")
Back to Journals » Therapeutics and Clinical Risk Management » Volume 19
Innovations in the Treatment of Dystrophic Epidermolysis Bullosa (DEB): Current Landscape and Prospects
Authors Hou PC, del Agua N , Lwin SM, Hsu CK, McGrath JA
Received 26 April 2023
Accepted for publication 2 June 2023
Published 14 June 2023 Volume 2023:19 Pages 455—473
DOI https://doi.org/10.2147/TCRM.S386923
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Garry Walsh
Ping-Chen Hou,1,* Nathalie del Agua,2,3,* Su M Lwin,4 Chao-Kai Hsu,1– 3 John A McGrath1,3,4
1Department of Dermatology, National Cheng Kung University Hospital, College of Medicine, National Cheng Kung University, Tainan, Taiwan; 2Institute of Clinical Medicine, College of Medicine, National Cheng Kung University, Tainan, Taiwan; 3International Center for Wound Repair and Regeneration (iWRR), National Cheng Kung University, Tainan, Taiwan; 4St John’s Institute of Dermatology, School of Basic and Medical Biosciences, King’s College London, London, UK
*These authors contributed equally to this work
Correspondence: Chao-Kai Hsu, Department of Dermatology, National Cheng Kung University Hospital, 138 Sheng-Li Road, Tainan City, Taiwan, Tel +886-62353535 Ext.5415, Fax +886-62766180, Email [email protected] John A McGrath, St John’s Institute of Dermatology, School of Basic and Medical Biosciences, King’s College London, London, UK, Tel +44-2071886409, Fax +44-2071888050, Email [email protected]
Abstract: Dystrophic epidermolysis bullosa (DEB) is one of the major types of EB, a rare hereditary group of trauma-induced blistering skin disorders. DEB is caused by inherited pathogenic variants in the COL7A1 gene, which encodes type VII collagen, the major component of anchoring fibrils which maintain adhesion between the outer epidermis and underlying dermis. DEB can be subclassified into dominant (DDEB) and recessive (RDEB) forms. Generally, DDEB has a milder phenotype, while RDEB patients often have more extensive blistering, chronic inflammation, skin fibrosis, and a propensity for squamous cell carcinoma development, collectively impacting on daily activities and life expectancy. At present, best practice treatments are mostly supportive, and thus there is a considerable burden of disease with unmet therapeutic need. Over the last 20 years, considerable translational research efforts have focused on either trying to cure DEB by direct correction of the COL7A1 gene pathology, or by modifying secondary inflammation to lessen phenotypic severity and improve patient symptoms such as poor wound healing, itch, and pain. In this review, we provide an overview and update on various therapeutic innovations for DEB, including gene therapy, cell-based therapy, protein therapy, and disease-modifying and symptomatic control agents. We outline the progress and challenges for each treatment modality and identify likely prospects for future clinical impact.
Keywords: skin, blister, gene therapy, cell therapy, inflammation, fibrosis
Introduction
Epidermolysis bullosa (EB) is a clinically and genetically heterogeneous group of blistering skin disorders, in which minor trauma to the skin results in skin fragility.1,2 The different types of EB are caused by inherited pathogenic variants in any one of 16 causative genes, which mostly encode structural proteins at or close to the dermal-epidermal junction (DEJ).1,2 EB can be classified into four major types according to the cleavage plane within the skin, comprising EB simplex (separation in the basal epidermis), junctional EB (separation within the lamina lucida), dystrophic EB (DEB) (separation within the upper dermis below the lamina densa), and Kindler EB (separation at multiple planes).1,2 This review article focuses on the DEB type, which accounts for ~30% of the affected individuals.3
DEB is caused by pathogenic variants in the COL7A1 gene, which encodes type VII collagen (C7), the major component of anchoring fibrils (AF) that maintains dermal-epidermal cohesion.1,2 DEB can be sub-classified into either dominant (DDEB) or recessive (RDEB) forms.2 The clinical severity of DEB is highly variable (Figure 1). Generally, DDEB patients have milder phenotypes with either mild-to-moderate or sometimes localized skin blistering, often with dystrophic finger and toenails. RDEB can resemble DDEB in its localized or intermediate forms but oftentimes the clinical features are more severe, with widespread skin blistering, fibrosis and scarring, joint contractures, and systemic comorbidities such as anemia, osteoporosis, or malnutrition. All forms of DEB show an increased risk of developing cutaneous squamous cell carcinoma (SCC) which typically occurs in areas of maximal scarring or chronic ulcers and is more common in severe forms of RDEB.1,2 DEB, and particularly RDEB, can limit daily activities and shorten life expectancy.1,2 The pathobiology underlying DEB is complex and essentially stems from C7 deficiency which leads to a lack of functional AFs causing fragility in skin and some mucosae, with additional dysfunction of innate immunity as C7 is also normally expressed in lymphoid extracellular matrix.4 Skin wounds can exhibit frustrated wound healing, partially healing and then breaking down again, or sometimes simply not healing at all. The impaired wound healing initiates chronic inflammation in the subjacent dermis, which drives skin fibrosis and, in some instances, carcinogenesis.1,5 It is estimated that over half of severe RDEB cases develop squamous cell carcinoma by the age of 30 years.6 Living with RDEB significantly impacts on an individual’s physical and psychological morbidity, leading to substantial healthcare-related financial burdens and implications for both patients and their family members.7,8
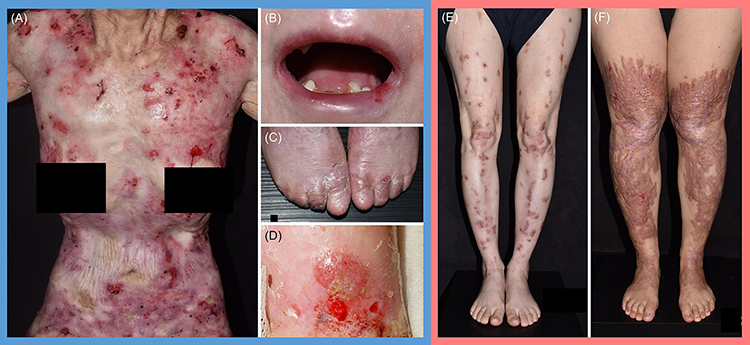 |
Figure 1 Clinical manifestations of DEB patients. (A) Generalized blistering and ulceration, (B) microstomia, (C) pseudosyndactyly, and (D) cutaneous squamous cell carcinoma, in RDEB. (E and F) Scattered, semi-confluent, erythematous or hyperpigmented prurigo-like papules and plaques of varying severity, as well as toenail dystrophy, in DDEB. |
Given the impaired quality of life and substantial health impact of DEB, the development of new and more effective therapies has been a major focus for several clinical teams and research groups worldwide. In broad terms, approaches to therapy have either adopted an “intention to cure” strategy, aiming to replace or fix the defective COL7A1 gene, or focused on reducing the inflammation in DEB tissue, the goal being to lessen disease severity, slow down disease progression, and improve key symptoms such as itch or pain. In this review, we scope the literature to document the various modalities that have emerged in pursuing better treatments for DEB. Schematic illustrations of approaches to therapy are depicted in Figures 2 and 3; a list of current clinical trials is shown in Supplementary Table 1.
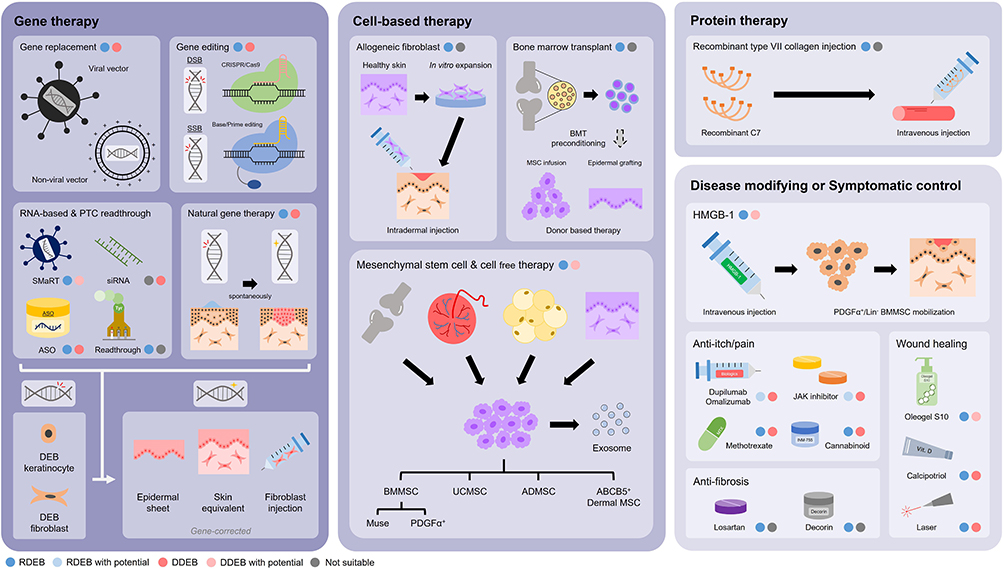 |
Figure 2 Schematic illustration of multimodal treatment options for DEB patients. Various treatment modalities for DEB include gene therapy, cell-based therapy, protein therapy, disease-modifying and symptomatic control agents. (Blue/Pink: has been tried in RDEB/DDEB; Light blue/Light pink: has not been tried in RDEB/DDEB but has potential for treatment; Grey: not suitable). Abbreviations: ABCB5, ATP-binding cassette subfamily B member 5; ADMSC, adipose tissue-derived mesenchymal stem cell; ASO, antisense oligonucleotide; BMMSC, bone marrow-derived mesenchymal stem cell; BMT, bone marrow transplant; C7, type VII collagen; DEB, dystrophic epidermolysis bullosa; DDEB, dominant dystrophic epidermolysis bullosa; DSB, double-strand break; HMGB-1, high mobility group box-1; MSC, mesenchymal stem cell; PTC, premature termination codon; RDEB, recessive dystrophic epidermolysis bullosa; siRNA, small interfering RNA; SMaRT, spliceosome-mediated RNA trans-splicing; SSB, single-strand break; UCMSC, umbilical cord-derived mesenchymal stem cell. |
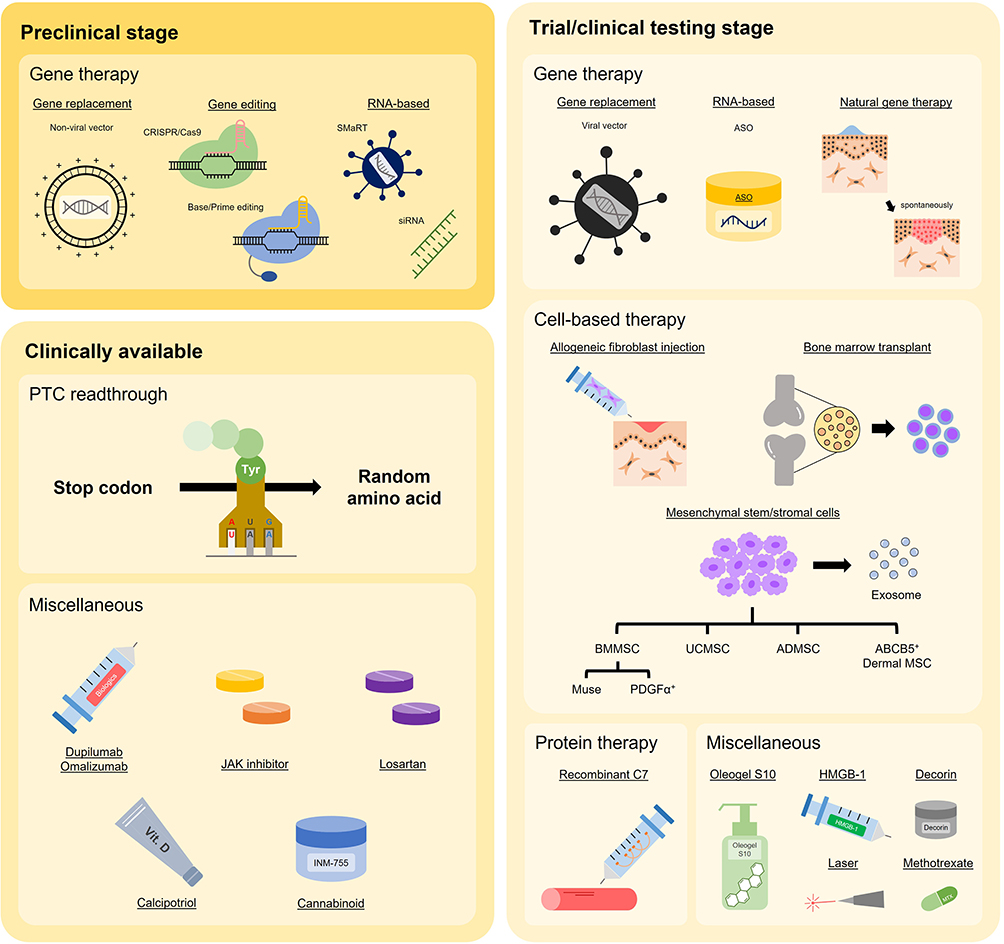 |
Figure 3 Schematic illustration of the clinical status (entering preclinical or clinical stage) of various DEB treatments. Abbreviations: ABCB5, ATP-binding cassette subfamily B member 5; ADMSC, adipose tissue-derived mesenchymal stem cell; ASO, antisense oligonucleotide; BMMSC, bone marrow-derived mesenchymal stem cell; BMT, bone marrow transplant; C7, type VII collagen; HMGB-1, high mobility group box-1; MSC, mesenchymal stem cell; PTC, premature termination codon; siRNA, small interfering RNA; SMaRT, spliceosome-mediated RNA trans-splicing; UCMSC, umbilical cord-derived mesenchymal stem cell. |
Gene Therapy
The molecular pathology of RDEB mostly involves bi-allelic loss-of-function variants in COL7A1, which has led to a major push to develop gene replacement approaches, mostly involving full-length COL7A1 cDNA. At genomic DNA level, most of the COL7A1 variants involve single-nucleotide substitutions which have also generated interest in gene editing applications, as well as other means of allele silencing or skipping of exons containing pathogenic variants, as well as ribosomal readthrough for nonsense variants. For DDEB, most pathogenic variants involve glycine residue substitutions within the C7 triple helix; gene therapy approaches have mostly therefore focused on allele silencing, exon skipping or gene editing.
Gene Replacement Therapy
Most gene replacement therapy techniques for RDEB transduce exogenous full-length wild-type cDNA displacing the mutant allele to reverse skin fragility in EB. COL7A1 cDNA is ~9 kb in size and an important consideration in the development of RDEB gene therapy was whether this would be too large for conventional viral vector delivery into skin cells. Thus, initial COL7A1 gene therapy in immunodeficient mice grafted with gene-modified skin equivalents (SEs) adopted a strategy of creating minigenes (removing cDNA encoding part of the C7 triple helix),9 although in subsequent studies full-length COL7A1 cDNA was introduced.10–16 Reconstituted SEs derived from transduced RDEB keratinocytes and fibroblasts showed functional correction, including C7 deposition, AF formation, and even established dermal-epidermal stability for months after engraftment onto immunodeficient mice,10–16 although only a few pre-clinical experiments advanced to human studies.14,15
In 2016, researchers from Stanford University developed a pioneering gene replacement therapy for patients with RDEB.17 Patient keratinocytes were transduced with a retroviral vector containing full-length COL7A1 cDNA and were cultured to create 35cm2 epidermal autografts (each about the size of a playing card), later called EB-101. These keratinocyte sheets, although somewhat fragile as they lacked a dermal equivalent, were transplanted onto six wounds in four adult subjects. All 24 grafts were well-taken, and nearly all (90%) demonstrated C7 localization at the DEJ and were still intact (87%) 3 months later. Efficacy generally declined after 1 year with no signs that the grafted skin had any growth advantage over the surrounding mutant skin.17 However, long-term graft follow-up showed that about 71% of the treated areas had over 50% wound closure after 2 years, and two patients even had overall durable skin graft coverage for over 4 years.18 In a recent update of the EB-101 trial (NCT01263379) by So et al,19 at year 5, 70% of the treated sites demonstrated at least 50% wound healing compared to baseline. Moreover, healing wounds were observed not to be painful or itchy. There were no serious side effects observed, including immunity against C7 or the development of SCC.19 The phase III, open-label clinical trial of the EB-101 for RDEB treatment has now been completed (NCT04227106). EB-101, developed by Abeona Therapeutics, has been granted Fast Track, Regenerative Medicine Advanced Therapy and Rare Pediatric Disease designations by the FDA (Food and Drug Administration) and Orphan Drug designation by both the FDA and the EMA (European Medicine Agency).
In addition to being synthesized by keratinocytes, C7 is also generated by dermal fibroblasts. In 2019, Lwin et al conducted a phase I trial (NCT02493816; Lenticol-F) to inject self-inactivating (SIN) lentiviral-mediated autologous gene-corrected fibroblasts into normal-appearing skin at a concentration of ~1×106 cells/cm2 and were able to show variable degrees of C7 restoration. While no AF regeneration was observed, synthesized copies of the COL7A1 cDNA were demonstrated in treated skin in one patient at 1 year.20 None of the patient group developed malignancy or autoimmune reactions in more than 1 year of follow-up. Similarly, Fibrocell Science Inc., subsequently acquired by Castle Creek Biosciences, reported a phase I/II clinical trial (NCT02810951) using similar cells, FCX-007 (later named dabocemagene autoficel, D-Fi), which showed that intradermal injections of autologous gene-corrected fibroblasts into RDEB chronic/recurrent wounds were safe and effective, wherein 80% (4/5) of the treated areas showed significant (over 70%) wound closure and linear staining of C7 at the DEJ 3 months after injection.21 A phase III intra-patient controlled open-label trial of D-Fi (NCT04213261) is in progress, having received Orphan Drug, Rare Pediatric Disease, Fast Track and Regenerative Advanced Therapy designations by the FDA. Practically, injecting fibroblasts into RDEB skin can be challenging given the scarred nature of the skin to be injected, and the transient but intense pain caused by injections into such sites.
SIN retroviral vectors have also been used to develop gene therapy products for clinical testing. Skin equivalents, comprising COL7A1-corrected keratinocytes and fibroblasts, have shown impressive grafting results in proof-of-principle transplantation studies in mice,15 and following development of a protocol for human testing,20 these are now being evaluated in a phase I/II study, EBGRAFT (NCT04186650) in up to 3 individuals with RDEB; results are awaited.
One of the major challenges with all the above approaches is that the gene therapy product needs to be used ex vivo in an individual’s cells to create a bespoke cell product for personal use. In contrast, taking a generic approach and moving closer to clinical application, topical gene therapy using a non-integrative replication-defective herpes simplex virus-1 (HSV-1) carrying two copies of the COL7A1 gene, named beremagene gerperpavec (B-VEC), has been developed by Stanford University and Krystal Biotech. In early 2022, a randomized, placebo-controlled, phase I/II clinical trial (NCT03536143) was reported,22 in which primary and secondary objectives of C7 expression, AF assembly, wound surface area reduction, duration of wound closure, and time to wound closure following B-VEC treatment were met. A few months later the results of a phase III study of B-VEC in RDEB (NCT04917874) were also published.23 Primary wound pairs were exposed to B-VEC and placebo in 31 patients. At 6 months, complete wound healing occurred in 67% of the wounds exposed to B-VEC compared with 22% of those exposed to placebo. Moreover, complete wound healing at 3 months occurred in 71% of the wounds treated with B-VEC compared with 20% of those exposed to placebo. There was also a reduction in pain severity during wound dressing changes with B-VEC compared to placebo. Over the two trials, minor side-effects associated with B-VEC included pruritus and chills. Because of the non-integrating nature of HSV, this form of gene therapy is not considered a permanent fix but a treatment that will need to be repeated. The half-life of C7 is at least 6–8 weeks and therefore repeat applications to the same site may only be needed every few months or so, notwithstanding that B-VEC is not a treatment for intact (unwounded) skin, nor does it currently target mucosal pathology or systemic disease associated with RDEB. Although nearly all the patients in the trials had RDEB, B-VEC was also tested in DDEB and shown to improve wound healing.23 Regarding immunogenicity, a history of previous HSV infection or antibody levels to HSV had no bearing on the trial data or any clinical significance. B-VEC was previously granted Fast Track and Regenerative Medicine Advanced Therapy designations by the FDA for the treatment of DEB and received full licensing approval for clinical use in the United States on 19 May 2023. Thus, B-VEC represents the first gene therapy treatment to be available for individuals with DEB.
Gene Editing
An alternative approach for C7 restoration, without the need for full-length COL7A1 cDNA transduction, is through gene editing. It is important to note that gene editing has not yet been clinically tested in patients with any form of DEB and therefore this section of the review focuses on a few landmark developments in the field.
Gene editing for RDEB started in 2013 when Osborn et al reported the use of TALENs to edit RDEB fibroblasts via homology-directed repair.24 These gene-corrected cells were expanded and reprogrammed into inducible pluripotent stem cells (iPSCs) and were used to generate C7-expressing SEs.24 A variety of gene-editing strategies using different nucleases (mainly TALEN and CRISPR/Cas9) have since been developed in preclinical studies, which demonstrated the success of generating gene-edited SEs with C7 expression that could be engrafted onto nude mice and which improved skin stability.25,26
To date, gene editing therapy that focuses on DDEB has only one preclinical study. Site-specific endonucleases for TALENs and CRISPR/Cas9 systems were designed to induce double-strand breaks in iPSCs derived from DDEB fibroblasts and achieved 90% gene correction.27 Four gene-edited iPSCs were then differentiated into keratinocytes and fibroblasts and showed C7 restoration.27 Recently, Neumayer et al28 developed a method to reprogram EB patient-derived fibroblasts into iPSCs and undertake CRISPR-based correction to generate skin composite grafts. Mouse xenografts showed no tumors and exhibited stable C7 re-expression thus offering promise for future clinical translation.
In addition to traditional nucleases, base editing is a relatively new technique of great treatment potential for DEB given that point mutations represent the major form (~80%) of genetic abnormality therein.29,30 Two types of base editors have been described so far, cytosine base editors (CBEs)31 and adenine base editors (ABEs), which can correct transition variants, that is, either C·G to A·T or A·T to C·G changes. Base editing is led by a synthetic single-guide RNA with editors consisting of either an adenosine deaminase or cytidine deaminase linked to a Cas9 nickase. The cytosine is converted to uracil, whereas adenine is converted to inosine, which is regarded as guanine by the DNA polymerase. The nickase creates a single-strand break, stimulating DNA repair to complete the editing.29 For DEB, base editing was first applied in 2020 by Osborn et al.32 They electroporated single guide RNA and ABEs in primary RDEB fibroblasts for COL7A1 gene correction, which significantly increased C7 expression with minimal off-target effects.32 Base editing of RDEB patient-derived fibroblasts using ABE8e, a new ABE developed through phage-assisted non-continuous and continuous evolution (PANCE/PACE), achieved a high accuracy (94.6%) of COL7A1 variant correction and successfully restored C7 expression in a generated 3D skin construct.33 Similarly, CBE has been applied to correct COL7A1 variants in RDEB-derived primary fibroblasts and iPSCs, with restoration of C7 and AFs, highlighting the future translational potential of this approach.34
Besides base editing, the advent of search-and-replace genome editing has expanded to encompass correction of a wider collection of COL7A1 variants using prime editors (PE), which contain a reverse transcriptase domain and a prime editing guide RNA to attain any site-specific edit.35 In a preclinical study by Hong et al, both BE (ABEmax) and PE (PE3) were used to edit the COL7A1 gene in primary RDEB fibroblasts.36 The PE3 demonstrated higher editing efficiency compared to ABEmax, while both approaches successfully generated gene-edited skin grafts with C7 and AF restoration.36
The need to demonstrate a lack of deleterious collateral damage to the genome, known as off-target effects, has created something of a roadblock to clinical translation but one which is likely to be surmountable before too long. It is also noteworthy that gene editing therapy, unlike gene replacement therapy, is mutation dependent and thus personalized, with significant cost and marketing implications for any manufacturer. Another matter is one akin to that facing gene replacement strategies, ie, whether to attempt ex vivo correction of cells with grafting/injection of the modified cell product, or to opt for in vivo gene editing, perhaps delivering the product directly onto wounds using a viral vector or a lipid nanoparticle-peptide delivery system. An additional challenge common to gene editing and gene replacement therapies is how to target unwounded skin (ie, not just open wounds), considering the relatively impermeable nature of stratified squamous epithelium and the skin barrier. For now, no clinical trials of gene editing, in any form, are listed for either RDEB or DDEB.
RNA-Based Therapy
RNA modification can bypass or manipulate mutated DNA sequences for gene correction. There are three known strategies used today: RNA trans-splicing, an antisense oligonucleotide (ASO), and small interfering RNA (siRNA). Of these, only ASO has so far progressed to clinical trial in DEB.
Spliceosome-mediated RNA trans-splicing (SMaRT) replaces the mutant mRNA with wild-type sequences through endogenous splicing molecules for gene correction.37 The RNA trans-splicing molecule (RTM) can be tailored for 5’, 3’, or internal exon replacement.38 Since SMaRT can correct mutated mRNA by replacing only a part of the entire gene, it is suitable for large genes such as COL7A1. Preclinical studies reported the successful gene correction of RDEB keratinocytes by either 5’ or 3’ replacement, which led to phenotypic reversion of RDEB keratinocytes with the restoration of C7 and AF-like structure in SEs.39–41 Despite the preclinical efficacies shown, SMaRT still carries pitfalls including viral delivery and off-target effects, as for DNA replacement therapy.42,43 Recently, a non-viral and non-invasive mode of delivery of 3’-RTMS6m was reported to be capable of replacing exons 65–118 in RDEB-derived immortalized keratinocytes and fibroblasts.44 Although the trans-splicing efficiency was low, the approach was still able to partially restore C7 expression in these cells and in SEs.44
Of more immediate clinical impact in DEB therapeutics, ASOs are short synthetic RNA strands complementary to mutant pre-mRNA which can interfere with the binding sites of splicing enzymes, leading to exon skipping.45 Such modification allows the generation of shorter, but still functional, proteins by removing the mutation-containing exons. This approach is especially practical for genes such as COL7A1, being composed mainly of in-frame exons and proteins with repetitive motifs.42,46 Indeed, of the 118 exons in COL7A1, 80 of these could be skipped in frame. In 2006, Goto et al first described the rescue of C7 in SEs generated from RDEB keratinocytes and fibroblasts after the injection of COL7A1 exon 70-targeted ASO.47 Thereafter, ASOs targeting exons 10, 13, 70, 73, 80, and 105 of COL7A1 have been designed with the resulting shortened C7s still proving to be functional.42,48–50 Of all the exons in COL7A1, exon 73 is a site for several pathogenic variants (both dominant and recessive), partly because it is the largest of all exons encoding the C7 triple helix but also because of some recurrent pathogenic variants therein. Developed by Freiburg University and ProQR, a topical 21-nucleotide ASO carbomer-based hydrogel targeting COL7A1 exon 73 (QR-313), was demonstrated to be safe and efficacious in preclinical studies, restoring C7 expression and improving epidermal-dermal cohesion.48 However, preliminary data from a clinical trial started in 2018 (NCT03605069) demonstrated low uptake and limited efficacy of QR-313 in improving RDEB wounds after topical application every other day for 8 weeks.51 As such, other attempts to skip exon 73, such as using PTW-002, developed by Phoenicis, are set to be tested in a clinical trial very soon (NCT05529134). The concept is that one would attempt to skip both mutant and wild-type exon 73, thus making is a suitable approach for both RDEB and DDEB harboring pathogenic variants in exon 73, with the generation of a slightly truncated but still functional C7 in both instances. One further expectation is that, with refinements in chemistry, it may be possible to deliver ASOs for COL7A1 systemically, ie, via intravenous injection, given the high targeting specificity of ASOs. Such work, however, is still in preclinical development.
As a further option, still at a preclinical stage, is the use of siRNAs, short double-stranded RNA sequences that are specifically designed to bind to the mRNA of interest, thereby silencing the gene.38 Pendaries et al designed an allele-specific siRNA targeting COL7A1 exon 87 in DDEB and demonstrated differential inhibition of the mutated allele.52 In terms of precision, such siRNA therapy can discriminate between two alleles even by a single-point polymorphism, but this approach infers a need for individual patient-tailored design of the siRNA, thus hindering universal application.53
Natural Gene Therapy: Revertant Mosaicism
Revertant mosaicism is a serendipitous event in which a pathogenic variant in a somatic cell is corrected or compensated by various genetic mechanisms, usually at a DNA level.54 Described in several forms of EB and other genetic skin diseases, revertant mosaicism has been observed in RDEB55 and mainly occurs in keratinocytes, although RDEB fibroblasts may also show similar natural gene therapy.56 Patches of revertant skin in RDEB usually measure 1–8cm in diameter and, when biopsied, show C7 at the DEJ and normal AF structures. A key challenge in translational research has been how to exploit this natural phenomenon for patient benefit.
In 2019, Matsumura et al reported that cultured epidermal autografts produced from clinically normal-appearing skin of an RDEB patient had long-standing engraftment for over 16 years.57 They were able to prove that the autograft had been derived from clinically revertant skin. Extending the study to three further patients, they found rapid healing of grafted sites within 2–4 weeks and long-term persistence of wound healing. One of the three patients had apparent revertant mosaicism in the donor skin and in the post-transplanted area. They concluded that cultured epidermal autografts from clinically normal skin are a potentially well-tolerated treatment for RDEB. In Japan, cultured epidermal autografts are covered by public healthcare insurance and DEB treatment is also included in the license (from 2019 onwards). The subsequent literature includes a report of an RDEB patient undergoing extensive skin grafting (almost 60% of body surface area) following expansion of revertant keratinocytes from a small initial skin biopsy.58
Premature Termination Codon Readthrough
Around 15–20% of RDEB cases carry nonsense pathogenic variants, which lead to premature termination codons (PTCs). One approach to therapy in such cases is to generate ribosomal readthrough to produce full-length RNA and subsequently generate protein. One group of compounds with the potential to induce such activity is aminoglycosides, including gentamicin. These drugs induce PTC readthrough through the interference with the decoding center of the ribosomes and the incorporation of a random amino acid via near-cognate tRNA.59 In vitro studies using gentamicin showed successful readthrough of nonsense mutations in RDEB keratinocytes, leading to C7 production.60 With encouraging preclinical results, gentamicin has since been applied topically, injected intradermally or administered systemically in RDEB patients. Woodley et al reported that 2-weeks of intravenous gentamicin promoted wound healing and regenerated C7 in four RDEB pediatric patients,61,62 without nephrotoxicity or ototoxicity.63,64 Gentamicin is a complex that consists of multiple congeners, and there are data to indicate that a sub-component, gentamicin X2, might possess even more potent and safe readthrough,65 and thus further refinement of such therapy is likely. Besides gentamicin, another non-aminoglycoside agent, PTC124 (Ataluren), also has readthrough capability although a preclinical study showed that it was unable to promote readthrough in the COL7A1 gene, and thus it has not been tested in individuals with RDEB.66 Amlexanox, an FDA-approved drug for recurrent aphthous ulcers, can also enhance readthrough and attenuate nonsense-mediated mRNA decay. Preclinical evidence has shown that amlexanox rescued COL7A1 mRNA transcripts and restored functional full-length C7 to a greater extent compared to gentamicin, and thus clinical testing is awaited.67
Cell-Based Therapy
Cell-based therapy encompasses a variety of therapies that include primary keratinocytes, fibroblasts, hematopoietic cells, and mesenchymal stem/stromal cells. Key considerations are whether such cells are autologous or allogeneic, whether they are truly stem cells or merely stromal cells, and how such cells are to be given to the patient with RDEB, ie, skin grafts, local injections, or intravenous infusions. Some of these approaches have already been covered in the gene therapy section above, but additional applications in patients are outlined below.
Fibroblasts
Besides keratinocytes, fibroblasts are the major source of C7 in skin. In animal studies, allogeneic fibroblasts were injected into the intact skin of RDEB mice, which led to C7 re-expression, but only at a dosage of 5×106 cells/cm2,16 and not at lower cell dosing of 1×106 cells/cm2.13 Based on the higher dosing, Wong et al reported clinical evidence of intradermal fibroblast injection in five RDEB patients:68 both allogeneic and haploidentical (from one parent) fibroblasts increased C7 expression for ~3–9 months. Several other clinical trials then injected allogeneic fibroblasts to either the base or the margin of RDEB chronic wounds at a dose of 2.5–5×106 cells/cm2, which promoted significant wound healing and, in some individuals, increased C7 expression, for 3–12 months.69–71 As the injected fibroblasts were not detectable 2 weeks after injection, the therapeutic effects were primarily attributed to the increased expression of endogenous mutant COL7A1 mRNA and C7 (ie, partially functional), likely through a paracrine effect of heparin-binding epidermal growth factor-like growth factor secreted by keratinocytes close to the injected fibroblasts.68,72 This observation also implies that RDEB patients with higher baseline C7 expression levels might potentially benefit more from such treatment. However, the painful process of intradermal injection into scarred skin proved to be intolerable for many patients.
Bone Marrow Transplantation
In murine models of RDEB, hematopoietic stem cells were shown to differentiate into epidermal cells and produce C7, and thereby prolong the life expectancy of RDEB mice.73,74 Based on these data, in 2010 Wagner et al reported a human case series using unfiltered HLA-matched bone marrow transplantation after immune-myeloablation in six RDEB patients.75 C7 restoration and the presence of donor cells were found in the recipients’ skin,75 and although considerable clinical improvement was noted in the majority, the procedure was still associated with considerable morbidity and mortality. Refinements to protocols, including use of reduced intensity conditioning, led to some reduction in mortality (to ~15%),76,77 but a lack of understanding about precisely how bone marrow transplantation was improving the skin (not all patients who improved had new C7 or AFs in their skin), as well as ongoing concerns regarding toxicity, led to a reduced popularity for such treatment.
Nevertheless, one consequence of an RDEB subject receiving a bone marrow transplantation was the development of immune tolerance in that individual to future cell products from the same donor. Infusion of MSCs from the identical donor following bone marrow transplantation, accompanied by post-transplant cyclophosphamide, was able to reduce acute rejection as well as promote wound healing in 8 pediatric RDEB patients.78 Cell tolerance was also extended to skin. Notably, epidermal grafting using the CELLUTOMETM Epidermal Harvesting System from the same donor after allogeneic hematopoietic cell transplantation demonstrated longer engraftment (up to 3 years) with no signs of rejection.79
Overall, despite some initial international uptake of bone marrow transplantation for RDEB therapy, most related transplant activity has now ceased in favor of other therapies.
Mesenchymal Stem Cell (MSCs)
Mesenchymal stem cells, also known as, mesenchymal stromal cells (MSCs) are characterized by their ability for self-renewal and multi-potency,80 yet in the context of DEB cell therapy, most MSCs used therapeutically are from unrelated donors with the anticipated clinical benefits arising from immunomodulatory or anti-inflammatory impact from the cells. Allogeneic MSCs possess little immunogenicity due to low expression of MHC class I molecules on their surface and lack of expression of MHC class II and costimulatory molecules; indeed, the safety of MSCs in humans has been demonstrated in several clinical trials.81 Thus, aggressive pre-conditioning regimens are not needed, and an antihistamine injection alone provides sufficient pre-medication. There are several sources of MSCs for potential application in DEB therapeutics, including bone marrow, adipose tissue, umbilical cord blood and amniotic fluid. MSCs exert therapeutic benefits through trophic effects (secreting cytokines and growth factors, including hepatocyte growth factor, insulin-like growth factor-2, nerve growth factor, and stromal-derived growth factor-1), immunomodulation of the local microenvironment, recruitment of reparative cells, and the promotion of neovascularization.80,82 Regarding wound healing, MSCs have been shown to modify several pertinent pathways, including Akt, ERK and STAT3.83
MSCs have either been injected intradermally or infused intravenously in patients with RDEB. Intradermal administration has the advantage of creating a high concentration of cells in a localized region which may facilitate interaction with host cells such as fibroblasts,83 whereas intravenous infusion has the potential for systemic impact. However, the fate of MSCs, in both skin and blood is uncertain. Regarding intravenous infusion, it is possible that most cells break up rapidly once in the bloodstream and fail to survive as intact, living cells. On the other hand, any MSCs that survive are destined to accumulate in the lungs and their fate beyond that, ie, whether they reach target tissues such as skin, is uncertain. In future, cell tracking studies will be necessary to reduce speculation about the destiny of MSCs in human cell therapy, both for RDEB and for other clinical settings.
Intradermal MSCs (bone marrow-derived) were assessed in two individuals with RDEB in 2010,84 with several clinical trials of intravenous MSCs (bone marrow or umbilical cord blood-derived) in both children and adults with RDEB following soon after.85–87 From these studies, the optimal dose and infusion schedule for the MSCs has yet to be determined. Nevertheless, it appears that 1–3×106 MSCs/kg given 2–3 times over 1 month, following repeat infusions every 6 months or so, might be recommended. The source of the MSCs does not seem to confer any clinical advantage. Side effects, beyond brief chills and transient hypotension, appear to be minimal. No major issues with allo-sensitization from repeated infusions using MSCs from multiple donors of different haplotypes have been documented. Several clinical trials have shown improved wound healing (Figure 4), reduced pain, and less itching, along with better sleep and improved quality of life scores, although none has resulted in new C7 or AF formation in the recipients’ skin.85–88 Collectively, it appears that intravenous MSC therapy works better in individuals with intermediate forms of RDEB rather than severe disease, and in children rather than adults, although considerable intra-individual variability in responses has been seen. Currently, there are no biomarkers associated with prediction of a favorable response to MSC therapy in RDEB.
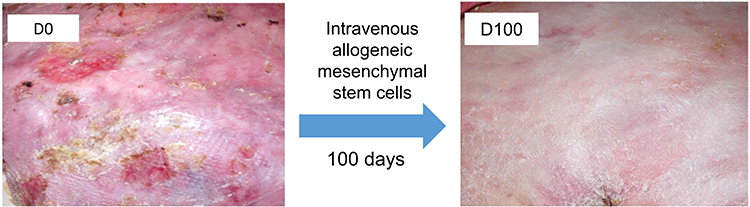 |
Figure 4 Treatment effect of intravenous allogeneic bone-marrow derived mesenchymal stem cells infusion in RDEB. The upper back of a 26-year-old patient with severe RDEB demonstrates improved wound healing and less inflammation 100 days after intravenous allogeneic mesenchymal stem cells infusion. |
MSCs are heterogeneous and include two specific sub-populations, MUSE cells (multilineage-differentiating stress enduring cells) and ABCB5+ (ATP-binding cassette subfamily B member 5) MSCs, that may have enhanced immunomodulatory, anti-inflammatory and survival characteristics over other MSCs; these cells have emerged as further therapeutic options for RDEB patients. MUSE cells represent a SSEA-3 (+) subpopulation of bone marrow cells that can differentiate into keratinocytes and fibroblasts in vitro. Clinical-grade MUSE cells, CL2020, developed by Life Science Institute (Tokyo, Japan) have been assessed in a phase I/II open-label trial with intravenous administration of 15 million cells to five DEB patients.89 Some clinical benefits were observed although optimization of MUSE cells infusion protocols is needed. ABCB5+ MSCs possess self-renewal and trilineage differentiation capacities and have been shown to extend lifespan in col7a1 knockout mice and to have greater anti-inflammatory activity,90 as well as the capacity to migrate to RDEB mouse skin and synthesize C7.91 A phase I/IIa clinical trial revealed that skin-derived allogeneic ABCB5+ MSCs given intravenously (2×106 cells/kg), 17 days apart on three occasions, significantly improved wound healing and quality of life for several months, as well as reducing pain and itch.91 A further double-blind, randomized, placebo-controlled phase III trial (NCT05464381) is currently under way. Nevertheless, it has yet to be proven that, following intravenous administration, either MUSE cells or ABCB5+ MSCs can be detected in human RDEB skin or that new C7 is generated at the DEJ.
Extracellular Vesicles (EVs)
A key question in allogeneic MSC cell therapy is whether it is truly necessary to use the whole cell or whether a subcomponent of the cell, such as the secreted extracellular vesicles (EVs), might be at least equally efficacious.92 EVs refer to a heterogeneous population of vesicular bodies of cellular origin that derive either from the endosomal compartment (exosomes) or from the plasma membrane (microvesicles).93 EVs carry a variety of cargos, including nucleic acids, proteins, and lipids, which can be taken up by other cells, both in the direct vicinity of the source cell and at distant sites in the body via biofluids, and which elicit a variety of phenotypic responses.94 McBride et al showed that bone marrow-derived MSCs release EVs containing COL7A1 mRNAs and C7 protein but whether systemic administration of EVs can impact on C7 expression in RDEB skin is unknown, although application of EVs directly to several types of chronic wounds is currently being assessed.95 With increased understanding of EVs, the development of cell-free therapy in RDEB might sustain the therapeutic efficacy without the challenge of manufacturing and transporting a living cell product. A phase I/IIa non-randomized trial (NCT04173650) that topically applies EVs from normal donors to chronic EB wounds may provide more clinical evidence in the future. Finally, in terms of cell products derived from MSCs, apart from EVs, mitochondria and apoptotic bodies may have the potential for transfer and stimulation of recipient tissue stem cells, respectively.96
Protein Therapy
Given that the key pathology in RDEB patients is a lack of C7, one direct strategy for restoring the deficiency has been to synthesize recombinant C7 and to deliver this intravenously such that it would reach wounded skin, various affected mucosae, and lymphoid extracellular matrix, thereby correcting the muco-cutaneous, systemic and innate immune pathologies associated with RDEB. Proof-of-principle that recombinant C7 could do this, and indeed assemble into AFs at the DEJ, was demonstrated in mice, without severe allergy or C7 autoantibodies appearing in skin.97–99 Following extensive safety data in humans, a clinical trial (NCT04599881) of intravenous recombinant C7 injections was started in 2019. The protocol tested different injection frequencies (4 weekly injections, followed by 7 biweekly injections and 12-week follow-up) at a dosage of 3mg/kg in 6 RDEB patients. Wound healing improved and some C7s were noted at the DEJ, but the overall improvements were described as “modest”.100 Publication of full clinical trial data are awaited, but no additional clinical trials are currently planned. Nevertheless, it remains plausible that intravenous recombinant C7 therapy might form the basis of a combination therapy with an additional local therapy, the potential advantage of the protein therapy being improvement of innate immune dysfunction and benefits in reducing infection in RDEB wounds, thus enhancing the chances of success for other local skin therapies.
High Mobility Group Box-1 (HMGB1)
Bone marrow contains a heterogeneous group of hematopoietic and non-hematopoietic stem cells, some of which may contribute to tissue repair. Within the non-hematopoietic collection of stem cells, there exists a sub-population of epithelial progenitors. These cells can be mobilized, either physiologically or artificially, by high mobility group box 1 (HMGB1). HMGB1 is a ubiquitous nuclear protein that modulates chromatin structure and gene transcription intracellularly, while extracellularly it acts as an alarmin to induce immune reactions in response to danger.101 Critical to stem cell mobilization for wound repair, the A box domain of HMGB1 has been shown to trigger the release of Lin−/PDGFRα+ bone marrow cells to the affected tissues: these cells can transdifferentiate into keratinocytes, fibroblasts and generate C7.102 Following murine studies, the StemRIM company in Osaka, Japan, developed a recombinant peptide of the A box for human use,103,104 to treat various diseases requiring tissue regeneration. The safety profile of the HMGB1 fragment (named Redasemtide) was established in a phase I randomized, double-blind, placebo-controlled trial (UMIN000018252) after intravenous infusion using six different concentrations (0.15–3mg/kg) in 48 healthy volunteers. Regarding RDEB, a phase II single-arm, non-randomized, uncontrolled clinical trial (UMIN000029962) was performed to assess the effects of 1mg/kg intravenous HMGB1 fragment administration in 9 RDEB patients. Preliminary results showed reduced blistering and improved wound healing in most subjects (unpublished data). The intravenous HMGB1 peptide has yet to be approved for clinical use in Japan, but current data indicate it may be helpful in intermediate forms of RDEB and perhaps DDEB. For more severe forms of RDEB, the approach may be to develop HMGB1 peptide-soaked nets for subcutaneous implantation which could then entrap the key progenitors, allowing them to be isolated and genetically modified for COL7A1 correction before autologous cell therapy, local or systemic, is returned to the patient.
Disease-Modifying or Symptomatic Control Treatments
In addition to the disease correction and other therapeutic approaches outlined above, considerable recent efforts have been made to try to improve the lives of people living with DEB by developing treatments that target the downstream consequences of the COL7A1 pathology. These approaches may involve drug repurposing or development of new therapies, potentially modifying disease severity and improving patient symptoms. Importantly, they are not intended to cure DEB but to improve quality of life.
Anti-Fibrosis: Losartan
Fibrosis and contractures are key clinical features in some forms of RDEB which can severely limit physical activities. The repetitive tissue damage and progressive scarring is largely mediated by TGF-β,105 and thus therapies that can reduce TGF-β signaling in RDEB skin have the potential to slow down disease progression and reduce some of the morbidity. Losartan, a well-characterized anti-hypertensive medication, is known to attenuate TGF-β signaling.105,106 It has been tested in a col7a1 hypomorphic mouse model, with its use delaying paw scarring and digit fusion, and thus providing a rationale for human therapy in RDEB.107 Recently, a case series treated six RDEB patients with daily oral losartan (0.7 mg/kg) for 6 weeks and demonstrated clinical benefits as well as improvements in skin histology108 A phase I/II REFLECT trial (EudraCT: 2015‑003670‑32) treated 29 pediatric RDEB patients with losartan for 10 months to prevent disease progression. Interim analysis of 18 treated cases showed positive safety data, while final evaluation is awaited.109 Interest in oral losartan for RDEB has sparked a few other small case reports108,110,111 but consensus opinion is that the larger trial data results should be awaited before clinical recommendations on the use of losartan are made.
Anti-Fibrosis: Decorin
A further means of reducing TGF-β activity in skin is to increase expression of decorin, a stromal proteoglycan which inhibits TGF-β through binding to its core protein, thus preventing TGF-β from interacting with its receptors. The clinical relevance of decorin was first noted in RDEB monozygotic twins, in whom one individual had a milder phenotype (despite the same COL7A1 pathology) with evidence of increased decorin expression in his skin compared to the more severely blistering sibling.105 To determine the therapeutic potential of decorin, a lentiviral vector harboring human decorin core protein cDNA was injected intraperitoneally into col7a1 hypomorphic mice.112 The increased decorin expression in these animals prolonged their survival period, postponed paw deformities/digit fusion, and reduced skin fibrosis.112 Moreover, intraperitoneal administration of decorin, fused with a skin-homing peptide, CRK (containing CendR domain), significantly reduced fibrotic gene expression, including those involved in TGF-β signaling, and attenuated fibrosis in RDEB mice skin.113 Regarding human RDEB, a topical gel containing recombinant decorin is currently under development by the company FIBRX Derm Inc for assessment in patients.
Anti-Itch: Biologics and Janus Kinase Inhibitors
Itch is a bothersome symptom that affects the life quality of many people with EB.114 One particularly itchy form of DEB, known as EB pruriginosa, is particularly troublesome and is hard to treat with conventional anti-inflammatory therapies.115,116 With the advent of new biologic agents to treat other more common inflammatory skin disorders such as atopic dermatitis, it was perhaps expected that some of these drugs would also be tried in itchy EB. One biologic that has been tested for EB pruritus is dupilumab, a monoclonal antibody targeting the IL-4 receptor, which thus reduces the activity of the cytokines IL-4 and IL-13. Of note, an increasing number of anecdotal reports have highlighted the potential value of targeting this pathway in EB pruriginosa117–125 (Figure 5). In total, approximately 16 patients with EB pruriginosa have received dupilumab, mostly with rapid and encouraging reductions in itch severity and skin inflammation. A recent study evaluated the circulating T cells by flow cytometry in four EB pruriginosa patients and showed skewing of Th2 immunity, which offers a rationale for dupilumab treatment.121 Besides upregulation of Th2 signaling, elevated IgE levels have also been demonstrated and anti-IgE therapy with omalizumab (300mg every 4 weeks) in some individuals has led to some lessening of skin inflammation and a moderate reduction in itch.126 Aside from biologics, oral baricitinib (2mg daily), a JAK1/2 inhibitor, has also been shown to reduce DEB itch.127 Similarly, another JAK inhibitor, tofacitinib, also rapidly reduced itch and skin inflammation in DEB.128 To date, no placebo-controlled clinical trials of biologics or JAK inhibitors have been performed in DEB and thus optimal drug selection and dosing in individual patients currently lack structured recommendations.
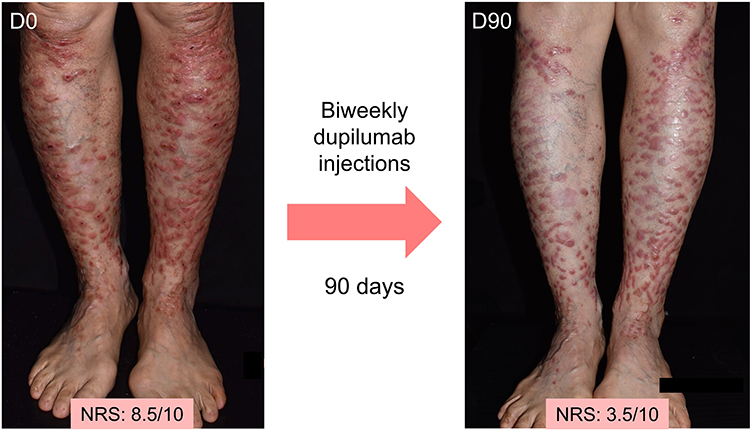 |
Figure 5 Treatment effect of intravenous biweekly dupilumab injections in DDEB. A 59-year-old patient with DDEB (EB pruriginosa) displays flattening and less erythema of the prurigo-like papules and nodules on both lower legs after biweekly dupilumab injections for 3 months. The itch numerical rating score also decreased from 8.5 to 3.5. Abbreviation: NRS, Numerical Rating Score. |
Anti-Pain/Itch: Cannabinoids
In addition to itch, pain is another prominent symptom for people with RDEB, particularly when skin dressings need to be changed. Although systemic and topical opioids often form part of routine care, recent interest in the EB community has emerged in exploring the potential of cannabinoids to provide pain relief. Cannabinoids bind to CB1 and CB2 receptors on the skin or nervous system to exert analgesic and anti-inflammatory effects. Previous reports showed that different forms of cannabinoids, including topical and sublingual cannabinoid-based medicines (CBM) oil, help alleviate pain and itch in 6 EB patients.129,130 The CBM oil is primarily composed of two kinds of cannabinoids, tetrahydrocannabinol (THC) and cannabidiol (CBD). The former is a CB1/CB2 partial agonist, and the latter mitigates THC-related side effects. A recent survey on CBM use in EB patients across five continents showed that CBM reduces pain and itch by 3 points in a numeric ranking score.131 Most patients in the cohort received CBM topically, orally, or through inhalation. Over 90% of the patients reported an improvement in overall symptoms and they were able to relax or sleep better after CBM use. Around 80% of the patients were able to reduce their use of other analgesics. Adverse events from CBM comprised dry mouth (44%), cough/wheezing (29%), and dry/red eyes (27%). Randomized, double-blind trials, using INM‐755 cream (NCT04908215) or sublingual Transvamix (containing 100mg/mL THC and 50mg/mL CBD, Netherlands Trial Register: NL9347)132 are underway to provide more robust evidence for CBM use in DEB.
Anti-Inflammation: Methotrexate, Small Molecules and Biologics
Methotrexate is a folate antagonist that is frequently used in common inflammatory skin diseases such as psoriasis. Its anti-inflammatory and anti-proliferative properties, and safety profile, are well established. Using microarray-based whole-transcriptome profiling in RDEB wound skin, several cytokine and chemokine signaling pathways such as JAK/STAT, IL-6, and IL-20 were found to be upregulated.133 In silico prediction for compounds that reverse gene expression signatures highlighted methotrexate as a leading candidate.133 Thus, reverse transcriptomics data predict that methotrexate may help alleviate systemic inflammation in RDEB, including reduction in itch.133 Clinical testing is awaited, but if successful, methotrexate may offer a cheaper and more widely available alternative to biologics or JAK inhibitors for some individuals. Similarly, immunohistochemical staining of IL-17A demonstrated five- to sevenfold increase in IL-17A+ cells in patients with DEB compared to healthy controls.134 Thus, small molecules and biologics135 targeting IL-17A, which are also commonly used to treat psoriasis, may be beneficial for some people with DEB.
Wound Healing Promotion: Oleogel-S10
Patients with EB have also listed improving wound healing as a major unmet need. To address this generic shortfall, Amyrt Pharma has developed Oleogel-S10, composed of 10% birch triterpene extracts, which have been shown to accelerate wound healing by modulating inflammation, stimulating keratinocyte migration, and imparting an antimicrobial effect.136,137 The active components have previously been used in burn and split-thickness skin graft wounds, and found to accelerate wound healing.136,137 In 2017, a prospective, placebo-controlled phase II trial in EB showed clinical improvements, where 5 of the 12 treated wounds showed faster re-epithelialization albeit without statistical significance.138 Recently, a phase III double-blind, randomized, vehicle-controlled trial (EASE) showed that topical Oleogel-S10, applied at least every 4 days, led to complete wound healing in 41% of the patients within 45 days compared to 29% in the vehicle arm.139 Adverse effects were minimal. However, the strength of the wound healing data for EB, including DEB, associated with Oleogel-S10 was not deemed strong enough by the FDA for product approval. In contrast, in June 2022 the EMA approved use of Oleogel-S10 to promote wound healing in EB.
Wound Healing Promotion: Calcipotriol
Calcipotriol is an active vitamin D3 analog that has been used clinically for several years to control the hyperproliferation of keratinocytes in patients with psoriasis.140 Regarding RDEB, Guttman-Gruber et al repurposed this drug to assess its wound-healing capability.141 Their preclinical results showed that at low dosages (100nM, ie, approximately 100-fold less than the concentration used for psoriasis), calcipotriol enhances the antimicrobial peptide cathelicidin (hCAP18), which promotes wound healing, and leads to improvements in antimicrobial defense.141 Subsequently, in a randomized, placebo-controlled clinical trial involving six RDEB patients, calcipotriol-treated lesions showed a significant reduction of the wounded areas (88%) compared to placebo (66%).142 Itch scores were also significantly reduced within 2–4 weeks of starting treatment.142 At present, there are no specific recommendations or formulations of calcipotriol for use in RDEB. It should be remembered that it is not appropriate to repurpose currently available calcipotriol creams because of the concentration differences relating to use in psoriasis or RDEB.
Wound Healing Promotion: Laser Therapy
RDEB wounds often display excessive granulation tissue, fibrin deposition, increased vascularity, and secondary infection, which may all contribute to delayed healing. In a bid to stimulate wound healing, different forms of lasers have been assessed in a small number of DEB patients. Photomodulation from some lasers may accelerate cell proliferation and collagen. One case study of a 4-year-old girl with RDEB showed that four sessions of low-level laser therapy (660nm, 100mW, 35J/cm2, 2J per point) over 10 days promoted wound healing.143 In another RDEB individual, use of a different form of laser, an ablative micro-fractionated 10,600-nm CO2 laser, was able to induce complete closure of a chronic wound after two laser sessions 4 weeks apart. Possible explanations for the good response included photo-microdebridement destroying wound biofilms, as well as synthesis of new collagen and growth factors.144 Pulsed dye laser, at a low fluence (595nm, 6J/cm2, 6ms, 7–10mm spot size with 10% overlap; frequency 8 weeks to 8 months), has also been shown to reduce erythema and even blistering in one DDEB patient.145 Although several of these responses were deemed impressive, patient numbers are low and control data around the natural history of DEB wound healing are lacking. For now, there are no formal recommendations on the use of lasers in DEB.
Conclusions
There is a desperate requirement to develop new and more effective therapies for all forms of DEB, with RDEB patients having the greatest burden of disease and unmet need. Nevertheless, several new therapies are being tested in patients which offer great hope for better clinical management. New treatments include both “intention to cure” approaches as well as interventions that target secondary inflammatory pathology or which directly try to improve patient symptoms. It is likely that optimal treatment of DEB will require combinations of therapy, concurrently or sequentially. The approval in May 2023 of the B-VEC topical COL7A1 gene therapy for RDEB wounds represents a substantial advance in therapeutics for this inherited blistering skin disease. Much still needs to be done, but there are realistic prospects for improving the quality of life for many individuals with DEB in the not-too-distant future.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Bardhan A, Bruckner-Tuderman L, Chapple IL, et al. Epidermolysis bullosa. Nat Rev Dis Primers. 2020;6(1):78. doi:10.1038/s41572-020-0210-0
2. Has C, Bauer JW, Bodemer C, et al. Consensus reclassification of inherited epidermolysis bullosa and other disorders with skin fragility. Br J Dermatol. 2020;183(4):614–627. doi:10.1111/bjd.18921
3. Petrof G, Papanikolaou M, Martinez AE, et al. The epidemiology of epidermolysis bullosa in England and Wales: data from the national epidermolysis bullosa database. Br J Dermatol. 2022;186(5):843–848. doi:10.1111/bjd.20958
4. Nyström A, Bornert O, Kühl T, et al. Impaired lymphoid extracellular matrix impedes antibacterial immunity in epidermolysis bullosa. Proc Natl Acad Sci U S A. 2018;115(4):E705–E714. doi:10.1073/pnas.1709111115
5. Tartaglia G, Cao Q, Padron ZM, South AP. Impaired wound healing, fibrosis, and cancer: the paradigm of recessive dystrophic epidermolysis bullosa. Int J Mol Sci. 2021;22(10):5104. doi:10.3390/ijms22105104
6. Fine JD, Johnson LB, Weiner M, Li KP, Suchindran C. Epidermolysis bullosa and the risk of life-threatening cancers: the national EB registry experience, 1986–2006. J Am Acad Dermatol. 2009;60(2):203–211. doi:10.1016/j.jaad.2008.09.035
7. Feinstein JA, Bruckner AL, Chastek B, Anderson A, Roman J. Clinical characteristics, healthcare use, and annual costs among patients with dystrophic epidermolysis bullosa. Orphanet J Rare Dis. 2022;17(1):367. doi:10.1186/s13023-022-02509-0
8. Angelis A, Mellerio JE, Kanavos P. Understanding the socioeconomic costs of dystrophic epidermolysis bullosa in Europe: a costing and health-related quality of life study. Orphanet J Rare Dis. 2022;17(1):346. doi:10.1186/s13023-022-02419-1
9. Chen M, O’Toole EA, Muellenhoff M, Medina E, Kasahara N, Woodley DT. Development and characterization of a recombinant truncated type VII collagen “minigene”. Implication for gene therapy of dystrophic epidermolysis bullosa. J Biol Chem. 2000;275(32):24429–24435. doi:10.1074/jbc.M003440200
10. Chen M, Kasahara N, Keene DR, et al. Restoration of type VII collagen expression and function in dystrophic epidermolysis bullosa. Nat Genet. 2002;32(4):670–675. doi:10.1038/ng1041
11. Baldeschi C, Gache Y, Rattenholl A, et al. Genetic correction of canine dystrophic epidermolysis bullosa mediated by retroviral vectors. Hum Mol Genet. 2003;12(15):1897–1905. doi:10.1093/hmg/ddg200
12. Goto M, Sawamura D, Ito K, et al. Fibroblasts show more potential as target cells than keratinocytes in COL7A1 gene therapy of dystrophic epidermolysis bullosa. J Invest Dermatol. 2006;126(4):766–772. doi:10.1038/sj.jid.5700117
13. Ortiz-Urda S, Lin Q, Green CL, Keene DR, Marinkovich MP, Khavari PA. Injection of genetically engineered fibroblasts corrects regenerated human epidermolysis bullosa skin tissue. J Clin Invest. 2003;111(2):251–255. doi:10.1172/JCI200317193
14. Siprashvili Z, Nguyen NT, Bezchinsky MY, Marinkovich MP, Lane AT, Khavari PA. Long-term type VII collagen restoration to human epidermolysis bullosa skin tissue. Hum Gene Ther. 2010;21(10):1299–1310. doi:10.1089/hum.2010.023
15. Titeux M, Pendaries V, Zanta-Boussif MA, et al. SIN retroviral vectors expressing COL7A1 under human promoters for ex vivo gene therapy of recessive dystrophic epidermolysis bullosa. Mol Ther. 2010;18(8):1509–1518 doi:10.1038/mt.2010.91.
16. Woodley DT, Krueger GG, Jorgensen CM, et al. Normal and gene-corrected dystrophic epidermolysis bullosa fibroblasts alone can produce type VII collagen at the basement membrane zone. J Invest Dermatol. 2003;121(5):1021–1028. doi:10.1046/j.1523-1747.2003.12571.x
17. Siprashvili Z, Nguyen NT, Gorell ES, et al. Safety and wound outcomes following genetically corrected autologous epidermal grafts in patients with recessive dystrophic epidermolysis bullosa. JAMA. 2016;316(17):1808–1817 doi:10.1001/jama.2016.15588.
18. Eichstadt S, Barriga M, Ponakala A, et al. Phase 1/2a clinical trial of gene-corrected autologous cell therapy for recessive dystrophic epidermolysis bullosa. JCI Insight. 2019;4(19):e130554 doi:10.1172/jci.insight.130554.
19. So JY, Nazaroff J, Iwummadu CV, et al. Long-term safety and efficacy of gene-corrected autologous keratinocyte grafts for recessive dystrophic epidermolysis bullosa. Orphanet J Rare Dis. 2022;17(1):377. doi:10.1186/s13023-022-02546-9
20. Lwin SM, Syed F, Di WL, et al. Safety and early efficacy outcomes for lentiviral fibroblast gene therapy in recessive dystrophic epidermolysis bullosa. JCI Insight. 2019;4(11):e126243. doi:10.1172/jci.insight.126243
21. Marinkovich M, Lane A, Sridhar K, Keene DR, Malyala A, Maslowski J. 591 A phase 1/2 study of genetically-corrected, collagen VII expressing autologous human dermal fibroblasts injected into the skin of patients with recessive dystrophic epidermolysis bullosa (RDEB). J Invest Dermatol. 2018;138(5):S100. doi:10.1016/j.jid.2018.03.599
22. Gurevich I, Agarwal P, Zhang P, et al. In vivo topical gene therapy for recessive dystrophic epidermolysis bullosa: a phase 1 and 2 trial. Nat Med. 2022;28(4):780–788. doi:10.1038/s41591-022-01737-y
23. Guide SV, Gonzalez ME, Bağcı IS, et al. Trial of beremagene geperpavec (B-VEC) for dystrophic epidermolysis bullosa. N Engl J Med. 2022;387(24):2211–2219. doi:10.1056/NEJMoa2206663
24. Osborn MJ, Starker CG, McElroy AN, et al. Talen-based gene correction for epidermolysis bullosa. Mol Ther. 2013;21(6):1151–1159. doi:10.1038/mt.2013.56
25. Izmiryan A, Ganier C, Bovolenta M, Schmitt A, Mavilio F, Hovnanian A. Ex vivo COL7A1 correction for recessive dystrophic epidermolysis bullosa using CRISPR/Cas9 and homology-directed repair. Mol Ther Nucleic Acids. 2018;12:554–567 doi:10.1016/j.omtn.2018.06.008.
26. Jacków J, Guo Z, Hansen C, et al. CRISPR/Cas9-based targeted genome editing for correction of recessive dystrophic epidermolysis bullosa using IPS cells. Proc Natl Acad Sci U S A. 2019;116(52):26846–26852 doi:10.1073/pnas.1907081116.
27. Shinkuma S, Guo Z, Christiano AM. Site-specific genome editing for correction of induced pluripotent stem cells derived from dominant dystrophic epidermolysis bullosa. Proc Natl Acad Sci U S A. 2016;113(20):5676–5681. doi:10.1073/pnas.1512028113
28. Neumayer G, Torkelson JL, Li S, et al. A scalable, GMP-compatible, autologous organotypic cell therapy for dystrophic epidermolysis bullosa. bioRxiv. 2023. doi:10.1101/2023.02.28.529447
29. Gaudelli NM, Komor AC, Rees HA, et al. Programmable base editing of A•T to G•C in genomic DNA without DNA cleavage. Nature. 2017;551(7681):464–471. doi:10.1038/nature24644
30. Rees HA, Liu DR. Base editing: precision chemistry on the genome and transcriptome of living cells. Nat Rev Genet. 2018;19(12):770–788. doi:10.1038/s41576-018-0059-1
31. Komor AC, Kim YB, Packer MS, Zuris JA, Liu DR. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature. 2016;533(7603):420–424. doi:10.1038/nature17946
32. Osborn MJ, Newby GA, McElroy AN, et al. Base editor correction of COL7A1 in recessive dystrophic epidermolysis bullosa patient-derived fibroblasts and IPSCs. J Invest Dermatol. 2020;140(2):338–347.e5. doi:10.1016/j.jid.2019.07.701
33. Sheriff A, Guri I, Zebrowska P, et al. ABE8e adenine base editor precisely and efficiently corrects a recurrent COL7A1 nonsense mutation. Sci Rep. 2022;12(1):19643. doi:10.1038/s41598-022-24184-8
34. Naso G, Gkazi SA, Georgiadis C, et al. Cytosine deaminase base editing to restore COL7A1 in dystrophic epidermolysis bullosa human: murine skin model. JID Innovations. 2023;3(3):100191. doi:10.1016/j.xjidi.2023.100191
35. Anzalone AV, Randolph PB, Davis JR, et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature. 2019;576(7785):149–157. doi:10.1038/s41586-019-1711-4
36. Hong SA, Kim SE, Lee AY, et al. Therapeutic base editing and prime editing of COL7A1 mutations in recessive dystrophic epidermolysis bullosa. Mol Ther. 2022;30(8):2664–2679. doi:10.1016/j.ymthe.2022.06.005
37. Bauer JW, Murauer EM, Wally V, Koller U. RNA trans-splicing for genodermatoses. Methods Mol Biol. 2013;961:441–455 doi:10.1007/978-1-62703-227-8_30.
38. Bornert O, Peking P, Bremer J, et al. RNA-based therapies for genodermatoses. Exp Dermatol. 2017;26(1):3–10. doi:10.1111/exd.13141
39. Murauer EM, Gache Y, Gratz IK, et al. Functional correction of type VII collagen expression in dystrophic epidermolysis bullosa. J Invest Dermatol. 2011;131(1):74–83. doi:10.1038/jid.2010.249
40. Mayr E, Ablinger M, Lettner T, et al. 5’RNA trans-splicing repair of COL7A1 mutant transcripts in epidermolysis bullosa. Int J Mol Sci. 2022;23(3):1732. doi:10.3390/ijms23031732
41. Murauer EM, Koller U, Hainzl S, Wally V, Bauer JW. A reporter-based screen to identify potent 3’ trans-splicing molecules for endogenous RNA repair. Hum Gene Ther Methods. 2013;24(1):19–27. doi:10.1089/hgtb.2012.180
42. Bremer J, Bornert O, Nyström A, et al. Antisense oligonucleotide-mediated exon skipping as a systemic therapeutic approach for recessive dystrophic epidermolysis bullosa. Mol Ther Nucleic Acids. 2016;5(10):e379. doi:10.1038/mtna.2016.87
43. Bremer J, van den Akker PC. Therapies for epidermolysis bullosa: delivery is key. Br J Dermatol. 2019;180(1):17–19. doi:10.1111/bjd.17324
44. Liemberger B, Bischof J, Ablinger M, et al. COL7A1 editing via RNA trans-splicing in RDEB-derived skin equivalents. Int J Mol Sci. 2023;24(5):4341. doi:10.3390/ijms24054341
45. Tarn WY, Cheng Y, Ko SH, Huang LM. Antisense oligonucleotide-based therapy of viral infections. Pharmaceutics. 2021;13(12):2015. doi:10.3390/pharmaceutics13122015
46. Rodrigues M, Yokota T. An overview of recent advances and clinical applications of exon skipping and splice modulation for muscular dystrophy and various genetic diseases. Methods Mol Biol. 2018;1828:31–55 doi:10.1007/978-1-4939-8651-4_2.
47. Goto M, Sawamura D, Nishie W, et al. Targeted skipping of a single exon harboring a premature termination codon mutation: implications and potential for gene correction therapy for selective dystrophic epidermolysis bullosa patients. J Invest Dermatol. 2006;126(12):2614–2620. doi:10.1038/sj.jid.5700435
48. Bornert O, Hogervorst M, Nauroy P, et al. QR-313, an antisense oligonucleotide, shows therapeutic efficacy for treatment of dominant and recessive dystrophic epidermolysis bullosa: a preclinical study. J Invest Dermatol. 2021;141(4):883–893.e6. doi:10.1016/j.jid.2020.08.018
49. Ham KA, Aung-Htut MT, Fletcher S, Wilton SD. Nonsequential splicing events alter antisense-mediated exon skipping outcome in COL7A1. Int J Mol Sci. 2020;21(20):7705. doi:10.3390/ijms21207705
50. Turczynski S, Titeux M, Tonasso L, Décha A, Ishida-Yamamoto A, Hovnanian A. Targeted exon skipping restores type VII collagen expression and anchoring fibril formation in an in vivo RDEB model. J Invest Dermatol. 2016;136(12):2387–2395. doi:10.1016/j.jid.2016.07.029
51. Marinkovich MP, Sridhar K, Karkala V, Y VK. 306 Topical QR-313, an antisense oligonucleotide, in the treatment of dystrophic epidermolysis bullosa. J Invest Dermatol. 2020;140(7):S37 doi:10.1016/j.jid.2020.03.312.
52. Pendaries V, Gasc G, Titeux M, Tonasso L, Mejía JE, Hovnanian A. SiRNA-mediated allele-specific inhibition of mutant type VII collagen in dominant dystrophic epidermolysis bullosa. J Invest Dermatol. 2012;132(6):1741–1743. doi:10.1038/jid.2012.11
53. Bornert O, Kühl T, Bremer J, van den Akker PC, Pasmooij AM, Nyström A. Analysis of the functional consequences of targeted exon deletion in COL7A1 reveals prospects for dystrophic epidermolysis bullosa therapy. Mol Ther. 2016;24(7):1302–1311. doi:10.1038/mt.2016.92
54. Jonkman MF, Scheffer H, Stulp R, et al. Revertant mosaicism in epidermolysis bullosa caused by mitotic gene conversion. Cell. 1997;88(4):543–551. doi:10.1016/S0092-8674(00)81894-2
55. Almaani N, Nagy N, Liu L, et al. Revertant mosaicism in recessive dystrophic epidermolysis bullosa. J Invest Dermatol. 2010;130(7):1937–1940. doi:10.1038/jid.2010.64
56. Twaroski K, Eide C, Riddle MJ, et al. Revertant mosaic fibroblasts in recessive dystrophic epidermolysis bullosa. Br J Dermatol. 2019;181(6):1247–1253. doi:10.1111/bjd.17943
57. Shinkuma S, Sawamura D, Fujita Y, et al. Long-term follow-up of cultured epidermal autograft in a patient with recessive dystrophic epidermolysis bullosa. Acta Derm Venereol. 2014;94(1):98–99. doi:10.2340/00015555-1592
58. Mochizuki R, Oka Y, Hashimoto S, et al. A case of recessive dystrophic epidermolysis bullosa treated with a cultured epidermal autograft. J Dermatol. 2021;48(4):e165–e166. doi:10.1111/1346-8138.15774
59. Bidou L, Allamand V, Rousset JP, Namy O. Sense from nonsense: therapies for premature stop codon diseases. Trends Mol Med. 2012;18(11):679–688. doi:10.1016/j.molmed.2012.09.008
60. Cogan J, Weinstein J, Wang X, et al. Aminoglycosides restore full-length type VII collagen by overcoming premature termination codons: therapeutic implications for dystrophic epidermolysis bullosa. Mol Ther. 2014;22(10):1741–1752. doi:10.1038/mt.2014.140
61. Woodley DT, Cogan J, Hou Y, et al. Gentamicin induces functional type VII collagen in recessive dystrophic epidermolysis bullosa patients. J Clin Invest. 2017;127(8):3028–3038. doi:10.1172/JCI92707
62. Woodley D, Kwong A, Cogan J, Hou Y. 1021 Intravenous gentamicin therapy for recessive dystrophic epidermolysis bullosa patients harboring nonsense mutations. J Invest Dermatol. 2019;139(5):S176. doi:10.1016/j.jid.2019.03.1097
63. Mosallaei D, Hao M, Antaya RJ, et al. Molecular and clinical outcomes after intravenous gentamicin treatment for patients with junctional epidermolysis bullosa caused by nonsense variants. JAMA Dermatol. 2022;158(4):366–374. doi:10.1001/jamadermatol.2021.5992
64. Martínez-Santamaría L, Maseda R, de Arriba MD, et al. Evaluation of systemic gentamicin as translational readthrough therapy for a patient with epidermolysis bullosa simplex with muscular dystrophy owing to PLEC1 pathogenic nonsense variants. JAMA Dermatol. 2022;158(4):439–443. doi:10.1001/jamadermatol.2022.0112
65. Friesen WJ, Johnson B, Sierra J, et al. The minor gentamicin complex component, X2, is a potent premature stop codon readthrough molecule with therapeutic potential. PLoS One. 2018;13(10):e0206158. doi:10.1371/journal.pone.0206158
66. McElroy SP, Nomura T, Torrie LS, et al. A lack of premature termination codon read-through efficacy of PTC124 (Ataluren) in a diverse array of reporter assays. PLoS Biol. 2013;11(6):e1001593. doi:10.1371/journal.pbio.1001593
67. Atanasova VS, Jiang Q, Prisco M, et al. Amlexanox enhances premature termination codon read-through in COL7A1 and expression of full length type VII collagen: potential therapy for recessive dystrophic epidermolysis bullosa. J Invest Dermatol. 2017;137(9):1842–1849. doi:10.1016/j.jid.2017.05.011
68. Wong T, Gammon L, Liu L, et al. Potential of fibroblast cell therapy for recessive dystrophic epidermolysis bullosa. J Invest Dermatol. 2008;128(9):2179–2189. doi:10.1038/jid.2008.78
69. Venugopal SS, Yan W, Frew JW, et al. A phase II randomized vehicle-controlled trial of intradermal allogeneic fibroblasts for recessive dystrophic epidermolysis bullosa. J Am Acad Dermatol. 2013;69(6):898–908.e7. doi:10.1016/j.jaad.2013.08.014
70. Petrof G, Martinez-Queipo M, Mellerio JE, Kemp P, McGrath JA. Fibroblast cell therapy enhances initial healing in recessive dystrophic epidermolysis bullosa wounds: results of a randomized, vehicle-controlled trial. Br J Dermatol. 2013;169(5):1025–1033. doi:10.1111/bjd.12599
71. Moravvej H, Abdollahimajd F, Naseh MH, et al. Cultured allogeneic fibroblast injection vs. fibroblasts cultured on amniotic membrane scaffold for dystrophic epidermolysis bullosa treatment. Br J Dermatol. 2018;179(1):72–79. doi:10.1111/bjd.16338
72. Nagy N, Almaani N, Tanaka A, et al. HB-EGF induces COL7A1 expression in keratinocytes and fibroblasts: possible mechanism underlying allogeneic fibroblast therapy in recessive dystrophic epidermolysis bullosa. J Invest Dermatol. 2011;131(8):1771–1774. doi:10.1038/jid.2011.85
73. Tolar J, Ishida-Yamamoto A, Riddle M, et al. Amelioration of epidermolysis bullosa by transfer of wild-type bone marrow cells. Blood. 2009;113(5):1167–1174. doi:10.1182/blood-2008-06-161299
74. Chino T, Tamai K, Yamazaki T, et al. Bone marrow cell transfer into fetal circulation can ameliorate genetic skin diseases by providing fibroblasts to the skin and inducing immune tolerance. Am J Pathol. 2008;173(3):803–814. doi:10.2353/ajpath.2008.070977
75. Wagner JE, Ishida-Yamamoto A, McGrath JA, et al. Bone marrow transplantation for recessive dystrophic epidermolysis bullosa. N Engl J Med. 2010;363(7):629–639. doi:10.1056/NEJMoa0910501
76. Uitto J, Bruckner-Tuderman L, McGrath JA, Riedl R, Robinson C. EB2017-progress in epidermolysis bullosa research toward treatment and cure. J Invest Dermatol. 2018;138(5):1010–1016. doi:10.1016/j.jid.2017.12.016
77. Geyer MB, Radhakrishnan K, Giller R, et al. Reduced toxicity conditioning and allogeneic hematopoietic progenitor cell transplantation for recessive dystrophic epidermolysis bullosa. J Pediatr. 2015;167(3):765–769.e1. doi:10.1016/j.jpeds.2015.05.051
78. Tolar J, Wagner JE. Allogeneic blood and bone marrow cells for the treatment of severe epidermolysis bullosa: repair of the extracellular matrix. Lancet. 2013;382(9899):1214–1223. doi:10.1016/S0140-6736(13)61897-8
79. Ebens CL, McGrath JA, Riedl JA, et al. Immune tolerance of allogeneic haematopoietic cell transplantation supports donor epidermal grafting of recessive dystrophic epidermolysis bullosa chronic wounds. Br J Dermatol. 2021;184(6):1161–1169. doi:10.1111/bjd.19503
80. Riedl J, Popp C, Eide C, Ebens C, Tolar J. Mesenchymal stromal cells in wound healing applications: role of the secretome, targeted delivery and impact on recessive dystrophic epidermolysis bullosa treatment. Cytotherapy. 2021;23(11):961–973. doi:10.1016/j.jcyt.2021.06.004
81. Golchin A, Farahany TZ, Khojasteh A, Soleimanifar F, Ardeshirylajimi A. The clinical trials of mesenchymal stem cell therapy in skin diseases: an update and concise review. Curr Stem Cell Res Ther. 2019;14(1):22–33. doi:10.2174/1574888X13666180913123424
82. Jo H, Brito S, Kwak BM, Park S, Lee MG, Bin BH. Applications of mesenchymal stem cells in skin regeneration and rejuvenation. Int J Mol Sci. 2021;22(5):2410. doi:10.3390/ijms22052410
83. McBride JD, Rodriguez-Menocal L, Candanedo A, Guzman W, Garcia-Contreras M, Badiavas EV. Dual mechanism of type VII collagen transfer by bone marrow mesenchymal stem cell extracellular vesicles to recessive dystrophic epidermolysis bullosa fibroblasts. Biochimie. 2018;155:50–58. doi:10.1016/j.biochi.2018.04.007
84. Conget P, Rodriguez F, Kramer S, et al. Replenishment of type VII collagen and re-epithelialization of chronically ulcerated skin after intradermal administration of allogeneic mesenchymal stromal cells in two patients with recessive dystrophic epidermolysis bullosa. Cytotherapy. 2010;12(3):429–431. doi:10.3109/14653241003587637
85. El-Darouti M, Fawzy M, Amin I, et al. Treatment of dystrophic epidermolysis bullosa with bone marrow non-hematopoeitic stem cells: a randomized controlled trial. Dermatol Ther. 2016;29(2):96–100. doi:10.1111/dth.12305
86. Petrof G, Lwin SM, Martinez-Queipo M, et al. Potential of systemic allogeneic mesenchymal stromal cell therapy for children with recessive dystrophic epidermolysis bullosa. J Invest Dermatol. 2015;135(9):2319–2321. doi:10.1038/jid.2015.158
87. Rashidghamat E, Kadiyirire T, Ayis S, et al. Phase I/II open-label trial of intravenous allogeneic mesenchymal stromal cell therapy in adults with recessive dystrophic epidermolysis bullosa. J Am Acad Dermatol. 2020;83(2):447–454. doi:10.1016/j.jaad.2019.11.038
88. Lee SE, Lee SJ, Kim SE, et al. Intravenous allogeneic umbilical cord blood-derived mesenchymal stem cell therapy in recessive dystrophic epidermolysis bullosa patients. JCI Insight. 2021;6(2):e143606. doi:10.1172/jci.insight.143606
89. Fujita Y, Nohara T, Takashima S, et al. Intravenous allogeneic multilineage-differentiating stress-enduring cells in adults with dystrophic epidermolysis bullosa: a phase 1/2 open-label study. J Eur Acad Dermatol Venereol. 2021;35(8):e528–e531. doi:10.1111/jdv.17201
90. Webber BR, O’Connor KT, McElmurry RT, et al. Rapid generation of Col7a1(-/-) mouse model of recessive dystrophic epidermolysis bullosa and partial rescue via immunosuppressive dermal mesenchymal stem cells. Lab Invest. 2017;97(10):1218–1224. doi:10.1038/labinvest.2017.85
91. Riedl J, Pickett-Leonard M, Eide C, et al. ABCB5+ dermal mesenchymal stromal cells with favorable skin homing and local immunomodulation for recessive dystrophic epidermolysis bullosa treatment. Stem Cells. 2021;39(7):897–903. doi:10.1002/stem.3356
92. Lai RC, Yeo RW, Lim SK. Mesenchymal stem cell exosomes. Semin Cell Dev Biol. 2015;40:82–88. doi:10.1016/j.semcdb.2015.03.001
93. Théry C, Witwer KW, Aikawa E, et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): a position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J Extracell Vesicles. 2018;7(1):1535750 doi:10.1080/20013078.2018.1535750.
94. O’Brien K, Breyne K, Ughetto S, Laurent LC, Breakefield XO. RNA delivery by extracellular vesicles in mammalian cells and its applications. Nat Rev Mol Cell Biol. 2020;21(10):585–606 doi:10.1038/s41580-020-0251-y.
95. Bray ER, Kirsner RS, Badiavas EV. Mesenchymal stem cell-derived extracellular vesicles as an advanced therapy for chronic wounds. Cold Spring Harb Perspect Biol. 2022;14(10):a041227 doi:10.1101/cshperspect.a041227.
96. Gebara N, Rossi A, Skovronova R, Aziz JM, Asthana A, Bussolati B. Extracellular vesicles, apoptotic bodies and mitochondria: stem cell bioproducts for organ regeneration. Curr Transplant Rep. 2020;7:105–113 doi:10.1007/s40472-020-00282-2.
97. Woodley DT, Keene DR, Atha T, et al. Injection of recombinant human type VII collagen restores collagen function in dystrophic epidermolysis bullosa. Nat Med. 2004;10(7):693–695 doi:10.1038/nm1063.
98. Hou Y, Guey LT, Wu T, et al. Intravenously administered recombinant human type VII collagen derived from Chinese hamster ovary cells reverses the disease phenotype in recessive dystrophic epidermolysis bullosa mice. J Invest Dermatol. 2015;135(12):3060–3067. doi:10.1038/jid.2015.291
99. Woodley DT, Wang X, Amir M, et al. Intravenously injected recombinant human type VII collagen homes to skin wounds and restores skin integrity of dystrophic epidermolysis bullosa. J Invest Dermatol. 2013;133(7):1910–1913 doi:10.1038/jid.2013.10.
100. Bruckner A, Tang J, Chung W, et al. Collagen 7 (C7) protein replacement therapy (PTR-01) durably reduces wound size and symptoms in patients with recessive dystrophic epidermolysis bullosa (RDEB). J Invest Dermatol. 2022;142(8):S50. doi:10.1016/j.jid.2022.05.303
101. Andersson U, Yang H, Harris H. High-mobility group box 1 protein (HMGB1) operates as an alarmin outside as well as inside cells. Semin Immunol. 2018;38:40–48. doi:10.1016/j.smim.2018.02.011
102. Tamai K, Yamazaki T, Chino T, et al. PDGFRalpha-positive cells in bone marrow are mobilized by high mobility group box 1 (HMGB1) to regenerate injured epithelia. Proc Natl Acad Sci U S A. 2011;108(16):6609–6614. doi:10.1073/pnas.1016753108
103. Goto T, Miyagawa S, Tamai K, et al. High-mobility group box 1 fragment suppresses adverse post-infarction remodeling by recruiting PDGFRα-positive bone marrow cells. PLoS One. 2020;15(4):e0230392. doi:10.1371/journal.pone.0230392
104. Kido T, Miyagawa S, Goto T, et al. The administration of high-mobility group box 1 fragment prevents deterioration of cardiac performance by enhancement of bone marrow mesenchymal stem cell homing in the delta-sarcoglycan-deficient hamster. PLoS One. 2018;13(12):e0202838. doi:10.1371/journal.pone.0202838
105. Odorisio T, Di Salvio M, Orecchia A, et al. Monozygotic twins discordant for recessive dystrophic epidermolysis bullosa phenotype highlight the role of TGF-β signalling in modifying disease severity. Hum Mol Genet. 2014;23(15):3907–3922. doi:10.1093/hmg/ddu102
106. Lim DS, Lutucuta S, Bachireddy P, et al. Angiotensin II blockade reverses myocardial fibrosis in a transgenic mouse model of human hypertrophic cardiomyopathy. Circulation. 2001;103(6):789–791. doi:10.1161/01.CIR.103.6.789
107. Nyström A, Thriene K, Mittapalli V, et al. Losartan ameliorates dystrophic epidermolysis bullosa and uncovers new disease mechanisms. EMBO Mol Med. 2015;7(9):1211–1228. doi:10.15252/emmm.201505061
108. Pourani MR, Vahidnezhad H, Mansouri P, et al. Losartan treatment improves recessive dystrophic epidermolysis bullosa: a case series. Dermatol Ther. 2022;35(7):e15515. doi:10.1111/dth.15515
109. Kiritsi D. Losartan for RDEB trial – results and international perspectives. EB2020 1st World Congress on Epidermolysis Bullosa; 2020 Jan 19–23; London, UK. Acta Derm Venereol. 2020;100:1–7 doi:10.2340/00015555-3586
110. Oldakovskiy V, Murashkin N, Lokhmatov M, et al. Our experience of using losartan for esophageal stenosis in children with dystrophic form of congenital epidermolysis bullosa. J Pediatr Surg. 2023;58(4):619–623. doi:10.1016/j.jpedsurg.2022.11.001
111. Relvas M, Figueiredo AC, Calado R, Calvão J, Ramos L. Losartan as therapy for recessive dystrophic epidermolysis bullosa: report of three cases. Dermatol Ther. 2022;35(9):e15678. doi:10.1111/dth.15678
112. Cianfarani F, De Domenico E, Nyström A, et al. Decorin counteracts disease progression in mice with recessive dystrophic epidermolysis bullosa. Matrix Biol. 2019;81:3–16. doi:10.1016/j.matbio.2018.12.001
113. Pemmari T, Ivanova L, May U, et al. Exposed CendR domain in homing peptide yields skin-targeted therapeutic in epidermolysis bullosa. Mol Ther. 2020;28(8):1833–1845. doi:10.1016/j.ymthe.2020.05.017
114. Papanikolaou M, Onoufriadis A, Mellerio JE, et al. Prevalence, pathophysiology and management of itch in epidermolysis bullosa. Br J Dermatol. 2021;184(5):816–825. doi:10.1111/bjd.19496
115. Kaushik A, Mahajan R, Karim A, Handa S, De D, Saikia B. Successful use of cyclosporine in epidermolysis bullosa pruriginosa. Dermatol Ther. 2020;33(6):e14489. doi:10.1111/dth.14489
116. Pallesen KAU, Lindahl KH, Bygum A. Dominant dystrophic epidermolysis bullosa pruriginosa responding to naltrexone treatment. Acta Derm Venereol. 2019;99(12):1195–1196. doi:10.2340/00015555-3304
117. Quintana BR, Durán EP, Pérez Cejudo JA. Successful control of recalcitrant pruritus in epidermolysis bullosa pruriginosa with dupilumab. Actas Dermosifiliogr. 2023. doi:10.1016/j.ad.2022.05.045
118. Wu PC, Dai YX, Li CL, Chen CC, Chang YT, Ma SH. Dupilumab zur behandlung von genodermatosen: eine systematische ìbersicht. J Dtsch Dermatol Ges. 2023;21(1):7–18 doi:10.1111/ddg.14924_g.
119. Yu L, Wang J, Bian L, et al. Dystrophic epidermolysis bullosa pruriginosa: successfully treated with dupilumab. Dermatitis. 2023;34(1):58–59. doi:10.1089/DERM.0000000000000954
120. Caroppo F, Milan E, Giulioni E, Belloni Fortina A. A case of dystrophic epidermolysis bullosa pruriginosa treated with dupilumab. J Eur Acad Dermatol Venereol. 2022;36(5):e365–e367. doi:10.1111/jdv.17887
121. Darbord D, Hickman G, Pironon N, et al. Dystrophic epidermolysis bullosa pruriginosa: a new case series of a rare phenotype unveils skewed Th2 immunity. J Eur Acad Dermatol Venereol. 2022;36(1):133–143. doi:10.1111/jdv.17671
122. Wang Y, Zhou M, Zhang L, Zheng S, Hong Y, Gao XH. Amelioration of dystrophic epidermolysis bullosa pruriginosa symptoms with dupilumab: a case report. Dermatol Ther. 2021;34(6):e15130. doi:10.1111/dth.15130
123. Clawson RC, Duran SF, Pariser RJ. Epidermolysis bullosa pruriginosa responding to dupilumab. JAAD Case Rep. 2021;16:69–71. doi:10.1016/j.jdcr.2021.07.036
124. Zhou AG, Little AJ, Antaya RJ. Epidermolysis bullosa pruriginosa treated with dupilumab. Pediatr Dermatol. 2021;38(2):526–527. doi:10.1111/pde.14493
125. Shehadeh W, Sarig O, Bar J, Sprecher E, Samuelov L. Treatment of epidermolysis bullosa pruriginosa-associated pruritus with dupilumab. Br J Dermatol. 2020;182(6):1495–1497. doi:10.1111/bjd.18855
126. Chen F, Guo Y, Zhou K, et al. The clinical efficacy and safety of anti-IgE therapy in recessive dystrophic epidermolysis bullosa. Clin Genet. 2022;101(1):110–115. doi:10.1111/cge.14062
127. Jiang X, Wang H, Lee M, Lin Z. Epidermolysis bullosa pruriginosa treated with baricitinib. JAMA Dermatol. 2021;157(10):1243–1244. doi:10.1001/jamadermatol.2021.3174
128. Chen KJ, Fang S, Ye Q, Jia M. Successful use of tofacitinib in epidermolysis bullosa pruriginosa. Clin Exp Dermatol. 2022;47(3):598–600. doi:10.1111/ced.14998
129. Chelliah MP, Zinn Z, Khuu P, Teng JMC. Self-initiated use of topical cannabidiol oil for epidermolysis bullosa. Pediatr Dermatol. 2018;35(4):e224–e227. doi:10.1111/pde.13545
130. Schräder NHB, Duipmans JC, Molenbuur B, Wolff AP, Jonkman MF. Combined tetrahydrocannabinol and cannabidiol to treat pain in epidermolysis bullosa: a report of three cases. Br J Dermatol. 2019;180(4):922–924. doi:10.1111/bjd.17341
131. Schräder NHB, Gorell ES, Stewart RE, et al. Cannabinoid use and effects in patients with epidermolysis bullosa: an international cross-sectional survey study. Orphanet J Rare Dis. 2021;16(1):377. doi:10.1186/s13023-021-02010-0
132. Schräder NHB, Duipmans JC, Renken RJ, et al. The C4EB study-transvamix (10% THC / 5% CBD) to treat chronic pain in epidermolysis bullosa: a protocol for an explorative randomized, placebo controlled, and double blind intervention crossover study. PLoS One. 2022;17(12):e0277512. doi:10.1371/journal.pone.0277512
133. Onoufriadis A, Proudfoot LE, Ainali C, et al. Transcriptomic profiling of recessive dystrophic epidermolysis bullosa wounded skin highlights drug repurposing opportunities to improve wound healing. Exp Dermatol. 2022;31(3):420–426. doi:10.1111/exd.14481
134. Haghighi Javid A, Li D, Technau-Hafsi K, Has C. IL-17A immune pattern across genetic acantholytic and blistering disorders. Clin Exp Dermatol. 2023;48(5):llad012 doi:10.1093/ced/llad012.
135. Castela E, Tulic MK, Rozières A, et al. EBS generalized severe is an inflammatory disease. Br J Dermatol. 2019;180:357–364. doi:10.1111/bjd.16897
136. Scheffler A. The wound healing properties of betulin from birch bark from bench to bedside. Planta Med. 2019;85(7):524–527. doi:10.1055/a-0850-0224
137. Schwieger-Briel A, Ott H, Kiritsi D, Laszczyk-Lauer M, Bodemer C. Mechanism of Oleogel-S10: a triterpene preparation for the treatment of epidermolysis bullosa. Dermatol Ther. 2019;32(4):e12983. doi:10.1111/dth.12983
138. Schwieger-Briel A, Kiritsi D, Schempp C, Has C, Schumann H. Betulin-based Oleogel to improve wound healing in dystrophic epidermolysis bullosa: a prospective controlled proof-of-concept study. Dermatol Res Pract. 2017;2017:5068969. doi:10.1155/2017/5068969
139. Kern JS, Sprecher E, Fernandez MF, et al. Efficacy and safety of Oleogel-S10 (birch triterpenes) for epidermolysis bullosa: results from the phase III randomized double-blind phase of the EASE study. Br J Dermatol. 2023;188(1):12–21. doi:10.1093/bjd/ljac001
140. Takahashi H, Ibe M, Kinouchi M, Ishida-Yamamoto A, Hashimoto Y, Iizuka H. Similarly potent action of 1,25-dihydroxyvitamin D3 and its analogues, tacalcitol, calcipotriol, and maxacalcitol on normal human keratinocyte proliferation and differentiation. J Dermatol Sci. 2003;31(1):21–28. doi:10.1016/S0923-1811(02)00136-6
141. Guttmann-Gruber C, Tockner B, Scharler C, et al. Low-dose calcipotriol can elicit wound closure, anti-microbial, and anti-neoplastic effects in epidermolysis bullosa keratinocytes. Sci Rep. 2018;8(1):13430. doi:10.1038/s41598-018-31823-6
142. Guttmann-Gruber C, Piñón Hofbauer J, Tockner B, et al. Impact of low-dose calcipotriol ointment on wound healing, pruritus and pain in patients with dystrophic epidermolysis bullosa: a randomized, double-blind, placebo-controlled trial. Orphanet J Rare Dis. 2021;16(1):473. doi:10.1186/s13023-021-02062-2
143. Minicucci EM, Barraviera SR, Miot H, Almeida-Lopes L. Low-level laser therapy for the treatment of epidermolysis bullosa: a case report. J Cosmet Laser Ther. 2010;12(4):203–205. doi:10.3109/14764172.2010.502460
144. Krakowski AC, Ghasri P. Case report: rapidly healing epidermolysis bullosa wound after ablative fractional resurfacing. Pediatrics. 2015;135(1):e207–e210. doi:10.1542/peds.2014-1779
145. Garza-Mayers AC, Su KA, Wiss K. Improved erythema and decreased blister formation in dominant dystrophic epidermolysis bullosa following treatment with pulsed dye laser. Pediatr Dermatol. 2022;39(6):1005–1006. doi:10.1111/pde.15126
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.