokin A, Ehrlich SD. Clustered regularly interspaced short palindrome repeats (CRISPRs) have spacers of extrachromosomal origin. Microbiology. 2005;151(8):2551–2561. doi:10.1099/mic.0.28048-0
53. Zhang D, Hussain A, Manghwar H, et al. Genome editing with the CRISPR‐Cas system: an art, ethics and global regulatory perspective. Plant Biotechnol J. 2020;18(8):1651–1669. doi:10.1111/pbi.13383
54. Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. A programmable dual-RNA–guided DNA endonuclease in adaptive bacterial immunity. science. 2012;337(6096):816–821. doi:10.1126/science.1225829
55. Mojica FJ, Díez-Villaseñor C, García-Martínez J, Almendros C. Short motif sequences determine the targets of the prokaryotic CRISPR defence system. Microbiology. 2009;155(3):733–740. doi:10.1099/mic.0.023960-0
56. Anzalone AV, Koblan LW, Liu DR. Genome editing with CRISPR–Cas nucleases, base editors, transposases and prime editors. Nat Biotechnol. 2020;38(7):824–844. doi:10.1038/s41587-020-0561-9
57. Pickar-Oliver A, Gersbach CA. The next generation of CRISPR–Cas technologies and applications. Nat Rev Mol Cell Biol. 2019;20(8):490–507. doi:10.1038/s41580-019-0131-5
58. Wang H, La Russa M, Qi LS. CRISPR/Cas9 in genome editing and beyond. Annu Rev Biochem. 2016;85:227–264. doi:10.1146/annurev-biochem-060815-014607
59. Kotowski M, Sharma S. CRISPR-based editing techniques for genetic manipulation of primary T cells. Methods Protoc. 2020;3(4):79. doi:10.3390/mps3040079
60. Long C, Amoasii L, Mireault AA, et al. Postnatal genome editing partially restores dystrophin expression in a mouse model of muscular dystrophy. Science. 2016;351(6271):400–403. doi:10.1126/science.aad5725
61. Ran FA, Cong L, Yan WX, et al. In vivo genome editing using Staphylococcus aureus Cas9. Nature. 2015;520(7546):186–191. doi:10.1038/nature14299
62. Kim E, Koo T, Park SW, et al. In vivo genome editing with a small Cas9 orthologue derived from Campylobacter jejuni. Nat Commun. 2017;8(1):14500. doi:10.1038/ncomms14500
63. Atsavapranee ES, Billingsley MM, Mitchell MJ. Delivery technologies for T cell gene editing: Applications in cancer immunotherapy. EBioMedicine. 2021;67:103354. doi:10.1016/j.ebiom.2021.103354
64. Yip BH. Recent advances in CRISPR/Cas9 delivery strategies. Biomolecules. 2020;10(6):839. doi:10.3390/biom10060839
65. Yu X, Liang X, Xie H, et al. Improved delivery of Cas9 protein/gRNA complexes using lipofectamine CRISPRMAX. Biotechnol Lett. 2016;38:919–929. doi:10.1007/s10529-016-2064-9
66. Yin H, Song CQ, Dorkin JR, et al. Therapeutic genome editing by combined viral and non-viral delivery of CRISPR system components in vivo. Nat Biotechnol. 2016;34(3):328–333. doi:10.1038/nbt.3471
67. Mout R, Ray M, Lee YW, Scaletti F, Rotello VM. In vivo delivery of CRISPR/Cas9 for therapeutic gene editing: progress and challenges. Bioconjug Chem. 2017;28(4):880–884. doi:10.1021/acs.bioconjchem.7b00057
68. Gao X, Tao Y, Lamas V, et al. Treatment of autosomal dominant hearing loss by in vivo delivery of genome editing agents. Nature. 2018;553(7687):217–221. doi:10.1038/nature25164
69. Alsaiari SK, Patil S, Alyami M, et al. Endosomal escape and delivery of CRISPR/Cas9 genome editing machinery enabled by nanoscale zeolitic imidazolate framework. J Am Chem Soc. 2018;140(1):143–146. doi:10.1021/jacs.7b11754
70. Sun W, Ji W, Hall JM, et al. Self‐assembled DNA nanoclews for the efficient delivery of CRISPR–Cas9 for genome editing. Angew Chem. 2015;127(41):12197–12201. doi:10.1002/ange.201506030
71. Lee K, Conboy M, Park HM, et al. Nanoparticle delivery of Cas9 ribonucleoprotein and donor DNA in vivo induces homology-directed DNA repair. Nat Biomed Eng. 2017;1(11):889–901. doi:10.1038/s41551-017-0137-2
72. Wang HX, Li M, Lee CM, et al. CRISPR/Cas9-based genome editing for disease modeling and therapy: challenges and opportunities for nonviral delivery. Chem Rev. 2017;117(15):9874–9906. doi:10.1021/acs.chemrev.6b00799
73. Seidel JA, Otsuka A, Kabashima K. Anti-PD-1 and anti-CTLA-4 therapies in cancer: mechanisms of action, efficacy, and limitations. Front Oncol. 2018;8:86. doi:10.3389/fonc.2018.00086
74. Tang F, Zheng P. Tumor cells versus host immune cells: whose PD-L1 contributes to PD-1/PD-L1 blockade mediated cancer immunotherapy? Cell Biosci. 2018;8:1–8. doi:10.1186/s13578-018-0232-4
75. Rohaan MW, van den Berg JH, Kvistborg P, Haanen JB. Adoptive transfer of tumor-infiltrating lymphocytes in melanoma: a viable treatment option. J Immunol Cancer. 2018;6(1):1–6. doi:10.1186/s40425-018-0391-1
76. Gomez-Eerland R, Nuijen B, Heemskerk B, et al. Manufacture of gene-modified human T-cells with a memory stem/central memory phenotype. Hum Gene Ther Methods. 2014;25(5):277–287. doi:10.1089/hgtb.2014.004
77. Fousek K, Watanabe J, Joseph SK, et al. CAR T-cells that target acute B-lineage leukemia irrespective of CD19 expression. Leukemia. 2021;35(1):75–89. doi:10.1038/s41375-020-0792-2
78. Albers JJ, Ammon T, Gosmann D, et al. Gene editing enables T-cell engineering to redirect antigen specificity for potent tumor rejection. Life Sci Alliance. 2019;2(2):e201900367. doi:10.26508/lsa.201900367
79. Morton LT, Reijmers RM, Wouters AK, et al. Simultaneous deletion of endogenous TCRαβ for TCR gene therapy creates an improved and safe cellular therapeutic. Mol Ther. 2018;28(1):64–74. doi:10.1016/j.ymthe.2019.10.001
80. Eyquem J, Mansilla-Soto J, Giavridis T, et al. Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature. 2017;543(7643):113–117. doi:10.1038/nature21405
81. Pettitt D, Arshad Z, Smith J, Stanic T, Holländer G, Brindley D. CAR-T cells: a systematic review and mixed methods analysis of the clinical trial landscape. Mol Ther. 2018;26(2):342–353. doi:10.1016/j.ymthe.2017.10.019
82. Weinkove R, George P, Dasyam N, McLellan AD. Selecting costimulatory domains for chimeric antigen receptors: functional and clinical considerations. Clin Transl Immunol. 2019;8(5):e1049. doi:10.1002/cti2.1049
83. Borowicz P, Chan H, Medina D, Gumpelmair S, Kjelstrup H, Spurkland A. A simple and efficient workflow for generation of knock‐in mutations in Jurkat T cells using CRISPR/Cas9. Scand J Immunol. 2020;91(4):e12862. doi:10.1111/sji.12862
84. Freen‐van Heeren JJ, Popović B, Guislain A, Wolkers MC. Human T cells employ conserved AU‐rich elements to fine‐tune IFN‐γ production. Eur J Immunol. 2020;50(7):949–958. doi:10.1002/eji.201948458
85. Nüssing S, House IG, Kearney CJ, et al. Efficient CRISPR/Cas9 gene editing in uncultured naive mouse T cells for in vivo studies. J Immunol. 2020;204(8):2308–2315. doi:10.4049/jimmunol.1901396
86. Klepsch V, Pommermayr M, Humer D, Brigo N, Hermann-Kleiter N, Baier G. Targeting the orphan nuclear receptor NR2F6 in T cells primes tumors for immune checkpoint therapy. Cell Commun Signal. 2020;18:1–2. doi:10.1186/s12964-019-0454-z
87. Tang N, Cheng C, Zhang X, et al. TGF-β inhibition via CRISPR promotes the long-term efficacy of CAR T cells against solid tumors. JCI Insight. 2020;5(4). doi:10.1172/jci.insight.133977
88. Su S, Zou Z, Chen F, et al. CRISPR-Cas9-mediated disruption of PD-1 on human T cells for adoptive cellular therapies of EBV positive gastric cancer. Oncoimmunology. 2017;6(1):e1249558. doi:10.1080/2162402X.2016.1249558
89. Chen X, Kozhaya L, Tastan C, et al. Functional interrogation of primary human T cells via CRISPR genetic editing. J Immunol. 2018;201(5):1586–1598. doi:10.4049/jimmunol.1701616
90. Hultquist JF, Hiatt J, Schumann K, et al. CRISPR–Cas9 genome engineering of primary CD4+ T cells for the interrogation of HIV–host factor interactions. Nat Protoc. 2019;14(1):1–27. doi:10.1038/s41596-018-0069-7
91. Okada M, Kanamori M, Someya K, Nakatsukasa H, Yoshimura A. Stabilization of Foxp3 expression by CRISPR-dCas9-based epigenome editing in mouse primary T cells. Epigenetics Chromatin. 2017;10:1–7. doi:10.1186/s13072-017-0129-1
92. Roth TL, Puig-Saus C, Yu R, et al. Reprogramming human T cell function and specificity with non-viral genome targeting. Nature. 2018;559(7714):405–409. doi:10.1038/s41586-018-0326-5
93. Jing Z, Zhang N, Ding L, et al. Safety and activity of programmed cell death-1 gene knockout engineered t cells in patients with previously treated advanced esophageal squamous cell carcinoma: an open-label, single-arm phase I study; 2018.
94. Lu Y, Xue J, Deng T, et al. Safety and feasibility of CRISPR-edited T cells in patients with refractory non-small-cell lung cancer. Nat Med. 2020;26(5):732–740. doi:10.1038/s41591-020-0840-5
95. Stadtmauer EA, Fraietta JA, Davis MM, et al. CRISPR-engineered T cells in patients with refractory cancer. Science. 2020;367(6481):eaba7365. doi:10.1126/science.aba7365
96. Iwai Y, Terawaki S, Honjo T. PD-1 blockade inhibits hematogenous spread of poorly immunogenic tumor cells by enhanced recruitment of effector T cells. Int Immunol. 2005;17(2):133–144. doi:10.1093/intimm/dxh194
97. Hamanishi J, Mandai M, Ikeda T, et al. Safety and antitumor activity of anti–PD-1 antibody, nivolumab, in patients with platinum-resistant ovarian cancer. J Clin Oncol. 2015;33(34):4015–4022. doi:10.1200/JCO.2015.62.3397
98. Qin S, Ren Z, Meng Z, et al. Camrelizumab in patients with previously treated advanced hepatocellular carcinoma: a multicentre, open-label, parallel-group, randomised, Phase 2 trial. Lancet Oncol. 2020;21(4):571–580. doi:10.1016/S1470-2045(20)30011-5
99. Garon EB, Hellmann MD, Rizvi NA, et al. Five-year overall survival for patients with advanced non‒small-cell lung cancer treated with pembrolizumab: results from the phase I KEYNOTE-001 study. J Clin Oncol. 2019;37(28):2518. doi:10.1200/JCO.19.00934
100. Hodi FS, O’day SJ, McDermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363(8):711–723. doi:10.1056/NEJMoa1003466
101. Song Y, Gao Q, Zhang H, et al. Treatment of relapsed or refractory classical Hodgkin lymphoma with the anti-PD-1, tislelizumab: results of a phase 2, single-arm, multicenter study. Leukemia. 2020;34(2):533–542. doi:10.1038/s41375-019-0545-2
102. Nishino M, Giobbie-Hurder A, Hatabu H, Ramaiya NH, Hodi FS. Incidence of programmed cell death 1 inhibitor–related pneumonitis in patients with advanced cancer: a systematic review and meta-analysis. JAMA Oncol. 2016;2(12):1607–1616. doi:10.1001/jamaoncol.2016.2453
103. Bae S, Park J, Kim JS. Cas-OFFinder: a fast and versatile algorithm that searches for potential off-target sites of Cas9 RNA-guided endonucleases. Bioinformatics. 2014;30(10):1473–1475. doi:10.1093/bioinformatics/btu048
104. Riese MJ, Moon EK, Johnson BD, Albelda SM. Diacylglycerol kinases (DGKs): novel targets for improving T cell activity in cancer. Front Cell Dev Biol. 2016;4:108. doi:10.3389/fcell.2016.00108
105. Zhao J, Lin Q, Song Y, Liu D. Universal CARs, universal T cells, and universal CAR T cells. J Hematol Oncol. 2018;11:1–9. doi:10.1186/s13045-018-0677-2
106. Metropulos AE, Munshi HG, Principe DR. The difficulty in translating the preclinical success of combined TGFβ and immune checkpoint inhibition to clinical trial. EBioMedicine. 2022;86:104380. doi:10.1016/j.ebiom.2022.104380
107. Wang H, Kaur G, Sankin AI, Chen F, Guan F, Zang X. Immune checkpoint blockade and CAR-T cell therapy in hematologic malignancies. J Hematol Oncol. 2019;12(1):1–20. doi:10.1186/s13045-019-0746-1
108. Sheykhhasan M, Manoochehri H, Dama P. Use of CAR T-cell for acute lymphoblastic leukemia (ALL) treatment: a review study. Cancer Gene Ther. 2022;29(8–9):1080–1096. doi:10.1038/s41417-021-00418-1
109. Yoon DH, Osborn MJ, Tolar J, Kim CJ. Incorporation of immune checkpoint blockade into chimeric antigen receptor T cells (CAR-Ts): combination or built-in CAR-T. Int J Mol Sci. 2018;19(2):340. doi:10.3390/ijms19020340
110. Guo X, Jiang H, Shi B, et al. Disruption of PD-1 enhanced the anti-tumor activity of chimeric antigen receptor T cells against hepatocellular carcinoma. Front Pharmacol. 2018;9:1118. doi:10.3389/fphar.2018.01118
111. Bailey SR, Vatsa S, Larson R, et al. Blocking IFNγ in CAR-T reduces checkpoint inhibitors and cell-mediated toxicity without compromising therapeutic efficacy in CD19+ malignancies. Blood. 2021;138:1723. doi:10.1182/blood-2021-146042
112. Zhang ZZ, Wang T, Wang XF, Zhang YQ, Song SX, Ma CQ. Improving the ability of CAR-T cells to hit solid tumors: Challenges and strategies. Pharmacol Res. 2022;175:106036. doi:10.1016/j.phrs.2021.106036
113. Klepsch V, Siegmund K, Baier G. Emerging next-generation target for cancer immunotherapy research: the orphan nuclear receptor NR2F6. Cancers. 2021;13(11):2600. doi:10.3390/cancers13112600
114. He QF, Xu Y, Li J, Huang ZM, Li XH, Wang X. CD8+ T-cell exhaustion in cancer: mechanisms and new area for cancer immunotherapy. Brief Funct Genomics. 2019;18(2):99–106. doi:10.1093/bfgp/ely006
115. Romano E, Romero P. The therapeutic promise of disrupting the PD-1/PD-L1 immune checkpoint in cancer: unleashing the CD8 T cell mediated anti-tumor activity results in significant, unprecedented clinical efficacy in various solid tumors. J Immunol Cancer. 2015;3(1):1–5. doi:10.1186/s40425-015-0059-z
116. Rouanne M, Roumiguié M, Houédé N, et al. Development of immunotherapy in bladder cancer: present and future on targeting PD (L) 1 and CTLA-4 pathways. World J Urol. 2018;36:1727–1740. doi:10.1007/s00345-018-2332-5
117. Chen Y, Tan J, Huang S, et al. Higher frequency of the CTLA‐4+ LAG‐3+ T‐cell subset in patients with newly diagnosed acute myeloid leukemia. Asia Pac J Clin Oncol. 2020;16(2):e12–8. doi:10.1111/ajco.13236
118. Corgnac S, Boutet M, Kfoury M, Naltet C, Mami-Chouaib F. The emerging role of CD8+ tissue resident memory T (TRM) cells in antitumor immunity: a unique functional contribution of the CD103 integrin. Front Immunol. 2018;9:1904. doi:10.3389/fimmu.2018.01904
119. DeBruin KA, Krassowska W. Modeling electroporation in a single cell. I. Effects of field strength and rest potential. Biophys J. 1999;77(3):1213–1224. doi:10.1016/S0006-3495(99)76973-0
120. Luecke S, Holleufer A, Christensen MH, et al. cGAS is activated by DNA in a length‐dependent manner. EMBO Rep. 2017;18(10):1707–1715. doi:10.15252/embr.201744017
121. Zierhut C, Yamaguchi N, Paredes M, Luo JD, Carroll T, Funabiki H. The cytoplasmic DNA sensor cGAS promotes mitotic cell death. Cell. 2019;178(2):302–315. doi:10.1016/j.cell.2019.05.035
122. Kim S, Kim D, Cho SW, Kim J, Kim JS. Highly efficient RNA-guided genome editing in human cells via delivery of purified Cas9 ribonucleoproteins. Genome Res. 2014;24(6):1012–1019. doi:10.1101/gr.171322.113
123. Michieletto D, Lusic M, Marenduzzo D, Orlandini E. Physical principles of retroviral integration in the human genome. Nat Commun. 2019;10(1):575. doi:10.1038/s41467-019-08333-8
124. Shen B, Zhang W, Zhang J, et al. Efficient genome modification by CRISPR-Cas9 nickase with minimal off-target effects. Nat Methods. 2014;11(4):399–402. doi:10.1038/nmeth.2857
125. Miller JB, Zhang S, Kos P, et al. Non‐viral CRISPR/Cas gene editing in vitro and in vivo enabled by synthetic nanoparticle co‐delivery of Cas9 mRNA and sgRNA. Angew Chem. 2017;129(4):1079–1083. doi:10.1002/ange.201610209
126. Schubert MS, Thommandru B, Woodley J, et al. Optimized design parameters for CRISPR Cas9 and Cas12a homology-directed repair. Sci Rep. 2021;11(1):19482. doi:10.1038/s41598-021-98965-y
127. Horii T, Arai Y, Yamazaki M, et al. Validation of microinjection methods for generating knockout mice by CRISPR/Cas-mediated genome engineering. Sci Rep. 2014;4(1):4513. doi:10.1038/srep04513
128. Raveux A, Vandormael-Pournin S, Cohen-Tannoudji M. Optimization of the production of knock-in alleles by CRISPR/Cas9 microinjection into the mouse zygote. Sci Rep. 2017;7(1):42661. doi:10.1038/srep42661
129. Boukany PE, Morss A, Liao WC, et al. Nanochannel electroporation delivers precise amounts of biomolecules into living cells. Nat Nanotechnol. 2011;6(11):747–754. doi:10.1038/nnano.2011.164
130. Kumar P, Nagarajan A, Uchil PD. Electroporation. Cold Spring Harb Protoc. 2019;2019(7):db–top096271. doi:10.1101/pdb.top096271
131. Seki A, Rutz S. Optimized RNP transfection for highly efficient CRISPR/Cas9-mediated gene knockout in primary T cells. J Exp Med. 2018;215(3):985–997. doi:10.1084/jem.20171626
132. Roth TL, Li PJ, Blaeschke F, et al. Pooled knockin targeting for genome engineering of cellular immunotherapies. Cell. 2020;181(3):728–744. doi:10.1016/j.cell.2020.03.039
133. Nguyen DN, Roth TL, Li PJ, et al. Polymer-stabilized Cas9 nanoparticles and modified repair templates increase genome editing efficiency. Nat Biotechnol. 2020;38(1):44–49. doi:10.1038/s41587-019-0325-6
134. D’Astolfo DS, Pagliero RJ, Pras A, et al. Efficient intracellular delivery of native proteins. Cell. 2015;161(3):674–690. doi:10.1016/j.cell.2015.03.028
135. Kholosy WM, Visscher M, Ogink K, et al. Simple, fast and efficient iTOP-mediated delivery of CRISPR/Cas9 RNP in difficult-to-transduce human cells including primary T cells. J Biotechnol. 2021;338:71–80. doi:10.1016/j.jbiotec.2021.07.006
136. Bošnjak B, Permanyer M, Sethi MK, et al. CRISPR/Cas9 genome editing using gold‐nanoparticle‐mediated laserporation. Adv Biosyst. 2018;2(11):1700184. doi:10.1002/adbi.201700184
137. Rahimmanesh I, Totonchi M, Khanahmad H. The challenging nature of primary T lymphocytes for transfection: effect of protamine sulfate on the transfection efficiency of chemical transfection reagents. Res Pharm Sci. 2020;15(5):437. doi:10.4103/1735-5362.297846
138. Afolabi L, Bi J, Yang X, Wan X. Suppression of the protein quality control system by TRIM30a sensitizes tumor to NK cell immune surveillance. Eur J Immunol. 2019;1(49):461.
139. Afolabi LO, Adeshakin AO, Sani MM, Bi J, Wan X. Genetic reprogramming for NK cell cancer immunotherapy with CRISPR/Cas9. Immunology. 2019;158(2):63–69. doi:10.1111/imm.13094
140. Bi J, Tian Z. NK cell exhaustion. Front Immunol. 2017;8:760. doi:10.3389/fimmu.2017.00760
141. Pomeroy EJ, Hunzeker JT, Kluesner MG, et al. A genetically engineered primary human natural killer cell platform for cancer immunotherapy. Mol Ther. 2020;28(1):52–63. doi:10.1016/j.ymthe.2019.10.009
142. Vivier E, Tomasello E, Baratin M, Walzer T, Ugolini S. Functions of natural killer cells. Nat Immunol. 2008;9(5):503–510. doi:10.1038/ni1582
143. Hermanson DL, Kaufman DS. Utilizing chimeric antigen receptors to direct natural killer cell activity. Front Immunol. 2015;6:195. doi:10.3389/fimmu.2015.00195
144. Rezvani K, Rouce RH. The application of natural killer cell immunotherapy for the treatment of cancer. Front Immunol. 2015;6:578. doi:10.3389/fimmu.2015.00578
145. Cortez VS, Ulland TK, Cervantes-Barragan L, et al. SMAD4 impedes the conversion of NK cells into ILC1-like cells by curtailing non-canonical TGF-β signaling. Nat Immunol. 2017;18(9):995–1003. doi:10.1038/ni.3809
146. Gao Y, Souza-Fonseca-Guimaraes F, Bald T, et al. Tumor immunoevasion by the conversion of effector NK cells into type 1 innate lymphoid cells. Nat Immunol. 2017;18(9):1004–1015. doi:10.1038/ni.3800
147. Vivier E, Ugolini S, Blaise D, Chabannon C, Brossay L. Targeting natural killer cells and natural killer T cells in cancer. Nat Rev Immunol. 2012;12(4):239–252. doi:10.1038/nri3174
148. Fang F, Xiao W, Tian Z. NK cell-based immunotherapy for cancer. In: Seminars in Immunology. Academic Press; 2017:37–54.
149. Sivori S, Meazza R, Quintarelli C, et al. NK cell-based immunotherapy for hematological malignancies. J Clin Med. 2019;8(10):1702. doi:10.3390/jcm8101702
150. Imai CM, Mihara K, Andreansky M, et al. Chimeric receptors with 4-1BB signaling capacity provoke potent cytotoxicity against acute lymphoblastic leukemia. Leukemia. 2004;18(4):676–684. doi:10.1038/sj.leu.2403302
151. Liu D, Tian S, Zhang K, et al. Chimeric antigen receptor (CAR)-modified natural killer cell-based immunotherapy and immunological synapse formation in cancer and HIV. Protein Cell. 2017;8(12):861–877. doi:10.1007/s13238-017-0415-5
152. Velasquez MP, Szoor A, Bonifant CL, et al. Two-pronged cell therapy for B-cell malignancies: engineering NK cells to target CD22 and redirect bystander T cells to CD19. Blood. 2016;128(22):4560. doi:10.1182/blood.V128.22.4560.4560
153. Chu Y, Hochberg J, Yahr A, et al. Targeting CD20+ aggressive B-cell non–Hodgkin lymphoma by anti-CD20 CAR mRNA-modified expanded natural killer cells in vitro and in NSG mice. Cancer Immunol Res. 2015;3(4):333–344. doi:10.1158/2326-6066.CIR-14-0114
154. Yilmaz A, Cui H, Caligiuri MA, Yu J. Chimeric antigen receptor-engineered natural killer cells for cancer immunotherapy. J Hematol Oncol. 2020;13(1):1–22. doi:10.1186/s13045-020-00998-9
155. Raulet DH, Gasser S, Gowen BG, Deng W, Jung H. Regulation of ligands for the NKG2D activating receptor. Annu Rev Immunol. 2013;31:413–441. doi:10.1146/annurev-immunol-032712-095951
156. Zhang T, Lemoi BA, Sentman CL. Chimeric NK-receptor–bearing T cells mediate antitumor immunotherapy. Blood. 2005;106(5):1544–1551. doi:10.1182/blood-2004-11-4365
157. Chang YH, Connolly J, Shimasaki N, Mimura K, Kono K, Campana D. A Chimeric Receptor with NKG2D Specificity Enhances Natural Killer Cell Activation and Killing of Tumor CellsNKG2D Receptor Enhances NK Cell Killing of Tumors. Cancer Res. 2013;73(6):1777–1786. doi:10.1158/0008-5472.CAN-12-3558
158. Muller N, Michen S, Tietze S, et al. Engineering NK cells modified with an EGFRvIII-specific chimeric antigen receptor to overexpress CXCR4 improves immunotherapy of CXCL12/SDF-1α-secreting glioblastoma. J Immunother. 2015;38(5):197–210. doi:10.1097/CJI.0000000000000082
159. Topfer K, Cartellieri M, Michen S, et al. DAP12-based activating chimeric antigen receptor for NK cell tumor immunotherapy. J Immunol. 2015;194(7):3201–3212. doi:10.4049/jimmunol.1400330
160. Altvater B, Landmeier S, Pscherer S, et al. 2B4 (CD244) signaling by recombinant antigen-specific chimeric receptors costimulates natural killer cell activation to leukemia and neuroblastoma cells. Clin Cancer Res. 2009;15(15):4857–4866. doi:10.1158/1078-0432.CCR-08-2810
161. Shimasaki N, Fujisaki H, Cho D, et al. A clinically adaptable method to enhance the cytotoxicity of natural killer cells against B-cell malignancies. Cytotherapy. 2012;14(7):830–840. doi:10.3109/14653249.2012.671519
162. Liu E, Tong Y, Dotti G, et al. Cord blood NK cells engineered to express IL-15 and a CD19-targeted CAR show long-term persistence and potent antitumor activity. Leukemia. 2018;32(2):520–531. doi:10.1038/leu.2017.226
163. Levin AM, Bates DL, Ring AM, et al. Exploiting a natural conformational switch to engineer an interleukin-2 ‘superkine’. Nature. 2012;484(7395):529–533. doi:10.1038/nature10975
164. Sekiba K, Yamagami M, Otsuka M, et al. Transcriptional activation of the MICA gene with an engineered CRISPR-Cas9 system. Biochem Biophys Res Commun. 2017;486(2):521–525. doi:10.1016/j.bbrc.2017.03.076
165. Kremer V, Ligtenberg MA, Zendehdel R, et al. Genetic engineering of human NK cells to express CXCR2 improves migration to renal cell carcinoma. J Immunol Cancer. 2017;5:1–3. doi:10.1186/s40425-016-0206-1
166. Adachi K, Kano Y, Nagai T, Okuyama N, Sakoda Y, Tamada K. IL-7 and CCL19 expression in CAR-T cells improves immune cell infiltration and CAR-T cell survival in the tumor. Nat Biotechnol. 2018;36(4):346–351. doi:10.1038/nbt.4086
167. Carlsten M, Levy E, Karambelkar A, et al. Efficient mRNA-based genetic engineering of human NK cells with high-affinity CD16 and CCR7 augments rituximab-induced ADCC against lymphoma and targets NK cell migration toward the lymph node-associated chemokine CCL19. Front Immunol. 2016;7:105. doi:10.3389/fimmu.2016.00105
168. Levy ER, Carlsten M. Childs RW. mRNA transfection to improve NK cell homing to tumors. Nat Killer Cells. 2016;2016:231–240.
169. Caruana I, Savoldo B, Hoyos V, et al. Heparanase promotes tumor infiltration and antitumor activity of CAR-redirected T lymphocytes. Nat Med. 2015;21(5):524–529. doi:10.1038/nm.3833
170. Putz EM, Mayfosh AJ, Kos K, et al. NK cell heparanase controls tumor invasion and immune surveillance. J Clin Invest. 2017;127(7):2777–2788. doi:10.1172/JCI92958
171. Blake SJ, Stannard K, Liu J, et al. Suppression of metastases using a new lymphocyte checkpoint target for cancer immunotherapyTargeting CD96 in cancer immunotherapy. Cancer Discov. 2016;6(4):446–459. doi:10.1158/2159-8290.CD-15-0944
172. Zhang Q, Bi J, Zheng X, et al. Blockade of the checkpoint receptor TIGIT prevents NK cell exhaustion and elicits potent anti-tumor immunity. Nat Immunol. 2018;19(7):723–732. doi:10.1038/s41590-018-0132-0
173. Andre P, Denis C, Soulas C, et al. Anti-NKG2A mAb is a checkpoint inhibitor that promotes anti-tumor immunity by unleashing both T and NK cells. Cell. 2018;175(7):1731–1743. doi:10.1016/j.cell.2018.10.014
174. Rautela J, Surgenor E, Huntington ND. Efficient genome editing of human natural killer cells by CRISPR RNP. BioRxiv. 2018;6:406934.
175. Li C, Guan X, Du T, et al. Inhibition of HIV-1 infection of primary CD4+ T-cells by gene editing of CCR5 using adenovirus-delivered CRISPR/Cas9. J Gen Virol. 2015;96(8):2381–2393. doi:10.1099/vir.0.000139
176. Wang D, Mou H, Li S, et al. Adenovirus-mediated somatic genome editing of Pten by CRISPR/Cas9 in mouse liver in spite of Cas9-specific immune responses. Hum Gene Ther. 2015;26(7):432–442. doi:10.1089/hum.2015.087
177. Kararoudi MN, Dolatshad H, Trikha P, et al. Generation of knock-out primary and expanded human NK cells using Cas9 ribonucleoproteins. J Vis Exp. 2018;2018(136):1.
178. Chu VT, Graf R, Wirtz T, et al. Efficient CRISPR-mediated mutagenesis in primary immune cells using CrispRGold and a C57BL/6 Cas9 transgenic mouse line. Proc Natl Acad Sci. 2016;113(44):12514–12519. doi:10.1073/pnas.1613884113
179. Andreu N, Phelan J, de Sessions PF, Cliff JM, Clark TG, Hibberd ML. Primary macrophages and J774 cells respond differently to infection with Mycobacterium tuberculosis. Sci Rep. 2017;7(1):42225. doi:10.1038/srep42225
180. Fehervari Z. Don’t eat me, activate me. Nat Immunol. 2015;16(11):1113.
181. Slee JB, Christian AJ, Levy RJ, Stachelek SJ. Addressing the inflammatory response to clinically relevant polymers by manipulating the host response using ITIM domain-containing receptors. Polymers. 2014;6(10):2526–2551. doi:10.3390/polym6102526
182. Inagaki K, Yamao T, Noguchi T, et al. SHPS-1 regulates integrin-mediated cytoskeletal reorganization and cell motility. EMBO J. 2000;19(24):6721–6731. doi:10.1093/emboj/19.24.6721
183. Barclay AN, Van den Berg TK. The interaction between signal regulatory protein alpha (SIRPα) and CD47: structure, function, and therapeutic target. Annu Rev Immunol. 2014;32:25–50. doi:10.1146/annurev-immunol-032713-120142
184. Chao MP, Tang C, Pachynski RK, Chin R, Majeti R, Weissman IL. Extranodal dissemination of non-Hodgkin lymphoma requires CD47 and is inhibited by anti-CD47 antibody therapy. J Am Soc Hematol. 2011;118(18):4890–4901.
185. Edris B, Weiskopf K, Volkmer AK, et al. Antibody therapy targeting the CD47 protein is effective in a model of aggressive metastatic leiomyosarcoma. Proc Natl Acad Sci. 2012;109(17):6656–6661. doi:10.1073/pnas.1121629109
186. Ho CC, Guo N, Sockolosky JT, et al. “Velcro” engineering of high affinity CD47 ectodomain as signal regulatory protein α (SIRPα) antagonists that enhance antibody-dependent cellular phagocytosis. J Biol Chem. 2015;290(20):12650–12663. doi:10.1074/jbc.M115.648220
187. Panni RZ, Linehan DC, DeNardo DG. Targeting tumor-infiltrating macrophages to combat cancer. Immunotherapy. 2013;5(10):1075–1087. doi:10.2217/imt.13.102
188. Mitchem JB, Brennan DJ, Knolhoff BL, et al. Targeting Tumor-Infiltrating Macrophages Decreases Tumor-Initiating Cells, Relieves Immunosuppression, and Improves Chemotherapeutic ResponsesMacrophages Regulate Tumor-Initiating Cells. Cancer Res. 2013;73(3):1128–1141. doi:10.1158/0008-5472.CAN-12-2731
189. Mout R, Ray M, Yesilbag tonga G, et al. Direct cytosolic delivery of CRISPR/Cas9-ribonucleoprotein for efficient gene editing. ACS nano. 2017;11(3):2452–2458. doi:10.1021/acsnano.6b07600
190. Mantovani A, Allavena P, Marchesi F, Garlanda C. Macrophages as tools and targets in cancer therapy. Nat Rev Drug Discov. 2022;16:1–22.
191. Ray M, Lee YW, Hardie J, et al. CRISPRed macrophages for cell-based cancer immunotherapy. Bioconjug Chem. 2018;29(2):445–450. doi:10.1021/acs.bioconjchem.7b00768
192. Luo YL, Xu CF, Li HJ, et al. Macrophage-specific in vivo gene editing using cationic lipid-assisted polymeric nanoparticles. ACS Nano. 2018;12(2):994–1005. doi:10.1021/acsnano.7b07874
193. Lee YW, Mout R, Luther DC, et al. In vivo editing of macrophages through systemic delivery of CRISPR‐Cas9‐ribonucleoprotein‐nanoparticle nanoassemblies. Adv Ther. 2019;2(10):1900041. doi:10.1002/adtp.201900041
194. Cantore A, Milani M, Annoni A, et al. Liver-directed gene therapy for hemophilia B with immune stealth lentiviral vectors. Blood. 2017;130:605.
195. Merad M, Sathe P, Helft J, Miller J, Mortha A. The dendritic cell lineage: ontogeny and function of dendritic cells and their subsets in the steady state and the inflamed setting. Annu Rev Immunol. 2013;31:563–604. doi:10.1146/annurev-immunol-020711-074950
196. Sun T, Nguyen A, Gommerman JL. Dendritic cell subsets in intestinal immunity and inflammation. J Immunol. 2020;204(5):1075–1083. doi:10.4049/jimmunol.1900710
197. Wculek SK, Cueto FJ, Mujal AM, Melero I, Krummel MF, Sancho D. Dendritic cells in cancer immunology and immunotherapy. Nat Rev Immunol. 2020;20(1):7–24. doi:10.1038/s41577-019-0210-z
198. Song XT. Genetic modification of dendritic cells with RNAi. Cancer Vaccines. 2014;2014:119–130.
199. Pulendran B, Davis MM. The science and medicine of human immunology. Science. 2020;369(6511):eaay4014. doi:10.1126/science.aay4014
200. Jost M, Jacobson AN, Hussmann JA, Cirolia G, Fischbach MA, Weissman JS. CRISPR-based functional genomics in human dendritic cells. Elife. 2021;10:e65856. doi:10.7554/eLife.65856
201. Zhang Y, Shen S, Zhao G, et al. In situ repurposing of dendritic cells with CRISPR/Cas9-based nanomedicine to induce transplant tolerance. Biomaterials. 2019;217:119302. doi:10.1016/j.biomaterials.2019.119302
202. Radbruch A, Muehlinghaus G, Luger EO, et al. Competence and competition: the challenge of becoming a long-lived plasma cell. Nat Rev Immunol. 2006;6(10):741–750. doi:10.1038/nri1886
203. Forthal DN. Functions of antibodies. Microbiol Spectr. 2014;2(4):2–4. doi:10.1128/microbiolspec.AID-0019-2014
204. Rodríguez-Pinto D. B cells as antigen presenting cells. Cell Immunol. 2005;238(2):67–75. doi:10.1016/j.cellimm.2006.02.005
205. Cheong TC, Compagno M, Chiarle R. Editing of mouse and human immunoglobulin genes by CRISPR-Cas9 system. Nat Commun. 2016;7(1):10934. doi:10.1038/ncomms10934
206. Pogson M, Parola C, Kelton WJ, Heuberger P, Reddy ST. Immunogenomic engineering of a plug-and-(dis) play hybridoma platform. Nat Commun. 2016;7(1):12535. doi:10.1038/ncomms12535
207. Wang X, Zhang W, Ding Y, Guo X, Yuan Y, Li D. CRISPR/Cas9-mediated genome engineering of CXCR4 decreases the malignancy of hepatocellular carcinoma cells in vitro and in vivo. Oncol Rep. 2017;37(6):3565–3571. doi:10.3892/or.2017.5601
208. Almeida JP, Lin AY, Langsner RJ, Eckels P, Foster AE, Drezek RA. In vivo immune cell distribution of gold nanoparticles in naive and tumor bearing mice. Small. 2014;10(4):812–819. doi:10.1002/smll.201301998
209. Chang TZ, Stadmiller SS, Staskevicius E, Champion JA. Effects of ovalbumin protein nanoparticle vaccine size and coating on dendritic cell processing. Biomater Sci. 2017;5(2):223–233. doi:10.1039/C6BM00500D
210. Johnson MJ, Laoharawee K, Lahr WS, Webber BR, Moriarity BS. Engineering of primary human B cells with CRISPR/Cas9 targeted nuclease. Sci Rep. 2018;8(1):12144. doi:10.1038/s41598-018-30358-0
211. Ashmore-Harris C, Fruhwirth GO. The clinical potential of gene editing as a tool to engineer cell-based therapeutics. Clin Transl Med. 2020;9(1):15. doi:10.1186/s40169-020-0268-z
212. Drake MJ, Bates P. Application of gene editing technologies to HIV-1. Curr Opin HIV AIDS. 2015;10(2):123. doi:10.1097/COH.0000000000000139
213. Palmer DC, Guittard GC, Franco Z, et al. Cish actively silences TCR signaling in CD8+ T cells to maintain tumor tolerance. J Exp Med. 2015;212(12):2095–2113. doi:10.1084/jem.20150304
214. June CH, O’Connor RS, Kawalekar OU, Ghassemi S, Milone MC. CAR T cell immunotherapy for human cancer. Science. 2018;359(6382):1361–1365. doi:10.1126/science.aar6711
215. Hu Y, Zhou Y, Zhang M, et al. CRISPR/Cas9-engineered universal CD19/CD22 dual-targeted CAR-T cell therapy for relapsed/refractory B-cell acute lymphoblastic leukemiaCRISPR/Cas9-engineered universal CAR-T therapy for ALL. Clin Cancer Res. 2021;27(10):2764–2772. doi:10.1158/1078-0432.CCR-20-3863
216. Cooper ML, Choi J, Staser K, et al. An “off-the-shelf” fratricide-resistant CAR-T for the treatment of T cell hematologic malignancies. Leukemia. 2018;32(9):1970–1983. doi:10.1038/s41375-018-0065-5
217. Sawasdikosol S, Burakoff S. A perspective on HPK1 as a novel immuno-oncology drug target. Elife. 2020;9:e55122. doi:10.7554/eLife.55122
218. Zhang XH, Tee LY, Wang XG, Huang QS, Yang SH. Off-target effects in CRISPR/Cas9-mediated genome engineering. Mol Ther. 2015;4:e264. doi:10.1038/mtna.2015.37
219. Mehta A, Merkel OM. Immunogenicity of Cas9 protein. J Pharm Sci. 2020;109(1):62–67. doi:10.1016/j.xphs.2019.10.003
220. Li T, Yang Y, Qi H, et al. CRISPR/Cas9 therapeutics: progress and prospects. Signal Transduct Target Ther. 2023;8(1):36. doi:10.1038/s41392-023-01309-7
221. Rath J. Safety and security risks of CRISPR/Cas9. Ethics Dumping: Case Studies from North-South Research Collaborations; 2018.
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.