Back to Journals » International Journal of Nanomedicine » Volume 11
Injectable, degradable, electroactive nanocomposite hydrogels containing conductive polymer nanoparticles for biomedical applications
Authors Wang Q, Wang Q, Teng W
Received 19 August 2015
Accepted for publication 28 November 2015
Published 5 January 2016 Volume 2016:11 Pages 131—145
DOI https://doi.org/10.2147/IJN.S94777
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 7
Editor who approved publication: Professor Lei Yang
Qinmei Wang,1 Qiong Wang,2 Wei Teng2
1Laboratory of Biomaterials, Key Laboratory on Assisted Circulation, Ministry of Health, Cardiovascular Division, First Affiliated Hospital, Sun Yat-sen University, Guangzhou, People’s Republic of China; 2Department of Prosthodontics, Hospital of Stomatology, Institute of Stomatological Research, Guanghua School of Stomatology, Sun Yat-sen University, Guangzhou, People’s Republic of China
Abstract: Injectable electroactive hydrogels (eGels) are promising in regenerative medicine and drug delivery, however, it is still a challenge to obtain such hydrogels simultaneously possessing other properties including uniform structure, degradability, robustness, and biocompatibility. An emerging strategy to endow hydrogels with desirable properties is to incorporate functional nanoparticles in their network. Herein, we report the synthesis and characterization of an injectable hydrogel based on oxidized alginate (OA) crosslinking gelatin reinforced by electroactive tetraaniline-graft-OA nanoparticles (nEOAs), where nEOAs are expected to impart electroactivity besides reinforcement without significantly degrading the other properties of hydrogels. Assays of transmission electron microscopy, 1H nuclear magnetic resonance, and dynamic light scattering reveal that EOA can spontaneously and quickly self-assemble into robust nanoparticles in water, and this nanoparticle structure can be kept at pH 3~9. Measurement of the gel time by rheometer and the stir bar method confirms the formation of the eGels, and their gel time is dependent on the weight content of nEOAs. As expected, adding nEOAs to hydrogels does not cause the phase separation (scanning electron microscopy observation), but it improves mechanical strength up to ~8 kPa and conductivity up to ~10-6 S/cm in our studied range. Incubating eGels in phosphate-buffered saline leads to their further swelling with an increase of water content <6% and gradual degradation. When growing mesenchymal stem cells on eGels with nEOA content ≤14%, the growth curves and morphology of cells were found to be similar to that on tissue culture plastic; when implanting these eGels on a chick chorioallantoic membrane for 1 week, mild inflammation response appeared without any other structural changes, indicating their good in vitro and in vivo biocompatibility. With injectability, uniformity, degradability, electroactivity, relative robustness, and biocompatibility, these eGels may have a huge potential as scaffolds for tissue regeneration and matrix for stimuli responsive drug release.
Keywords: injectability, electroactivity, nanocomposite hydrogels, reinforcement, tetraaniline, alginate, gelatin
Introduction
Recently, injectable electroactive biodegradable hydrogels have attracted much attention for biomedical applications in regenerative medicine and drug delivery due to the following advantages.1–7 Firstly, hydrogels can mimic the nature of tissue from the physical, chemical, electrical, and biological properties, and thus provide an appropriate microenvironment for cell growth and drug release. Secondly, the injectability endows hydrogels with many virtues such as improved patient compliance, facile administration via a minimally invasive mode, wide suitability for complex shapes of target sites, and high loading capacity for cells and therapeutic molecules.8,9 Thirdly, the electroactivity exerts great influence on cell activities and endows hydrogels with electrical stimuli response. It has been demonstrated that electroactive materials can promote the adhesion, migration, proliferation, and differentiation of cells including cardiomyocytes, neurons, endothelial cells, fibroblasts, mesenchymal stem cells (MSCs), and preosteoblasts with or without electrical stimuli.1,6,7,10–12 Furthermore, the electroactive materials can resist the oxidative damage to tissue during tissue regeneration by scavenging free radicals.13,14
In general, such hydrogels can be prepared by introducing conducting composites into injectable degradable hydrogels through chemical/physical conjugation or simple mechanical mixing.1–3,5,6,10–16 Among the used conducting composites, carbon nanotubes and oligomers of conducting polymers are the most popular due to their superb properties. Carbon nanotubes can impart both high mechanical and electrical conductivity properties to hydrogels, but the difficulty in batch production with uniform and repeated structure and properties, the weak interactions with hydrophilic hydrogels and the tendency to aggregate due to their hydrophobicity, and the unsatisfactory biocompatibility/toxicity limit their biomedical applications.2,17 Furthermore, it is not easy to functionalize carbon nanotubes because they do not have any functional groups. By contrast, aniline oligomers such as tetraaniline or pentaaniline, the most often used conducting oligomers, have superiority in easy synthesis, low cost, stability, facile processing, good biocompatibility, similar electrochemical behaviors to conducting polymers, and are readily removed from the body by renal excretion due to the low molecular weight (Mw).12 And additionally, they possess terminal amine groups for functionalization. Therefore, many electroactive hydrogels (eGels) containing aniline oligomers have been developed, where aniline oligomers were chemically conjugated onto the precursor molecules of hydrogels as a side chain or one part of a main chain. For example, Liu et al synthesized an electroactive gelatin-based copolymer of aniline pentamer-graft-gelatin (AP-g-GA), and then used 1-ethyl-3-(3-dimethyl aminopropyl) carbodiimide to crosslink AP-g-GA to form hydrogels.16 The authors found that the hydrogel containing 1.9% AP showed the best biocompatibility and cell adhesion ability. Li et al prepared a series of injectable eGels by genipin crosslinking gelatin-graft-polyaniline (GP) under physiological conditions.3 The conductivity of GP hydrogels was ~10−4 S/cm, and the hydrogels were non-toxic and could support the adhesion and proliferation of MSCs and C2C12 myoblast cells. But unfortunately, polyaniline is undegradable.
Although much progress has been made, it is well known that the weak strength is one of the main drawbacks of injectable hydrogels for biomedical applications, and simply introducing conductive polymers into hydrogels makes a limited contribution to the mechanical strength. In practice, most studies have focused on obtaining electroactivity, while the mechanical properties of hydrogels have been seldom explored. It is reported that incorporating functional nanoparticles into hydrogels can endow them with desirable properties including reinforcement and electroactivity.2,18 For reinforcement, well distributed and strong interactions of nanoparticles with hydrogels are vitally important. Therefore, incorporating conductive polymer nanoparticles into hydrogels may be an alternative efficient strategy to obtain both robust and eGels. To the best of our knowledge, this field has received little attention thus far.
In this work, an injectable hydrogel was synthesized by using oxidized alginate (OA) and electroactive tetraaniline-graft-OA nanoparticles (nEOAs) to crosslink gelatin, where nEOAs were expected to impart electroactivity to hydrogels and simultaneously reinforce them. The gel formation process, injectability (characterized by gel time), structure and morphology, mechanical strength, swelling and degradable behaviors, electroactivity, and in vitro and in vivo biocompatibility of hydrogels with different nEOA contents were investigated.
Experimental methods
Materials
Gelatin (type A, Bloom 300), sodium alginate (medium viscosity grade) and N-phenyl-p-phenylenediamine were purchased from Sigma-Aldrich Co. (St Louis, MO, USA). OA, with an oxidation degree (OD, the percentage of oxidized structure unit in sodium alginate) of 58.10% and weight-average Mw of ~2.64×104 g/mol (data from gel permeation chromatograph), was prepared through periodate oxidation of sodium alginate in a water–ethanol mixture following the published method.19,20 The fertilized chicken eggs were obtained from the Institute of Livestock Science, South China Agricultural University (Guangzhou, People’s Republic of China). Sprague Dawley rats were purchased from the laboratory animal center of Sun Yat-sen University. MSCs were obtained from neonatal Sprague Dawley rats (1–3 days old), their isolation, purification, culture, and passage were performed according to the standard method.21 All animal care and experimental procedures were conducted according to institutional animal care and use guidelines, and approved by the Animal Ethics Committee of Sun Yat-sen University. The other reagents were purchased from Guangzhou Chemical Reagent Factory (Guangzhou, People’s Republic of China). All chemicals were used as received unless specified.
Preparation of electroactive nEOAs
Electroactive EOA copolymer was first prepared according to our previously reported method.19 Briefly, phenyl/amino-capped tetraaniline was synthesized by chemical oxidation-coupling reaction of N-phenyl-p-phenylenediamine followed by reduction reaction. Then fresh tetraaniline was grafted onto OA to form an amphiphilic brush copolymer of EOA through the reaction between the aldehyde groups on OA and amine groups on tetraaniline. The mass content of grafted tetraaniline in EOA (mTA%) was determined by element analysis,19 which could be adjusted in the range of 0%~40% by changing the feed molar ratio of aldehyde groups on OA and amine groups on tetraaniline. The concentration of aldehyde groups in EOA available for further chemical reactions was measured by the hydroxylamine hydrochloride method,22 and Mw of EOA was characterized by gel permeation chromatograph, which was described in detail in our previously published paper.19 When the feeding molar ratio of aldehyde groups on OA and amine groups on tetraaniline is 100%, EOA with mTA% of 35.40% and Mw of 2.15×104 g/mol was obtained. nEOAs were prepared by simply dissolving EOA in water under stirring. The self-assembly process of EOA in water was studied by 1H nuclear magnetic resonance (NMR) on a Varian INOVA500NB spectrometer through monitoring their signal changes with time, and the pH response of nEOAs was studied by 1H NMR, dynamic light scattering (DLS) analysis on a Zeta Potential Analyzer (Brookhaven Instruments Corp., Holtsville, NY, USA), and transmission electron microscopy (TEM) observation on a JEM-2010 HR TEM (JEOL, Tokyo, Japan) using phosphotungstic acid staining method following our previously reported method.19
Synthesis of eGels
OA (OD, 58.10%) and nEOAs (WTA%, 35.40%) were dissolved in 0.1 mol/L sodium tetraborate decahydrate to get a mixed crosslinker solution with a concentration of 0.20 g/mL, and gelatin was dissolved in distilled water at 37°C to obtain a 0.15 g/mL solution. Then according to the feeding composition presented in Table 1, the crosslinker solution was added to gelatin solution under magnetic stirring at 37°C to form a hydrogel. The scheme for hydrogel preparation was presented in Figure 1A. The gel time, which was defined as the time taken for the formed gel to stop the stir bar according to Balakrishnan and Jayakrishnan23 and Mo et al,24 was recorded using a stopwatch. The gel properties were adjusted by the weight content of nEOAs in eGels.
 | Table 1 Feeding composition and some properties of hydrogels prepared from OA/nEOA crosslinking gelatin |
 | Figure 1 Preparation of electroactive hydrogels. |
Morphology observation of eGels
The hydrogels were prepared by solution casting method in our home-made mold (Figure 1B), with a reaction container which is composed of two symmetrical semi-cylinders of blue silastic. The crosslinker solution of (nEOAs + OA) and gelatin solution were mechanically mixed in the mold at 37°C for 20 seconds. Thereafter, the reaction was kept overnight at 37°C, and the formed hydrogels were taken out, frozen in liquid nitrogen for 40 seconds, cracked, and lyophilized. Their cross-sections were observed under a scanning electron microscope (TM-1000; Hitachi Ltd., Tokyo, Japan) at 15 kV in secondary electron mode after a standard treatment.
Rheology analysis
All rheological tests were performed at room temperature on an ARG2 rheometer (TA Instruments, New Castle, DE, USA) following the reported method.9 To confirm the gel formation and to determine the gel time, the time sweep rheology spectrum was first performed on eGel-7 (7 means the weight percentage of nEOAs in eGel) by immediately placing 1 mL precursor solution on the lower plate of the rheometer at the fixed oscillation stress of 0.5 Pa, and then the strain sweep was conducted at a frequency of 1.0 Hz. For frequency sweep, eGel discs (40×1 mm) were prepared as mentioned in the “Morphology observation of eGels” section, then incubated with phosphate-buffered saline (PBS) at 37°C for 1 hour, and the sweep was conducted at 10% strain.
Water-uptake capacity of hydrogels
The water-uptake capability of hydrogel was characterized by incubating it in PBS. Briefly, eGels containing 0.225 g gelatin were prepared in a 10 mL screw-capped test tube at 37°C as mentioned above, and 10 minutes later, 5 mL of PBS was added and the tubes were incubated at 37°C. At predetermined time intervals, eGels were taken out, carefully blotted-up by filter papers, and weighed, followed by continuous incubation. The test was stopped when equilibrium swelling was reached. The water content in eGel was:
eGel(%)=(Ws−Wd)/Ws×100%, | (1) |
where Ws and Wd are the mass of the swollen and dry hydrogels, respectively. The weight at each time point was the average value of five hydrogels during incubation.
In vitro degradation of eGels
The biodegradation of eGels was evaluated by monitoring the change in mass with time. eGels were prepared in our home-made molds as mentioned above. Then they were immersed in 0.5 mol·L−1 PBS (pH =7.4) and incubated at 37°C. At predetermined time intervals, they were taken out of the solution, washed with distilled water, dried by lyophilization, and weighed. The weight at each time point was the average value of three hydrogels during degradation.
Electroactivity of eGels
The electroactivity of eGels was evaluated by three experiments. The first one is to explore its reversible doping/dedoping. In brief, a thin film of eGels was coated on a glass slide, then it was alternately immersed in 1 mol/L of NaOH or HCl, and the color was photographed. The second one is to determine the cyclic voltammograms (CV) of eGels on an IM6E Electrochemical Workstation (Zahner Elektrik, Kronach, Germany) using Hg/Hg2Cl2 and Pt as the reference and counter electrode, respectively. The working electrode was a glassy carbon electrode, and the eGel with nEOA weight content (WEOA%) of 7% was evenly coated on its surface. The CV was obtained in 1 mol·L−1 HCl at a scan rate of 100 mV·sec−1. The third one is to determine the conductivity of eGels using the standard Van Der Pauw four-probe method.19 The eGel films (5 mm thick) were prepared in our home-made molds as mentioned in the section of “Morphology observation of eGels” section, doped by 1 mol/L HCl, and then thoroughly dried in a vacuum oven for 1 week. Thereafter, the conductivity of dried films was measured on a conductivity meter (SDY-4; Guangzhou Institute of semiconducting material, Guangzhou, People’s Republic of China).
In vitro cell compatibility of eGels
The effects of eGels with different nEOA contents on the cell compatibility were evaluated by cell growth curves and cell morphology observation.21,25 MSCs between passage 2 and 5 were chosen as a model cell due to their multipotential differentiation and wide applications in cell therapy as a stem cell. Briefly, 200 μL of thoroughly mixed hydrogel precursor solutions (eGel-0, eGel-7, eGel-14, eGel-21) were added into six-well plates respectively, and the plates were incubated at 37°C for 24 hours to allow gel formation. All eGels were sterilized with ultraviolet light for 45 minutes before use. Thereafter, MSCs were seeded in six-well plates with eGels at a density of 4×104 cells/cm2 and routinely cultured in complete media (Hyclone Dulbecco’s Modified Eagle’s Medium/F12 1:1 supplemented with 10% fetal bovine serum, 100 μg/mL streptomycin and 100 U/mL penicillin). At designated time points, 10% alamarBlue (Invitrogen, Thermo Fisher Scientific, Waltham, MA, USA) was added, and cells were incubated for another 4 hours. Thereafter, the medium was taken out for absorbance analysis at 570 and 600 nm on an enzyme-linked immunosorbent assay plate reader (Model 550; Bio-Rad Laboratories Inc., Hercules, CA, USA). Simultaneously, in order to fluorescently label nucleus and F-actin of the cytoskeleton for density and morphology observation, some cells were stained by 4′,6-diamidino-2-phenylindole and rhodamine phalloidin (Biotium Inc., Hayward, CA, USA) respectively following the supplier’s protocols, and then imaged under a fluorescence microscope. The other cells were continuously cultured in a fresh medium for the next analysis. Cells cultured in wells without eGels were used as a control. The absorbance of medium in blank wells (no cell seeding) with or without eGels was used for background subtraction for corresponding eGel groups or the control, respectively. The cell viability was expressed as the percentage of the absorbance of the test group (cells growing on eGels) relative to control.
In vivo biocompatibility of eGels in a chorioallantoic membrane (CAM)
The in vivo biocompatibility was evaluated using a CAM of the developing chicken embryo as a model following the reported method.26,27 Fertilized chicken eggs were incubated at 37°C with ~60% humidity. On day 7 post-fertilization, eggs were first sterilized by immersing in the benzalkonium bromide solution, and then a small window with diameter of ~1.5 cm at the top of the chicken egg shell was made by cutting the shell with a grinding wheel. Thereafter, CAM was carefully exposed without damaging the embryonic structure, and 30 μL of the sterilized eGel precursor solutions with different nEOA concentrations was gently added onto the CAM surfaces using a pipette. After that, the window was sealed by a transparent tape, and the egg was returned to the incubator. One week later, 5 mL of ice-cold methanol/acetone (v/v =1:1) was added into the eggs through the window to fix the vessels. After 20 minutes, CAM was separated and washed thoroughly. Then some CAMs were immediately observed under a stereomicroscope (AZ100; Nikon Corporation, Tokyo, Japan) for gross evaluation, and the others were fixed in 10% formalin, embedded in paraffin, sectioned, and stained with hematoxylin and eosin (H and E) for histopathological evaluation. The stained slides were observed under an inverted phase contrast microscope (CKX41; Olympus Corporation, Tokyo, Japan), and the inflammation and tissue response were evaluated. Non-treated CAMs were used as controls.
Statistical analysis
Each experiment was performed at least three times. Data were expressed as mean ± standard deviation. A one-way analysis of variance (ANOVA) was used for statistical analysis. P<0.05 is considered statistically significant.
Results and discussion
Self-assembly behavior of electroactive EOA with pH and time
The details of synthesis of EOA with different tetraaniline contents were reported in our previous publication.19 It is worth noting that we have proved that EOA can spontaneously self-assemble into spherical nanoparticles in water with hydrophobic tetraaniline as core and hydrophilic anionic OA as shell. Such nanoparticle structure may be beneficial to crosslinking and electroactivity because large aldehyde groups for crosslinking are exposed thoroughly in the outer shell, and the aggregated tetraaniline in the inner core may make electron migration easier than the single scattered tetraaniline. Furthermore, we have demonstrated that nEOAs are biocompatible, biodegradable, and electroactive.19 In this study, OA with the OD of ~58.1% (weight-average Mw, ~2.64×104 g/mol) and EOA with tetraaniline mass content of 35.4% were used for all experiments, and the self-assembly behavior of this EOA with pH and time was studied. As expected, TEM observation reveals that in the aqueous solution (pH 7), EOA exists in the form of nanoparticles (Figure 2A), and the diameter of nanoparticles is 332.4 nm with polydispersity of 0.107 and zeta potential of −54.68±1.78 mV (DLS results).This strongly negative zeta potential makes the colloid solution of nEOAs very stable. Because tetraaniline locates in the core of nEOAs and the self-assembly of EOA will lead to the shielding of signals from tetraaniline, we used 1H NMR to monitor the signal changes of EOA solution with time to evaluate their self-assembly rate. The 1H NMR spectra (Figure 2B) show that, in the freshly prepared EOA solution, proton signals from tetraaniline at 6.88, 7.00, 7.26, 7.50 ppm (multiplet) for 17 hydrogens in benzene ring, and at 7.85 ppm (singlet) for 1 hydrogen in secondary amine appear, but no more than 30 minutes later, all these signals disappear, indicating the finish of self-assembly of EOA. Therefore, it is suggested that when preparing EOA solution for hydrogel preparation, it should be incubated for at least 30 minutes to ensure the formation of nanoparticles.
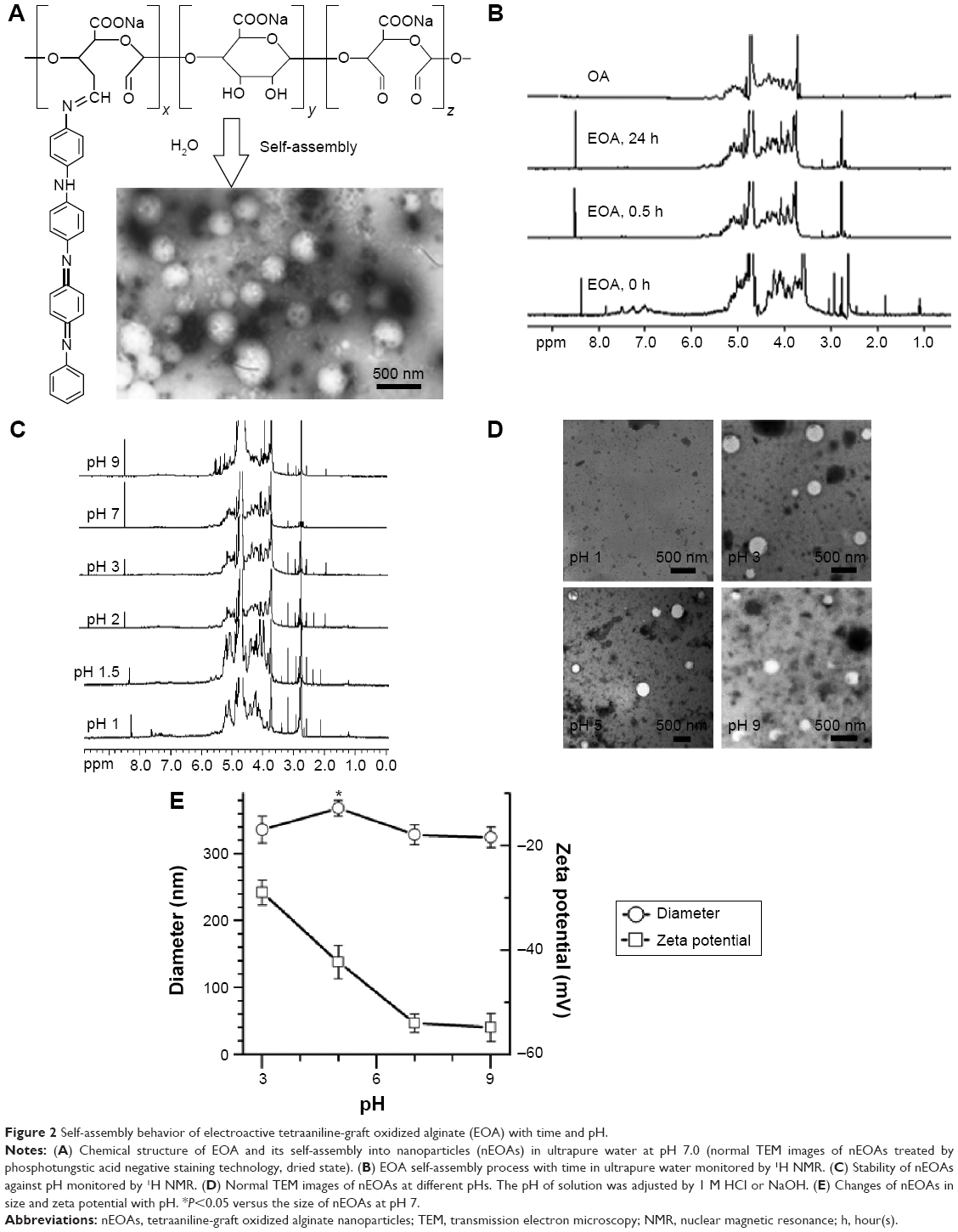 | Figure 2 Self-assembly behavior of electroactive tetraaniline-graft oxidized alginate (EOA) with time and pH. |
Due to different pH environments in vivo, we further investigated the stability of nEOAs at different pHs by using TEM, 1H NMR, and DLS. In 1H NMR spectra of EOA solutions (Figure 2C), the signals from tetraaniline at 6.88, 7.00, 7.26, 7.50, and 7.85 ppm disappear at pH >2, suggesting the self-assembly of EOA as discussed above. TEM images (Figure 2D) further evidenced this. In our examined pH, EOA exists in the form of nanoparticles at pH 3, 5, 7, 9, suggesting their high stability against pH, which is very important for them to keep their structure in vivo for some time to function even with pH change. Finally, the changes in diameter and zeta potential of nEOAs with pH were analyzed by DLS (Figure 2E). With the increase of pH, the zeta potential of nEOAs decreases from ~−29 mV to ~−54 mV, which is due to the gradual deprotonation of carboxyls in OA (pKa ~4.0). But surprisingly, with the increase of pH, a small change in diameter takes place. For example, the diameter of nEOAs is 335.9 nm at pH 3, then slightly increases to the maximum of 368.1 nm at pH 5, and decreases to 324.5 nm at pH 9 (polydispersity for all samples <0.2). This phenomenon may be caused by two factors. The first one is the deprotonation of carboxyls of OA in the shell with pH increase. Such deprotonation drives the increase in electrostatic repulsion and the decrease in hydrogen bonding interactions between OA chains in the shell, and both lead to the afflux of water and thus the swelling of nanoparticles. However, because the thickness of the shell is only ~10% of the nanoparticle,19 the change in shell size due to the increase of pH may be limited. The other size-dependent factor is the deprotonation of tetraaniline. This deprotonation drives the increase in hydrophobic and hydrogen bonding interactions between tetraaniline, which compresses the tetraaniline core to a lesser size. However, the change in size is not large probably due to the conjugation structure of four benzene rings. Although nEOA is pH-sensitive, that is the shell/core structure changes with pH variation, the joint influences of these opposite driving forces on nanoparticles keep their size relatively stable. This property may be especially beneficial for in vivo applications because the relatively stable size may assure the minimal disturbance to ambient tissue. Additionally, with dual (pH and electrical) sensitivity, nEOAs may have extra superiority in drug release. For example, in addition to a drug delivery system based on electrical stimulation, they might be used as a generalized pH-sensitive drug delivery system such as a conducting polypyrrole nanoparticle system developed by Samanta et al,28 which can release any charged drug preferentially at both acidic and basic pH by varying the pH, the charge of the drug and by adding small amounts of charged amphiphiles. To date, little attention has been paid to the pH sensitivity of conducting polymer nanoparticles. Therefore, the application of nEOAs in drug delivery will be explored in our future work.
Synthesis of eGel
The eGel was prepared by using the mixture of OA and nEOAs to crosslink gelatin (Figure 1A). Briefly, in our eGels, there are two kinds of crosslinking: one is chemical crosslinking, which is realized by Schiff’s base formation between the ε-amino groups of lysine or hydroxylysine side groups in gelatin and the aldehyde groups in OA/nEOAs; the other is the physical crosslinking, which is realized by the borate complexation with diols in OA/nEOAs and by the hydrogen bonding interactions between carboxyls, amine groups, hydroxyls, and amides in EOA/OA/gelatin. The details of aforementioned crosslinking mechanism were reported in the publications of Balakrishnan et al.9,23,29 In this study, (nEOAs + OA) at a concentration of 0.20 g/mL in 0.1 M borax and gelatin at a concentration of 0.15 g/mL in water were mixed to get hydrogels, and the feeding component was presented in Table 1. Such hydrogel system was chosen because Balakrishnan et al demonstrated that the hydrogel made from OA in 0.1 M borax + gelatin in water had good in vitro and in vivo biocompatibility and was suitable for cartilage regeneration, drug release, and wound dressing as an injectable adhesive biomimetic scaffold.9,23,29–30 To further improve its properties for new and wider clinical applications, we integrated electroactive degradable conducting polymer nanoparticle of nEOAs within this hydrogel. As presented in Table 1, adding nEOAs can easily adjust the gel time of hydrogels: the higher the weight content of nEOA in eGels, the longer the gel time. For pure OA, the gel time is 30 seconds, but for pure nEOAs, the gel time is longer than 8 hours (data not shown). Obviously, the lengthy gel time is not suitable for clinical applications. The long gel time of nEOAs may be attributed to the following reasons. One is the decrease in available aldehyde groups (-CHO) for crosslinking reactions. For example, on EOA, although those –CHO consumed for tetraaniline graft were only 25.5% of the original –CHO on OA, calculated by the following equation:
| (2) |
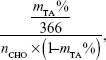
where mTA% is the mass content of grafted tetraaniline in EOA, 366 is the Mw of tetraaniline, and nCHO is the moles of aldehyde group per gram of OA, those –CHO available for further chemical reactions were only 29.8% of all the remaining –CHO and 22.0% of the original –CHO on OA respectively (determined by hydroxylamine hydrochloride method). The other reason is that polyaniline is a radical scavenger, which may interfere with the crosslinking reaction to some extent.3,13,14 These results suggest that eGels with different nEOA content may meet different demands for clinical applications as an injectable hydrogel.
Morphology of eGels
Scanning electron microscopy (SEM) images show that all eGels have a uniform and porous interior structure, and pores are interconnected (Figure 3). The pore size is dependent on the concentration of nEOAs in hydrogels. When the mass percentage of nEOAs in hydrogels is 0% (eGel-0), 7% (eGel-7) or 21% (eGel-21), the average pore size is ~30, ~20, <10 μm, respectively. This may be ascribed to dual crosslinkers of nEOA and OA. On the outer surface of nEOA, there are large reactive aldehyde groups which can “catch” large gelatin molecules, and thus the nanoparticle-crosslinked gelatin forms one network. Similarly, OA-crosslinked gelatin forms the other network. These two networks interpenetrate each other and are tangled due to the chemical linking between aldehyde in nEOA/OA and amine groups in gelatin and hydrogen bonding interactions among nEOA/OA/gelatin, and thus a stronger and more compact hydrogel is formed compared with pure EOA-crosslinked gelatin. Furthermore, the higher the concentration of nEOAs, the more compact the network forms. This more compact network may be beneficial to improve the mechanical properties and conductivity. It should be noted that although nEOAs were used as a crosslinker, one cannot observe a phase separation between nanoparticles and the network in SEM images of hydrogels, indicating the excellent dispersibility and the perfect integration of nEOAs in hydrogels, which favors the improvement of mechanical properties.2,31
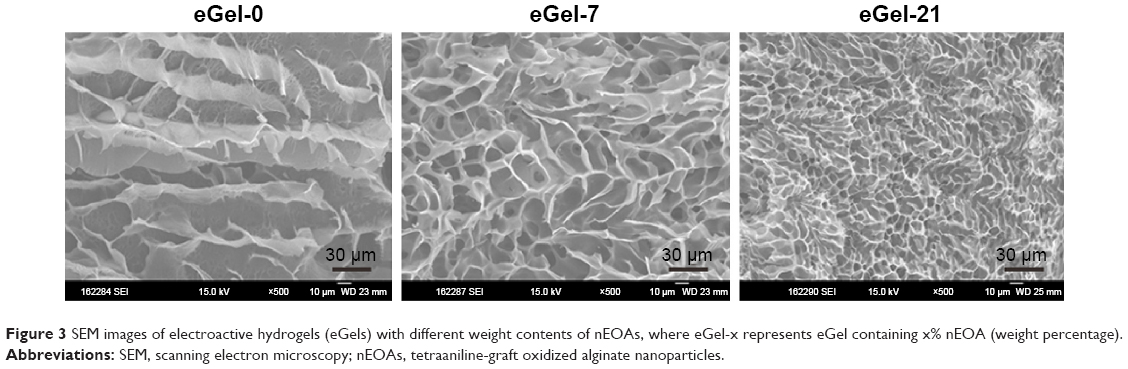 | Figure 3 SEM images of electroactive hydrogels (eGels) with different weight contents of nEOAs, where eGel-x represents eGel containing x% nEOA (weight percentage). |
Rheology analysis of eGels
The mechanical properties and formation process of eGels were analyzed by using a rheometer. To confirm the hydrogel formation and determine the gel time, we first performed the time-sweep-rheology spectra (Figure 4A, the inset is a partially magnified view) by using eGel-7 as a model material. The time point where the storage modulus (G′) is equal to the loss modulus (G′′) is defined as the gel time of hydrogel. And at this time point, the hydrogel forms. It can be seen that both G′ and G′′ increase with time. However, the rising speed of G′ is much higher than that of G′′: G′ rapidly increases to 377 Pa, while G′′ slowly rises to 18 Pa in 900 seconds. At 75 seconds, G′ equals to G′′ (inset in Figure 4A). Therefore, the gel time of eGel-2.5 is 75 seconds. After this gel time point, G′ exceeds G′′, denoting the elastic portion (solid response) is predominant in the material, and the crosslinking reaction is still going on. It should be noted that gel time from rheology analysis is longer than that from the stir bar method (45 seconds, Table 1). Such discrepancy is caused by the different mixing manners in two methods: nearly static in the former and continuous magnetic stirring in the latter. Figure 4B presents the strain-sweep-rheology spectra of eGel-7. When the strain is less than 30%, eGel-7 behaves the viscoelasticity. For this reason, frequency sweep was carried out at 10% strain (Figure 4C). For each eGel, G′ (the index of material stiffness) is higher than G′′, which is a typical gel response. In our studied range, G′ increases with the weight content of nEOAs, confirming that nEOAs can improve the mechanical strength of eGels. When the weight content of nEOAs is 0 (eGel-0), G′ is ~924 Pa, and this value is comparable with the reported value of the same system.9 When nEOA content increases to 7% (eGel-7) and 21% (eGel-21), G′ is 2.1- and 8.7-fold higher than that of eGel-0, respectively. The difference among G′ of three samples is statistically significant (P<0.01, one-way ANOVA). Such increase may be attributed to the augmentation in crosslinking density due to chemical crosslinking of nEOAs with gelatin and the reinforcement function of nEOAs.2,18,32 The increase in crosslinking density is understandable because the extra nanoparticle crosslinker of nEOAs besides OA is added. For the reinforcement from nanoparticles, there are two key factors determining their reinforcing effects, their distribution in the matrix and interfacial adhesion.2,31 Theoretically, nEOAs have potential to behave excellently in the above two aspects. Firstly, nEOA has highly hydrophilic anionic OA as shell with zeta potential of ~−54 mV, which endows it with excellent dispersibility and stability (no aggregation) in the precursor solution of hydrogels as discussed above. Thus the precursor solution composed of OA, nEOAs, and gelatin is a homogeneous solution, ensuring the uniform structure of subsequently formed hydrogels and the good distribution of nEOAs in hydrogels. This uniform structure is confirmed in SEM images of eGels (Figure 3), where the aggregation of nEOAs cannot be found. Secondly, nEOAs obviously have strong interfacial adhesion with OA-crosslink-gelatin hydrogel matrix because in OA shell, there are large aldehyde groups for chemical crosslinking, and large carboxyl and hydroxyl groups for physical crosslinking such as hydrogen bonding interactions with gelatin/OA. Additionally, adding nEOAs produces a more integrated and compact hydrogel network (Figure 3), which is beneficial for increasing the mechanical strength. Taken together, nEOAs can controllably improve the mechanical strength by nano-reinforcement, and the range of mechanical strength is comparable to most soft tissues,33 indicating their potential applications in soft tissue regeneration and drug delivery.
 | Figure 4 Rheology behavior of electroactive hydrogels (eGels). |
Electrochemical properties and conductivity of hydrogels
We performed three experiments to evaluate the electroactivity of eGels. First, a doping/dedoping test was carried out. As shown in Figure 1B, normally prepared eGel-7 is black. However, after alternately immersing eGel-7 in 1 M HCl or NaOH, its color repeatedly reversibly changes from green to blue (Figure 5A), indicating its reversible transition from electroactive form (green) to insulative form (blue), confirming the hydrogel has the reversible doping/dedoping ability. The electrochemistry of hydrogel in 1 mol·L−1 HCl was further studied by cyclic voltammetry. As shown in Figure 5B, for pure OA-crosslinked hydrogel (eGel-0), no redox peaks appear; while for nEOA/OA-crosslinked hydrogel (eGel-7), one pair of redox peaks appears, indicating its reversibility of redox and the effective charge transportation through the crosslinked hydrogel. The redox peaks of eGel-7 are assigned to the conversion between leucoemeraldine state and emeraldine state of the conjugated tetraaniline, and its mean redox potential is 0.49 V, similar to the nEOA solution previously reported by our group,19 suggesting that integrated nEOAs contribute to the electroactivity of hydrogel. Finally, the conductivity of dried eGels with different nEOA contents doped by 1 mol/L HCl was determined by the standard method of four probes, and the results are presented in Table 1. With nEOA content increasing, the conductivity of eGel increases. This increase can be easily understood because higher nEOA content means higher tetraaniline content, which supplies more electrons and a better, more compact network structure for conduction. The conductivity of hydrogel without nEOAs (eGel-0) is ~5.66×10−8 S/cm, which is ascribed to ionic conductivity such as large carboxylic anions, amine cations, and boron cations in hydrogels, and is comparable to glutaraldehyde-crosslink-chitosan polysaccharide hydrogel (~3.13×10−8 S/cm).34 The conductivity of hydrogels with nEOA weight content from 7% to 21% was in the range of 6.37×10−7 and 7.52×10−6 S/cm, which has a similar magnitude to the other degradable conducting hydrogels containing conducting oligomers such as aniline tetramer and pentamer.12,16,34 But obtaining the same conductivity needs lower tetraaniline content in our system than in other similar systems, which may be caused by the aggregation of tetraaniline in nEOAs. Compared with pure tetraaniline which has a conductivity of ~10−3 s·cm−1, our system has much lower conductivity. This decrease in conductivity of eGel is reasonable due to the introduction of gelatin and OA with less conductivity. In eGel, both electrical conductivity from tetraaniline and ionic conductivity contribute to the conductivity of hydrogels, but the former is predominant. All of these results suggest that eGel has adjustable electroactivity, and their conductivity range may be exactly sufficient for in vivo applications due to the microcurrent characteristics.12,15,35
 | Figure 5 Electrochemical properties of eGel-14. |
Fluid uptake capacity of eGels
The fluid uptake affects hydrogel properties such as mechanical properties and volume. In general, excessive fluid uptake is not expected because it will lead to poor mechanical strength and large volume expansion (excessive swelling). The excessive swelling may induce compression on surrounding tissues, which will bring about pain and other harmful effects.4 We evaluated the fluid uptake capacity of eGels by incubating them in PBS at 37°C. All eGels can absorb and hold water, and the equilibrium is reached within 24 hours (Figure 6A). Table 1 shows that in our studied concentration range, at equilibrium, introducing nEOA slightly decreases the fluid uptake of eGels, and this decrease is enhanced with nEOA content in eGel. Such change may be ascribed to the opposite driving forces for swelling. The increase in driving force limits expansion due to higher crosslinking density in hydrogel via additional crosslinking of nEOAs with gelatin. As shown in Figure 2B, higher nEOA content in eGels causes higher crosslinking density and smaller pore size, leading to a decrease in space for water to enter. The other increase in driving force for expansion (water uptake) is due to higher electrostatic repulsion between carboxylic anions from added nEOAs. The synergic actions of the two factors lead to the slight decrease in water uptake capacity of eGels with increasing nEOA content. At equilibrium, the water content in eGel is in the range of 88%~91%, which is slightly higher than the original (~85%). Such a degree of swelling may be suitable for in vivo applications, ensuring that hydrogels tightly adhere to the surrounding tissues without over-squeezing them.
 | Figure 6 Behavior of electroactive hydrogels (eGels) in PBS. |
In vitro degradation of eGels
One of the drawbacks of traditional conductive polymers such as polyaniline is their non-degradability, which brings the risk of chronic inflammation because they remain in the body for a long period. It is worth noting that we have already proved that nEOAs in PBS can be degraded, and that their degradation rate decreases with the increase of the grafted tetraaniline weight content in EOA,19 and Balakrishnan and Jayakrishnan have demonstrated that OA crosslinking gelatin is degradable.23 Therefore, we suspect that eGels were degradable, and their in vitro degradation in PBS was studied by monitoring their weight change with time. Figure 6B shows that the degradation rate of eGel depends on the nEOA content in hydrogel: the higher the EOA content, the lower the degradation rate. The degradation pattern agrees with the fluid uptake capacity and mechanical strength of eGels, namely, high mechanical strength and less water uptake lead to slower degradation. Furthermore, eGels disappear at weight ratio (Wt/W0) of ~35%, suggesting that eGels are hydrolyzed through both the surface and interior due to the large amount of water in eGels. Taken together, eGel is degradable, and its degradation rate can be controlled by changing the nEOA concentration in hydrogels for different applications. Furthermore, the hydrolytic susceptibility of imine bonds between gelatin and OA/nEOAs and the degradability of gelatin/OA/nEOAs would enable the eGels to be fully degraded into tetraaniline, low-MW polysaccharides and polypeptides, and finally all these degradation products can be removed from the body via metabolism.12
Cell compatibility of eGels
We chose MSCs as model cells for cytocompatibility evaluation, and the results will be extremely valuable for future clinical applications of test materials because as a stem cell, MSCs are sensitive to toxicity, and they have been widely applied in cell therapy. We first used alamarBlue assay to evaluate the growth of MSCs on eGels. As presented in Figure 7A, cells on tissue culture polystyrene, eGel-7, and eGel-14 have a similar lag and log phase of growth, suggesting they are nontoxic to MSCs and do not affect their growth. By contrast, cells on eGel-0 exhibit a slightly slower growth probably due to its relatively weak mechanical strength, and cells on eGel-21 are markedly less than the other groups presumably because of the undesirable side effects (such as toxicity) of large amounts of tetraaniline in the gel. Such phenomenon is observed in reported biomaterials containing aniline oligomers. For example, Baheiraei et al demonstrated that blending of biodegradable electroactive polyurethane containing AP and polycaprolactone had good cell compatibility and could support cell growth similar to tissue culture plates; however, the blending polymer with lower AP content gave better results.10 Liu et al synthesized a biodegradable eGel based on AP-g-GA crosslinked by EDC, and they cited that although electrical conductivity could enhance the cell proliferation, increasing AP content from 1.9% to 10.5% increased the cytotoxicity of AP-g-GA and decreased cell proliferation on hydrogels, which was ascribed to the toxicity of high content of AP.16 Hu et al evidenced that for AP crosslinking chitosan, polymers with AP weight content of 4.9% had the most significant improvement in proliferation and differentiation of PC-12 cells compared to those with AP content of 2.4% and 9.5%.11 These results suggest that tetraaniline content is a key influential factor in biocompatibility of tetraaniline containing biomaterials, and the optimal value should be carefully determined via experiments for specific biomedical applications. The growth of cells on different eGels was further assessed by 4′,6-diamidino-2-phenylindole staining of cell nucleus, and the results are in accordance with alamarBlue assay. Here we only present images of eGel-7 and eGel-14 groups as examples (Figure 7B). For eGel-7 group, cell number increases with incubation: on the fourth day, cells approach ~100% confluence, and on the fifth day, cells are very crowded; for eGel-14 group, it has similar cell density to eGel-7; and for eGel-21, the growth of MSCs is inhibited 3 days later (data not shown), which may be caused by the exposure of tetraaniline due to hydrogel degradation. With high nEOA content in eGel-21, more tetraaniline is released compared to eGel-14 and eGel-7, leading to the different cell response. Additionally, we observed the morphology and cytoskeletal architecture of MSCs on different substrates via rhodamine-phalloidin fluorescent staining on the fourth day (Figure 7C). It can be seen that MSCs spread well on tissue culture polystyrene, eGel-0, eGel-7, and eGel-14, no obvious difference in morphology has been discovered, and they all exhibit a typical spindle-like appearance. All these data suggest that eGel-7 and eGel-14 have good cell compatibility.
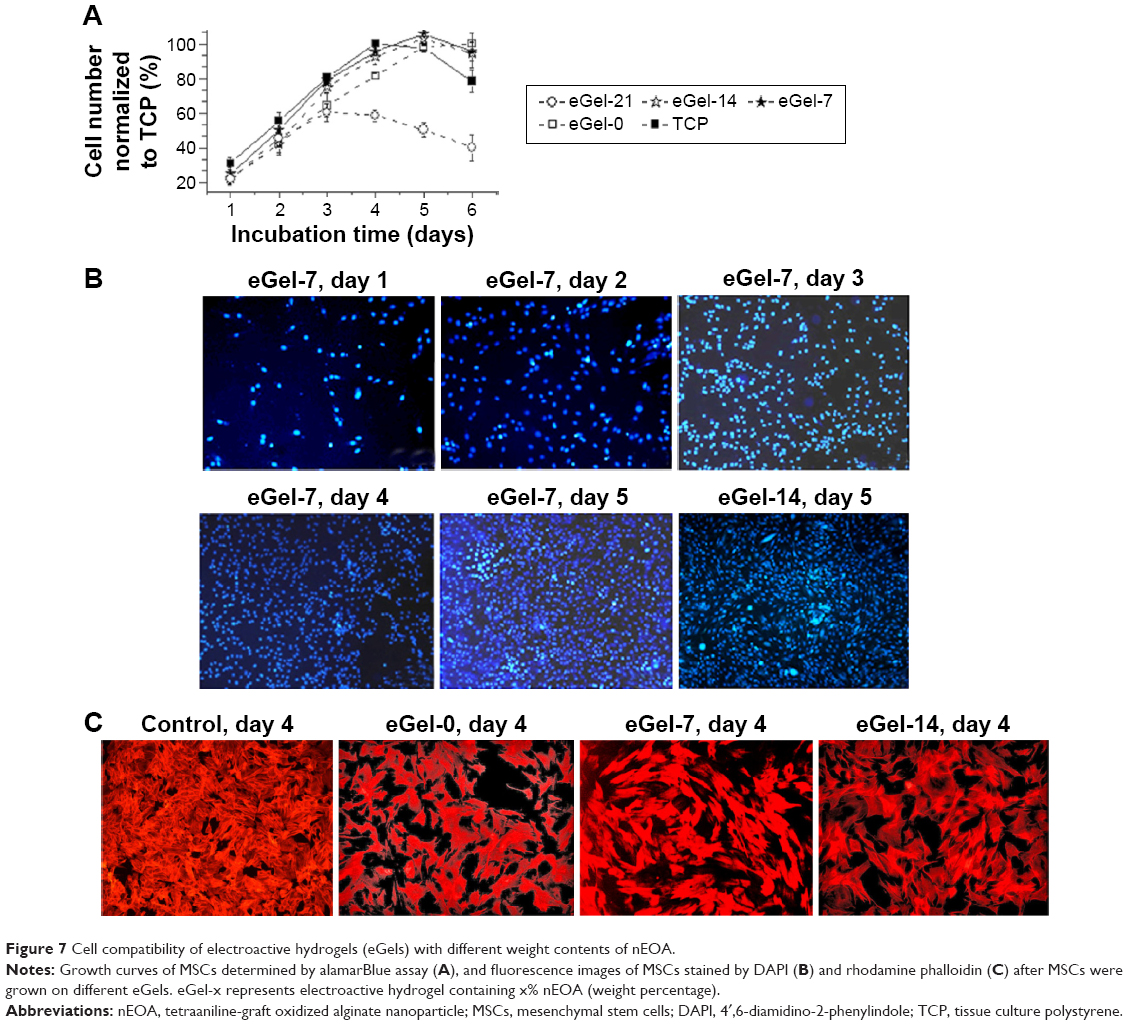 | Figure 7 Cell compatibility of electroactive hydrogels (eGels) with different weight contents of nEOA. |
In vivo tissue response to hydrogels
To explore the in vivo compatibility of eGels, the tissue response to them was assessed. We used CAM as an in vivo model because such a model has a similar tissue response as mammalian models, but it is low cost, simple, and rapid.26,27 Furthermore, this in vivo model allows a continuous visual inspection of the test site through the shell window. Therefore, CAM model has been extensively used to test the biocompatibility of biomaterials for initial screening for a more complex mammalian model. CAM is composed of an ectoderm constituted by a multilayer of epitheliums, a mesodermal stroma, and an endoderm constituted by a single layer of epitheliums. Basically, CAM is sensitive to toxic materials, and toxic materials often induce inflammation and scarring.26,27 In our case, after adding eGels on CAM surfaces, no embryos died during the 1-week incubation. Compared with the control (saline-treated group), all eGel groups at EOA content ≤14% have integrated and clear blood vessel systems without obvious bleeding and tissue damage at the gross level (Figure 8A, B), and none of these groups caused any other structural changes in CAMs except mild inflammation with the histological evaluation (H and E staining) (Figure 8C). Detailedly, as shown in Figure 8C, all eGel groups have loose stroma and normal ecto/endoderm similar to the control, while their blood vessels are mildly dilated and congested with inflammatory cells (blue stained nucleus) and a few inflammatory cells migrated into the stroma, indicating a mild tissue response induced by these eGels. In contrast, eGel-21 induces moderate tissue response such as ectoderm thickening and local aggregation of high density inflammatory cells in its histological sections (data not shown). These results are consistent with the cell experiment, that is, the increase of nEOA (namely tetraaniline) content decreases the biocompatibility of eGels. Taken together, the in vitro and in vivo experiments demonstrate that eGel-0, eGel-7 and eGel-14 have good compatibility in CAM, which is due to the intrinsic biocompatibility of alginate and gelatin with biological cues for cell adhesion and growth, and encapsulated tetraaniline in alginate without large direct exposure to cells or tissues. Further in vivo experiments of eGels in a mammalian model will be explored in our laboratory.
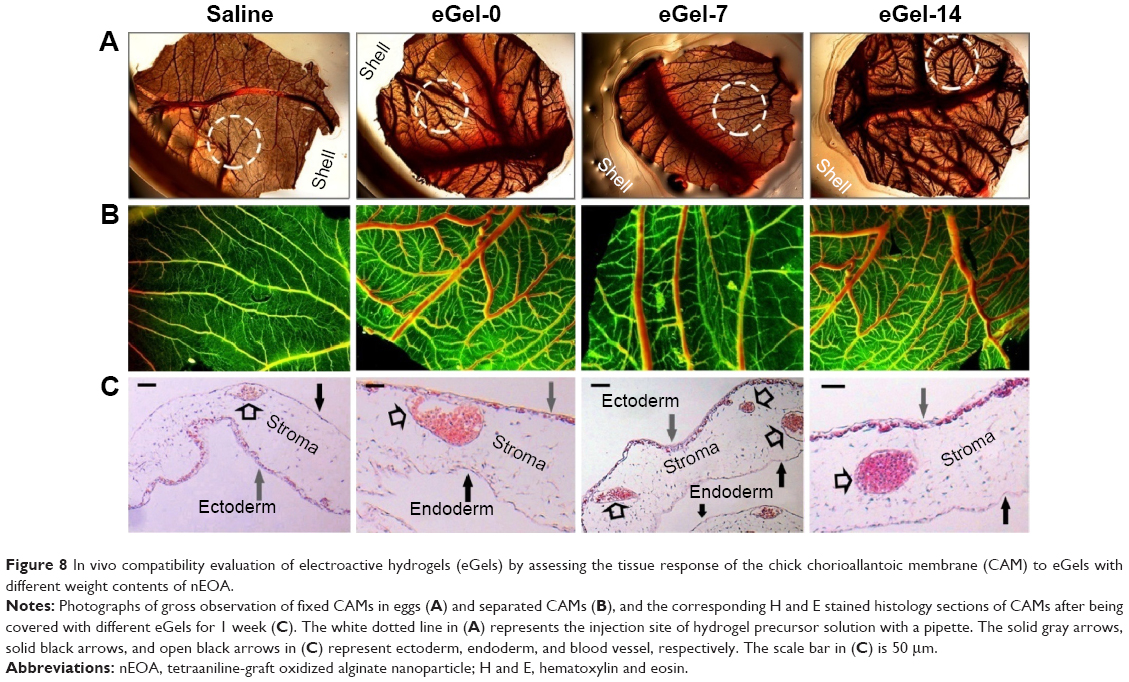 | Figure 8 In vivo compatibility evaluation of electroactive hydrogels (eGels) by assessing the tissue response of the chick chorioallantoic membrane (CAM) to eGels with different weight contents of nEOA. |
Conclusion
We have developed a new way to prepare an injectable, electroactive, and robust hydrogel by incorporating degradable conductive polymer nanoparticles with high hydrophilicity and reactivity into hydrogel matrix. This nanoparticle can be facilely prepared from the self-assembly of the amphiphilic brush copolymer synthesized by grafting conducting tetraaniline (side chain) onto degradable OA (main chain). With highly hydrophilic OA as shell, nEOAs have large aldehyde and carboxylic groups on their surfaces, which endow nEOAs with well distributed and strong interface adhesion within hydrogel matrix of OA crosslinking gelatin and thus reinforce the hydrogel. With tetraaniline as core, nEOAs impart electroactivity to the hydrogel. Furthermore, by adjusting the weight content of nEOAs, the mechanical strength, swelling, electroactivity, degradability, and biocompatibility of hydrogels can be controlled to some extent. With dual (pH and electricity) stimuli-responsive nature, these nanoparticle-reinforced hydrogels may find extensive applications in tissue engineering and drug delivery. Future work will focus on the stimuli-responsive behaviors of hydrogels and their biomedical applications.
Acknowledgments
We would like to thank Professor Huilan Rao from Cancer Center, Sun Yat-sen University, People’s Republic of China for her help in histological analysis, Mr Ruoyao Zhou from Okemos High School, USA for his help in English checking of this manuscript, and Professor Wenge Yao from Guangdong University of Finance, People’s Republic of China for her help in statistical analysis. This work was supported by the National Science Foundation of China (81101153), and the Science and Technology Plan Funds of Guangdong (2009B030801117, 2010B050700012, 2013B051000017).
Disclosure
The authors report no conflicts of interest in this work.
References
Cui H, Liu Y, Cheng Y, et al. In vitro study of electroactive Tetraaniline-Containing thermosensitive hydrogels for cardiac tissue engineering. Biomacromolecules. 2014;15(4):1115–1123. | ||
Gaharwar AK, Peppas NA, Khademhosseini A. Nanocomposite hydrogels for biomedical applications. Biotechnol Bioeng. 2014;111(3):441–453. | ||
Li L, Ge JJ, Guo B, et al. In situ forming biodegradable electroactive hydrogels. Polym Chem. 2014;5:2880–2890. | ||
Liu Y, Meng H, Konst S, et al. Injectable dopamine-modified poly(ethylene glycol) nanocomposite hydrogel with enhanced adhesive property and bioactivity. ACS Appl Mater Interfaces. 2014;6(19):16982–16992. | ||
Wu Y, Guo B, Ma PX. Injectable Electroactive Hydrogels Formed via Host-Guest Interactions. ACS Macro Lett. 2014;3(11):1145–1150. | ||
Cui H, Shao J, Wang Y, et al. PLA-PEG-PLA and its electroactive tetraaniline copolymer as multi-interactive injectable hydrogels for tissue engineering. Biomacromolecules. 2013;14(6):1904–1912. | ||
Mihardja SS, Sievers RE, Lee RJ. The effect of polypyrrole on arteriogenesis in an acute rat infarct model. Biomaterials. 2008;29(31):4205–4210. | ||
Radhakrishnan J, Krishnan UM, Sethuraman S. Hydrogel based injectable scaffolds for cardiac tissue regeneration. Biotechnol Adv. 2014;32(2):449–461. | ||
Balakrishnan B, Joshi N, Jayakrishnan A, Banerjee R. Self-crosslinked oxidized alginate/gelatin hydrogel as injectable, adhesive biomimetic scaffolds for cartilage regeneration. Acta Biomater. 2014;10(8):3650–3663. | ||
Baheiraei N, Yeganeh H, Ai J, et al. Synthesis, characterization and antioxidant activity of a novel electroactive and biodegradable polyurethane for cardiac tissue engineering application. Mater Sci Eng C Mater Biol Appl. 2014;44:24–37. | ||
Hu J, Huang L, Zhuang X, et al. Electroactive aniline pentamer Cross-Linking chitosan for stimulation growth of electrically sensitive cells. Biomacromolecules. 2008;9(10):2637–2644. | ||
Guo B, Glavas L, Albertsson A. Biodegradable and electrically conducting polymers for biomedical applications. Progress in Polymer Science. 2013;38(9):1263–1286. | ||
Cui H, Cui L, Zhang P, et al. In situ electroactive and antioxidant supramolecular hydrogel based on Cyclodextrin/Copolymer inclusion for tissue engineering repair. Macromol Biosci. 2014;14(3):440–450. | ||
Saikia JP, Banerjee S, Konwar BK, et al. Biocompatible novel starch/polyaniline composites: characterization, anti-cytotoxicity and antioxidant activity. Colloids Surf B Biointerfaces. 2010;81(1):158–164. | ||
Baheiraei N, Yeganeh H, Ai J, et al. Preparation of a porous conductive scaffold from aniline pentamer-modified polyurethane/PCL blend for cardiac tissue engineering. J Biomed Mater Res A. 2015;103(10):3179–3187. | ||
Liu YD, Hu J, Zhuang XL, et al. Synthesis and characterization of novel biodegradable and electroactive hydrogel based on aniline oligomer and gelatin. Macromol Biosci. 2012;12(2):241–250. | ||
Gulati N, Gupta H. Two faces of carbon nanotube: toxicities and pharmaceutical applications. Crit Rev Ther Drug Carrier Syst. 2012;29(1):65–88. | ||
Schexnailder P, Schmidt G. Nanocomposite polymer hydrogels. Colloid Polym Sci. 2009;287:1–11. | ||
Wang Q, He W, Huang JQ, et al. Synthesis of water soluble, biodegradable, and electroactive polysaccharide crosslinker with aldehyde and carboxylic groups for biomedical applications. Macromol Biosci. 2011;11(3):362–372. | ||
Balakrishnan B, Lesieur S, Labarre D, Jayakrishnam A. Periodate oxidation of sodium alginate in water and in ethanol-water mixture: A comparative study. Carbohydr Res. 2005;340(7):1425–1429. | ||
Huang Z, Teng W, Liu L, et al. Efficient cytosolic delivery mediated by polymersomes facilely prepared from a degradable, amphiphilic, and amphoteric copolymer. Nanotechnology. 2013;24(26):265104. | ||
Zhao H, Heindel ND. Determination of degree of substitution of formyl groups in polyaldehyde dextran by the hydroxylamine hydrochloride method. Pharm Res. 1991;8(3):400–402. | ||
Balakrishnan B, Jayakrishnan A. Self-cross-linking biopolymers as injectable in situ forming biodegradable scaffolds. Biomaterials. 2005;26(18):3941–3951. | ||
Mo X, Iwata H, Matsuda S, Ikada Y. Soft tissue adhesive composed of modified gelatin and polysaccharides. J Biomater Sci Polym Ed. 2000;11(4):341–351. | ||
Bidez PR 3rd, Li S, Macdiarmid AG, et al. Polyaniline, an electroactive polymer, supports adhesion and proliferation of cardiac myoblasts. J Biomater Sci Polymer Ed. 2006;17(1–2):199–212. | ||
Valdes TI, Kreutzer D, Moussy F. The chick chorioallantoic membrane as a novel in vivo model for the testing of biomaterial. J Biomed Mater Res. 2002;62(2):273–282. | ||
Da-Lozzo EJ, Moledo RC, Faraco CD, et al. Curcumin/xanthan–galactomannan hydrogels: Rheological analysis and biocompatibility. Carbohydr Polym. 2013;93(1):279–284. | ||
Samanta D, Meiser JL, Zare RN. Polypyrrole nanoparticles for tunable, pH-sensitive and sustained drug release. Nanoscale. 2015;7(21):9497–9504. | ||
Balakrishnan B, Mohanty M, Umashankar PR, Jayakrishnan A. Evaluation of an in situ forming hydrogel wound dressing based on oxidized alginate and gelatin. Biomaterials. 2005;26(32):6335–6342. | ||
Balakrishnan B, Mohanty M, Fernandez AC, Mohanan PV, Jayakrishnan A. Evaluation of the effect of incorporation of dibutyryl cyclic adenosine monophosphate in an in situ-forming hydrogel wound dressing based on oxidized alginate and gelatin. Biomaterials. 2006;27(8):1355–1361. | ||
Xiao Y, He L, Che J. An effective approach for the fabrication of reinforced composite hydrogel engineered with SWNTs, polypyrrole and PEGDA hydrogel. Journal of Materials Chemistry. 2012;22(16):8076–8082. | ||
Wang C, Shen H, Tian Y, et al. Bioactive nanoparticle-gelatin composite scaffold with mechanical performance comparable to cancellous bones. ACS Appl Mater Interfaces. 2014;6(15):13061–13068. | ||
Engler AJ, Sen S, Sweeney HL, Discher DE. Matrix elasticity directs stem cell lineage specification. Cell. 2006;126(4):677–689. | ||
Guo BL, Finne-Wistrand A, Albertsson AC. Facile synthesis of degradable and electrically conductive polysaccharide hydrogels. Biomacromolecules. 2011;12(7):2601–2609. | ||
Rowlands AS, Cooper-White JJ. Directing phenotype of vascular smooth muscle cells using electrically stimulated conducting polymer. Biomaterials. 2008;29(34):4510–4520. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.