")
Back to Journals » Vascular Health and Risk Management » Volume 10
Inhibition of hepatic microsomal triglyceride transfer protein – a novel therapeutic option for treatment of homozygous familial hypercholesterolemia
Authors Vuorio A, Tikkanen MJ, Kovanen P
Received 4 December 2013
Accepted for publication 30 January 2014
Published 6 May 2014 Volume 2014:10 Pages 263—270
DOI https://doi.org/10.2147/VHRM.S36641
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Alpo Vuorio,1,2 Matti J Tikkanen,3 Petri T Kovanen4
1Health Center Mehiläinen, Vantaa, Finland; 2Finnish Institute of Occupational Health, Lappeenranta, Finland; 3Heart and Lung Center, Helsinki University Central Hospital, Folkhälsan Research Center, Biomedicum, Helsinki, Finland; 4Wihuri Research Institute, Biomedicum, Helsinki, Finland
Abstract: Familial hypercholesterolemia (FH) is an autosomal dominant disease caused by mutations in the low-density lipoprotein (LDL)-receptor gene (LDLR). Patients with homozygous FH (hoFH) have inherited a mutated LDLR gene from both parents, and therefore all their LDL-receptors are incapable of functioning normally. In hoFH, serum LDL levels often exceed 13 mmol/L and tendon and cutaneous xanthomata appear early (under 10 years of age). If untreated, this extremely severe form of hypercholesterolemia may cause death in childhood or in early adulthood. Based on recent data, it can be estimated that the prevalence of hoFH is about 1:500,000 or even 1:400,000. Until now, the treatment of hoFH has been based on high-dose statin treatment combined with LDL apheresis. Since the LDL cholesterol-lowering effect of statins is weak in this disease, and apheresis is a cumbersome treatment and not available at all centers, alternative novel pharmaceutical therapies are needed. Lomitapide is a newly introduced drug, capable of effectively decreasing serum LDL cholesterol concentration in hoFH. It inhibits the microsomal triglyceride transfer protein (MTTP). By inhibiting in hepatocytes the transfer of triglycerides into very low density lipoprotein particles, the drug blocks their assembly and secretion into the circulating blood. Since the very low density lipoprotein particles are precursors of LDL particles in the circulation, the reduced secretion of the former results in lower plasma concentration of the latter. The greatest concern in lomitapide treatment has been the increase in liver fat, which can be, however, counteracted by strictly adhering to a low-fat diet. Lomitapide is a welcome addition to the meager selection of drugs currently available for the treatment of refractory hypercholesterolemia in hoFH patients.
Keywords: microsomal triglyceride transfer protein inhibitor, familial hypercholesterolemia, LDL-cholesterol, metabolism, lomitapide
Introduction
Familial hypercholesterolemia (FH) is an autosomal dominant disease caused by mutations in the low-density lipoprotein (LDL)-receptor gene (LDLR). More than 1,200 different mutations are known in the LDLR gene.1 Some mutations cause only partial loss of function (in LDLR defective patients), and some mutations lead to a total loss of function (in LDLR negative patients). Since hepatic LDL-receptors are the main regulators of the plasma LDL-cholesterol level, the genetic loss of their function causes lifelong elevation of plasma LDL-cholesterol level.2
In heterozygous FH (heFH), one parent of the affected child is an FH-heterozygote and usually the other parent is not. In such families, every child, regardless of sex, is at risk of inheriting either a normal or mutated copy of the LDLR gene. Since one copy of the mutated gene is sufficient to cause the disease and the penetrance of the mutation is 100%, in a family, one-half of the children – on average – have the disease. In small families, all, none, or any number of the children can be heFH patients. In a heFH patient, one-half of the hepatic LDL-receptor population is mutated, and another one-half functions normally. Consequently, the plasma LDL-cholesterol level is roughly twice the level in the normal population (or in the non-FH parent).
Both parents of a hoFH child are FH heterozygotes. In such families, on average, 25% of the children are homozygotes, 50% heterozygotes, and 25% normal. The same variability in the actual numbers of these genotypes in a single family also applies here (as noted for a heFH family). The homozygotes have inherited a mutated LDLR gene from both parents, and therefore the LDL-receptor dependent hepatic clearance of circulating LDL particles is absent or near-absent. Therefore, the hepatic regulation of LDL clearance must depend on other less efficacious mechanisms, with resulting extremely high plasma LDL-cholesterol levels, which can reach levels more than five-fold that observed in the unaffected population. Interestingly, this extreme form of hypercholesterolemia is expressed already in utero, as exemplified by the observed ten-fold increase in the level of plasma LDL-cholesterol in the hoFH fetus at week 20 of gestation.3
Prevalence of hoFH
The homozygous form of familial hypercholesterolemia (hoFH) is a very rare condition, and the estimate is about one birth out of 1 million births when the prevalence of heterozygous familial hypercholesterolemia (heFH) is assumed to be one case per 500 persons.4 New data show that this traditional estimate may be an underestimate and that heFH prevalence is rather in the order of 1:200–250.5 This means that the prevalence of hoFH could actually be about 1:500,000 or even 1:400,000. There are some populations in which FH is exceptionally frequent because of a founder effect. To these populations belong South African Afrikaners,6,7 Lebanese Christians,8,9 and French Canadians,10 in which the prevalence of hoFH is estimated to be one out of 30,000, 100,000, and 275,000 births, respectively.
Phenotype and genotype of hoFH
In hoFH, serum low-density cholesterol (LDL) levels are usually more than 13 mmol/L (most commonly in the range of 15–30 mmol/L), and tendon and cutaneous xanthomata develop early (under 10 years of age).4,7,10–14 Mabuchi et al15 have reported that patients with hoFH have serum LDL-cholesterol levels about 2 times higher than those of their heterozygous parents. In the Lebanese study of an hoFH population (N=80), the LDL-cholesterol level was, on average, only 11.4 mmol/L.9
However, the majority of these Lebanese patients had been treated for years by LDL apheresis, rendering it likely that their LDL-cholesterol levels had been higher before the initiation of this mode of treatment. In the patients who received lipid-lowering therapy at the highest tolerated dose, the diagnostic level of LDL-cholesterol for hoFH is about 7 mmol/L.14 Usually, diagnosis in hoFH can be rather easily confirmed clinically, ie, by searching for the presence of xanthomata and by establishing the mode of inheritance of the hypercholesterolemia. Genetic testing is of great help not only for diagnostic purposes, but also in terms of determining the severity of the disease. As expected, it has been shown that the patients with hoFH caused by LDLR defective mutations have better prognosis compared to those who have LDLR negative mutations.11 This underlines the importance of genetic testing of hoFH patients participating in clinical treatment trials.
Considering the huge number of LDLR mutations worldwide, it is understandable that most patients with hoFH have two different LDLR mutations.16 Patients with identical mutations in both alleles are called true homozygotes, while those with different mutations are called compound heterozygotes. Double-negative true homozygosity is more severe than compound heterozygosity with one negative and one defective allele, which again is more severe than true double-defective homozygosity.
In addition to LDL-cholesterol, also lipoprotein (a) [Lp(a)] is elevated in hoFH. Kraft et al17 showed that patients with hoFH had almost two-fold higher Lp(a) levels than those with heFH. Interestingly, Lp(a) is an independent risk factor for coronary heart disease (CHD) in FH.18 Additionally, there are other genetic risk factors modifying the serum LDL-cholesterol levels and response to therapy, and ultimately the development of CHD in hoFH.19
While mutations in the LDLR gene result in FH, a similar phenotype is sometimes caused by mutations in other genes, such as apolipoprotein-B gene (apoB),20 and proprotein convertase subtilisin/kexin type 9 gene (PCSK9).21 Mutations in these three genes constitute the molecular spectrum of autosomal dominant hypercholesterolemia (ADH).21 Autosomal recessive hypercholesterolemia (ARH) results from a rare mutation in the LDL-receptor adapter protein gene (LDLRAP1).22
The extremely high cholesterol level in hoFH causes atherosclerotic changes already early in childhood and usually premature symptomatic CHD before the age of 20 years, often a combination of stenosis of the coronary ostia and of the aortic root in the supravalvular area.4,16 If untreated, death may occur in childhood. However, the rate of atherosclerosis progression in hoFH may vary from patient to patient.9 There are extreme cases of children with hoFH who suffered an acute myocardial infarction (AMI) already at a very young age. Thus, one report from Germany describes a child with hoFH who suffered an AMI at the age of 7 years, and another report describes an even younger child with hoFH who died because of an AMI at the age of 4 years.23,24 By aid of computed tomographic coronary angiography, hoFH was found to affect especially the coronary ostiae,25 but such changes may be stress test negative. In addition to premature CHD, aortic (supravalvular) stenosis is critical for the prognosis of patients with hoFH, and therefore these patients must be followed up regularly by echocardiography.26
Established modes of treatment of hoFH
Statins
Hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase inhibitor (statin) treatment is used for hoFH, either as monotherapy or as an adjuvant therapy to LDL apheresis. Early studies with lovastatin (originally called mevinolin) showed only little benefit in the treatment of hoFH.27,28 Later, when more effective statins became available, somewhat greater reductions in LDL-cholesterol concentrations have been achieved. Accordingly, such statins have been tested using the following regimens: simvastatin up to 160 mg/d in three divided doses;19 atorvastatin, 80 mg/d;29 and rosuvastatin, 80 mg/d.30 The achieved decreases in LDL-cholesterol have been: 31% with simvastatin; 28% with atorvastatin; and 26% with rosuvastatin.
Interestingly, when the daily dose of rosuvastatin was increased from 40–80 mg or that of atorvastatin from 80–120 mg/d or to 160 mg/d, no additional benefit in terms of LDL-cholesterol decrease was observed. These results contrast with those observed in non-FH and heFH patients, in whom a dose-response is a rule. This difference can be explained by the total absence or near-absence of hepatic LDL-receptors in hoFH, ie, the inhibition of hepatic cholesterol synthesis does not lead to any significant compensatory increase in functional LDL-receptors on hepatocytes. Regarding safety issues in these high-dose statin trials, it is no table that no serious adverse events occurred during simvastatin or atorvastatin treatment, and only one serious adverse effect (epistaxis) was considered to be related to the high-dose rosuvastatin treatment.
Since among the receptor-negative hoFH patients no upregulation of LDL-receptors by statin is possible, other statin-dependent mechanisms, such as inhibition of cholesterol synthesis, must be operative.19,31 In receptor-defective hoFH patients, the inhibition of cholesterol synthesis by a statin probably increases the residual activity of LDL-receptors, which together contributed to the somewhat greater, albeit minimal relative lowering of the LDL-cholesterol levels. Considering the limited LDL cholesterol-lowering efficiency of predominantly statin therapy in hoFH, it is surprising that this treatment appears to delay morbidity and increase longevity in patients with hoFH.32 Thus, the increasing longevity due to modern therapies makes it possible that a hoFH patient reaches childbearing age and becomes a parent.
Because hoFH is a life-threatening condition, statin therapy is started already very early in childhood by a pediatrician.16,33 If the reduction of LDL-cholesterol level remains small, additional lipid lowering therapies should be added.
LDL apheresis
LDL apheresis has been the cornerstone of treatment in hoFH despite the fact that it only transiently reduces plasma LDL-cholesterol levels, the values returning to preapheresis levels relatively soon (in a few days) after apheresis.34 Its use is recommended by guidelines based on lowering of not only LDL-cholesterol but also of Lp(a).35–38 In general, LDL apheresis is recommended for hoFH children over 7 years of age and adults whose serum total cholesterol remains over 9 mmol/L.39 Three recent follow-up studies in hoFH children support an early start of LDL apheresis to aggressively lower LDL-cholesterol levels.33,40,41 Importantly, this drastic lowering LDL-cholesterol achieved by LDL apheresis increases longevity and decreases morbidity in hoFH.42 According to current understanding, statin therapy should be started even earlier than LDL apheresis in hoFH children.33
Liver transplantation
The first liver transplantation was performed over 30 years ago to “cure” an hoFH child.43 The serum LDL-cholesterol of this child decreased significantly because of the normally functioning LDL-receptors in the transplanted liver, but despite close-to-normal LDL-cholesterol levels, the child died because of an allograft rejection. Since that time, the prognosis of patients with liver transplantation has remarkably improved because of new immunosuppressive medications, which help to prevent allograft rejection.44 The experience in implementing this extreme mode of treatment to cure the metabolic defect in an hoFH patient is based on only a few case reports.45–47
Inhibition of microsomal triglyceride transfer protein
Background
The microsomal triglyceride transfer protein (MTTP) is obligatory for the formation of very-low-density lipoprotein (VLDL) and chylomicron particles. VLDL is formed in hepatocytes and chylomicrons in enterocytes.48 MTTP is localized in the endoplasmic reticulum of these cells. In the rare autosomal recessive disorder abetalipoproteinemia, patients have inherited a mutated nonfunctional gene for MTTP from both parents, and, consequently, their hepatocytes and enterocytes fail to produce apoB-containing lipoproteins, ie, VLDL and chylomicrons, respectively.49,50 Patients with untreated abetalipoproteinemia suffer from gastrointestinal, neurological, ophthalmological, and hematological abnormalities. The phenotype is caused by the decreased absorption of dietary fats and fat-soluble vitamins. Heterozygous patients for the mutation in the MTTP gene (the parents of a patient with abetalipoproteinemia) do not show signs or symptoms of the condition. They have normal levels of apoB-containing lipoproteins and even a prolonged life expectancy.51
Preclinical studies on MTTP inhibition
The discovery of a mutation in the MTTP gene causing abetalipoproteinemia was essential for the initiation and ensuing development of drugs to inhibit MTTP.52–55 In 1998, an MTTP inhibitor (compound 9) was tested in homozygous Watanabe-heritable hyperlipidemic (WHHL) rabbits,56 an animal model of human hoFH,57 and was shown to normalize plasma lipoprotein levels without alterations in liver transaminase levels. Some years later, WHHL rabbits were treated by the MTTP inhibitor for 4 weeks, using doses that ranged from 3–12 mg/kg.58 The study showed that treatment with such an inhibitor decreases plasma cholesterol and triglyceride concentrations, while in the liver the triglyceride content increased and the vitamin E content decreased.
Clinical trial in patients with moderate hypercholesterolemia
Lomitapide (AEGR-733 and BMS-201038, Aegerion Pharmaceuticals, Inc., Cambridge, MA, USA) is the first MTTP inhibitor developed beyond Phase II. It is taken orally and has been tested in a placebo-controlled 12-week Phase II study among 84 patients with moderate hypercholesterolemia.59 Monotherapy with 5 mg/d, 7.7 mg/d, and 10 mg/d of lomitapide led to dose-dependent decreases in serum LDL-cholesterol by 19%, 26%, and 30%, respectively. Combination therapy with ezetimibe further increased the responses. The efficacy of combining ezetimibe in the regimen is based on the ability of ezetimibe to inhibit the Niemann-Pick C1-like 1 (NPC1L1) lipid transporter, which mediates cholesterol uptake from the intestine. It also facilitates reuptake of cholesterol from biliary canaliculi and so augments cholesterol accumulation in hepatocytes.60 Thus, ezetimibe has the potential to inhibit this reuptake process, and at least, in theory, to inhibit cholesterol accumulation in the liver.
Nine out of 28 patients in the lomitapide monotherapy group and four out of 28 patients in the lomitapide–ezetimibe combination therapy group discontinued the trial because of adverse effects, mainly elevated liver enzyme levels and gastrointestinal symptoms. Liver transaminase elevations were reversible, and none of the patients had elevations in bilirubin levels. Lomitapide caused up to a 9% decrease of high-density lipoprotein (HDL) cholesterol from the baseline. This may have been due to reduced fat intake during the trial. The authors suggested that lomitapide had reduced apolipoprotein A-1 (apoA-1) secretion from the liver and intestine, or both. It is also known that patients with abetalipoproteinemia have an increased apoA-1 catabolism.61 In this trial, serum triglycerides remained unchanged, possibly because of the low dose of lomitapide chosen to minimize intestinal and liver fat accumulation. In addition, the diet contained less than 20% fat, which probably explains why gastrointestinal adverse effects were quite rare.
Clinical trials in patients with hoFH
The favorable results in WHHL rabbits and the known devastating prognosis of untreated hoFH, together encouraged clinical scientists to carry out lomitapide studies in patients with hoFH. The benefit–risk ratio of MTTP inhibition was assumed to be in these high-risk patients more favorable than in any other group of hypercholesterolemic patients.62
The first human study with lomitapide was an open-label dose-escalation trial (Table 1) carried out in six patients (three men and three women) with hoFH.63 Genetic analysis confirmed that five of the patients had a receptor-negative form of hoFH, and one was receptor-defective. The patients were advised to consume a diet containing less than 10% of energy from total dietary fat. For the evaluation of hepatic fat content, MRI of the liver was carried out at baseline, 4 weeks after each dose increment and 4 weeks after drug withdrawal. A kinetic study of apoB-containing lipoproteins was carried out in three patients.
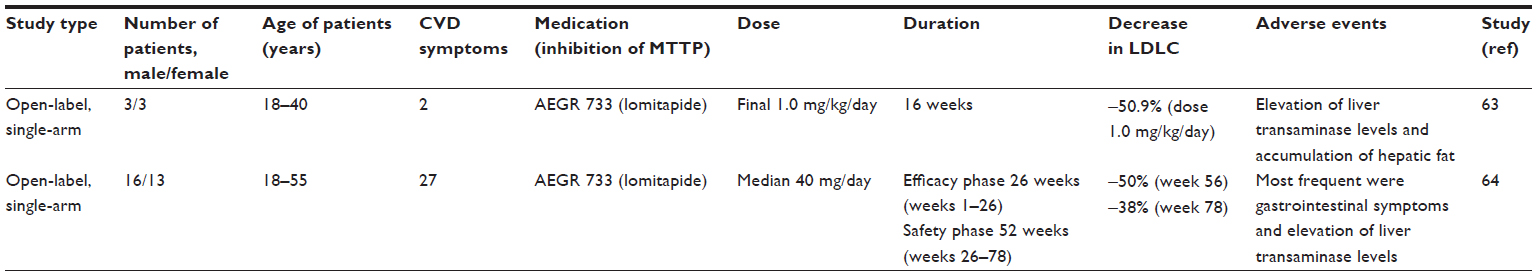 | Table 1 Trials of cholesterol lowering with lomitapide in homozygous familial hypercholesterolemia |
The patients received lomitapide at four different doses for 4 weeks, the mean doses administered being 2.0, 6.7, 20.1, and 67.0 mg/d, corresponding to 0.03, 0.1, 0.3, and 1.0 mg per kg body weight per day. At the highest dose used, significant decreases were observed in serum LDL cholesterol, apoB, and triglycerides (51%, 56%, and 65%, respectively; P<0.001 for all). In contrast, the concentrations of serum HDL cholesterol, apoA-I or Lp(a) did not change. The lack of lowering of Lp(a) despite LDL cholesterol reduction is unknown. Kinetic studies indicated that treatment for 4 weeks with the highest dose of lomitapide had decreased the rate of production LDL apoB by about 70% from the baseline.
Five of six study participants reported gastrointestinal adverse effects. By the end of the 4-week period with the largest lomitapide dose, a trend toward decrease in body weight was observed. Increases in liver transaminases occurred in four of the six patients, and the changes were dose-dependent in some patients. Most importantly, liver fat was increased substantially in four of the six patients. Two patients did not have elevations of transaminases and their hepatic fat content increased less than 10%. Both transaminase levels and liver fat content returned to baseline levels within 4 weeks after cessation of the treatment in all but one patient in whom it took 14 weeks.
Also, 29 men and women with hoFH, aged 18–55 years, participated in a multicenter trial carried out in the USA, Canada, South Africa, and Italy (Table 1).64 The diagnosis of hoFH was based either on clinical criteria or genetic analysis. The patients were treated with increasing doses (5–60 mg/d) of lomitapide, the maximum dose depending on tolerability. The average daily dose achieved with lomitapide was 40 mg. Also, 23 out of 29 patients completed the whole study including both the efficacy phase (until week 26) and safety phase (until week 78). The primary end point was the change of serum LDL-cholesterol from baseline at the end of 26 weeks. Serum LDL-cholesterol was reduced by 50% (P<0.0001) from baseline at 26 weeks. The reduction in LDL attributable to the drug was similar, whether or not they were on apheresis. At the end of week 78, the mean concentration of serum LDL-cholesterol was 38% lower than at baseline (P<0.0001). These changes may have been influenced by reductions in the daily dose of lomitapide, which was necessary because of the elevated levels of transaminases in some patients.
Gastrointestinal symptoms were the most frequent adverse effects in both hoFH trials. In the long-term study, adverse gastrointestinal effects were very frequent (93%) and resulted in discontinuation of three patients. In addition, four patients had serum transaminase levels over five times the upper limit of normal, but these changes were reversible after decreasing lomitapide dose or discontinuing this medication temporarily. No bilirubin increases were observed. There was a mean increase during the first 26 weeks in the liver fat content from 1%–9%, which did not progress further after this. Liver fat accumulation is regarded as the greatest safety concern during lomitapide treatment.65
Summary and future prospects
The original finding essential for the development of the MTTP inhibitors and, ultimately, lomitapide occurred already in 1993 when the genetic defect behind human abetalipoproteinemia was discovered.66 The homozygous WHHL-rabbit, the animal model for hoFH, offered a unique possibility to test different MTTP inhibitor molecules,56 which then led to the development of lomitapide. Lomitapide has been shown to be useful as a monotherapy in hoFH patients, and, as clearly observed in recent studies also a valuable add-on therapy to LDL apheresis.63,64,67
Since it is unethical to carry out placebo-controlled studies in hoFH because of the extreme severity of this disease, possible effects on cardiovascular prognosis must be evaluated on the basis of retrospective studies of LDL-cholesterol lowering by drugs alone or in combination with LDL apheresis.32,40,41,68
The increase in liver fat during lomitapide treatment can be explained, in theory, either as a metabolic effect of lomitapide, or it could represent an off-target effect leading to hepatotoxicity.65,69 Moreover, it is conceivable that genetic variation in the molecular mechanisms causing increased liver fat content may partly explain the interindividual variation in the rate of liver fat accumulation. The Juxtapid Risk Evaluation Program emphasizes careful initial monitoring of alkaline phosphatase, bilirubin, alanine aminotransferase and aspartate aminotransferase, followed by regular monitoring of the two transaminase levels.70,71 Moreover, if magnetic resonance spectroscopy is available in a medical unit taking care of an hoFH patient, the degree of liver fat accumulation should be monitored.
Besides lomitapide, another drug preventing the production of apoB-containing lipoproteins by the liver has been developed. Mipomersen, this new drug for the treatment of hoFH, is now available in the USA and has been studied in hoFH patients.13 It is an injectable antisense oligonucleotide capable of binding apoB messenger RNA and so preventing apoB production.72 In hoFH patients with the defective LDLR mutation, PCSK9 inhibitors capable of upregulating the residual activity of the LDL receptor is also an option.73
In summary, the role of lomitapide in the treatment of hoFH has great potential as an add-on therapy to LDL apheresis, statin treatment, and ezetimibe treatment. Clearly, longer-term follow-up studies are still needed. At this time, lomitapide provides us a novel tool in our attempts to decrease mortality and morbidity among the patients with the most severe form of genetic hypercholesterolemia, those with hoFH.
Disclosure
Alpo Vuorio has received lecture honoraria from Aegerion Ltd.
Matti Tikkanen has received consultancy fees from Pfizer, Amgen, and Aegerion Ltd. Matti Tikkanen holds a grant with Amgen, and has a trial grant pending with Pfizer. Matti Tikkanen holds stock/stock options with the Orion company. Matti Tikkanen is currently a chairman of the committee creating and updating the Finnish National Guidelines for Dyslipidemia.
Petri Kovanen has received consultancy fees from Amgen and Aegerion Ltd. Petri Kovanen has received payment for lectures from Raisio and Unilever. Petri Kovanen holds stock/stock options with the Orion company. Petri Kovanen is a member of the committee creating and updating the Finnish National Guidelines for Dyslipidemia.
The authors have no other conflicts of interest in this work.
References
Usifo E, Leigh SE, Whittall RA, et al. Low-density lipoprotein receptor gene familial hypercholesterolemia variant database: update and pathological assessment. Ann Hum Genet. 2012;76(5):387–401. | |
Brown MS, Kovanen PT, Goldstein JL. Regulation of plasma cholesterol by lipoprotein receptors. Science. 1981;212(4495):628–635. | |
Brown MS, Kovanen PT, Goldstein JL, et al. Prenatal diagnosis of homozygous familial hypercholesterolaemia. Expression of a genetic receptor disease in utero. Lancet. 1978;1(8063):526–529. | |
Goldstein JL, Hobbs HH, Brown MS. Familial hypercholesterolemia. In: Scriver CR, Beaudet AL, Sly WS, et al, editors. The Metabolic and Molecular Bases of Inherited Disease. 8th ed. New York: McGraw-Hill; 2001:2863–2913. | |
Nordestgaard BG, Chapman MJ, Humphries SE, et al; European Atherosclerosis Society Consensus Panel. Familial hypercholesterolemia is underdiagnosed and undertreated in the general population: guidance for clinicians to prevent coronary heart disease: consensus statement of the European Atherosclerosis Society. Eur Heart J. 2013;34(45):3478a–3490a. | |
Leitersdorf E, Van der Westhuyzen DR, Coetzee GA, Hobbs HH. Two common low density lipoprotein receptor gene mutations cause familial hypercholesterolemia in Afrikaners. J Clin Invest. 1989;84(3):954–961. | |
Seftel HC, Baker SG, Sandler MP, et al. A host of hypercholesterolaemic homozygotes in South Africa. Br Med J. 1980;281(6241):633–636. | |
Lehrman MA, Schneider WJ, Brown MS, et al. The Lebanese allele at the low density lipoprotein receptor locus. Nonsense mutation produces truncated receptor that is retained in endoplasmic reticulum. J Biol Chem. 1987;262(1):401–410. | |
Fahed AC, Safa RM, Haddad FF, et al. Homozygous familial hypercholesterolemia in Lebanon: a genotype/phenotype correlation. Mol Genet Metab. 2011;102(2):181–188. | |
Moorjani S, Roy M, Gagné C, et al. Homozygous familial hypercholesterolemia among French Canadians in Québec Province. Arteriosclerosis. 1989;9(2):211–216. | |
Haitas B, Baker SG, Meyer TE, Joffe BI, Seftel HC. Natural history and cardiac manifestations of homozygous familial hypercholesterolaemia. Q J Med. 1990;76(279):731–740. | |
Bertolini S, Cassanelli S, Garuti M, et al. Analysis of LDL receptor gene mutations in Italian patients with homozygous familial hypercholesterolemia. Arterioscler Thromb Vasc Biol. 1999;19(2):408–418. | |
Raal FJ, Santos RD, Blom DJ, et al. Mipomersen, an apolipoprotein B synthesis inhibitor, for lowering of LDL cholesterol concentrations in patients with homozygous familial hypercholesterolemia: a randomised, double-blind, placebo-controlled trial. Lancet. 2010;375(9719):998–1006. | |
Raal FJ, Santos RD. Homozygous familial hypercholesterolemia: current perspectives on diagnosis and treatment. Atherosclerosis. 2012;223(2):262–268. | |
Mabuchi H, Nohara A, Noguchi T, et al; Hokuriku FH Study Group. Molecular genetic epidemiology of homozygous familial hypercholesterolemia in the Hokuriku district of Japan. Atherosclerosis. 2011;214(2):404–407. | |
Naoumova RP, Thompson GR, Soutar AK. Current management of severe homozygous hypercholesterolaemias. Curr Opin Lipidol. 2004;15(4):413–422. | |
Kraft HG, Lingenhel A, Raal FJ, Hohenegger M, Utermann G. Lipoprotein(a) in homozygous familial hypercholesterolemia. Arterioscler Thromb Vasc Biol. 2000;20(2):522–528. | |
Holmes DT, Schick BA, Humphries KH, Frohlich J. Lipoprotein(a) is an independent risk factor for cardiovascular disease in heterozygous familial hypercholesterolemia. Clin Chem. 2005;51(11):2067–2073. | |
Raal FJ, Pilscher GJ, Illingworth DR, et al. Expanded-dose simvastatin is effective in homozygous familial hypercholesterolaemia. Atherosclerosis. 1997;135(2):249–256. | |
Innerarity TL, Weisgraber KH, Arnold KS, et al. Familial defective apolipoprotein B-100: low density lipoproteins with abnormal receptor binding. Proc Natl Acad Sci U S A. 1987;84(19):6919–6923. | |
Abifadel M, Varret M, Rabès JP, et al. Mutations in PCSK9 cause autosomal dominant hypercholesterolemia. Nat Genet. 2003;34(2):154–156. | |
Garcia CK, Wilund K, Arca M, et al. Autosomal recessive hypercholesterolemia caused by mutations in a putative LDL receptor adaptor protein. Science. 2001;292(5520):1394–1398. | |
Kästner M, Höher M, Vogt M, Lang D. Myokardinfarkt bei einem 7jährigen Kind mit familiärer Hypercholesterinämie. [Myocardial infarction in a seven-and-a-half year old child with familial hypercholesterolemia]. Monatsschr Kinderheilkunde. 1998;146(12):1176–1180. German. | |
Widhalm K, Binder CB, Kreissl A, et al. Sudden death in a 4-year-old boy: a near-complete occlusion of the coronary artery caused by an aggressive lipoprotein receptor mutation (W556R) in homozygous familial hypercholesterolemia. J Pediatr. 2011;158(1):167. | |
Santos RD, Miname MH, Martinez LR, et al. Non-invasive detection of aortic and coronary atherosclerosis in homozygous familial hypercholesterolemia by 64 slice multi-detector row computed tomography angiography. Atherosclerosis. 2008;197(2):910–915. | |
Kawaguchi A, Yutani C, Yamamoto A. Hypercholesterolemic valvulopathy: an aspect of malignant atherosclerosis. Ther Apher Dial. 2003;7(4):439–443. | |
Uauy R, Vega GL, Grundy SM, Bilheimer DM. Lovastatin therapy in receptor-negative homozygous familial hypercholesterolaemia: lack of effect on low-density lipoprotein concentrations or turnover. J Pediatr. 1988;113(2):387–392. | |
Thompson GR, Ford J, Jenkison M, Trayner I. Efficacy of mevinolin as adjuvant therapy for refractory familial hypercholesterolemia. Q J Med. 1986;60(232):803–811. | |
Raal FJ, Pappu AS, Illingworth DR, et al. Inhibition of cholesterol synthesis by atorvastatin in homozygous familial hypercholesterolaemia. Atherosclerosis. 2000;150(2):421–428. | |
Marais AD, Raal FJ, Stein EA, et al. A dose-titration and comparative study of rosuvastatin and atorvastatin in patients with homozygous familial hypercholesterolaemia. Atherosclerosis. 2008;197(1):400–406. | |
Marais AD, Firth JC, Naoumova RP, Thompson GR. The role of HMG-CoA reductase inhibitors in homozygous familial hypercholesterolaemia. In: Gotto Jr AM, Paoletti R, Smith LC, Catapano AL, Jackson AS, editors. Drugs Affecting Lipid Metabolism: Risk Factors and Future Directions. New York, Springer; 1996:181–186. | |
Raal FJ, Pilcher GJ, Panz VR, et al. Reduction in mortality in subjects with homozygous familial hypercholestrolemia associated with advances in lipid-lowering therapy. Circulation. 2011;124(20):2202–2207. | |
Kolansky DM, Cuchel M, Clark BJ, et al. Longitudinal evaluation and assessment of cardiovascular disease in patients with homozygous familial hypercholesterolemia. Am J Cardiol. 2008;102(11):1438–1443. | |
Kroon AA, van’t Hof MA, Demacker PN, Stalenhoef AF. The rebound of lipoproteins after LFL-apheresis. Kinetics and estimation of mean lipoprotein levels. Atherosclerosis. 2000;152(2):519–526. | |
National Institute for Health and Clinical Excellence. Identification and Management of Familial Hypercholesterolaemia. NICE Clinical Guideline 71. London: National Institute for Health and Clinical Excellence; 2008. Available from: http://www.nice.org.uk/nicemedia/pdf/CG071NICEGuideline.pdf. Accessed October 20, 2013. | |
Græsdal A, Bogsrud MP, Holven KB, et al. Apheresis in homozygous familial hypercholesterolemia: the results of a follow-up of all Norwegian patients with homozygous familial hypercholesterolemia. J Clin Lipidol. 2012;6(4):331–339. | |
Harada-Shiba M, Arais H, Oikawa S, et al. Guidelines for the management of familial hypercholesterolemia. J Atheroscler Thromb. 2012;19(12):1043–1060. | |
Schwartz J, Winters JL, Padmanabhan A, et al. Guidelines on the use of therapeutic apheresis in clinical practice-evidence-based approach from the Writing Committee of the American Society for Apheresis: the sixth special issue. J Clin Apher. 2013;28(3):145–284. | |
Thompson GR, Barbir M, Davies D, et al. Efficacy criteria and cholesterol targets for LDL apheresis. Atherosclerosis. 2010;208(2):317–321. | |
Palcoux JB, Atassi-Dumont M, Lefevre P, et al. Low-density lipoprotein apheresis in children with familial hypercholesterolemia: follow-up to 21 years. Ther Apher Dial. 2008;12(3):195–201. | |
Hudgins L, Kleinman B, Scheuer A, White S, Gordon BR. Long-term safety and efficacy of low-density lipoprotein apheresis in childhood for homozygous familial hypercholesterolemia. Am J Cardiol. 2008;102(9):1199–1204. | |
Thompson GR. The evidence-base for the efficacy of lipoprotein apheresis in combating cardiovascular disease. Atheroscler Supp. 2013;14(1):67–70. | |
Starzl TE, Bilheimer DW, Bahnson HT, et al. Heart–liver transplantation in a patient with familial hypercholesterolemia. Lancet. 1984;1(8391):1382–1383. | |
Malatack JJ. Liver transplantation as treatment for familial homozygous hypercholesterolemia: too early or too late. Pediatr Transplant. 2011;15(2):123–125. | |
Maiorana A, Nobili V, Calandra S, et al. Preemptive liver transplantation in a child with familial hypercholesterolemia. Pediatr Transplant. 2011;15(2):E25–E29. | |
Küçükkartallar T, Yankol Y, Kanmaz T, Topalogğlu S, Acarli K, Kalayoglu M. Liver transplantation as a treatment option for three siblings with homozygous familial hypercholesterolemia. Pediatr Transplant. 2011;15(3):281–284. | |
Kakaei F, Nikeghbalian S, Kazemi K, et al. Liver transplantation for homozygous familial hypercholesterolemia: two case reports. Transplantation Proc. 2009;41(7):2939–2941. | |
Hussain MM, Shi J, Dreizen P. Microsomal triglyceride transfer protein and its role in apoB-lipoprotein assembly. J Lipid Res. 2003;44(1):22–32. | |
Wetterau JR, Aggerbeck LP, Bouma ME, et al. Absence of microsomal triglyceride transfer protein in patients with abetalipoproteinemia. Science. 1992;258(5084):999–1001. | |
Berriot-Varoqueaux N, Aggerbeck LP, Samson-Bouma M, Wetterau JR. The role of the microsomal triglyceride transfer protein in abetalipoproteinemia. Annu Rev Nutr. 2000;20:663–697. | |
Glueck CJ, Gartside PS, Mellies MJ, Steiner PM. Familial hypobeta-lipoproteinemia: studies in 13 kindreds. Trans Assoc Am Physicians. 1977;90:184–203. | |
Jamil H, Gordon DA, Eustice DC, et al. An inhibitor of the microsomal triglyceride transfer protein inhibits apoB secretion from HepG2 cells. Proc Natl Acad Sci U S A. 1996;93(21):11991–11995. | |
Bakillah A, Nayak N, Saxena U, Medford RM, Hussain MM. Decreased secretion of ApoB follows inhibition of ApoB-MTP binding by a novel antagonist. Biochemistry. 2000;39(16):4892–4899. | |
Ksander G, deJesus R, Yuan A, et al. Diaminoindanes as microsomal triglyceride transfer protein inhibitors. J Med Chem. 2001;44(26):4677–4687. | |
Chang G, Ruggeri RB, Harwood HJ Jr. Microsomal triglyceride transfer protein (MTP) inhibitors: discovery of clinically active inhibitors using high-throughput screening and parallel synthesis paradigms. Curr Opin Drug Discov Devel. 2002;5(4):562–570. | |
Wetterau JR, Gregg RE, Harrity TW, et al. An MTP inhibitor that normalizes atherogenic lipoprotein levels in WHHL rabbits. Science. 1998;282(5389):751–754. | |
Kita T, Brown MS, Watanabe Y, Goldstein JL. Deficiency of low density lipoprotein receptors in liver and adrenal gland of the WHHL rabbit, an animal model of familial hypercholesterolemia. Proc Natl Acad Sci U S A. 1981;78(4):2268–2272. | |
Shiomi M, Ito T. MTP inhibitor decreases plasma cholesterol levels in LDL receptor-deficient WHHL rabbits by lowering the VLDL secretion. Eur J Pharmacol. 2001;431(1):127–131. | |
Samaha FF, McKenney J, Bloedon LT, Sasiela WJ, Rader DJ. Inhibition of micosomal triglyceride transfer protein alone or with ezetimibe in patients with moderate hypercholesterolemia. Natl Clin Pract Cardiovasc Med. 2008;5(8):497–505. | |
Temel RE, Tang W, Ma Y, et al. Hepatic Niemann-Pick C1-like 1 regulates biliary cholesterol concentration and is a target of ezetimibe. J Clin Invest. 2007;117(7):1968–1978. | |
Ikewaki K, Rader DJ, Zech LA, Brewer HB Jr. In vivo metabolism of apolipoprotein A-I and E in patients with abetalipoproteinemia: implications for the roles of apolipoproteins B and E in HDL metabolism. J Lipid Res. 1994;35(10):1809–1819. | |
Cuchel M, Rader DJ. Microsomal transfer protein inhibition in humans. Curr Opin Lipidol. 2013;24(3):246–250. | |
Cuchel M, Bloedon LT, Szapary PO, et al. Inhibition of microsomal triglyceride transfer protein in familial hypercholesterolemia. N Engl J Med. 2007;356(2):148–156. | |
Cuchel M, Meagher EA, du Toit Theron H, et al; Phase 3 HoFH Lomitapide Study investigators. Efficacy and safety of a microsomal triglyceride transfer protein inhibitor in patients with homozygous familial hypercholesterolaemia: a single-arm, open-label, phase 3 study. Lancet. 2013;381(9860):40–46. | |
Raal FJ. Lomitapide for homozygous familial hypercholesterolaemia. Lancet. 2013;381(9860):7–8. | |
Sharp D, Blinderman L, Combs KA, et al. Cloning and gene defects in microsomal triglyceride transfer protein associated with abetalipoproteinaemia. Nature. 1993;365(6441):65–69. | |
Robinson JG. Management of familial hypercholesterolemia: a review of the recommendations from the National Lipid Association Expert Panel on Familial Hypercholesterolemia. J Manag Care Pharm. 2013;19(2):139–149. | |
Sachais BS, Katz J, Ross J, Rader DJ. Long-term effects of LDL apheresis in patients with severe hypercholesterolemia. J Clin Apher. 2005;20(4):252–255. | |
Marais AD, Blom DJ. Recent advances in the treatment of homozygous familial hypercholesterolaemia. Curr Opin Lipidol. 2013;24(4):288–294. | |
Aegerion Pharmaceuticals Inc. FDA approves Aegerion Pharmaceuticals’ JUXTAPID™ (lomitapide) capsules for homozygous familial hypercholesterolemia (HoFH) [press release]. Cambridge, MA: Aegerion Pharmaceuticals Inc.; 2012 [Dec 24]. Available from: http://ir.aegerion.com/releasedetail.cfm?ReleaseID=728650. Accessed February 17, 2013. | |
Goldberg AC. Emerging low-density lipoprotein therapies: Microsomal triglyceride transfer protein inhibitors. J Clin Lipidol. 2013;7(Suppl 3):S16–S20. | |
Plakogiannis R, Cioce L, Fisher EA, Underberg JA. The role of mipomersen therapy in the treatment of familial hypercholesterolemia. Clin Investig (Lond). 2012;2(10):1033–1037. | |
Stein EA, Honarpur N, Wasserman SM, Xu F, Scott R, Raal FJ. Effect of the proprotein convertase subtilisin/kexin 9 monoclonal antibody, AMG 145, in homozygous familial hypercholesterolemia. Circulation. 2013;128(19);128(19):2113–2120. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.