Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 15
Innate Immunity and Cell Surface Receptors in the Pathogenesis of COPD: Insights from Mouse Smoking Models
Authors De Cunto G ![]() , Cavarra E
, Cavarra E ![]() , Bartalesi B
, Bartalesi B ![]() , Lucattelli M
, Lucattelli M ![]() , Lungarella G
, Lungarella G ![]()
Received 16 January 2020
Accepted for publication 3 May 2020
Published 20 May 2020 Volume 2020:15 Pages 1143—1154
DOI https://doi.org/10.2147/COPD.S246219
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Richard Russell
Giovanna De Cunto, Eleonora Cavarra, Barbara Bartalesi, Monica Lucattelli, Giuseppe Lungarella
Department of Molecular and Developmental Medicine, University of Siena, Siena, Italy
Correspondence: Giuseppe Lungarella Email [email protected]
Abstract: Chronic obstructive pulmonary disease (COPD) is mainly associated with smoking habit. Inflammation is the major initiating process whereby neutrophils and monocytes are attracted into the lung microenvironment by external stimuli present in tobacco leaves and in cigarette smoke, which promote chemotaxis, adhesion, phagocytosis, release of superoxide anions and enzyme granule contents. A minority of smokers develops COPD and different molecular factors, which contribute to the onset of the disease, have been put forward. After many years of research, the pathogenesis of COPD is still an object of debate. In vivo models of cigarette smoke-induced COPD may help to unravel cellular and molecular mechanisms underlying the pathogenesis of COPD. The mouse represents the most favored animal choice with regard to the study of immune mechanisms due to its genetic and physiological similarities to humans, the availability of a large variability of inbred strains, the presence in the species of several genetic disorders analogous to those in man, and finally on the possibility to create models “made-to-measure” by genetic manipulation. The review outlines the different response of mouse strains to cigarette smoke used in COPD studies while retaining a strong focus on their relatability to human patients. These studies reveal the importance of innate immunity and cell surface receptors in the pathogenesis of pulmonary injury induced by cigarette smoking. They further advance the way in which we use wild type or genetically manipulated strains to improve our overall understanding of a multifaceted disease such as COPD. The structural and functional features, which have been found in the different strains of mice after chronic exposure to cigarette smoke, can be used in preclinical studies to develop effective new therapeutic agents for the different phenotypes in human COPD.
Keywords: cigarette smoking, emphysema, airway remodelling, vascular remodelling, smoking cessation, persistent inflammation
Introduction
A first comprehensive review of all animal models of emphysema relating to the pathogenesis of this disease has reported in 2002.1 In this review, the reader can find information on practically all of the models of experimental emphysema published to that date in the English literature. Because cigarette smoke (CS) represents the most important risk factor for the development of Chronic Obstructive Pulmonary Disease (COPD), animal models of CS-induced disease have recently been developed and widely used. Studies have mainly focused on small laboratory animals, and in particular on the mouse as the animal choice for the relatively low cost, its rapid reproductive cycle and large litter sizes, the availability of antibodies and probes for this species, the availability of many inbred strains and their mutants, and finally the fact that the mouse genome shows a large degree of homology with the human genome and the mouse genes can be easily manipulated. The use of inbred mice eliminates problems of genetic variability that complicate human studies. The development of animal models of COPD that accurately recapitulates the critical features of human disease is valuable in efforts to develop effective treatments. Several hallmarks of COPD, such as chronic inflammation, airway remodelling, emphysema, pulmonary hypertension (PH) and impaired lung function may be seen in smoking laboratory animals. Similar to humans, the treatment with corticosteroids and other anti-inflammatory agents is not able to resolve these features. Unfortunately, this animal does not fully model the human condition.
This species as an animal choice for studying COPD has some limitations that have to be taken into consideration before planning experimental studies.2 Briefly, unlike humans, mice are obligatory nose breathers. Few animal species smoke cigarettes the way humans do.3 Thus, when exposed to cigarette smoke, this results in a very different pattern of particle filtration in the nares and upper respiratory tract from that experienced by humans who are mouth breathers.3 Various smoking machines are frequently used in mouse studies. Breathing smoke generated by a machine, however, is more like passive smoke exposure than active smoking.2,3 This is more of a problem for “whole-body exposures,” but also pertains to “nose-only devices”. For this reason, mice exposed to CS with the different current methodologies, namely by “nose only”,4 or by “whole body”,5 model in both cases a “second-hand smoking”.2,3
Exposure to Cigarette Smoke (CS)
CS is delivered by a smoking machine either by a “nose only,”4 or by a “whole body” methodology.5 Neither cigarette types, nor the number of cigarettes/day nor the duration of the treatment has been standardized and each is a personal methodological choice. Generally, mice are exposed either to research-grade cigarettes from the University of Kentucky (Lexington, KY), or to commercially available Marlboro Red, which have the advantage to be available everywhere. However, studies carried out with these different methodological approaches provided new knowledge on the complex pathogenic mechanisms involved in COPD.
Strain Susceptibility to CS-Induced Lung Emphysema
Like humans, not all inbred strains of mice develop emphysema following CS-exposure. In strains prone to develop pulmonary changes, the emphysematous lesion is mild and never reaches the anatomical degree that characterizes that observed in human smokers or in other mouse models (ie papain- or elastase-induced emphysema). The different susceptibility for the development of emphysema has been reported for the first time in three mouse strains, which show significant individual variations in antioxidant defenses when acutely exposed to CS.6,7 ICR mice increase their lung antioxidant defenses when acutely exposed to CS, while C57 Bl/6J and DBA/2 mice did not.6 C57 Bl/6J and DBA/2 mice developed lung changes at 7 months after CS-exposure, while ICR mice did not.7 The following study, carried out on other strains of mice (ie NZWLac/J, A/J, SJ/L, and AKR/J) contributed to the identification of other resistant or susceptible strains for the development of emphysema.8 However, the genetic contribution to that variability has not been investigated in the last study.
Sex Differences in CS-Induced Emphysema and Airway Remodelling
Female smokers have an increased risk of developing COPD and disease progression compared with male smokers with a similar smoke history.9–12 The causes and mechanism(s) for sexual dimorphism in the risk for COPD have so far received little attention. Like humans, female mice exposed for long term to CS develop a greater burden of COPD changes in lungs including airway resistance with airway wall thickness, and oxidative stress compared to those of males.13,14 These differences were attenuated in female mice by ovariectomy. Oxidative stress has been shown to be an important pathogenic factor in COPD15 and recent studies demonstrated that women have more elevated circulating oxidative stress markers than men in both healthy non-smokers and smokers without the disease.16,17 Very recently it has been reported that after cigarette smoke exposure, parenchymal tissues from female mice failed to induce antioxidant-related genes in response to smoke exposure, and this effect was restored by ovariectomy.18 After smoke exposure, differential increases in Mmp12 and Cxcl2 gene expression correlated with an increase in foamy macrophages in parenchymal tissues of female than in male mice.
The data reported in this paper demonstrate that the altered oxidative milieu and the inflammation (shown as important factors in the contribution of COPD) are significantly increased in female than in male mice after long-term exposure to CS.18 The increased lymphoid follicles in the airways in female COPD lung may be due to increased BAFF gene and protein expression related to female sex hormones and increased oxidative milieu generated by long-term exposure to CS.18 Actually, the biological role of lymphoid follicles and their potential contribution to the disease is still an object of debate and further studies are required to elucidate their capability of altering the microbiome in the lung tissue, or of inducing autoimmunity. Further studies in male and female smoking mice may represent a reasonable way to study the potential contributions of female sex hormones and increased oxidative milieu generated by chronic exposure in the pathogenesis of COPD.
Genetic Contribution to Strain Susceptibility to Cigarette Smoking
The reason for the different strain sensitivity has been attributed in the following studies to an upregulated Nrf2 pathway caused by an increased expression of peroxiredoxin 1.19 The ablation of the Nrf2 gene in the ICR resistant mice leads to the development of significant emphysema after CS-exposure.20 Thus, low activity levels of Nrf2-mediated antioxidant pathways in C57Bl/6J and DBA/2 mice may act as a major determinant of their susceptibility to CS.20 Unlike C57Bl/6J and DBA/2 strains, the ICR mice showed a significant increase in glutathione in erythrocytes and a significant decrease in most of the oxidized forms of cysteine, cysteinyl glycine, homocysteine and glutathione in plasma after the same exposition to CS.21 Thus, CS-exposure induces systemic oxidative stress only in some mouse strains, which are susceptible to develop emphysema.21 Additional evidence for a significant role for an oxidative damage in the development of CS-induced alveolar injury derives from studies carried out by Petrache et al on C57 Bl/6 transgenic mice overexpressing human Cu/Zn superoxide dismutase demonstrating a beneficial effect on air space enlargements caused by ceramide-induced superoxide production and apoptosis.22,23
Genetic studies in smoking mice suggest that a variety of molecular factors and pathways may influence the strain susceptibility to develop a particular lesion.24 Different mouse strains5,25-27 can develop after chronic exposure to CS specific pathological features similar to those associated with major phenotypes of human disease. In these strains, the various aspects of the human disease, that include chronic inflammation, emphysema, airway remodelling and impaired lung function, have been successfully recapitulated and provide a valuable platform to investigate mechanisms and new therapeutic targets. At the present time, mouse strains that develop emphysema associated or not with goblet cell metaplasia of airways epithelium4 (Figure 1A and B) pulmonary fibrosis25 (Figure 1C and D), PH26 (Figure 1E and F), or peribronchiolar and/or peribronchial fibrosis27 (Figure 1G and H) are available for these studies.
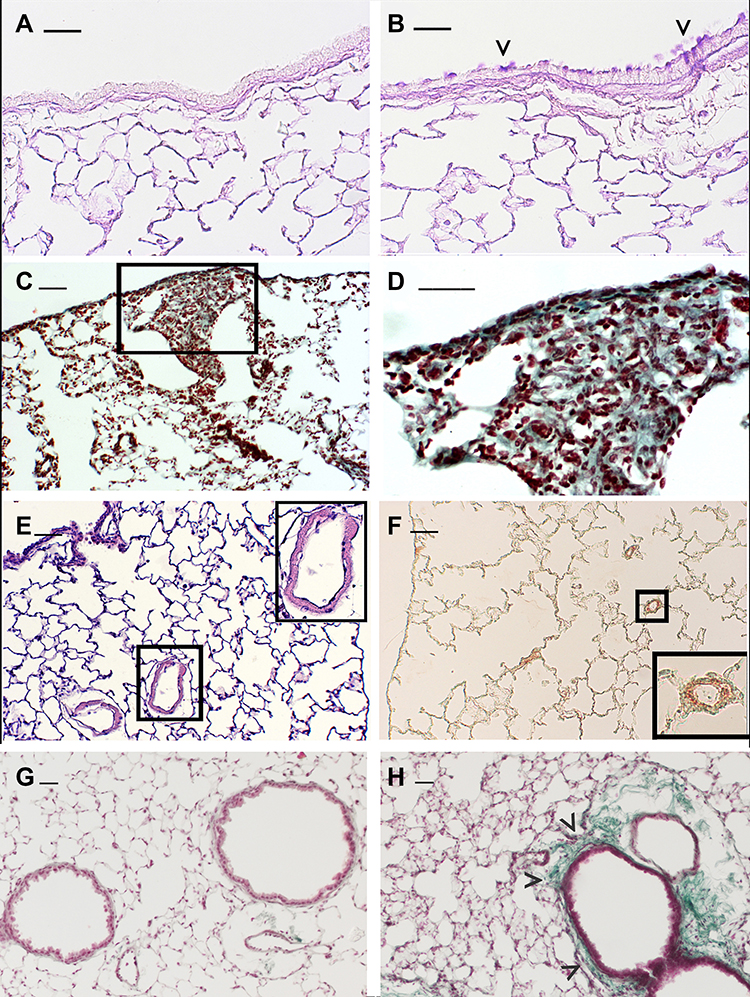 |
Figure 1 Emphysema-associated pulmonary lesions which can be observed in different mouse strains after chronic exposure of CS. Representative histologic sections from lungs of C57 Bl/6J mice at 6 months after air- (A) or CS-exposure (B). Mice practically do not have goblet cells in their bronchi and bronchioles. Note the appearance of clusters of goblet cells in airways after chronic exposure to CS. (C and D) DBA/2 mice at 6 months after CS exposure show evident areas of fibrosis (sea-green stain in the black square) associated with disseminated foci of pulmonary emphysema. This histological picture firstly described in smoking DBA/2 mice25 was subsequently described also in man as “Combined Emphysema –Fibrosis Syndrome” (CPFE)110 In (D), high magnification of the lung parenchyma present in the black square of (C). (E) Histologic section from distal airways from FVBPAR−2-TgN mice showing muscularisation of small (≤80 mm) intrapulmonary vessels that precedes the development of PH (~45% increase) and right ventricular hypertrophy.26 (F) Note in the excessive thickening of a-SMA-positive layers in small intrapulmonary vessels. (in insets: higher magnification of lung parenchyma present in black squares). (G and H) Lung sections from an air-exposed (G) and a CS-exposed (H) C57 Bl/6J mouse at 10 months from the start of the exposure. Distal airways of the air-exposed mice show a normal appearance. Peribronchiolar region from a mouse at 10 months after CS exposure is thickened by an evident fibrotic reaction (sea-green stain) (arrowheads). (A and B): PAS staining; (E): haematoxylin and eosin staining; (C, D, G and H): Masson’s Trichrome staining; (F): Immunostaining with anti-a-SMA antibodies. Scale bars = 40 μm. These images are property of the authors. |
Very recently Rader et al28 demonstrated in 34 inbred strains of mice a broad range and degree of susceptibility to CS-induced lung changes with no response in CBA/J to extreme susceptibility in A/J mice. They identified the Abi3bp gene as a novel candidate gene contributing to emphysema susceptibility. This gene encodes for the tumor suppressor ABI3BP (also identified as TARSH, or eratin) mainly expressed in the lung.
It is intriguing that this gene, that controls growth and differentiation of stem and tumor cells, promotes senescence in some cells may also protect from CS-induced emphysema potentially by promoting growth and survival of lung epithelium and by blocking its senescence. The role of Abi3bp gene in emphysema needs more investigation in humans and experimental animals to determine the real contribution of this gene to emphysema susceptibility.
In addition to general limitations identified from the authors (ie only female mice were used, inability to detect any regions that met strictly multiple testing correction thresholds, emphysema is only one of the pathological entities of COPD), the moderate functional and anatomical changes that follow CS-exposure in animals are certainly another important limiting factor.
However, ABI3BP may be an important determinant of disease course.
Actually, COPD is a heterogeneous disease, which includes emphysema, chronic bronchitis with mucus hypersecretion, bronchiolar and vascular remodelling, and in some cases areas of fibrotic changes, in which emphysema and fibrosis may coexist. Therefore, there is a large variation in COPD symptoms with regard to the severity of bronchitis, and the rate of decline in FEV1 among individuals.29 The pathogenesis of COPD is still subject of investigation, and several pathogenic mechanisms are involved in the development of pulmonary changes that characterize the disease. As mentioned above, these mechanisms include protease/antiprotease and oxidant/antioxidant imbalances,29–31 cell apoptosis,23,32 cellular senescence33 and abnormal immune responses.34
Role of Immunity in the Pathogenesis of Pulmonary Lesions
Many elements of the innate and adaptive immune responses are abnormal in COPD.35,36 The innate immune response was for a long time considered important in COPD pathogenesis; however, some experimental evidence supports a role of acquired responses that requires the proliferation of T cells as central regulators of the inflammatory network.37 However, more recent data obtained in different labs showed that a severe reduction in the number and function of peripheral T cells does not modify pulmonary changes induced by CS-exposure.38–41 Thus, studies carried out in smoking mice indicate that that innate immunity represents a leading actor in the early development of lung changes and that adaptive immune response is implicated only in later stages of the disease. It was also found that TCR components are downregulated in pulmonary CD8 cells from COPD patients.42 Nevertheless, the dysfunction of the antigen-specific response of these cells in COPD may predispose to recurrent infections in the late stage of the disease. Actually, studies carried out both in humans and animals indicate that innate inflammatory cells activated by different stimuli on cell surfaces are necessary to develop pulmonary changes in smoking mice.43–51 Of interest, the aberrant tissue repair in COPD patients and in smoking mice is accompanied by chronic neutrophilic inflammation in pulmonary structures.52–54
Whether and how T cells actually contribute to COPD pathogenesis in humans still remains undefined.
Multiple susceptibility genes for COPD have been identified, namely genes that encode for the receptor for advanced glycation end products (RAGE), Toll-like receptors (TLRs) 2 and 4.55–57 These results provided new information on pathways involved CS-induced airway inflammation suggesting a relevant role for pattern recognition receptors (PRRs) in the pathophysiology of COPD.
Proinflammatory Responses Induced by Activation of PRRs by Pathogen- (PAMPs) or Damage-Associated Molecular Patterns (DAMPs)
A role for DAMPs has been put forward in the pathophysiology of COPD.58 Studies carried out in patients and experimental animals indicate that CS can induce epithelial necroptosis and DAMP release (ie AGE, endogenous ATP, HMGB1, MyD88, etc.)46,59 that initiates airway inflammation in COPD. In mice, CS-induced neutrophilic airway inflammation and emphysema can be statistically lowered upon inhibition of RAGE, or purinergic receptor subtypes (such as P2Y2R or P2X7R) as demonstrated in RAGE, or P2Y2R and P2X7 knockout mice.44,45,60 (Figure 2). Thus, CS-exposure induces DAMPs release that results in innate immune responses and in the release of IL-1β, MCP-1, as well as MIP-2 and KC, the mouse homolog for human IL-8, which regulate migration and infiltration of neutrophils and monocytes/macrophages. These cells promote an overload of oxidants and proteases that lead to epithelial damage and cell death.
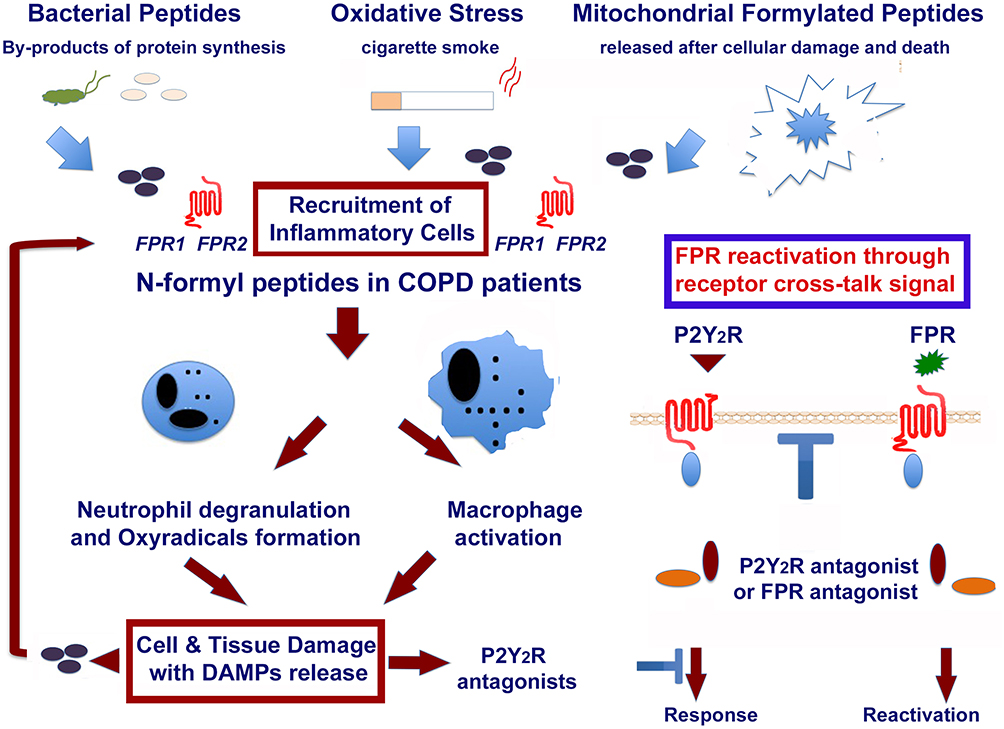 |
Figure 2 Role of formylated peptides and FPRs in the initiation and progression of COPD in smokers. Formyl peptides are present in tobacco leaves. They are active components of mainstream and side stream cigarette smoke. Formyl peptides are also actively secreted by pathogens or passively released from dying host cells after tissue injury. The major component of formyl peptides, N-Formyl-L-methionyl-L-leucyl-L-phenylalanine (FMLP), can promote by itself the recruitment of inflammatory cells. FMLP engagement of its high-affinity receptor FPR1 can lead neutrophil degranulation and release of superoxide anion, as well as macrophage activation and polarization. These inflammatory cells cause an overload of oxidants and proteases, which lead to epithelial cell death and DAMPs release (ie endogenous ATP, AGE, mitochondrial formyl peptides, HMGB1, MyD88, etc.) in the microenvironment. This may result in the activation of other PPRs, which amplify the inflammatory response. The reactivation of desensitized FPRs by P2Y2 ligation is also reported in the figure. This image is the property of the authors. |
Although the activation of the abovementioned receptors has been associated with the development of emphysematous lesions, their importance for the maintenance of tissue-damaging innate cells in the lung is not known. In addition, the exact role of these PPRs in the initiation and progression of the disease remain to be defined.
Formyl Peptide Receptors (FPRs) and COPD. A Suggestive Hypothesis
It is well known that neutrophils are increased in the lungs of COPD patients.61,62 The neutrophil number and their products (eg proteases, oxyradicals) correlate with the development and severity of the disease.63–67 In particular, neutrophil elastase contributes as a major actor to the development of lung emphysema68–70 and other pathological features of COPD.23,71–75
Neutrophil ability to sense pro-inflammatory stimuli is ascribed to a variety of surface receptors, many of which belong to the G protein-coupled receptors (GPCR). Formyl peptide receptor 1 (FPR1) was firstly described on polymorphonuclear leukocytes. After interaction with the specific ligand, it triggers several functions, such as chemotaxis, lysosomal granule release, oxygen-free radical production, and phagocytosis.76,77
The principal ligands for FPR1 are bacterial78 and mitochondrial79 formylated peptides actively secreted by invading pathogens or passively released from dead and dying host cells after tissue injury. Of interest, N-Formyl-L-methionyl-L-leucyl-L-phenylalanine (FMLP) the major component of formylated peptides is present in tobacco leaves and is also an active component of mainstream and side stream CS.80 FMLP and its high-affinity receptor FPR1 on neutrophils and macrophages can modulate a variety of cellular responses associated with inflammation: shape change, chemotaxis, adhesion, phagocytosis, release of superoxide anions, and granule contents.81 When intratracheally instilled FMLP induces morphological alterations in mice resembling those present in human COPD or in animals exposed to CS.51,82,83 Further support for an important pathogenic role of formyl peptides and their receptors is that COPD patients exhibit an altered microbiome during the course of the disease. This may explain the increased susceptibility of patients to microbial- and viral-induced exacerbations.
These studies strongly suggested a role for formyl peptides in the initiation and progression of disease in smokers (Figure 2).
In this context, we demonstrated that the ablation of fpr-1 gene in mice, or treatment with specific FPRs antagonists, prevents lung recruitment of inflammatory cells leading to a complete protection from CS-induced pulmonary lesions43 (Figures 2 and 3). These receptors are over-expressed in patients with COPD84 and have been recently involved, together with mitochondrial formylated peptides, also in acute lung inflammation and injury.85
 |
Figure 3 Studies on cell surface receptors and COPD were carried out in genetically modified mice. Genetic ablation of the formyl-peptide receptor-1 (fpr-1) gene in mice, or treatment with specific antagonists of FPRs prevents recruitment of inflammatory cells in the lung leading to a complete protection from smoking-induced lung emphysema and airway remodelling (84). CS-induced neutrophilic inflammation and emphysema can be also statistically lowered upon inhibition of RAGE, or purinergic receptor subtypes (such as P2Y2R or P2X7R) as demonstrated in RAGE (60), or P2Y2R and P2X7 knockout mice (44, 45). After chronic CS-exposure, par2 gene overexpression in FVB mice leads to emphysematous changes associated with PH and RVH (26). The absence of proteinase-activated receptor-1 signaling in C57 Bl/6 mice confers protection form FMLP-induced goblet cell metaplasia (51). This image is the property of the authors. |
Formyl Peptides, FPRs and Smoking Cessation
Human and animal studies demonstrated that, once COPD is initiated, the pulmonary inflammation continues86–88 and emphysema cannot be reversed following smoking cessation. Current therapies do not adequately halt inflammation and are not able to break the “vicious cycle” that is the cause of persistent inflammation in COPD patients.
More recently we observed that also in mice chronically exposed to CS, lung inflammation persists after smoking cessation.89 This results in a progressive alveolar loss and pulmonary remodelling characterized by the presence of goblet cell metaplasia of airways epithelium and peri-bronchiolar fibrosis.89 A marked increase of neutrophils and macrophages in lung parenchyma characterizes the chronic persistent inflammation we observed in these animals. We also observed a persistent activation of NF-kB associated with a nuclear localization of the p65 (RelA) component of the complex that is able to modulate gene transcription of pro- or anti-inflammatory cytokines and chemokines (ie KC, MIP-1beta, IL-6 and IL-10) or enzymes (such as MMP-12, Arginase 1) and factors (ie FGF-1, PAR-1 and IL-13). All these factors have been implicated in the pathogenesis of COPD.90–94
In this context, by using a “curative” experimental design that is currently used in pre-clinical studies,95 we observed that selective inhibitors of FPRs and FPR-1 can prevent, after smoking cessation, deterioration of lung structures,89 which lead to airspace enlargement and airway remodelling. These changes constitute the pathological basis of airflow obstruction in COPD patients.96,97
As mentioned above, formylated peptides derived from bacteria and mitochondria are ligands for FPR1. They are released during infection, by bacteria (PAMPs) and dying host cells (DAMPs). Thus, FPR1 is involved in both infective and sterile inflammation, which characterize the course of the disease.
In lung inflammation, bacteria colonize very often the lower respiratory tract of smoking patients98 representing a further source of formyl peptides99 that may amplify the effects of formylated peptides introduced with CS and/or released from mitochondria after cellular damage.
Such a study provides a compelling rationale for FPRs inhibition as a novel therapeutic strategy for treating COPD patients after smoking cessation.
Cross Talk Among Surface Receptors in Inflammatory Cells
Neutrophils and other cells such as monocytes, macrophages and dendritic cells express several G-protein-coupled receptors (GPCRs), which cross regulate each other. This fact may offer an explanation of why CS-induced neutrophilic airway inflammation and emphysema can be significantly lowered upon inhibition of various receptors such as RAGE, or purinergic receptors. Ligation of FPRs transfers neutrophils to a desensitized state that renders neutrophils non-responsive to additional stimulation with the same agonist, or other low-affinity receptor agonists (homologous desensitization) and to a variety of ligands for other unrelated receptors (heterologous desensitization).100 It has been recently clarified a cross-talk mechanism in neutrophils, by which signals generated by the receptor for ATP (P2Y2) reactivate desensitized formyl peptide receptors (FPRs) so that these ligand-bound inactive FPRs resume signalling.101 This mechanism unidirectional in nature results in reactivation of FPRs and, in amplification of the neutrophil response to PAMPS and DAMPS (Figure 2).
Complete understanding of the cross talk among other surface receptors may provide new knowledge for the development of new therapies for inflammatory diseases including COPD.
Proteases and Their Receptors in COPD
Proteases identified in the lung were initially involved in extracellular matrix destruction. Proteases from neutrophils, macrophages and some lymphocyte subsets are involved in host defense, but when present in excess they can cause tissue injury, organ dysfunction, and lung diseases (eg emphysema, asthma and pulmonary fibrosis). Indeed, recent studies suggest that serine- or metallo-proteases exert a lot of regulatory functions by promoting chemokine and cytokine activation and degradation, shedding of cell surface receptors, proteolysis of cytokine binding proteins, and activation of different specific cell surface receptors, named proteinase-activated receptors (PARs).102 These receptors, belonging to the GPCRs family are found widely throughout the body and are activated through proteolytic cleavage by a variety of serine proteinases. Among these receptors, PAR1 and PAR2 exert both physiological and pathological effects in the respiratory system and in particular in the development of PH and mucus hypersecretion, which can be variably associated with parenchymal lesions (emphysema and small airway disease) characterizing human COPD. These receptors and their activating serine proteases have been recently implicated in vascular and bronchial remodelling that develops after CS exposure (Figure 3). FVB transgenic mice overexpressing PAR2 show after chronic exposure to CS emphysematous changes associated with PH and right ventricular hypertrophy (RVH).26 These changes in FVB wild type mice do not develop in association with emphysema. A recent study also suggests a role for PAR1 in airways remodelling (goblet cell metaplasia and peribronchiolar fibrosis) caused by formylated peptides.51 These lesions develop in C57 Bl/6 wild type mice but do not appear in mice knocked out for par1 gene. Further investigation on protease function in lung inflammation will have important implications in health and disease.
Functional Studies on Laboratory Mouse
In general, mouse models of human diseases present several advantages with respect to other species due to the high similarity of their genome with the human genome and to their capacity to reproduce fast. However, depending on the functional study to be performed, their small size may constitute a serious limitation to these studies. This is the case for functional lung studies in the mouse. These limitations have been addressed in numerous articles devoted to functional studies on mice.103–105 Howsoever difficult it may be, these limitations can be reduced by coupling functional studies done in vivo on the intact animal with functional studies done ex vivo or in vitro on lungs or pulmonary tissues taken from the laboratory mouse.
These combined experimental approaches have been used to study lung function in mice with pulmonary changes caused by CS exposure27 as well as their comorbidities, such as PH and RVH26 or skeletal muscle dysfunction which contributes to exercise limitation in emphysema patients.106
Conclusions and Future Directions
The laboratory mouse has been used for over one hundred years to study the cause of human diseases and more recently, by using genetics, to understand mammalian physiology and development, and human disease processes.107 Mouse models can be used to collect phenotype data that cannot be collected from human subjects in order to validate candidate genes in human genetic association studies and to study the influence of single genes in physiological or pathological processes. While smaller than the human genome, the mouse genome closely mirrors that of the human.108 The overall organization of the mouse and human genomes is remarkably syntenic109 and about 17,000 mouse protein-coding genes have a known direct human ortholog (http://www.informatics.jax.org).
These considerations may explain why inbred strains of mouse, their genetic manipulated or spontaneous mutants are widely used to study human pulmonary diseases.
In our opinion, future research directed to identifying the basis of COPD exacerbations and clarifying the pathogenic mechanisms should include simultaneous characterization in humans of multiple phenotypes (such as susceptibility, severity, rate of progression, propensity to exacerbations, and emphysema vs chronic bronchitis) because different genes may be related to a different aspect of the disease, and may or not may be the same as those determining susceptibility to disease.
In future researches, mouse models are essential components of a human genetic program because they can be used to study a defined phenotype and to identify and select novel candidate genes of probable biological relevance for the pathophysiology of COPD.
Author Contributions
All authors contributed to conception and design, data acquisition and interpretation, drafting and revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Snider GLMartorana PA, Lucey ECLungarella G. Animal models of emphysema. In: Voelkel N MacNee W, editors. Chronic Obstructive Lung Disease. Hamilton, London: BC Decker Inc.; 2002:237–256.
2. Rahman I, De Cunto G, Sundar IK, Lungarella G. Vulnerability and genetic susceptibility to cigarette smoke-induced emphysema in mice. Am J Respir Cell Mol Biol. 2017;57(3):270–271. doi:10.1165/rcmb.2017-0175ED.
3. Rennard SI. Cigarette smoke in research. Am J Respir Cell Mol Biol. 2004;31:479–480. doi:10.1165/rcmb.F284
4. Shapiro SD, Goldstein NM, Houghton AM, Kobayashi DK, Kelley D, Belaaouaj A. Neutrophil elastase contributes to cigarette smoke-induced emphysema in mice. Am J Pathol. 2003;163:2329–2335. doi:10.1016/S0002-9440(10)63589-4
5. Bartalesi B, Cavarra E, Fineschi S, Lucattelli M, Martorana PA, Lungarella G. Different lung responses to cigarette smoke in two strains of mice sensitive to oxidants. Eur Respir J. 2005;25:15–22. doi:10.1183/09031936.04.00067204
6. Cavarra E, Lucattelli M, Gambelli F, et al. Human SLPI inactivation after cigarette smoke exposure in a new in vivo model of pulmonary oxidative stress. Am J Physiol Lung Cell Mol Physiol. 2001;281:L412–L417. doi:10.1152/ajplung.2001.281.2.L412
7. Cavarra E, Bartalesi B, Lucattelli M, et al. Effects of cigarette smoke in mice with different levels of proteinase inhibitor and sensitivity to oxidants. Am J Respir Crit Care Med. 2001;164:886–890. doi:10.1164/ajrccm.164.5.2010032
8. Guerassimov A, Hoshino Y, Takubo Y, et al. The development of emphysema in cigarette smoke-exposed mice is strain dependent. Am J Respir Crit Care Med. 2004;170(9):974–980. doi:10.1164/rccm.200309-1270OC
9. Martinez FJ, Curtis JL, Sciurba F; National Emphysema Treatment Trial Research Group. Sex differences in severe pulmonary emphysema. Am J Respir Crit Care Med. 2007;176(3):243–252. doi:10.1164/rccm.200606-828OC
10. de Torres JP, Cote CG, Lopez MV, et al. Sex differences in mortality in patients with COPD. Eur Respir J. 2009;33(3):528–535. doi:10.1183/09031936.00096108
11. Gonzalez AV, Suissa S, Ernst P. Gender differences in survival following hospitalisation for COPD. Thorax. 2011;66:38–42. doi:10.1136/thx.2010.141978
12. Foreman MG, Zhang L, Murphy J; COPDGene Investigators. Early-onset chronic obstructive pulmonary disease is associated with female sex, maternal factors, and African American race in the COPD Gene Study. Am J Respir Crit Care Med. 2011;184:414–420. doi:10.1164/rccm.201011-1928OC
13. Tam A, Churg A, Wright JL, et al. Sex differences in airway remodeling in a mouse model of chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2016;193:825–834. doi:10.1164/rccm.201503-0487OC
14. Tam A, Bates JH, Churg A, Wright JL, Man SF, Sin DD. Sex-related differences in pulmonary function following 6 months of cigarette exposure: implications for sexual dimorphism in mild COPD. PLoS One. 2016;11(10):e0164835. doi:10.1371/journal.pone.0164835
15. Yao H, Rahman I. Current concepts on oxidative/carbonyl stress, inflammation and epigenetics in pathogenesis of chronic obstructive pulmonary disease. Toxicol Appl Pharmacol. 2011;254:72–85. doi:10.1016/j.taap.2009.10.022
16. Brunelli E, Domanico F, La Russa D, Pellegrino D. Sex differences in oxidative stress biomarkers. Curr Drug Targets. 2014;15:811–815. doi:10.2174/1389450115666140624112317
17. Hakim IA, Harris R, Garland L, Cordova CA, Mikhael DM, Sherry Chow HH. Gender difference in systemic oxidative stress and antioxidant capacity in current and former heavy smokers. Cancer Epidemiol Biomarkers Prev. 2012;21:2193–2200. doi:10.1158/1055-9965.EPI-12-0820
18. Tam A, Tanabe N, Churg A, et al. Sex differences in lymphoid follicles in COPD airways. Respir Res. 2020;21:46. doi:10.1186/s12931-020-1311-8
19. Rangasamy T, Misra V, Lee H, Singh A, Biswal S. Differences in Nrf2 activity between emphysema resistant (ICR) and susceptible (C57Bl/6J) mice strains in response to acute cigarette smoke exposure. Proc Am Thor Soc. 2006;3:A129.
20. Rangasamy T, Cho CY, Thimmulappa RK, et al. Genetic ablation of Nrf2 enhances susceptibility to cigarette smoke-induced emphysema in mice. J Clin Invest. 2004;114:1248–1259.
21. Rossi R, Giustarini D, Fineschi S, De Cunto G, Lungarella G, Cavarra E. Differential thiol status in blood of different mouse strains exposed to cigarette smoke. Free Radic Res. 2009;43:538–545. doi:10.1080/10715760902893332
22. Petrache I, Medler TR, Richter AT, et al. Superoxide dismutase protects against apoptosis and alveolar enlargement induced by ceramide. Am J Physiol Lung Cell Mol Physiol. 2008;295:L44–L53. doi:10.1152/ajplung.00448.2007
23. Petrache I, Natarajan V, Zhen L, et al. Ceramide upregulation causes pulmonary cell apoptosis and emphysema-like disease in mice. Nat Med. 2005;11:491–498. doi:10.1038/nm1238
24. Cavarra E, Fardin P, Fineschi S, et al. Early response of gene clusters is associated with mouse lung resistance or sensitivity to cigarette smoke. Am J Physiol Lung Cell Mol Physiol. 2009;296:L418–L429. doi:10.1152/ajplung.90382.2008
25. Lucattelli M, Bartalesi B, Cavarra E, et al. Is neutrophil elastase the missing link between emphysema and fibrosis? Evidence from two mouse models. Respir Res. 2005;6:
26. De Cunto G, Cardini S, Cirino G, Geppetti P, Lungarella G, Lucattelli M. Pulmonary hypertension in smoking mice over-expressing protease-activated receptor-2. Eur Respir J. 2011;37:823–834. doi:10.1183/09031936.00060210
27. De Cunto G, Brancaleone V, Riemma MA, et al. Functional contribution of sphingosine-1-phosphate to airway pathology in cigarette smoke exposed mice. Br J Pharmacol. 2020;177:267–281. doi:10.1111/bph.14861
28. Rader J, Gregory DA, Leme SA, et al. Variable susceptibility to cigarette smoke-induced emphysema in 34 inbred strains of mice implicates Abi3bp in emphysema susceptibility. Am J Respir Cell Mol Biol. 2017;57:365–375.
29. Sandford AJ, Chagani T, Weir TD, Connett JE, Anthonisen NR, Paré PD. Susceptibility genes for rapid decline of lung function in the lung health study. Am J Respir Crit Care Med. 2001;163:469–473. doi:10.1164/ajrccm.163.2.2006158
30. Owen CA. Roles for proteinases in the pathogenesis of chronic obstructive pulmonary disease. Internet J COPD. 2008;3:253–268. doi:10.2147/COPD.S2089
31. MacNee W, Rahman I. Is oxidative stress central to the pathogenesis of chronic obstructive pulmonary disease? Trends Mol Med. 2001;7:55–62. doi:10.1016/S1471-4914(01)01912-8
32. Wickenden JA, Clarke MC, Rossi AG, et al. Cigarette smoke prevents apoptosis through inhibition of caspase activation and induces necrosis. Am J Respir Cell Mol Biol. 2003;29:562–570. doi:10.1165/rcmb.2002-0235OC
33. Yao H, Yang SR, Edirisinghe I, et al. Disruption of p21 attenuates lung inflammation induced by cigarette smoke, LPS, and fMLP in mice. Am J Respir Cell Mol Biol. 2008;39:7–18. doi:10.1165/rcmb.2007-0342OC
34. Tuder RM, Petrache I. Pathogenesis of chronic obstructive pulmonary disease. J Clin Invest. 2012;122:2749–2755. doi:10.1172/JCI60324
35. Cosio MG, Guerassimov A. Chronic obstructive pulmonary disease. Inflammation of small airways and lung parenchyma. Am J Respir Crit Care Med. 1999;160:S21–S25. doi:10.1164/ajrccm.160.supplement_1.7
36. Saetta M, Turato G, Maestrelli P, Mapp CE, Fabbri LM. Cellular and structural bases of chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2001;163:1304–1309. doi:10.1164/ajrccm.163.6.2009116
37. Maeno T, Houghton AM, Quintero PA, Grumelli S, Owen CA, Shapiro SD. CD8+ T cells are required for inflammation and destruction in cigarette smoke-induced emphysema in mice. J Immunol. 2007;178:8090–8096. doi:10.4049/jimmunol.178.12.8090
38. D’Hulst AI, Maes T, Bracke KR, et al. Cigarette smoke-induced pulmonary emphysema in scid-mice. Is the acquired immune system required? Respir Res. 2005;6:e147. doi:10.1186/1465-9921-6-147
39. Motz GT, Eppert BL, Wesselkamper SC, Flury JL, Borchers MT. Chronic cigarette smoke exposure generates pathogenic T cells capable of driving COPD-like disease in Rag2-/- mice. Am J Respir Crit Care Med. 2010;181:1223–1233. doi:10.1164/rccm.200910-1485OC
40. Eppert BL, Wortham BW, Flury JL, Borchers MT. Functional characterization of T cell populations in a mouse model of chronic obstructive pulmonary disease. J Immunol. 2013;190:1331–1340. doi:10.4049/jimmunol.1202442
41. De Cunto G, Lunghi B, Bartalesi B, et al. Severe reduction in number and function of peripheral T cells does not afford protection toward emphysema and bronchial remodeling induced in mice by cigarette smoke. Am J Pathol. 2016;186:1814–1824. doi:10.1016/j.ajpath.2016.03.002
42. Grundy S, Plumb J, Lea S, Kaur M, Ray D, Singh D. Down regulation of T cell receptor expression in COPD pulmonary CD8 cells. PLoS One. 2013;8:e71629. doi:10.1371/journal.pone.0071629
43. Cardini S, Dalli J, Fineschi S, Perretti M, Lungarella G, Lucattelli M. Genetic ablation of the Fpr1 gene confers protection from smoking- induced lung emphysema in mice. Am J Respir Cell Mol Biol. 2012;47(3):332–339. doi:10.1165/rcmb.2012-0036OC
44. Cicko S, Lucattelli M, Müller T, et al. Purinergic receptor inhibition prevents the development of smoke-induced lung injury and emphysema. J Immunol. 2010;185:688–697. doi:10.4049/jimmunol.0904042
45. Lucattelli M, Cicko S, Müller T, et al. P2X7 receptor signaling in the pathogenesis of smoke- induced lung inflammation and emphysema. Am J Respir Cell Mol Biol. 2011;44:423–429. doi:10.1165/rcmb.2010-0038OC
46. Lommatzsch M, Cicko S, Müller T, et al. Extracellular adenosine triphosphate and chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2010;181:928–934. doi:10.1164/rccm.200910-1506OC
47. Fineschi S, De Cunto G, Facchinetti F, et al. Receptor for advanced glycation end products contributes to postnatal pulmonary development and adult lung maintenance program in mice. Am J Respir Cell Mol Biol. 2013;48:164–171. doi:10.1165/rcmb.2012-0111OC
48. Stogsdill MP, Stogsdill JA, Bodine BG, et al. Conditional overexpression of receptors for advanced glycation end-products in the adult murine lung causes airspace enlargement and induces inflammation. Am J Respir Cell Mol Biol. 2013;49:128–134. doi:10.1165/rcmb.2013-0013OC
49. Robinson AB, Stogsdill JA, Lewis JB, Wood TT, Reynolds PR. RAGE and tobacco smoke: insights into modeling chronic obstructive pulmonary disease. Front Physiol. 2012;3:301. doi:10.3389/fphys.2012.00301
50. Lazar Z, Müllner N, Lucattelli M, et al. NTPDase1/CD39 and aberrant purinergic signalling in the pathogenesis of COPD. Eur Respir J. 2016;47:254–263. doi:10.1183/13993003.02144-2014
51. Atzori L, Lucattelli M, Scotton CJ, et al. Absence of proteinase-activated receptor-1 signaling in mice confers protection form f-MLP-induced goblet cell metaplasia. Am J Respir Cell Mol Biol. 2009;41:680–687. doi:10.1165/rcmb.2007-0386OC
52. Curtis JL, Freeman CM, Hogg JC. The immunopathogenesis of chronic obstructive pulmonary disease: insights from recent research. Proc Am Thorac Soc. 2007;4:512–521. doi:10.1513/pats.200701-002FM
53. Tzortzaki EG, Siafakas NM. A Hypothesis for the initiation of COPD. Eur Respir J. 2008;34:310–315. doi:10.1183/09031936.00067008
54. Van der Vaart H, Postma DS, Timens W, Ten Hacken NHT. Acute effects of cigarette smoke on inflammation and oxidative stress: a review. Thorax. 2004;59:713–721. doi:10.1136/thx.2003.012468
55. Repapi E, Sayers I, Wain LV, et al. Genome-wide association study identifies five loci associated with lung function. Nat Genet. 2010;42(1):36–44. doi:10.1038/ng.501
56. Artigas MS, Wain LV, Repapi E, et al. Effect of five genetic variants associated with lung function on the risk of chronic obstructive lung disease, and their joint effects on lung function. Am J Respir Crit Care Med. 2011;184:786–795. doi:10.1164/rccm.201102-0192OC
57. Budulac SE, Bozen HM, Hiemstra PS, et al. Toll-like receptor (TLR2 and TLR4) polymorphisms and chronic obstructive pulmonary disease. PLoS One. 2012;7:e43124. doi:10.1371/journal.pone.0043124
58. Pouwels SD, Heijink IH, Van Oosterhout AJM, Nawijn MC. A specific DAMP profile identifies susceptibility to smoke-induced airway inflammation. Eur Respir J. 2014;43:1183–1186. doi:10.1183/09031936.00127813
59. Ferhani N, Letuve S, Kozhich A, et al. Expression of high-mobility group box 1 and of receptor for advanced glycation end products in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2010;181:917–927. doi:10.1164/rccm.200903-0340OC
60. Waseda K, Miyahara N, Taniguchi A, et al. Emphysema requires the receptor for advanced glycation end products triggering on structural cells. Am J Respir Cell Mol Biol. 2015;52:482–491. doi:10.1165/rcmb.2014-0027OC
61. Rutgers SR, Timens W, Kaufmann HF, van der Mark TW, Koeter GH, Postma DS. Comparison of induced sputum with bronchial wash, bronchoalveolar lavage and bronchial biopsies in COPD. Eur Respir J. 2000;15:109–115. doi:10.1183/09031936.00.15110900
62. Stansecu D, Sanna A, Veriter C, Kostinev S, Calcagni PG, Fabbri LM. Airways obstruction, chronic expectoration and rapid decline in FEV1 in smokers are associated with increased levels of sputum neutrophils. Thorax. 1996;51:267–271. doi:10.1136/thx.51.3.267
63. Thompson AB, Daughton D, Robbins RA, Ghafouri MA, Ohlerking M, Rennard SI. Intraluminal airway inflammation in chronic bronchitis: characterization and correlation with clinical parameters. Am J Respir Crit Care Med. 1989;140:1527–1537.
64. Pilette C, Colinet B, Kiss R, et al. Increased galectin-3 expression and intraepithelial neutrophils in small airways in severe chronic obstructive pulmonary disease. Eur Respir J. 2007;29:914–922. doi:10.1183/09031936.00073005
65. Donaldson GC, Seemungal TA, Patal IS, et al. Airway and systemic inflammation and decline in lung function in patients with COPD. Chest. 2005;128:1995–2004. doi:10.1378/chest.128.4.1995
66. Parr DG, White AJ, Bayley DL, Guest PJ, Stockley RA. Inflammation in sputum relates to the progression of disease in COPD: a prospective study. Respir Res. 2006;7e:136. doi:10.1186/1465-9921-7-136
67. Yoshioka A, Betsuyaku T, Nishimura M, Miyamoto K, Kondo T, Kawakami Y. Excessive neutrophil elastase in bronchoalveolar lavage fluid from patients with sub-clinical emphysema. Am J Respir Crit Care Med. 2005;152:2127–2132. doi:10.1164/ajrccm.152.6.8520785
68. Stockley RA. Neutrophils in the pathogenesis of COPD. Chest. 2002;121:151S–155S. doi:10.1378/chest.121.5_suppl.151S
69. Snider GL, Hayes JA, Franzblau C, Kagan HM, Stone PJ, Khorty A. Relationship between elastolytic activity and experimental emphysema inducing properties of papain preparations. Am Rev Respir Dis. 1974;110:254–257.
70. Damiano VV, Tsang A, Kucich U, et al. Immunolocalization of elastase in human emphysematous lungs. J Clin Invest. 1986;78:482–493. doi:10.1172/JCI112600
71. Smallman LA, Hill SL, Stockley RA. Reduction of ciliary beat frequency in vitro by sputum from patients with bronchiectasis: a serine proteinase effect. Thorax. 1984;39:663–667. doi:10.1136/thx.39.9.663
72. Aoshiba K, Yasuda K, Yasui S, Tamaoki J, Nagai A. Serine proteases increase oxidative stress in lung cells. Am J Physiol Lung Cell Mol Physiol. 2001;281:L556–L564. doi:10.1152/ajplung.2001.281.3.L556
73. Nakajoh M, Fukushima T, Suzuki K, et al. Retinoic acid inhibits elastase-induced injury in human lung epithelial cells. Am J Respir Cell Mol Biol. 2002;28:298–304.
74. Shao MX, Nadel JA. Neutrophil elastase induces MUC5AC mucin production in human airway epithelial cells. Proc Natl Acad Sci USA. 2005;102:767–772. doi:10.1073/pnas.0408932102
75. Takeyabu K, Augusti C, Ueki IF, Lausier J, Cardell LO, Nadel JA. Neutrophil-dependent goblet cell degranulation: role of membrane bound elastase and adhesion molecules. Am J Physiol. 1998;19:L294–L302.
76. Boulay F, Tardif M, Brouchon L, Vignais P. Synthesis and use of a novel N-formyl peptide derivative to isolate a human N-formyl peptide receptor cDNA. Biochem Biophys Res Commun. 1990;168:1103–1109. doi:10.1016/0006-291X(90)91143-G
77. International Union of Basic and Clinical Pharmacology. LXXIII. Nomenclature for the formyl peptide receptor (FPR) family. Pharmacol Rev. 2009;61:119–161. doi:10.1124/pr.109.001578
78. Migeotte I, Communi D, Parmentier M. Formyl peptide receptors: a promiscuous subfamily of G protein-coupled receptors controlling immune responses. Cytokine Growth Factor Rev. 2006;17(6):501–519. doi:10.1016/j.cytogfr.2006.09.009
79. Rabiet MJ, Huet E, Boulay F. Human mitochondria-derived N formylated peptides are novel agonists equally active on FPR and FPRL1, while Listeria monocytogenes-derived peptides preferentially activate FPR. Eur J Immunol. 2005;35:2486–2495. doi:10.1002/eji.200526338
80. Hasday JD, Bascom R, Costa JJ, Fitzgerald T, Dubin W. Bacterial endotoxin is an active component of cigarette smoke. Chest. 1999;115(3):829–835. doi:10.1378/chest.115.3.829
81. Dorward DA, Lucas CD, Chapman GB, Haslett C, Dhaliwal K, Rossi A. The role of formylated peptides and formyl peptide receptor 1 in governing neutrophil function during acute inflammation. Am J Pathol. 2015;185:1172–1184. doi:10.1016/j.ajpath.2015.01.020
82. Cavarra E, Martorana PA, Gambelli F, de Santi MM, van Even P, Lungarella G. Neutrophil recruitment into the lungs is associated with increased lung elastase burden, decreased lung elastin, and emphysema in alpha 1 proteinase inhibitor-deficient mice. Lab Invest. 1996;75(2):273–280.
83. Cavarra E, Martorana PA, de Santi MM, Bartalesi B, Cortese S, Lungarella G. Neutrophil influx into the lungs of beige mice is followed by elastolytic damage and emphysema. Am J Respir Cell Mol Biol. 1999;20:264–269.
84. Stockley RA, Grant RA, Llewellyn-Jones CG, Hill SL, Burnett D. Neutrophil formyl-peptide receptors: relationship to peptide-induced responses and emphysema. Am J Respir Crit Care Med. 1994;149:464–468. doi:10.1164/ajrccm.149.2.8306047
85. Dorward DA, Lucas CD, Doherty MK, et al. Novel role for endogenous mitochondrial formylated peptide-driven formyl peptide receptor 1 signalling in acute respiratory distress syndrome. Thorax. 2017;72(10):928–936. doi:10.1136/thoraxjnl-2017-210030
86. Willemse BW, Ten Hacken NH, Rutgers B, Lesman-Leegte IG, Postma DS, Timens W. Effect of 1-year smoking cessation on airway inflammation in COPD and asymptomatic smokers. Eur Respir J. 2005;26:835–845. doi:10.1183/09031936.05.00108904
87. Gamble E, Grootendorst DC, Hattotuwa K, et al. Airway mucosal inflammation in COPD is similar in smokers and ex-smokers: a pooled analysis. Eur Respir J. 2007;30(3):467–471. doi:10.1183/09031936.00013006
88. Lapperre TS, Postma DS, Gosman MM, et al. Relation between duration of smoking cessation and bronchial inflammation in COPD. Thorax. 2006;61:115–121. doi:10.1136/thx.2005.040519
89. De Cunto G, Bartalesi B, Cavarra E, Balzano E, Lungarella G, Lucattelli M. Ongoing lung inflammation and disease progression in mice after smoking cessation. Beneficial effects of formyl-peptide receptors blockade. Am J Pathol. 2018;188(10):2195–2206. doi:10.1016/j.ajpath.2018.06.010
90. Pahl HL. NF-kB activators and target genes of Rel/NF-kB transcription factors. Oncogene. 1999;18(49):6853–6866. doi:10.1038/sj.onc.1203239
91. Hol J, Wilhelmsen L, Haraldsen G. The murine IL-8 homologues KC, MIP-2, and LIX are found in endothelial cytoplasmic granules but not in Weibel-Palade bodies. J Leukoc Biol. 2010;87(3):501–508. doi:10.1189/jlb.0809532
92. Menten P, Wuyts A, Van Damme J. Macrophage inflammatory protein-1. Cytokine Growth Factor Rev. 2002;6:455–481. doi:10.1016/S1359-6101(02)00045-X
93. Lunghi B, De Cunto G, Cavarra E, Bartalesi B, Lungarella G, Lucattelli M. Smoking p66Shc knocked out mice develop respiratory bronchiolitis with fibrosis but not emphysema. PLoS One. 2015;10:e 0119797. doi:10.1371/journal.pone.0119797
94. Yu C, Wang F, Jin C, et al. Role of fibroblast growth factor type 1 and 2 in carbon tetrachloride-induced hepatic injury and fibrogenesis. Am J Pathol. 2003;163(4):1653–1662. doi:10.1016/S0002-9440(10)63522-5
95. Martorana PA, Beume R, Lucattelli M, Wollin L, Lungarella G. Roflumilast fully prevents emphysema in mice chronically exposed to cigarette smoke. Am J Respir Crit Care Med. 2005;172:848–853. doi:10.1164/rccm.200411-1549OC
96. Cosio M, Ghezzo H, Hogg JC, et al. The relations between structural changes in small airways and pulmonary-function tests. N Engl J Med. 1978;298(23):1277–1281. doi:10.1056/NEJM197806082982303
97. Hasegawa M, Nasuhara Y, Onodera Y, et al. Airflow limitation and airway dimensions in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2006;173(12):1309–1315. doi:10.1164/rccm.200601-037OC
98. Sethi S. Bacterial infections and the pathogenesis of COPD. Chest. 2000;117(5):286S–291S. doi:10.1378/chest.117.5_suppl_1.286S
99. Matheson M, Rynell AC, McClean M, Berend N. Cigarette smoking increases neutrophil formyl methionyl leucyl phenylalanine receptor numbers. Chest. 2003;123:1642–1646. doi:10.1378/chest.123.5.1642
100. Fu H, Bylund J, Karlsson A, Pellme S, Dahlgren C. The mechanism for activation of the neutrophil NADPH-oxidase by the peptides formyl–Met–Leu–Phe and Trp–Lys–Tyr–Met–Val–Met differs from that for interleukin-8. Immunology. 2004;112:201–210. doi:10.1111/j.1365-2567.2004.01884.x
101. Önnheim K, Christenson K, Gabl M, et al. A novel receptor cross-talk between the ATP receptor P2Y2 and formyl peptide receptors reactivates desensitized neutrophils to produce superoxide. Exp Cell Res. 2014;323:209–217. doi:10.1016/j.yexcr.2014.01.023
102. Lungarella G, Cavarra E, Fineschi S, Lucattelli M. Dual role for proteases in lung inflammation. In: Vergnolle N, Chignard M, editors. Proteases and Their Receptors in Inflammation. Basel: Spriger Basel; 2011:123–144.
103. Vlahos R, Bozinovski S. Recent advances in pre-clinical models of COPD. Clin Sci. 2014;126:253–265. doi:10.1042/CS20130182
104. Rinaldi M, Maes K, De Vleeschlauwer S, et al. Long-term nose-only cigarette smoke exposure induced emphysema and mild skeletal muscle dysfunction in mice. Dis Model Mech. 2012;5:333–341. doi:10.1242/dmm.008508
105. Jobse BN, Rhem RG, Wang IQ, Counter WB, Stampfli MR, Labiris NR. Detection of lung dysfunction using ventilation and perfusion SPECT in a mouse model of chronic cigarette smoke exposure. J Nucl Med. 2013;54:616–623. doi:10.2967/jnumed.112.111419
106. Sullo N, Roviezzo F, Matteis M, et al. Skeletal muscle oxidative metabolism in an animal model of pulmonary emphysema. Formoterol and skeletal muscle dysfunction. Am J Respir Cell Mol Biol. 2013;48:198–203. doi:10.1165/rcmb.2012-0167OC
107. Schofield PN, Hoehndorf R, Gkoutos GV. Mouse genetic and phenotypic resources for human genetics. Hum Mutat. 2012;33:826–836. doi:10.1002/humu.22077
108. Waterston RH, Lindblad-Toh K, Birney E; Mouse Genome Sequencing Consortium. Initial sequencing and comparative analysis of the mouse genome. Nature. 2002;420:520–562. doi:10.1038/nature01262
109. Paigen K. One hundred years of mouse genetics: an intellectual history. I. The classical period (1902–1980). Genetics. 2003;163:1–7.
110. Cottin V, Nunes H, Brillet PY, et al. Combined pulmonary fibrosis and emphysema: a distinct underrecognised entity. Eur Respir J. 2005;26:586–593. doi:10.1183/09031936.05.00021005
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.