")
Back to Journals » International Journal of Nanomedicine » Volume 10 » Issue 1
Inhibition of colon cancer cell growth by nanoemulsion carrying gold nanoparticles and lycopene
Authors Huang RS, Wei Y, Stephen Inbaraj B, Chen B
Received 11 December 2014
Accepted for publication 22 January 2015
Published 8 April 2015 Volume 2015:10(1) Pages 2823—2846
DOI https://doi.org/10.2147/IJN.S79107
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 6
Editor who approved publication: Dr Thomas Webster
Rwei-Fen S Huang,1,2,* Yi-Jun Wei,1,2,* Baskaran Stephen Inbaraj,3 Bing-Huei Chen1,3,4
1Graduate Institute of Nutrition and Food Science, 2Department of Nutritional Science, 3Department of Food Science, 4Graduate Institute of Medicine, Fu Jen University, Taipei, Taiwan
*These authors contributed equally to this work
Abstract: Lycopene (LP), an important functional compound in tomatoes, and gold nanoparticles (AN), have received considerable attention as potential candidates for cancer therapy. However, the extreme instability and poor bioavailability of LP limits its in vivo application. This study intends to develop a nanoemulsion system incorporating both LP and AN, and to study the possible synergistic effects on the inhibition of the HT-29 colon cancer cell line. LP–nanogold nanoemulsion containing Tween 80 as an emulsifier was prepared, followed by characterization using transmission electron microscopy (TEM), dynamic light scattering (DLS) analysis, ultraviolet spectroscopy, and zeta potential analysis. The particle size as determined by TEM and DLS was 21.3±3.7 nm and 25.0±4.2 nm for nanoemulsion and 4.7±1.1 nm and 3.3±0.6 nm for AN, while the zeta potential of nanoemulsion and AN was -32.2±1.8 mV and -48.5±2.7 mV, respectively. Compared with the control treatment, both the combo (AN 10 ppm plus LP 12 µM) and nanoemulsion (AN 0.16 ppm plus LP 0.4 µM) treatments resulted in a five- and 15-fold rise in early apoptotic cells of HT-29, respectively. Also, the nanoemulsion significantly reduced the expressions of procaspases 8, 3, and 9, as well as PARP-1 and Bcl-2, while Bax expression was enhanced. A fivefold decline in the migration capability of HT-29 cells was observed for this nanoemulsion when compared to control, with the invasion-associated markers being significantly reversed through the upregulation of the epithelial marker E-cadherin and downregulation of Akt, nuclear factor kappa B, pro-matrix metalloproteinase (MMP)-2, and active MMP-9 expressions. The TEM images revealed that numerous nanoemulsion-filled vacuoles invaded cytosol and converged into the mitochondria, resulting in an abnormally elongated morphology with reduced cristae and matrix contents, demonstrating a possible passive targeting effect. The nanoemulsion containing vacuoles were engulfed and internalized by the nuclear membrane envelop for subsequent invasion into the nucleoli. Taken together, LP–nanogold nanoemulsion could provide synergistic effects at AN and LP doses 250 and 120 times lower than that in the combo treatment, respectively, demonstrating the potential of nanoemulsion developed in this study for a possible application in colon cancer therapy.
Keywords: gold–lycopene nanoemulsion, apoptosis, migration, invasion, colon cancer cell HT-29, passive targeting
Introduction
According to the worldwide statistics released on cancer by the World Health Organization in November 2014, colon cancer was the third leading cause of cancer deaths among men and the second leading cause among women in 2012, contributing to 694,000 deaths out of an overall 8.2 million deaths from all cancer types. More specifically, according to a published report by the Ministry of Health in Taiwan, colon cancer was the third leading cause of cancer death in Taiwan in 2012. Colon cancers can be divided into familial adenomatous polyposis, hereditary nonpolyposis colorectal cancer, sporadic colon cancer, and colitis-associated colorectal cancer.1 Research has shown that patients with inflammatory bowel diseases such as Crohn’s disease and ulcerative colitis represent a high-risk group for colon cancer.1 This kind of colon cancer is one of the most frequently seen malignant tumors due to the high degree of invasion and migration to the other organs like the liver, lung, bone, and brain.2 Clinically, the most effective colon cancer therapy is an operation in combination with chemotherapy, radiotherapy, immunotherapy, Chinese herbs, and so on. Nevertheless, the survival rate is only about 20%–40% 5 years after the operation, which can be attributed to reoccurrence and the migration of colon tumors.3 The risk factors associated with colon cancer occurrence have been shown to include dietary habits, family disease history, inflammatory bowel disease, Streptococcus virus, and smoking.3
The major cause of cancer death has been demonstrated to be due to cancer cell migration, not the tumor itself. Migration includes the following steps: 1) detachment from the original tumor and migration (intravasation) into the blood or lymphatic system; 2) orientation and recognition of the migration target and position and extravasation from the blood or lymphatic system; and 3) establishment of a new base for new tumor growth. The major route to initiate the invasion and migration of cancer cells is through epithelial–mesenchymal transition (EMT), which involves conversion of the epithelial cells to mesenchymal cells, a change in cell morphology and structure, as well as an increase in adhesion and migration substance.4,5
In the past two decades nanotechnology has emerged as a new and promising technique for the prevention, diagnosis, and treatment of cancer. Though the targeted delivery of drugs could be achieved by conventional carriers, the use of nanoparticles with an optimum size, surface characteristics, and dosage could enhance the solubility of lipophilic drugs and lead to the enhanced permeability and retention (EPR) effect for passive targeting, enable multiple payloads, and minimize side effects.6 Initially, the nanomaterials were believed to be biologically inert, but growing literature reports have highlighted the toxicity and potential risks of their use, especially for nanoparticles <10 nm.7 Of the various nanomaterials, gold possess unique physical and chemical properties through its conjugation with a variety of drugs to serve as carrier for drug delivery, contrast agents for imaging enhancement, and for topical thermal therapy.8 More importantly, the incorporation of gold nanoparticles (AN) with specific cancer cell receptor ligands, such as folate, and the chemotherapeutic drug, irinotecan, can enhance the specificity and efficiency of cancer chemotherapy through active targeting.9 For instance, Paciotti et al10 reported that AN carrying tumor necrosis factor (TNF)-α induced cancer-specific cytotoxicity and reduced the host toxicity of colonic xanthograph mice. Nevertheless, high doses of AN may also exert toxic effects such as the promotion of human fibroblast cell migration.11 For improvements in cancer therapy efficiency, and reductions of host toxicity in vivo, nanoparticles have been modified by the incorporation of liposomes, polymeric materials, and dendrimers, with lipid-based nanoassemblies being the least toxic.12,13 In view of this, it is possible to enhance cancer therapy through the conjugation of AN with lipid-based nanoemulsion containing lycopene (LP). Additionally, one of the major advantages of AN is that they can be manufactured into sizes that range from 1–150 nm,8 but the effect of various sized AN on colon cancer cell growth remains uncertain.
Similarly, LP, a pivotal biological compound present in tomatoes, has received considerable attention in the past two decades due to its protective effect against chronic diseases such as coronary heart disease, skin cancer, and prostate cancer.14 Increased intake of the functional component, LP, has been reported to reduce risks of several cancers due to its efficiency in triggering the cell cycle arrest of hepatoma cells, breast cancer cells, and lung cancer cells, as well as the antiproliferation of colon cancer cells and leukemia cells, and the antimetastasis of hepatoma cells.14–16 Accumulating evidence suggests the potentiality of LP for targeting cancer therapy as an anticancer agent. However, the extreme instability and poor bioavailability of LP may limit its application in cancer therapy.17 Thus, developing a carrier system to deliver a nontoxic dose of LP for bioavailability enhancement and specific targeting of certain organs should be a vital strategy for cancer therapy. In a recent study, Chen et al17 reported that through the preparation of LP micelles and LP chylomicron, the absolute bioavailability of LP in rats could be greatly enhanced. More importantly, LP chylomicrons are available in the market and have been successfully used for the treatment of patients with prostauxe symptoms in a Phase III clinical trial conducted in both the United States and Taiwan.18 Nanoemulsions are simple ternary systems composed of an oil phase, water phase, and surfactant, with cosurfactant being used in some preparations.19 Such nanoemulsions are spontaneously formed systems possessing physicochemical properties such as transparency, optical isotropy, low viscosity, and thermodynamic stability.19 In the present study, for the preparation of LP–nanogold nanoemulsion, oil-containing LP paste from tomato extract was used as the oil phase, Tween 80 was used as a surfactant, and an aqueous AN solution was used as the water phase.
As the therapeutic options for colon cancer patients in developing invasive and metastatic capacities via multistage EMT are limited, colon-specific nanoparticles may allow for the local delivery of an effective dose of bioactive compound to the colon to improve pharmacotherapy.13 This study aims to explore the inhibition mechanism of the colon cancer cell line, HT-29, as affected by various treatments including AN or LP alone, a combination of AN plus LP, as well as the nanoemulsion of AN plus LP. Also, the possible cell uptake mechanism and passive targeting effect of the nanoemulsion developed in this experiment will be elucidated.
Materials and methods
Materials
Human colon cancer cell line HT-29 (American Type Culture Collection [ATCC]: HTB-38™) and human lung fibroblast cell line MRC-5 (ATCC: CCL-171) were purchased from the Taiwan Food Industry Development and Research Institute (Hsinchu City, Taiwan). Cell culture medium McCoy’s 5A is composed of 10% fetal bovine serum (FBS), 0.22% sodium bicarbonate, and 100 units/mL penicillin–streptomycin, with a pH adjusted to 7.4. Matrigel™ was purchased from BD (Franklin Lakes, NJ, USA). Minimum essential medium (MEM) is composed of 10% FBS, 0.15% sodium bicarbonate, 100 units/mL penicillin–streptomycin, 0.1 mM nonessential amino acids, and 1.0 mM sodium pyruvate, with a pH adjusted to 7.4. Both McCoy’s 5A medium and MEM were procured from Sigma-Aldrich Co. (St Louis, MO, USA). Sodium chloride and potassium chloride were from J.T. Baker (Phillipsburg, NJ, USA).
Dimethyl sulfoxide (DMSO) and MTT formazan powder were from Sigma-Aldrich Co. The Annexin V (AV)-FITC apoptosis detection kit was from Strong Biotech (Taipei, Taiwan). Ammonium persulfate, glycine, 2-mercaptoethanol, phenylmethylsulfonyl fluoride (PMSF), tetramethylene diamine, bromophenol blue, and MTT were from Sigma-Aldrich Co. Methanol, trisbase, isopropanol, and ethanol were from J.T. Baker. Sodium dodecyl sulfate (SDS) was from SERVA Electrophoresis GmbH (Heidelberg, Germany). The polyvinylidene fluoride (PVDF) membrane was from Whatman plc (GE Healthcare UK Ltd, Little Chalfont, UK). Bovine serum albumin (BSA) standard was from Thermo Fisher Scientific (Waltham, MA, USA). Protein assay dye reagent and acrylamide were from Bio-Rad Laboratories, Inc. (Hercules, CA, USA). Restained protein ladder was from Bioman Scientific Co., Ltd. (Taipei, Taiwan). Tween® 20 was from AMRESCO LLC (Solon, OH, USA). β-actin and procaspase-8 were from EMD Millipore (Billerica, MA, USA), Procaspase-3 and Bcl-2 were from BD. Procaspase-9 was from Bioscience (Irvine, CA, USA). Bax, Vimentin, β-catenin, Akt 1+2+3 antibody, and PARP were from GeneTex (Taipei, Taiwan). The AV-FITC apoptosis detection kit was from Strong Biotech. Nuclear factor kappa B (NFkB), antirabbit immunoglobulin G (IgG) antibody, matrix metalloproteinase (MMP-2, MMP-9), and E-cadherin were from Cell Signaling Technology, Inc. (Danvers, MA, USA).
The instruments used included a hemocytometer, CO2 incubator, laminar flow, inverted microscope, Western blot and electrophoresis equipments, pH meter, temperature-controlled water bath, microcentrifuge, enzyme-linked immunosorbent assay (ELISA) reader, UVP photographic gel system, flow cytometry, liquid nitrogen reservoir, and a −80°C refrigerator.
Preparation of AN
AN with sizes of 1–3 nm, 3–5 nm, and 10–15 nm were provided by Gold NanoTech, Inc. (Taipei, Taiwan), with the concentrations being controlled at 15 ppm, 80 ppm, and 90 ppm, respectively. These nanoparticles were prepared by a molecular beam epitaxy process, as previously reported.20 Briefly, bulk gold material was cut and ground, followed by vaporization with an ultrahigh vacuum (10−8 Pa) to its atomic state and condensation in the presence of inert gas to form AN. The concentration of AN was determined using an inductively coupled plasma mass spectrometer (PerkinElmer-SCIEX ELAN 6100 DRC; PerkinElmer, Inc., Waltham, MA, USA).20
Preparation of LP–nanogold nanoemulsion
AN with a size of 3–5 nm were used for the preparation of LP nanogold nanoemulsion based on a method described by Chen et al.17 From the stock LP paste containing 42.9 mg of LP per g, the LP–nanogold nanoemulsion was prepared by mixing 40 mg of paste containing 1.7 mg of LP with 1.2 g of Tween 80, after which the mixture was stirred manually for homogeneity. Next, 7.6 mL of AN (80 ppm) were added, followed by 3.2 mL of deionized water; this was subsequently sonicated for 1 hour to obtain LP–nanogold nanoemulsion (12 mL) with a final LP and gold concentration of 141.6 μg/mL and 51.0 μg/mL, respectively. The loading of AN was determined based on the direct comparison of the absorption intensity corresponding to the typical surface plasmon resonance (SPR) at 520 nm with that of the calibration curve prepared using four standard AN solutions (20 μg/mL, 40 μg/mL, 60 μg/mL, and 80 μg/mL) obtained from the stock AN solution (80 μg/mL), as supplied by the manufacturer. To study the effect of LP and the AN+LP combo on HT-29 cells, LP paste was dissolved in DMSO to prepare a stock solution containing LP at 1.10 mM. Although LP dissolved in physiological fluid could better mimic the in vivo physiological condition, several organic solvents such as DMSO and tetrahydrofuran have been successfully and widely used in cell-related studies.21,22
Characterization of AN and LP–nanogold nanoemulsion
The particle size of both AN and LP–nanogold nanoemulsion were determined by taking a portion of the sample in a precision quartz cell (Hellma Analytics, Müllheim, Germany) and analyzing it in a dynamic light scattering (DLS) instrument (90 plus model) from Brookhaven Instruments Corporation (Holtsville, NY, USA). For optical measurements by ultraviolet (UV)-visible spectroscopy, a Beckman DU-640 spectrophotometer (Beckman Coulter, Inc., Brea, CA, USA) was used to measure absorbance in the wavelength range of 300–800 nm with a resolution of 1 nm. The images of AN and LP–nanogold nanoemulsion were visualized in a JEM 2100 F model JEOL transmission field emission electron microscope (JEOL, Tokyo, Japan). About 20 μL of a AN sample was dropped onto a carbon-coated 200 mesh copper grid from Electron Microscopy Sciences (Hatfield, PA, USA) and allowed to stand for 1 minute. Then, the excess sample was removed carefully using filter paper and dried overnight for transmission electron microscopy (TEM) imaging at 120 kV. For the TEM imaging of LP–nanogold nanoemulsion, a negative staining method was adopted for the sample preparation, owing to the absence of any metal-related compounds to provide the required contrast. After dropping 20 μL of LP–nanogold nanoemulsion sample on a copper grid and removing the excess sample, 20 μL of 2% phosphotungstic acid (Sigma-Aldrich Co.) prepared in pH 6.5-adjusted deionized water was added in order to negatively stain, so that the nanoemulsion particles could appear in white against a dark background. After 1 minute, the excess stain was removed with filter paper and dried overnight for imaging. In addition, a SZ100 model HORIBA nanoparticle analyzer (HORIBA Ltd., Kyoto, Japan) was used for measuring the zeta potential of AN and LP–nanogold nanoemulsion by diluting it 25 times with deionized water. A method based on Chen et al17 was modified and used for the determination of the encapsulation efficiency of LP in the LP–nanogold nanoemulsion. Briefly, 1 mL of sample diluted in dihydrogen phosphate buffer solution (25 mM; pH 5.5) was poured into a stirred ultrafiltration cell containing a filter membrane with a molecular weight cutoff of 30,000 Da. Next, N2 gas was flushed into the cell (35–40 psi) to accelerate the penetration of free LP through the membrane and prevent oxidation. After filtration, the membrane filter containing the encapsulated LP was treated with 10 mL of dichloromethane for the complete breakdown of nanoemulsion, followed by centrifuging (855 g) at 4°C for 10 minutes, collecting the bottom layer and repeatedly mixing with 10 mL of dichloromethane until the bottom layer became colorless. The LP extracts were then pooled, evaporated to dryness under N2, dissolved in dichloromethane, filtered through a 0.22 μm membrane filter, and 20 μL was IJNected into a high-performance liquid chromatography (HPLC)–diode array detector for the determination of LP.17
Stability determination of LP nanoemulsion
Based on a method reported for the determination of nanoemulsion stability by Chen et al,17 the basic LP nanoemulsion formulation without adding AN was heated at 100°C for 4 hours or stored at 4°C and 25°C separately over a period of 3 months. During this storage period, 0.1 g of the sample was collected every week for the determination of particle size distribution and polydispersity by DLS. Likewise, 0.8 g of nanoemulsion was adjusted to a pH of 2.0, 3.5, 6.0, 6.8, and 7.4 with a Na2HPO4/citric acid buffer solution, separately, followed by storing for 1 hour, 2 hours, 4 hours, 6 hours, 12 hours, or 24 hours at 25°C, and then eventually measuring the concentrations of LP in all samples using an HPLC method reported by Hsu et al.23 In addition, the particle size distribution of all nanoemulsion samples was measured by DLS.
Effect of AN, AN+LP (combo), and LP–AN nanoemulsion on colon cancer cells
Cell culture
The human colon cancer cell line HT-29 was incubated in McCoy’s 5A medium and the pH was adjusted to 7.4, after which the cells were transferred to a steam-saturated incubator at 37°C under 5% CO2 for subculture every 2–3 days. Similarly, the human lung fibroblast cell line MRC-5 was incubated in MEM medium and the pH was adjusted to 7.4, after which cells were transferred to a steam-saturated incubator at 37°C under 5% CO2 for subculture every 3–4 days.
Cell morphology
Both HT-29 and MRC-5 cells were seeded in six-well plates (3×105 cells/well), separately. After 24 hours of incubation, the cells were adhered to the plates and then different doses of AN, LP, or their combination were added: 1) 10 ppm AN (3–5 nm); 2) 12 μM LP; 3) 10 ppm AN (3–5 nm) plus 12 μM LP; 4) AN (0.02 ppm)–LP (0.04 μM) nanoemulsion; 5) AN (0.04 ppm)–LP (0.1 μM) nanoemulsion; 6) AN (0.1 ppm)–LP (0.2 μM) nanoemulsion; 7) AN (0.12 ppm)–LP (0.3 μM) nanoemulsion; and 8) AN (0.16 ppm)–LP (0.4 μM) nanoemulsion. In addition to the aforementioned treatments, the blank emulsion without LP and AN was prepared following the procedure described earlier, and added at a concentration of 1.4 μL/mL of medium. After incubation for 24 hours, the supernatant was removed and the residue was washed twice with phosphate buffered saline (PBS). Cell morphology was then observed and photographed by a microscope.
Experimental treatments
HT-29 cells were seeded in 96-well plates, with each well containing 2×104 cells and triplicate experiments were performed. Cell culture medium, McCoy’s 5A (100 μL), was added to each well and the cells were cultured in an incubator under 5% CO2 at 37°C. After 24 hours of incubation, the cells were adhered to the plates and then different doses of AN and/or LP were added: 1) 1–15 ppm of AN (1–3 nm); 2) 5–50 ppm of AN (3–5 nm); 3) 550 ppm of AN (10–15 nm); 4) 2–20 μM of LP; 5) 0.002 ppm AN–0.004 μM LP nanoemulsion; 6) 0.02 ppm AN–0.04 μM LP nanoemulsion; 7) 0.04 ppm AN–0.1 μM LP nanoemulsion; 8) 0.1 ppm AN–0.2 μM LP nanoemulsion; 9) 0.12 ppm AN–0.3 μM LP nanoemulsion; 10) 0.16 ppm AN–0.4 μM LP nanoemulsion; 11) 0.2 ppm AN–0.6 μM LP nanoemulsion; 12) 0.24 ppm AN–0.7 μM LP nanoemulsion; 13) 0.3 ppm AN–0.8 μM LP nanoemulsion; and 14) 0.32 ppm AN–0.9 μM LP nanoemulsion. In addition, five concentrations (1.4 μL/mL, 3.4 μL/mL, 6.9 μL/mL, 10.3 μL/mL, and 14 μL/mL) of blank emulsion without AN and LP was prepared and added to the medium. After incubation for 24 hours, the culture medium was removed and the cells were harvested for the following analysis.
MTT assay
The harvested cells were washed with PBS twice, followed by adding 200 μL of MTT solution (0.5 mg MTT/mL) for the reaction for 30–60 minutes in the dark at 37°C. Then, the MTT solutions were sucked into glass bottles and 150 μL of DMSO was added to each well and stood at 37°C for 10 minutes to dissolve the purple crystal. The absorbance was measured at 570 nm with an ELISA reader. The cell viability was determined using the following formula:
Cell viability (%) = Treatment/control ×100. | (1) |
Apoptosis analysis
The AV-FLUOS kit was used to measure the apoptotic cells with membrane phosphatidylserine translocation (Hoffmann-La Roche Ltd., Basel, Switzerland). Cells were suspended in incubation buffer that contained 1 mg/L propidium iodide (PI) and a 1:50 dilution of AV-FLUOS labeling solution. The mixed solution was incubated for 15 minutes on ice, and green AV and red PI fluorescence intensities were analyzed on an EPICS XLMCL flow cytometer (Beckman Coulter, Inc., Fullerton, CA, USA). Cells with AV-positive and PI-negative fluorescence were defined as apoptotic. Cells with AV-negative and PI-positive fluorescence (PI permeable) were defined as necrotic.
Wound healing analysis
HT-29 cells were seeded in 24-well plates with each well containing 3×105 cells, and then Mycos’s medium was added for incubation. After cell adhesion, a wound was formed by scratching with a 10 μL tip and the residual cells were washed twice with PBS. Then, a photograph was taken by an optical microscope at 0 hours and after successive incubation for 24 hours, which was subjected to ImageJ software analysis. The cell migration area of both sides could be calculated based on the difference between the whole area and the middle wound area. The migration rate over the 24-hour period was calculated by dividing the cell migration area after 24 hours of treatment by that at 0 hours.
Western blot
For protein extraction, the medium was removed and washed with PBS once, followed by adding lysis buffer (10 M protease inhibitor cocktail and 1 M PMSF), scratching cells with a scraper, transferring them into a microcentrifuged tube, reacting on ice for 15 minutes, centrifuging at 13,200 rpm for 30 minutes at 4°C, collecting the supernatant, and storing it at −20°C. The protein concentration was measured by Bio-Rad assay (Bio-Rad Laboratories, Inc.) with BSA as the standard. For protein quantitation, a solution of protein assay dye reagent and deionized water (1:4, v/v) was prepared. Then, various concentrations of BSA standards of 12.5 ppm, 62.5 ppm, 125 ppm, 250 ppm, 375 ppm, and 500 ppm were prepared, after which the BSA standards and diluted protein samples were added to 96-well plates and reacted with 200 μL of protein assay dye solution for 5 minutes. Then, the plate was placed on an ELISA reader for absorbance measurement at 595 nm. The protein concentrations in the samples were calculated based on the linear regression equation of the BSA standard curve obtained by plotting the concentration against absorbance.
For protein electrophoresis, 60 μg of protein extract was mixed with gel-loading buffer containing Tris-HCl (pH 6.8), 10% SDS, 5% β-mercaptoethanol, 30% glycerol and 1.2 mg bromophenol blue. Then, the mixture was heated in a dry bath at 105°C for 10 minutes, followed by cooling on ice, and adding buffer solution and 8% SDS–polyacrylamide for protein separation with the upper gel volt set at 80 V and the lower gel volt set at 100 V for 90 minutes. After electrophoresis, gels were collected and placed on a clip with a wet sponge, filter paper, gel slice, and PVDF membrane, which was presoaked with methanol and moved to transfer buffer for activation. After clipping tightly, gels were placed into an electrophoresis tank and added with the transfer buffer solution containing 25 mM Tris base, 192 mM glycine, 4% SDS, and 20% methanol, with the pH controlled at 8.3. Following the current of 200 mA that flowed for 2 hours, the proteins were separated on gels and successfully transferred to the PVDF membrane, which was soaked in blocking buffer containing 5% skim milk powder for reaction for 30 minutes twice to remove the background noise. The PVDF membrane was then washed with TBS-T (Tris-buffered saline and Tween 20) buffer containing 20 mM of Tris-HCl, 150 mM of NaCl, and 0.1% Tween 20 (pH 7.6) for 5 minutes three times to remove the unattached protein, followed by adding the primary antibody actin diluted 2,000 times (1:2,000) with TBS-T buffer containing 5% BSA for protein conjugation. Proteins were probed with human monoclonal antibody directed against procaspase-3 (1:4,000), procaspase-8 (1:2,000), procaspase-9 (1:1,000), Bcl-2 (1:1,000), PARP (1:1,000), Bax (1:100–1:1,000), MMP-2 (1:1,000), MMP-9 (1:1,000), E-cadherin (1:10,000), Vimentin (1:10,000), β-catenin (1:1,000), NFkB (1:1,000), and Akt 1+2+3 antibody (1:5,000). After shaking at 4°C for 18 hours, the PVDF membrane was washed with TBS-T buffer three times for 5 minutes each. Again, the PVDF membrane was collected and soaked in secondary antibody containing antirabbit IgG antibody and antimouse antibody with a dilution ratio of 1:2,000 and 1:3,000 in blocking buffer, respectively. Following the reaction that proceeded at room temperature for 1 hour for protein conjugation, the PVDF membrane was washed with TBS-T buffer for a total of three times for 5 minutes each and detected by an enhanced chemiluminescence method for automatic image expression by a film developer and by a UVP BioSpectrum® system for protein intensity.
Cellular uptake examination by TEM
The ultrastructural characteristics of untreated and treated HT-29 cells were visualized by TEM. Cell cultures were prefixed with 2.5% glutaraldehyde and 2% paraformaldehyde at 4°C for 1–2 hours. The cells were then postfixed with 1% osmium tetroxide, dehydrated sequentially with 30%, 50%, 70%, 90%, and 100% ethanol, as well as 100% acetone, and finally embedded in Spurr’s resin for polymerization in an oven. Then, ultrathin sections were cut, followed by staining with 4% uranyl acetate and 0.2% lead citrate and mounting on a carbon-coated copper grid for TEM imaging at 120 kV.
Statistical analysis
Triplicate experiments were carried out and the data were subjected to statistical analysis by the SAS 9.3 software system.24 The data were expressed as the mean ± standard deviation, and the statistical difference (P<0.05) of the mean comparison among various treatments was performed using Student’s t-test.
Results
Characterization of AN and LP–nanogold nanoemulsion
The TEM images revealed the shape of the AN to be roughly spherical in shape with the mean particle size based on the particle distribution histogram obtained directly from the image, which was 4.7±1.1 nm (Figure 1Aa). The particle size, as measured by DLS, also showed a similar particle size of 3.3±0.6 nm (Figure 1Ab), which conformed to the average particle size range of 3–5 nm, as specified by the manufacturer. In addition, a narrow particle size distribution with a low polydispersity value of 0.068 was observed, indicating that good homogeneity in nanoparticle dispersion does occur in deionized water. The UV-visible measurement showed a well-defined absorption peak at 520 nm, which can be attributed to the typical SPR in AN (Figure 1Ac). The high negative zeta potential value of −48.5±2.7 mV implied that the surface charge of AN was negative and a stable suspension was formed in aqueous medium.
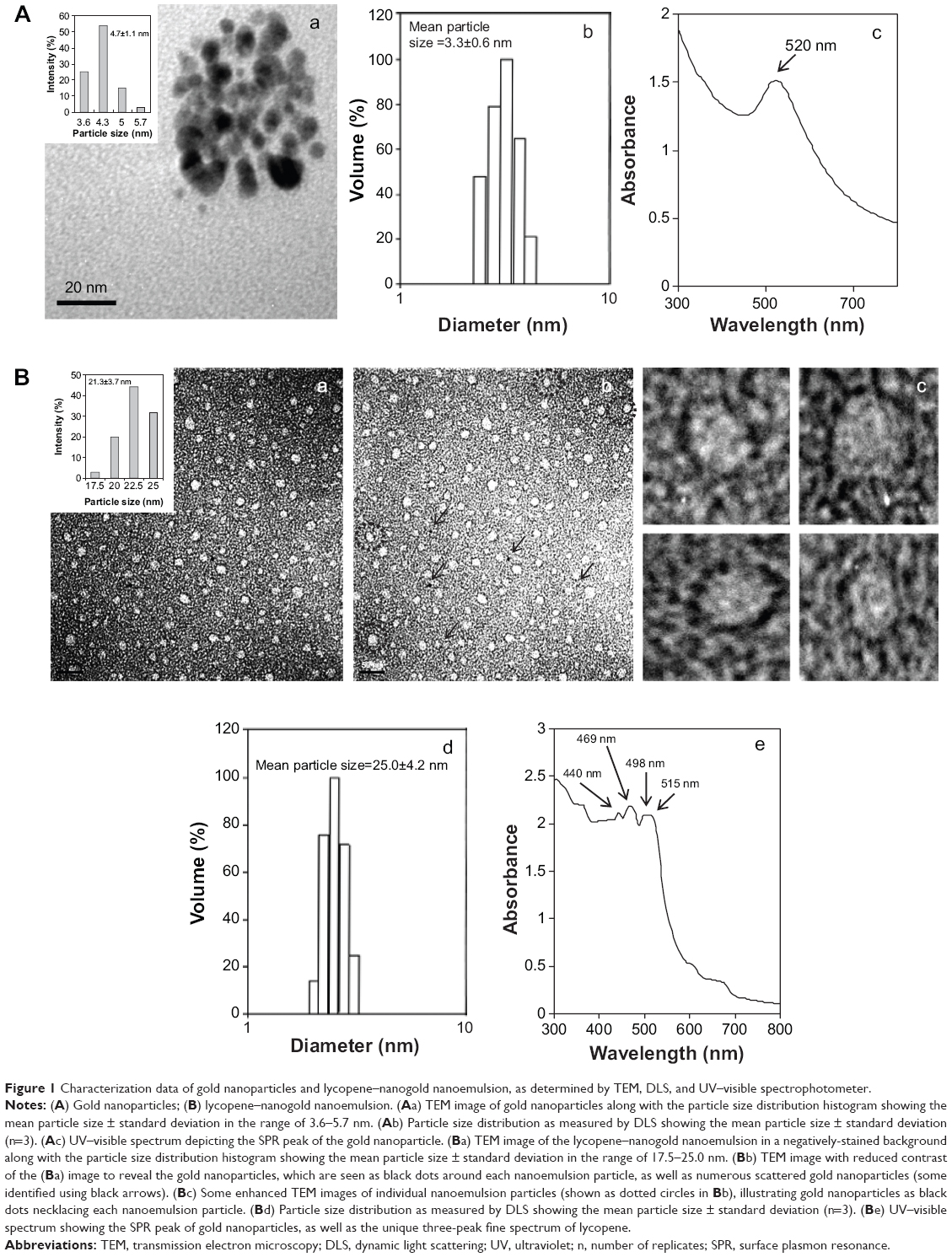 | Figure 1 Characterization data of gold nanoparticles and lycopene–nanogold nanoemulsion, as determined by TEM, DLS, and UV–visible spectrophotometer. |
Figure 1Ba–c depict the TEM images of LP–nanogold nanoemulsion captured after negatively staining with phosphotungstic acid, as described previously. Apparently, the LP–nanoemulsion appeared as approximately round-shaped particles against a dark background with an average particle size (based on the particle size distribution histogram) estimated to be 21.3±3.7 nm (Figure 1Ba). To visualize the distribution of AN in LP–nanogold nanoemulsion, the image contrast in Figure 1Ba was substantially reduced, and it is evident that AN were distributed in the nanoemulsion (some indicated by arrows) (Figure 1Bb). Interestingly, the AN were also arranged around each nanoemulsion particle, forming a pattern of a necklace, as clearly seen from the enlarged view of four representative single nanoemulsion particles (shown in Figure 1Bc). It is probable that the van der Waals’ force of attraction may be responsible for this observed arrangement of AN around each nanoemulsion particle, as LP is a nonpolar lipophilic compound. The mean particle size of LP–nanogold nanoemulsion, as determined by DLS, was 25.0±4.2 nm with a narrow particle size distribution and low polydispersity (0.048), suggesting that a highly homogeneous nanoemulsion was successfully prepared (Figure 1Bd). The UV-visible spectrum showed an SPR peak (515 nm) corresponding to AN, and a triple-absorption peak (440 nm, 469 nm, and 498 nm) characteristic of LP (Figure 1Be). The zeta potential analysis revealed a high negative value of −32.2±1.8 mV, suggesting that LP–nanogold nanoemulsion with high stability was prepared based on a previous report that the zeta potential should be higher than +25 mV or lower than −25 mV to attain a high degree of stability.17 Theoretically, the surface charge of LP nanoemulsion should be nearly neutral or slightly negative, as the nanoemulsion preparation involved nonpolar LP and nonionic surfactant Tween 80. Obviously, Tween 80 contributes a low negative surface charge because of its tendency to shift the share plane position in the electrical double layer.17 However, an obtained high negative value should be due to the presence of AN. Moreover, the presence of ionic contaminants and free fatty acids added as substrates during Tween 80 manufacture may also contribute to this high negative value.17 The encapsulation efficiency of LP in nanoemulsion was determined to be about 80%.
Stability of LP nanoemulsion
Based on DLS analysis, no significant difference in particle size (19.5–20.5 nm) was observed for the LP nanoemulsion samples stored at 4°C and 25°C for 3 months or heated at 100°C for 4 hours, demonstrating the high stability of this LP nanoemulsion. It could be noted that there is a particle size difference of about 5 nm between LP nanoemulsion (20.5 nm) and LP–nanogold nanoemulsion (25.0 nm), which may be due to the incorporation of AN with particle sizes of 3.3–4.7 nm, as mentioned earlier. Also, the mean values of LP as determined by HPLC analysis ranged from 126.2–148.6 μg/mL during storage or heating. Similarly, only a minor change in the level of LP was found when the nanoemulsion samples were stored at a pH of 2.0, 3.5, 6.0, 6.8, and 7.4 for 1 hour, 2 hours, 4 hours, 6 hours, 12 hours, or 24 hours.
Dose effects of AN, LP, AN+LP and LP–nanogold nanoemulsion on the cytotoxicity of HT-29 cells
The dose effects of AN with different sizes on the cytotoxicity of human colonic adenocarcinoma HT-29 cells are shown in Figure 2A, revealing that the half maximal inhibitory concentration (IC50) for AN sizes of 1–3 nm, 3–5 nm, and 10–15 nm was 13 ppm, 24 ppm, and 34 ppm, respectively. Consistent with the previous evidence on high host toxicity for 1–3 nm AN and low efficiency for 10–15 nm AN, the particle size of AN at 3–5 nm was selected for subsequent experiments.20 The IC50 for LP alone was 16.8 μM (Figure 2B), and thus a low dose below IC50 of AN or LP alone or in combination was then tested for its efficiency in inducing the cytotoxicity of HT-29 cells. As shown in Figure 2C, AN at 10 ppm in combination with LP at 8 μM or 12 μM (AN+LP combo treatment) exerted a synergistic effect, as shown by a reduction of 50–60% viability of the HT-29 cells. However, with LP–nanogold nanoemulsion treatment at a lower dose of AN (0.16 ppm) plus LP (0.4 μM), a significantly higher cytotoxicity toward HT-29 cells was observed, as evidenced by an 80% reduction in cellular viability. Therefore, the effective doses of AN or LP alone, in combination, or in nanoemulsion treatments were selected for the subsequent mechanistic studies.
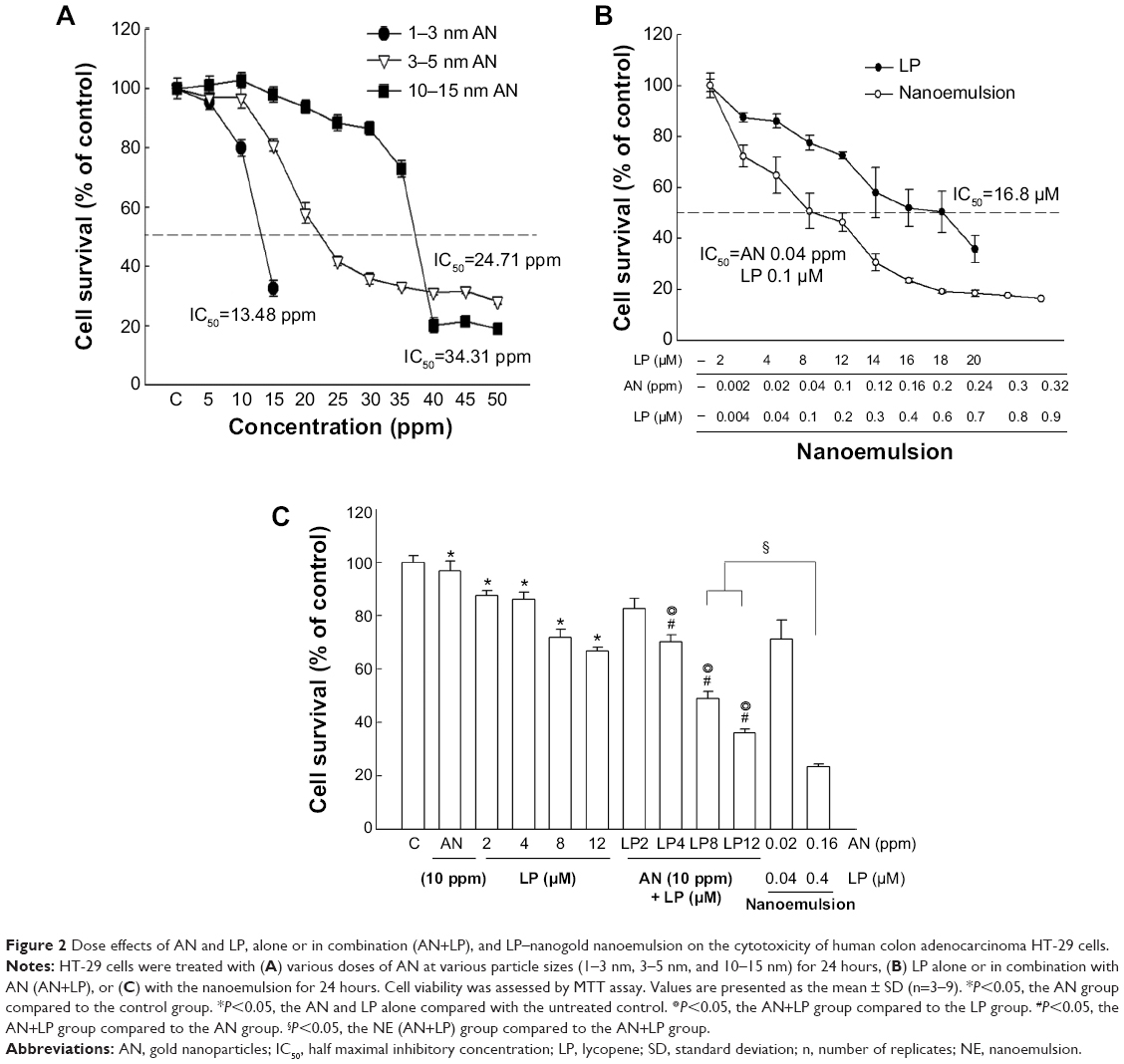 | Figure 2 Dose effects of AN and LP, alone or in combination (AN+LP), and LP–nanogold nanoemulsion on the cytotoxicity of human colon adenocarcinoma HT-29 cells. |
Effects of AN, LP, AN+LP, and nanoemulsion treatments on the apoptosis of HT-29 cells
Figure 3A shows the morphological changes of HT-29 cells as affected by the various treatments including AN or LP alone, in combination, and nanoemulsion. Exposure to all the treatments induced apoptotic morphological alterations in cell shrinkage and rounded cellular appearance as compared to the untreated controls. Comparatively, the nanoemulsion treatment resulted in a more severe shrinking in cell size and more apoptotic cellular aggregations. Such altered apoptotic morphology in response to nanoemulsion exposure should not be caused by the emulsion containing only the emulsifier and medium, as the emulsion alone did not show any cell morphological changes.
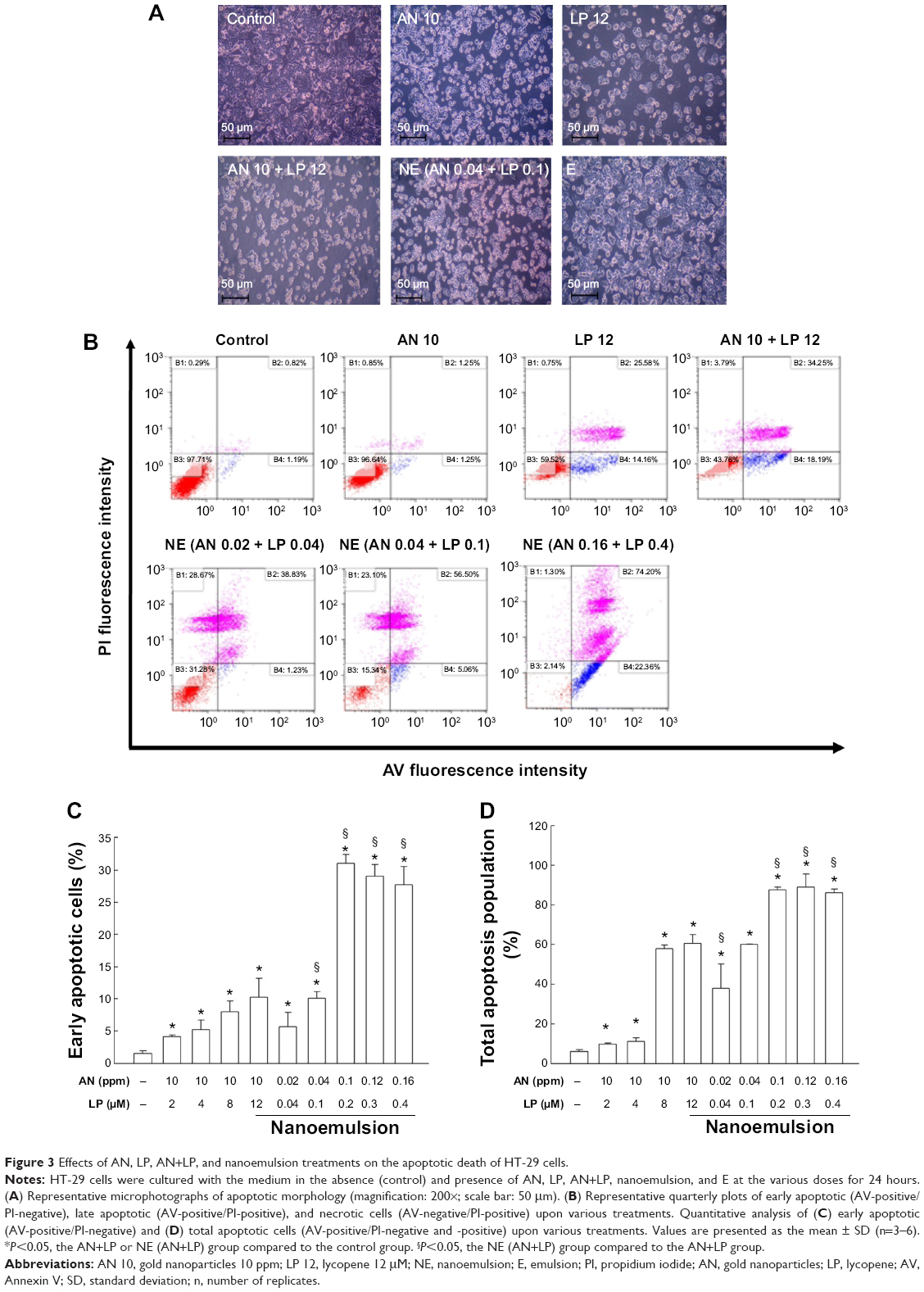 | Figure 3 Effects of AN, LP, AN+LP, and nanoemulsion treatments on the apoptotic death of HT-29 cells. |
To verify if such cytotoxic effects of the combo or nanoemulsion treatments are specific to cancer cells, normal human epithelial fibroblast MRC-5 cells were exposed to the same treatments with the same doses. All of the treatments and the emulsion treatment did not show any significant morphologic change in MRC-5 cells as compared to the untreated controls (Figure S1), revealing that the cytotoxic effects of AN+LP in combo and the nanoemulsion treatments were specific to HT-29 cells.
Using AV and PI to stain the apoptotic cells (AV-positive) and necrotic cells (PI-positive), the plots of the early apoptotic (AV+, PI−), late apoptotic (AV+, PI+), as well as the necrotic cells (AV−, PI+) during various treatments, are presented in Figure 3B, with the quantitative data presented in Figure 3C–D. The treatment of AN+LP (10 ppm +12 μM) was shown to induce a 15-fold rise of early apoptotic cells as compared with the untreated control (P<0.05). Upon the combo treatment with the AN dose fixed at 10 ppm, the early apoptotic cells increased following a rise in LP from 2–12 μM. However, with the nanoemulsion treatment at a much lower dose (by 100-fold) than the combo treatment, a 19-fold increase in early apoptotic cells as compared with the untreated control (P<0.05) was shown. Apparently, the apoptotic effect of nanoemulsion (AN 0.16 ppm + LP 0.4 μM) toward HT-29 cells was significantly higher than that of the combo treatment at various doses (Figure 3C). By taking into account the late apoptotic cells, the nanoemulsion treatment (AN 0.16 ppm + LP 0.4 μM) generated an 85% rise in the total apoptotic population of HT-29 cells, which was significantly higher than the combo treatment (AN 10 ppm + LP 12 μM), showing a 60% apoptosis of HT-29 cells (P<0.05).
It is worth pointing out that the high inhibition efficiency of the nanoemulsion treatment (AN 0.16 ppm + LP 0.4 μM) toward HT-29 cells should not be due to the emulsion containing only the emulsifier and medium, as the emulsion alone (1.4 μL) did not cause early apoptotic cell death, but only induced a portion (12%) of late apoptotic cell death (Figure S2).
Effects of AN, LP, AN+LP, and nanoemulsion treatments on apoptotic death signaling of HT-29 cells
We next investigated the apoptotic signaling mechanisms involved in apoptosis, as affected by various AN and LP treatments. Figure 4 shows the Western blot outcomes of the apoptotic mediator caspase 8 and the executioner caspase 3. Accordingly, the reduced procaspase levels also indicate activation of death signals through proteolytic cleavage on procaspase enzymes. Compared to control treatment, the exposure of HT-29 cells to nanoemulsion at most doses significantly reduced procaspase 3 and 8 levels (Figure 4A and B). Approximately a 90% reduction of procaspase 3 and 8 levels was observed after treatment of HT-29 cells with the nanoemulsion containing AN (0.16 ppm) plus LP (0.4 μM). However, with the combo treatment of AN (10 ppm) plus LP (12 μM), only a slight decline in the levels of procaspases 3 and 8 was shown when compared to control, implying that the nanoemulsion treatment could activate the death signal to a greater extent than the combo treatment. A similar outcome was shown for the PARP-1 level (Figure 4C). It is possible that the nanoemulsion (AN 0.16 ppm + LP 0.4 μM) could penetrate into the nucleus of HT-29 cells for PARP-1 activation by engulfing into nuclear envelope folds, whereas the combo treatment failed to activate PARP-1.
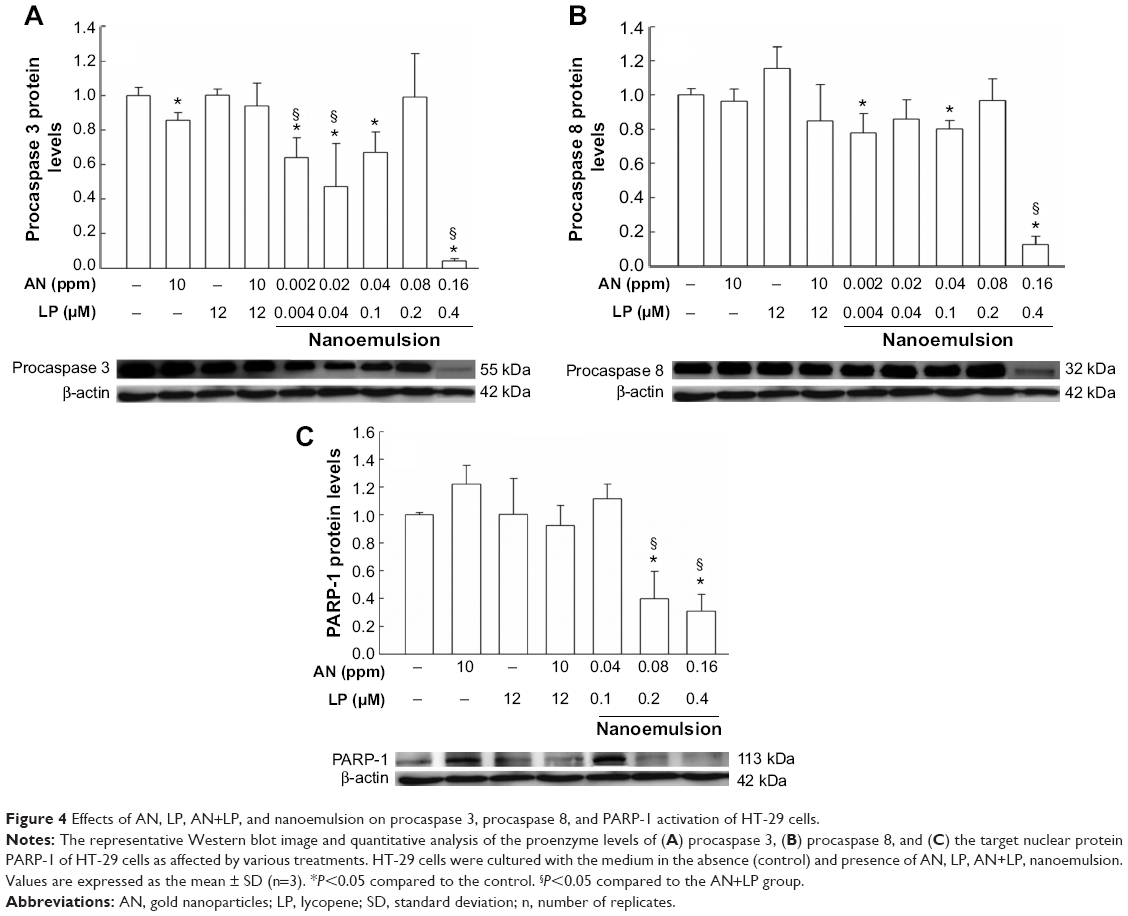 | Figure 4 Effects of AN, LP, AN+LP, and nanoemulsion on procaspase 3, procaspase 8, and PARP-1 activation of HT-29 cells. |
Effects of AN, LP, AN+LP, and nanoemulsion treatments on the mitochondria-associated apoptotic death signaling of HT-29 cells
As the mitochondria-associated death signals are alternative mediators of caspase 3 for the transduction of apoptotic death signaling, the antiapoptotic protein Bcl-2 and proapoptotic protein Bax were also assayed. The data in Figure 5A revealed that a 95% reduction of Bcl-2 was caused by the nanoemulsion treatment (AN 0.16 ppm + LP 0.4 μM), but only a 40% reduction was caused by the combo treatment (AN 10 ppm + LP 12 μM) when compared to control. Apparently, the nanoemulsion treatment possessed much higher antiapoptotic activity toward HT-29 cells than did the combo treatment. Similarly, compared to control, both the nanoemulsion (AN 0.16 ppm + LP 0.4 μM) and combo (AN 10 ppm + LP 12 μM) treatments showed a higher Bax level by 2.8- and 2.5-fold, respectively (Figure 5B). Surprisingly, a nanoemulsion treatment containing AN (0.08 ppm) plus LP (0.2 μM) was found to have a higher Bax level than that containing AN (0.16 ppm) plus LP (0.4 μM). Obviously, both Bcl-2 and Bax are the major contributors for the release of mitrochondria-associated signaling into the cytosolic compartment. For the procaspase 9 level, both the nanoemulsion (AN 0.16 ppm + LP 0.4 μM) and combo (AN 10 ppm + LP 12 μM) treatments were lower than control by 98% and 20%, respectively (Figure 5C).
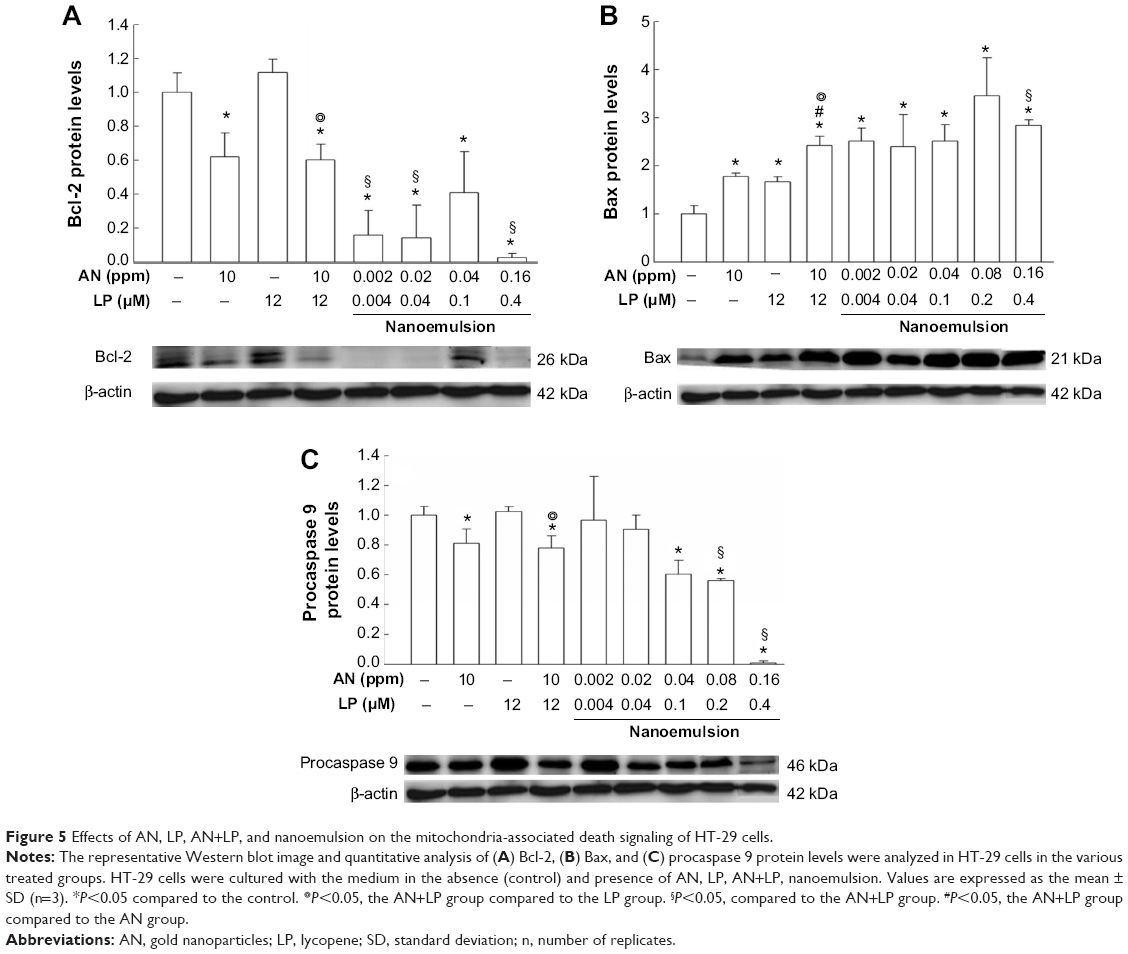 | Figure 5 Effects of AN, LP, AN+LP, and nanoemulsion on the mitochondria-associated death signaling of HT-29 cells. |
Effects of AN, LP, AN+LP, and nanoemulsion treatments on the migration of HT-29 cells
Figure 6A–F show the representative image photographs and quantitative migration data of the HT-29 cells as affected by the AN, LP, AN+LP, blank emulsion, and nanoemulsion treatments. Treatment of HT-29 cells with low doses of AN (0.02–0.16 ppm) or LP (0.1–0.8 μM) showed only a slight change in the migration rate of HT-29 cells (Figure 6B and C). However, with a higher dose of AN at 10 ppm, LP at 12 μM, and combo (AN 10 ppm + LP 12 μM), the migration rate was reduced by 89%, 49%, and 82%, respectively (Figure 6D). Comparatively, the nanoemulsion treatment containing AN 0.04 ppm plus LP 0.1 μM also showed a large drop in the migration rate of HT-29 cells by 85%, but with the blank emulsion volume at 1.4–14 μL, the migration rate only decreased by 5%–39% (Figure 6E). Most importantly, following a rise in the dose of AN and LP in the nanoemulsion, a further drop of the migration rate to zero was observed, demonstrating the high efficiency of the nanoemulsion in retarding the migration of HT-29 cells (Figure 6F). This phenomenon was supported by photographic images of HT-29 cells, as affected by various treatments in Figure 6A.
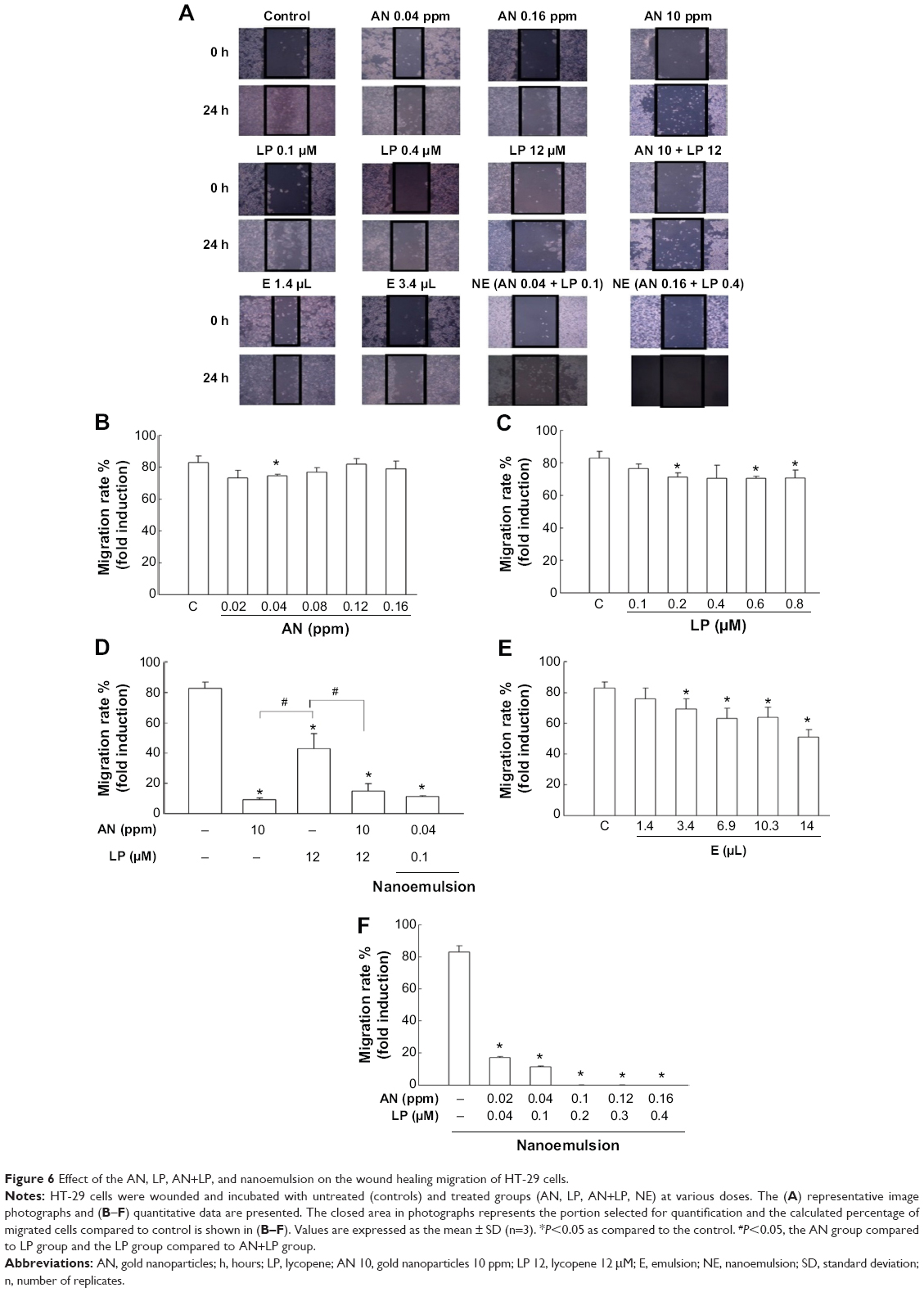 | Figure 6 Effect of the AN, LP, AN+LP, and nanoemulsion on the wound healing migration of HT-29 cells. |
Effects of AN, LP, AN+LP, and nanoemulsion treatments on invasion-associated signaling of HT-29 cells
Figure 7A–E shows the expressions of migration and invasion-associated signaling of HT-29 cells as affected by the AN, LP, AN+LP, and nanoemulsion treatments. It has been well established that the loss of the epithelial marker E-cadherin could induce mesenchymal cell transition (Vimentin marker), upregulate the proteolytic enzymes MMPs (MMP-2 and MMP-9), and initiate EMT progression so that cancer cells were detached from the tissues for migration and invasion. Compared to control, the nanoemulsion treatment (AN 0.04 ppm + LP 0.1 μM) significantly enhanced the expression of the epithelial marker E-cadherin (Figure 7A), downregulated its upstream master molecule Akt expression (Figure 7D) as well as NFκB expression, without altering EMT transition markers vimentin and β-catenin. Similarly, compared to control, the combo treatment (AN 10 ppm + LP 12 μM) could significantly elevate the expressions of the epithelial marker E-cadherin (Figure 7A), vimentin (Figure 7B), β-catenin (Figure 7C), and NFkB (Figure 7E), but reduced Akt expression (Figure 7D). In addition, the AN (10 ppm) alone could promote mesenchymal transition and inflammation through the upregulation of E-cadherin, vimentin, β-catenin, and NFkB (Figure 7A–C and E), whereas the LP alone elevated β-catenin and reduced Akt expressions (Figure 7C and D). Compared to the LP alone, the AN (10 ppm) alone could enhance the expressions of E-cadherin, vimentin, β-catenin, and NFkB to a larger extent (Figure 7A–C and E).
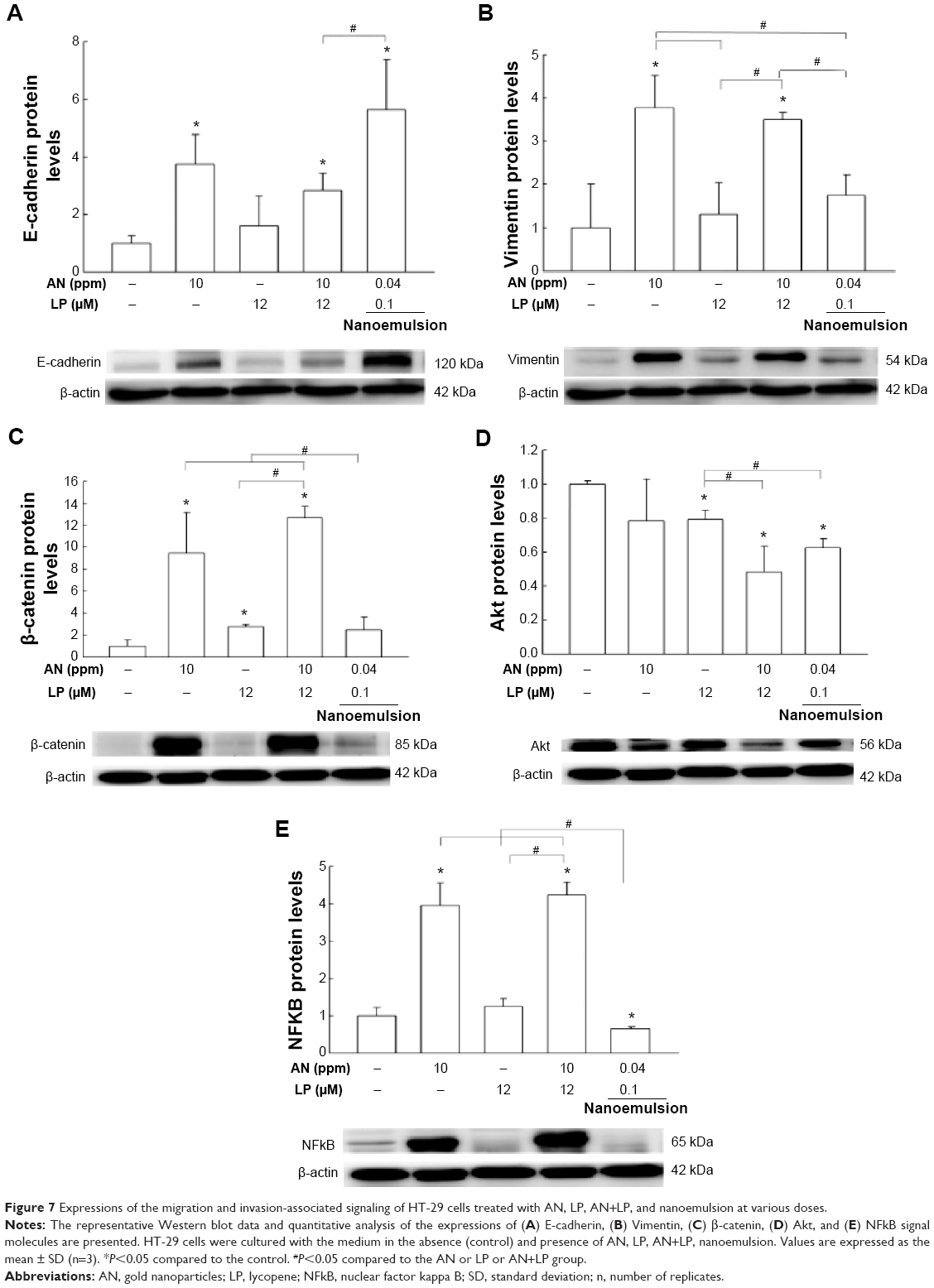 | Figure 7 Expressions of the migration and invasion-associated signaling of HT-29 cells treated with AN, LP, AN+LP, and nanoemulsion at various doses. |
During the EMT process, the connection between cells can be destroyed, resulting in a decline in cell adhesion and an increase in migration, while more MMPs can be secreted to act on the extracellular matrix, leading to the proliferation and angiogenesis of tumor cells.25 The expressions of MMP-2 and MMP-9 as affected by various treatments are shown in Figure 8A–E. Compared to control, the levels of pro-MMP-2 for the treatments of AN, combo (AN 10 ppm + LP 12 μM), and nanoemulsion (AN 0.04 ppm + LP 0.1 μM) were reduced by 0.4-, 0.5-, and 0.4-fold, respectively (Figure 8A and B). However, the pro-MMP-2 levels for the LP treatment alone, as well as the active MMP-2 levels for all the four treatments except control did not show any significant change (Figure 8A–C). On the contrary, for active MMP-9 expression, a larger decrease of 0.8-, 0.5-, 0.9-, and 0.7-fold was observed for the treatments of AN, LP, combo (AN 10 ppm + LP 12 μM), and nanoemulsion (AN 0.04 ppm + LP 0.1 μM), respectively (Figure 8D and E). This outcome suggested that all the four treatments could inhibit adhesion and migration of the HT-29 cells.
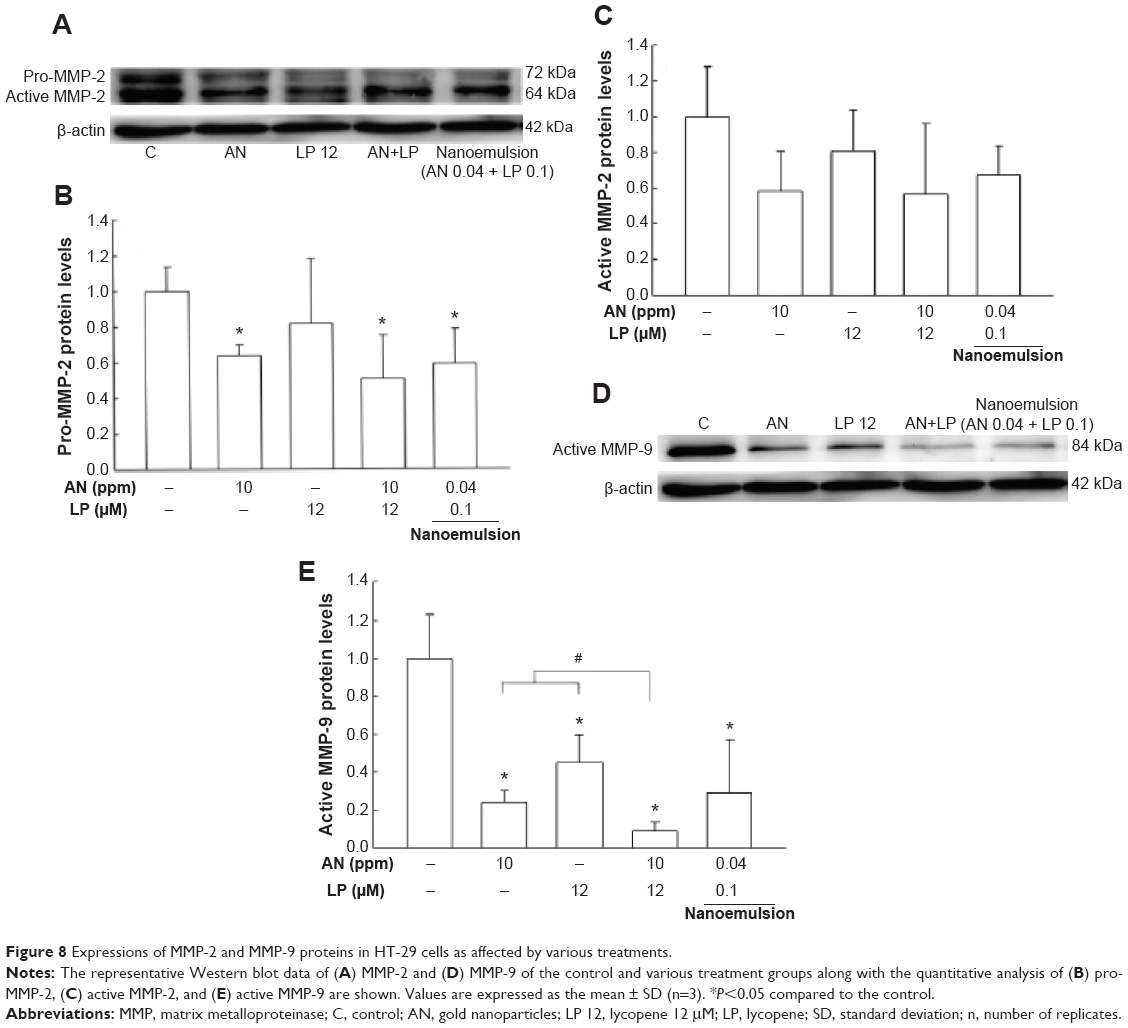 | Figure 8 Expressions of MMP-2 and MMP-9 proteins in HT-29 cells as affected by various treatments. |
Possible molecular and compartment targets of the nanoemulsion treatment on the migration and apoptosis of HT-29 cells
To further elucidate why the nanoemulsion was more effective in inducing apoptosis and reducing migration of the HT-29 cells than the AN or LP alone, TEM analysis was conducted to observe how the various treatments influenced the cellular microcompartments and molecular targets. As shown in Figure 9A, the untreated control HT-29 cells exhibited a normal nuclear organism with an intact nuclear double layer of the membrane envelope, nucleoli, and homogeneous euchromatin fibers (Figure 9Aa and b). In addition, the round and normal shape of the mitochondria with clear layers of cristae and few vacuoles resided in the cytosol for the control treatment, with the plasma membrane protruding as filaments, as shown in Figure 9Aa, possibly for cell-to-cell communication (Figure 9Aa and b). The HT-29 cells treated with AN alone at 10 ppm also possessed a similar morphology and shape (Figure 9B).
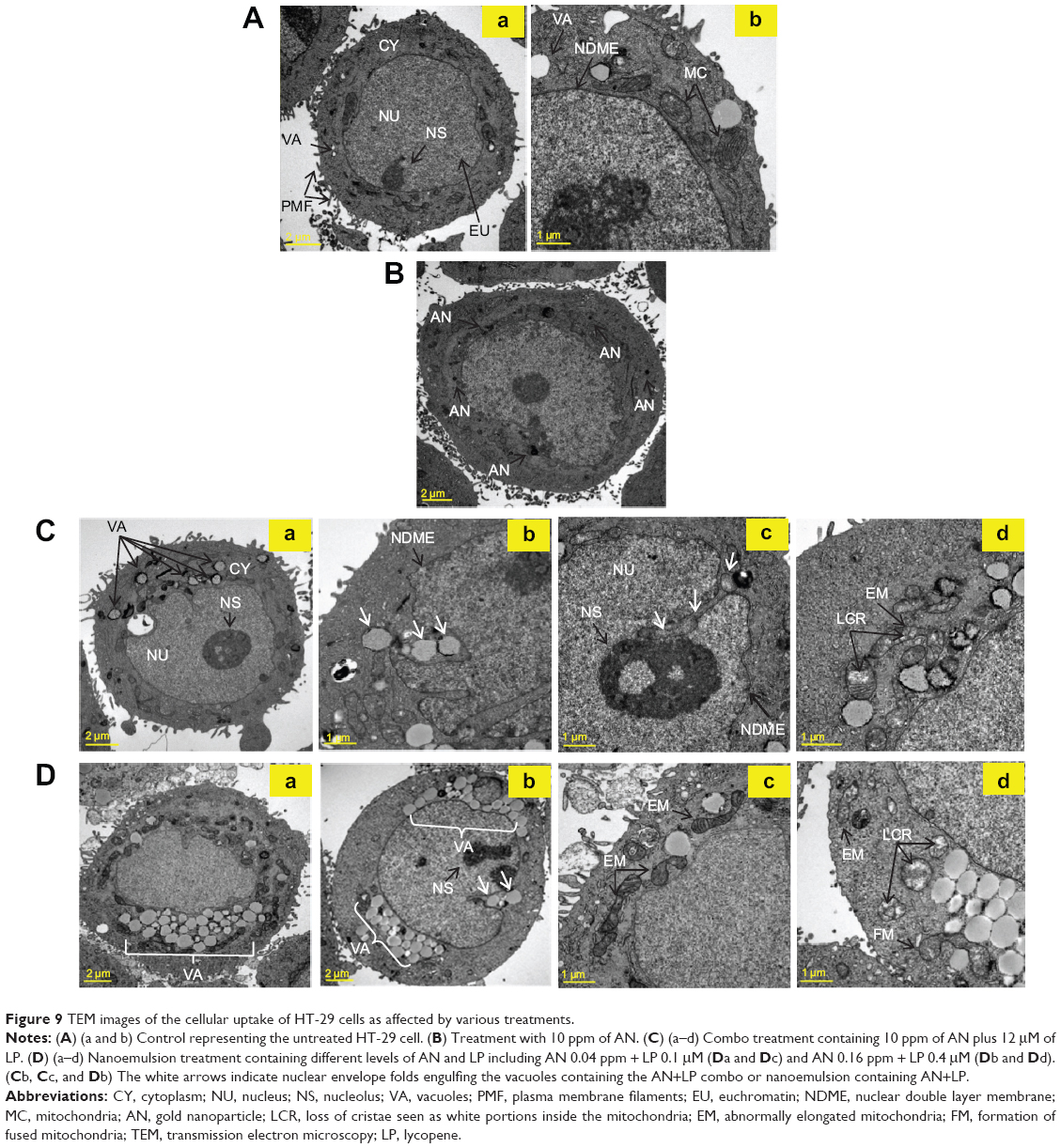 | Figure 9 TEM images of the cellular uptake of HT-29 cells as affected by various treatments. |
For cells treated with the combo group (AN 10 ppm + LP 12 μM) for 24 hours, several vacuoles surrounding the AN appeared in the cytosol (Figure 9Ca). Then, the AN-containing vacuoles were engulfed inside the nuclear envelop folds formed in the HT-29 cells for subsequent invasion into nucleoli, as evident in Figure 9Cb and c. As shown in Figure 9Cd, the AN-containing vacuoles were attached and emerged into the mitochondria, which displayed abnormally elongated morphology with reduced cristae and matrix contents. Compared to the combo treatment (Figure 9Ca), the nanoemulsion treatment at a lower dose could generate more vacuoles accumulated in the cytosolic compartment (Figure 9Da and b). The nuclear internalization of AN-containing vacuoles was also observed in Figure 9Db. Furthermore, the nanoemulsion treatment resulted in the mitochondria becoming more aberrantly elonged (Figure 9Dc), accompanied by a loss of cristae and matrix contents, as well as the formation of fused mitochondria (Figure 9Dd). As cristae are folds of the inner membrane of a mitochondrion that can provide an increase in surface area, the loss of cristae can retard both the electron transport chain and the chemiosmosis processes, leading to impairment of adenosine triphosphate production.
Discussion
Effect of size and dose of AN on HT-29 cell growth
It has been well documented that the size of AN has a great impact on cell toxicity. For instance, a size of 1.2 nm and 1.4 nm of AN was shown to induce cell apoptosis and necrosis, respectively, whereas a size of 15 nm did not show cell toxicity.26 However, in a similar study, Wen and Majetich27 reported a high toxicity for 8 nm AN and a medium toxicity for 37 nm AN. Likewise, a size of 4.8 nm AN was reported to possess high cell toxicity, while 46.6 nm AN exhibited low cell toxicity.28 More recently Huang et al29 studied the size-dependent localization and penetration of AN in breast cancer cells and found that after 3 hours and 24 hours of incubation, the accumulation of AN (2 nm and 6 nm) in the cytoplasm and cell nucleus followed a time-dependent increase, with the accumulation percentage in the nucleus being 15.5% for 2 nm AN and 16.6% for 6 nm AN, but a size of 15 nm AN did not show any accumulation. This outcome implied that the smaller the size of the AN, the more susceptible the penetration into the nucleus. In addition, the authors also studied the distribution of AN in various organs of breast tumor-bearing BALB/c mice with intravenous Injection of tiopronin-coated AN, and reported that the clearance times of 2 nm, 6 nm, and 15 nm AN from the blood were 24 hours, 8 hours, and 10 minutes, respectively.29 Also, both 2 nm and 6 nm AN were mainly accumulated in the kidney, while 15 nm AN deposited in the liver and spleen, with the penetration efficiency into the tumor cells being higher for 2 nm and 6 nm AN than for 15 nm AN. In a similar study dealing with the effect of various sized AN on the cytotoxicity of leukemia cell, k562, the following order was observed: 1–3 nm >5–6 nm >15–20 nm.20 Thus, the size of 3–5 nm AN used in our experiment should be able to penetrate into HT-29 cells to induce apoptosis or necrosis.
Effect of LP on HT-29 cell growth
As shown in the previous section, the IC50 was 16.79 μM for LP treatment with various doses. After LP treatment with 2 μM, 4 μM, 8 μM, and 12 μM for 24 hours, the early apoptosis of HT-29 cells rose by 1%, 4%, 11%, and 16%, whereas the late apoptosis increased by 5%, 4%, 20%, and 23%, and necrosis raised by 8%, 5%, 21%, and 24%, respectively. Taken together, the number of total apoptotic cells (early apoptosis plus late apoptosis) rose by 6%, 8%, 31%, and 39%, respectively, after LP treatment with 2 μM, 4 μM, 8 μM, and 12 μM for 24 hours. Obviously, with the high-dose (12 μM) treatment of LP, the proportion of HT-29 cells undergoing late apoptosis or necrosis was higher than early apoptosis. However, a slightly higher inhibition effect was reported by Tang et al30 as a 47% suppression of the HT-29 cells was observed after 24 hours of incubation with the 10 μM LP standard. The difference in antiproliferation effect may be accounted for by the source of LP used in cell culture (ie, in our experiment, LP paste from tomato was used as the raw material, while in Tang et al’s study,30 LP standard was employed). Also, in a similar study, LP was shown to suppress leptin-mediated cell invasion and MMP-7 expression in HT-29 cells, even at physiological concentrations ranging from 0.5 μM to 2 μM.31
Effect of combo treatment (AN plus LP) and nanoemulsion on HT-29 cell growth
With the combination of 10 ppm AN and 12 μM LP, a synergistic effect was shown when compared to AN or LP alone, as evidenced by a rise in the proportion of early apoptotic, late apoptotic, and necrotic cells. The same synergistic effect was found for LP–nanogold nanoemulsion. Comparatively, LP nanoemulsion could induce the early apoptosis of HT-29 cells to a greater extent than AN (10 ppm) alone and the combo treatment. However, there was no significant difference in late apoptotic and necrotic cells between LP–nanogold nanoemulsion and the combo treatment. In addition to size, the incubation time also plays a vital role for cell apoptosis and necrosis. For instance, Tsai et al20 studied the effect of 5–6 nm AN (5 ppm) on leukemia cell, k562, and reported that the proportion of cell apoptosis or necrosis rose following an increase in incubation time. A similar outcome was observed in our experiment, as shown by an increase of 20% in cell necrosis and 24% in cell apoptosis after treatment of HT-29 cells with 3–5 nm AN for 24 hours.
Effect of emulsifier on HT-29 cells
Due to the presence of the emulsifier in LP–nanogold nanoemulsion, it is imperative to elucidate if it can induce cell toxicity. The IC50 was shown to be 6.1 μL/mL medium after treatment with blank emulsion (without LP and AN) at 1.4 μL/mL, 3.4 μL/mL, 6.9 μL/mL, 10.3 μL/mL, and 14 μL/mL medium, separately, for 24 hours, accompanied by only a slight rise of 23% in early cell apoptosis/necrosis and 3%–5% in late cell apoptosis/necrosis (Figure S2). Apparently, the emulsifier (Tween 80) used for the preparation of nanoemulsion should not cause any significant cell death. For instance, with the same emulsifier dose at 1.4 μL/mL medium, LP–nanogold nanoemulsion induced a higher toxicity toward HT-29 cells than blank emulsion, implying that the cytotoxicity of nanoemulsion was mainly due to the presence of AN and LP.
Protein expression associated with apoptosis of HT-29 cells
As shown in Figures 4 and 5, with AN treatment alone, the expressions of procaspases 3 and 9, as well as Bcl-2 declined significantly, while the expressions of Bax and PARP-1 climbed when compared to control. A similar tendency was shown for the combo treatment (AN+LP) with the exception of PARP-1 expression. Comparatively, the expressions of procaspases 8 and 9 and Bcl-2 were significantly lower, and Bax significantly higher, for the combo treatment than for LP alone. But for the AN–LP nanoemulsion treatment, a low dose of AN (0.002 ppm) plus LP (0.004 μM) could result in lower expressions of procaspases 3 and 8 and Bcl-2, as well as higher expression of Bax than control. However, when compared to the combo treatment (AN+LP), the expressions of procaspase 3 and Bcl-2 were lower for this AN–LP nanoemulsion. A similar trend was observed for the nanoemulsion containing 0.02 ppm AN plus 0.04 μM LP. Nonetheless, with the exception of 0.08 ppm AN plus 0.2 μM LP, following a rise in dose of AN and LP in the nanoemulsion, the expressions of procaspases 3, 8, and 9, as well as Bcl-2 were significantly lower than control, but that of Bax was significantly higher. The most pronounced effect was observed for the nanoemulsion containing 0.16 ppm AN plus 0.4 μM LP, as shown by a large decline in the expressions of procaspases 3, 8, and 9, as well as Bcl-2 and PARP-1 when compared to control and the other treatments, while Bax expression was significantly higher than the control, AN or LP alone, and the combo treatment.
Accordingly, cell apoptosis can be divided into receptor-linked apoptosis and DNA damage-induced apoptosis, with the former including many death receptors (DR) such as Fas/CD95, TNF, DR3, DR4, DR5, and so on. Among the various apoptosis-associated proteins, caspase represents the most important one as it can be responsible for signal transduction, finally leading to apoptosis. The study associated with regulation in retarding early apoptosis often refers to various protein kinases prior to the initiation of caspase 3, which is regarded as an irreversible step toward apoptosis.32 Generally, caspase is synthesized in the cell in procaspase form, containing procaspases 2, 8, 9, and 10 as the initiator, and procaspases 3, 7, and 6 as the executioner, which can cleave death substrates such as PARP, lamin, CAD, and actin for apoptosis execution. Structurally, procaspases can be further divided into caspase recruitment domain including procaspases 1, 2, 4, 5, and 9, and death effector domain (DED) including procaspases 8 and 10. Proteins containing DED such as Fas-associated death domain are able to recruit procaspase 8 for the initiation of caspase 8 which, in turn, activates caspase 3 for the cleavage of protein substrate to induce apoptosis. However, the initiation of procaspase 9 is mainly due to the interaction between apoptotic protease activation factor 1 (Apaf-1) and cytochrome C.33 For apoptosis-regulated proteins, mitochondria can be regarded as a target cell for both the Bcl-2 family (inhibit apoptosis) and Bax family (promote apoptosis), as the hydrophobic peptide at the C-end of the former (Bcl-2) can be inserted into the outer membrane of the mitochondria to prevent a decline in mitochondria potential, and the latter (Bax) to lower membrane potential. Thus, the mitochondria are important regulators of the cell death signal route.34 In a similar study, Tsai et al20 also reported that AN could activate caspase 3 to induce apoptosis after the treatment of leukemia cell, k562, with AN (5–6 nm) for 24 hours. More recently, Choudhury et al35 further pointed out that AN could penetrate into lung cancer cell, A549, for conjugation with micro-organelle to induce apoptosis with arrest at G0/G1. Also, with AN treatment, both Bax and P53 expressions were upregulated, while both Bcl-2 and PARP-1 expressions were downregulated.35 In our experiment with the treatment of the nanoemulsion containing AN (0.16 ppm) plus LP (0.4 μM), the expressions of procaspases 8, 3, and 9, as well as PARP-1 and Bcl-2 declined, while the Bax expression rose. Thus, it was postulated that the AN–LP nanoemulsion could accelerate apoptosis through the activation of caspases 8, 9, and 3, and through regulation by the mitochondria. Nevertheless, the indicator associated with late apoptosis needs to be further explored.
Migration of HT-29 cells
As indicated before, with the treatments of AN alone (10 ppm) and the nanoemulsion of AN (0.04 ppm) plus LP (0.1 μM) for 24 hours, the cell migration was reduced by 74% and 72%, respectively. It is worth pointing out that the dose of 0.04 ppm AN in the nanoemulsion was 250-fold lower than that of the AN (10 ppm) alone, while the dose of 0.1 μM LP in the nanoemulsion was 120-fold lower than that of LP (12 μM) alone. This finding further demonstrated that a synergistic effect did occur for the AN–LP nanoemulsion. Most importantly, the doses of AN and LP in the nanoemulsion could be reduced greatly to inhibit cell migration. In a study dealing with the effect of various doses of AN (80 nm) combined with the antioxidant ginkgolide A (GA-GNP) on vascular smooth muscle cell proliferation and migration in vitro, and neointimal hyperplasia in a mouse model, Weakley et al36 reported that GA-GNP could reduce the expression of superoxide anions and inhibit the proliferation and migration of vascular smooth muscle cells through the downregulation of ERK1/2 which, in turn, remedied topical arterial damage and reduced inner membrane proliferation and subsequent narrowing.
Malignant colon cancer develops as a multistep process involving a series of aberrant genetic, metabolic, and signaling changes in cancerous cells that evolve toward more aggressive, poorly differentiated phenotypes and acquired metastatic capacity via EMT.37 Alteration of EMT by downregulation of E-cadherin, an epithelial cell marker, can cause cancer cells to lose their epithelial junction and begin migration. Mesenchymal cancer cells, as marked by the expression of Vimentin and E-cadherin, express MMP to enable the digestion of matrix proteins for invasion and metastatic spread.38 It is notable that the expression of the epithelial cell adhesion factor, E-cadherin, was significantly higher for the nanoemulsion containing 0.04 ppm AN plus 0.1 μM LP than the AN alone and the combo treatment, demonstrating a synergistically protective effect against the migration development of colon cancer cells through reinforcement of the epithelial marker for membrane attachment. Unlike the AN alone and combo treatments, which generated higher expressions of mesenchymal protein Vimentin than control by 3.7- and 3.4-fold, respectively, the nanoemulsion treatment did not significantly enhance Vimentin and β-catenin expressions, but they did suppress pro-MMP-2 and active MMP-9 expressions of the untreated control cells. This finding collectively supports the suppressive effect of the nanoemulsion on the EMT development of colon cancer cells. In a previous study only dealing with the effect of LP on tumor growth and progression in a mouse xenograft model of colon cancer, Tang et al39 reported that LP could enhance the expressions of E-cadherin and P21/CIP1/WAF1 through the inhibition of proliferating cell nuclear antigen and β-catenin in the nucleus, as well as expressions of COX-2, prostaglandin E2, and phosphorylated ERK1/2. In addition, LP was shown to be effective in inhibiting tumor growth and progression in a mouse xenograft model of colon cancer, with the inhibition being inversely proportional to the MMP-9 concentration in mouse blood.39 The response of mesenchymal stem cells toward fibronectin–gold nanocomposites was studied by Hung et al,40 illustrating that this treatment could enhance cell proliferation, decrease reactive oxygen species production, and increase expressions of MMP-9 and endothelial nitric oxide synthase which, in turn, affected the migration of human umbilical mesenchymal stem cells. Additionally, AN (15 nm) was found to be efficient in inhibiting HePG2-CM-induced proliferation and migration of epithelial cells through the downregulation of vascular endothelial growth factor (VEGF) after incubation of human umbilical vein endothelial cells in HePG2-CM medium.40 In the present study, we demonstrated that the AN–LP nanoemulsion relative to the LP only treatment exhibited a higher suppressive effect in the migration of the EMT markers of HT-29 cells, as evident by the enhanced expression of E-cadherin and the decreased expressions of Akt and NFkB. Nonetheless, the detailed mechanism of the AN–LP nanoemulsion in inhibiting the migration of cancer cells needs to be further explored.
Mechanism of signal transduction initiation
For NFkB expression, only the treatments of AN alone and in combination were significantly higher than the control. NFkB is a vital protein that can promote tumor progression, with the most common dimer forms being P50/P65 and P52/P65. Thus, the specific antibody, P65, was selected as an indicator. It has been well documented that IkB, a downregulated protein conjugated with NFkB, and which is responsible for the inhibition of NFkB function in the cytoplasm, can undergo phosphorylation to lose its function due to ubiquitination after receiving external transduction, resulting in NFkB release for migration into the nucleus, and conjugation with a gene-regulated promoter for activation.41 A previous study illustrated that in addition to inflammation, NFkB can also be associated with cancer progression, including cell apoptosis, cell cycle regulation, differentiation, and migration.42 Nevertheless, the role of NFkB in cell apoptosis and cancer progression is controversial. For instance, Fas as a tumor-inhibiting protein can conjugate with NFkB on many FAS promoter sites for regulation of FAS transcription. In other words, a typical NFkB can activate FAS for transcription, while a spare NFkB can inhibit FAS function. Thus, the inhibition of NFkB in cancer progression is probably achieved through the retardation of damage of the host immune cells caused by Fas-induced cell apoptosis.43 Additionally, it has been well established that NFkB can affect colon cancer progression through the regulation of many associated genes such as cyclin D1, Bcl-XL, VEGF, IL-8, COX-2, and MMP-9.44 Most importantly, the current drugs used for colon cancer therapy have been demonstrated to be able to regulate NFkB expression which, in turn, inhibit cancer cell proliferation and migration.44 In a previous study, Hsu and Huang45 reported that during folic acid deficiency or supplementation, the NFkB activation could be regulated through the PI3K/Akt and Hh transduction route which, in turn, affected inflammation of the colon cancer cell line, HCT116, through the further regulation of cytokine TNF-α expression. In some other related studies, K-Ras mutation was shown to account for 40% and B-Raf mutation for 10% in colorectal cancer.46 Furthermore, both K-Ras and B-Raf could activate signal transduction route of MAPKs/ERK47 which, in turn, affected tumor progression through the regulation of cell survival, proliferation, and apoptosis.48 Also, K-Ras was shown to activate the P13k/Akt transduction route,49 with the mutation of K-Ras and P13K accounting for 22% in colorectal cancer.50 A similar outcome was observed by Tang et al30 who demonstrated that LP was not only effective at suppressing Akt activation and nonphosphorylated β-catenin levels in colon cancer cell line, HT-29, but that it also enhanced the phosphorylation level of β-catenin leading to proteosomal degradation through the downregulation of the Wntl/β-catenin pathway. Similarly, Lin et al51 illustrated that LP could not only enhance the expression and stability of E-cadherin, but it also inhibited the phosphorylation of Akt and the expression of MMP-7, a MMP modulated by extracellular growth factors and proinflammatory cytokines in cancer cells for degradation of the extracellular matrix and creation of a microenvironment to promote tumor angiogenensis and metastasis. As the Akt transduction route can activate partial antiapoptosis molecules,52 it was postulated that the antiproliferation effect of colon cancer cell line, HT-29, by the nanoemulsion (AN 0.04 ppm + LP 0.1 μM) observed in our study should be due to the regulation of NFkB expression through the PI3K/Akt transduction route.
Targeting effect of nanoemulsion
Nanoparticle size has been shown to play a vital role in cell uptake efficiency. In a study dealing with cell type-dependent uptake, as well as the localization and cytotoxicity of 1.9 nm AN, Coulter et al8 reported that AN uptake preferentially occurred in tumor cells, resulting in an increased expression of cleaved caspases, but that AN cytotoxicity remained low. Furthermore, the authors observed small electron-dense AN in an unaggregated state inside the nuclear membrane, which should enhance DNA damage and the formation of lethal double-strand breaks.8 It was also suggested that the clathrin-mediated endocytosis is the main uptake route for particles up to 200 nm, after which caveolae-mediated internalization dominates.8,53 More specifically, the optimal size for clathrin-mediated endocytosis was shown to be in the range of 25–30 nm, as reduced uptake was found for small (<10 nm) or large (>100 nm) particles.8,54
The penetration of nanoemulsion into the cytoplasm and its subsequent engulfing inside the nuclear envelope folds, as shown in Figure 9D, indicated that this nanoemulsion may be used as a delivery system for passive targeting through the EPR effect. Among the various organelles, the nucleus is the most important one for the proliferation and apoptosis of cells. Most importantly, controlling the processes governed by the nucleus has been a primary goal for nuclear-targeted cancer therapy.55 According to literature reports, the endothelial cells found in the blood vessels of normal tissues may have gaps between them that are about 7–10 nm in size, but these gaps may increase to a few hundred nanometers in the tumor microvasculature to allow for nanoparticle accumulation through the EPR effect.56,57 Thus, the nanoemulsion should be able to diffuse from the extracellular matrix into the cytoplasm and nucleus for antitumor efficiency. In a review paper dealing with the impact of nanotechnology on drug delivery, Farokhzad and Langer56 pointed out that tumor tissue accumulation is a passive process requiring long circulation to facilitate the time-dependent extravasations of drug delivery systems through the leaky tumor microvasculature for accumulation. Nevertheless, more in vivo studies are needed to be carried out to elucidate the underlying mechanism in anti-colon tumor growth by the nanoemulsion developed in our study.
Acknowledgment
The authors wish to thank Mr Yen-Sheng Wu from Tzong Jao Hang’s Electron Microscope Laboratory, School of Medicine, Fu Jen Catholic University, Taipei, Taiwan for technical assistance in recording the transmission electron microscopic images.
Disclosure
The authors report no conflicts of interest in this work.
References
Cunningham D, Atkin W, Lenz HJ, et al. Colorectal cancer. Lancet. 2010;375(9719):1030–1047. | ||
Yamada T, Mori Y, Hayashi R, et al. Suppression of intestinal polyposis in Mdr1-deficient ApcMin/+ mice. Cancer Res. 2003;63(5):895–901. | ||
Winawer SJ, Zauber AG, Gerdes H, et al. Risk of colorectal cancer in the families of patients with adenomatous polyps. National Polyp Study Workgroup. N Engl J Med. 1996;334(2):82–87. | ||
Thiery JP, Sleeman JP. Complex networks orchestrate epithelial-mesenchymal transitions. Nat Rev Mol Cell Biol. 2006;7(2):131–142. | ||
Garber K. Epithelial-to-mesenchymal transition is important to metastasis, but questions remain. J Natl Cancer Inst. 2008;100(4):232–233, 239. | ||
Siddiqui IA, Adhami VM, Chamcheu JC, Mukhtar H. Impact of nanotechnology in cancer: emphasis on nanochemoprevention. Int J Nanomedicine. 2012;7:591–605. | ||
Oberdörster G. Nanotoxicology: in vitro-in vivo dosimetry. Environ Health Perspect. 2012;120(1):A13; author reply A13. | ||
Coulter JA, Jain S, Butterworth KT, et al. Cell type-dependent uptake, localization, and cytotoxicity of 1.9 nm gold nanoparticles. Int J Nanomedicine. 2012;7:2673–2685. | ||
Lu W, Zhang G, Zhang R, et al. Tumor site-specific silencing of NF-kappaB p65 by targeted hollow gold nanosphere-mediated photothermal transfection. Cancer Res. 2010;70(8):3177–3188. | ||
Paciotti GF, Myer L, Weinreich D, et al. Colloidal gold: a novel nanoparticle vector for tumor directed drug delivery. Drug Deliv. 2004; 11(3):169–183. | ||
Leu JG, Chen SA, Chen HM, et al. The effects of gold nanoparticles in wound healing with antioxidant epigallocatechin gallate and α-lipoic acid. Nanomedicine. 2012;8(5):767–775. | ||
Torchilin VP. Passive and active drug targeting: drug delivery to tumors as an example. Handb Exp Pharmacol. 2010;(197):3–53. | ||
Zhao C, Feng Q, Dou Z, et al. Local targeted therapy of liver metastasis from colon cancer by galactosylated liposome encapsulated with doxorubicin. PLoS One. 2013;8(9):e73860. | ||
Mein JR, Lian F, Wang XD. Biological activity of lycopene metabolites: implications for cancer prevention. Nutr Rev. 2008;66(12):667–683. | ||
Erhardt JG, Meisner C, Bode JC, Bode C. Lycopene, beta-carotene, and colorectal adenomas. Am J Clin Nutr. 2003;78(6):1219–1224. | ||
Lian F, Smith DE, Ernst H, Russell RM, Wang XD. Apo-10′-lycopenoic acid inhibits lung cancer cell growth in vitro, and suppresses lung tumorigenesis in the A/J mouse model in vivo. Carcinogenesis. 2007;28(7):1567–1574. | ||
Chen YJ, Inbaraj BS, Pu YS, Chen BH. Development of lycopene micelle and lycopene chylomicron and a comparison of bioavailability. Nanotechnology. 2014;25(15):155102. | ||
Chen BH [webpage on the Internet]. Tomato extract provides lycopene with improved bioavailability. Bristol, UK: Nanotechweb.org, IOP Publishing; 2014. Available from: http://www.nanotechweb.org/cws/article/lab/56668. Accessed. | ||
Akhter S, Jain GK, Ahmad FJ, et al. Investigation of nanoemulsion system for transdermal delivery of domperidone: ex-vivo and in vivo studies. Curr Nanosci. 2008;4:381–390. | ||
Tsai YY, Huang YH, Chao YL, et al. Identification of the nanogold particle-induced endoplasmic reticulum stress by omic techniques and systems biology analysis. ACS Nano. 2011;5(12):9354–9369. | ||
Ford NA, Elsen AC, Zuniga K, Lindshield BL, Erdman JW. Lycopene and apo-12′-lycopenal reduce cell proliferation and alter cell cycle progression in human prostate cancer cells. Nutr Cancer. 2011;63(2): 256–263. | ||
Makon-Sébastien N, Francis F, Eric S, et al. Lycopene modulates THP1 and Caco2 cells inflammatory state through transcriptional and nontranscriptional processes. Mediators Inflamm. 2014;2014:507272. | ||
Hsu BY, Pu YS, Inbaraj BS, Chen BH. An improved high performance liquid chromatography-diode array detection-mass spectrometry method for determination of carotenoids and their precursors phytoene and phytofluene in human serum. J Chromatogr B Analyt Technol Biomed Life Sci. 2012;899:36–45. | ||
Statistical Analysis System. SAS Procedures and SAS/Graph User’s Guide, Version 6. Cary, NC: SAS Institute, Inc.; 2011. | ||
Min C, Eddy SF, Sherr DH, Sonenshein GE. NF-kappaB and epithelial to mesenchymal transition of cancer. J Cell Biochem. 2008;104(3): 733–744. | ||
Pan Y, Neuss S, Leifert A, et al. Size-dependent cytotoxicity of gold nanoparticles. Small. 2007;3(11):1941–1949. | ||
Wen T, Majetich SA. Ultra-large-area self-assembled monolayers of nanoparticles. ACS Nano. 2011;5(11):8868–8876. | ||
Zhang XD, Wu D, Shen X, et al. Size-dependent radiosensitization of PEG-coated gold nanoparticles for cancer radiation therapy. Biomaterials. 2012;33(27):6408–6419. | ||
Huang K, Ma H, Liu J, et al. Size-dependent localization and penetration of ultrasmall gold nanoparticles in cancer cells, multicellular spheroids, and tumors in vivo. ACS Nano. 2012;6(5):4483–4493. | ||
Tang FY, Shih CJ, Cheng LH, Ho HJ, Chen HJ. Lycopene inhibits growth of human colon cancer cells via suppression of the Akt signaling pathway. Mol Nutr Food Res. 2008;52(6):646–654. | ||
Lin MC, Wang FY, Kuo YH, Tang FY. Cancer chemopreventive effects of lycopene: suppression of MMP-7 expression and cell invasion in human colon cancer cells. J Agric Food Chem. 2011;59(20): 11304–11318. | ||
Bevers EM, Comfurius P, Dekkers DW, Harmsma M, Zwaal RF. Regulatory mechanisms of transmembrane phospholipid distributions and pathophysiological implications of transbilayer lipid scrambling. Lupus. 1998;7 Suppl 2:S126–S131. | ||
McConkey DJ. Biochemical determinants of apoptosis and necrosis. Toxicol Lett. 1998;99(3):157–168. | ||
Kluck RM, Bossy-Wetzel E, Green DR, Newmeyer DD. The release of cytochrome c from mitochondria: a primary site for Bcl-2 regulation of apoptosis. Science. 1997;275(5303):1132–1136. | ||
Choudhury D, Xavier PL, Chaudhari K, et al. Unprecedented inhibition of tubulin polymerization directed by gold nanoparticles inducing cell cycle arrest and apoptosis. Nanoscale. 2013;5(10):4476–4489. | ||
Weakley SM, Wang X, Mu H, et al. Ginkgolide A-gold nanoparticles inhibit vascular smooth muscle proliferation and migration in vitro and reduce neointimal hyperplasia in a mouse model. J Surg Res. 2011; 171(1):31–39. | ||
Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100(1): 57–70. | ||
Huber MA, Kraut N, Beug H. Molecular requirements for epithelial-mesenchymal transition during tumor progression. Curr Opin Cell Biol. 2005;17(5):548–558. | ||
Tang FY, Pai MH, Kuo YH, Wang XD. Concomitant consumption of lycopene and fish oil inhibits tumor growth and progression in a mouse xenograft model of colon cancer. Mol Nutr Food Res. 2012;56(10): 1520–1531. | ||
Hung HS, Tang CM, Lin CH, et al. Biocompatibility and favorable response of mesenchymal stem cells on fibronectin-gold nanocomposites. PLoS One. 2013;8(6):e65738. | ||
Ghosh S, Karin M. Missing pieces in the NF-kappaB puzzle. Cell. 2002;109 Suppl:S81–S96. | ||
Aggarwal BB. Nuclear factor-kappaB: the enemy within. Cancer Cell. 2004;6(3):203–208. | ||
Liu F, Bardhan K, Yang D, et al. NF-κB directly regulates Fas transcription to modulate Fas-mediated apoptosis and tumor suppression. J Biol Chem. 2012;287(30):25530–25540. | ||
Bassères DS, Baldwin AS. Nuclear factor-kappaB and inhibitor of kappaB kinase pathways in oncogenic initiation and progression. Oncogene. 2006;25(51):6817–6830. | ||
Hsu SH, Huang RFS. Inflammatory-associated nuclear factor-B and invasion-related signaling pathway in human colorectal carcinoma cells. Nutr Sci J. 2012;38:29–39. | ||
Fransén K, Klintenäs M, Osterström A, Dimberg J, Monstein HJ, Söderkvist P. Mutation analysis of the BRAF, ARAF and RAF-1 genes in human colorectal adenocarcinomas. Carcinogenesis. 2004;25(4): 527–533. | ||
Yuen ST, Davies H, Chan TL, et al. Similarity of the phenotypic patterns associated with BRAF and KRAS mutations in colorectal neoplasia. Cancer Res. 2002;62(22):6451–6455. | ||
Fang JY, Richardson BC. The MAPK signalling pathways and colorectal cancer. Lancet Oncol. 2005;6(5):322–327. | ||
Repasky GA, Chenette EJ, Der CJ. Renewing the conspiracy theory debate: does Raf function alone to mediate Ras oncogenesis? Trends Cell Biol. 2004;14(11):639–647. | ||
Parsons DW, Wang TL, Samuels Y, et al. Colorectal cancer: mutations in a signalling pathway. Nature. 2005;436(7052):792. | ||
Lin MC, Wang FY, Kuo YH, Tang FY. Cancer chemopreventive effects of lycopene: suppression of MMP-7 expression and cell invasion in human colon cancer cells. J Agric Food Chem. 2011;59(20): 11304–11318. | ||
Meier F, Schittek B, Busch S, et al. The RAS/RAF/MEK/ERK and PI3K/AKT signaling pathways present molecular targets for the effective treatment of advanced melanoma. Front Biosci. 2005;10:2986–3001. | ||
Rejman J, Oberle V, Zuhorn IS, Hoekstra D. Size-dependent internalization of particles via the pathways of clathrin- and caveolae-mediated endocytosis. Biochem J. 2004;377(Pt 1):159–169. | ||
Zhang S, Li J, Lykotrafitis G, Bao G, Suresh S. Size-dependent endocytosis of nanoparticles. Adv Mater. 2009;21:419–424. | ||
Dam DH, Lee JH, Sisco PN, et al. Direct observation of nanoparticle-cancer cell nucleus interactions. ACS Nano. 2012;6(4):3318–3326. | ||
Farokhzad OC, Langer R. Impact of nanotechnology on drug delivery. ACS Nano. 2009;3(1):16–20. | ||
Frank D, Tyagi C, Tomar L, et al. Overview of the role of nanotechnological innovations in the detection and treatment of solid tumors. Int J Nanomedicine. 2014;9:589–613. |
Supplementary materials
 | Figure S1 Representative microphotographs of MRC-5 cells exposed to AN (10 ppm), LP (12 μM), AN+LP (10 ppm, 12 μM), nanoemulsion (AN 0.16 ppm + LP 0.4 μM), and blank E (1.4 μL). |
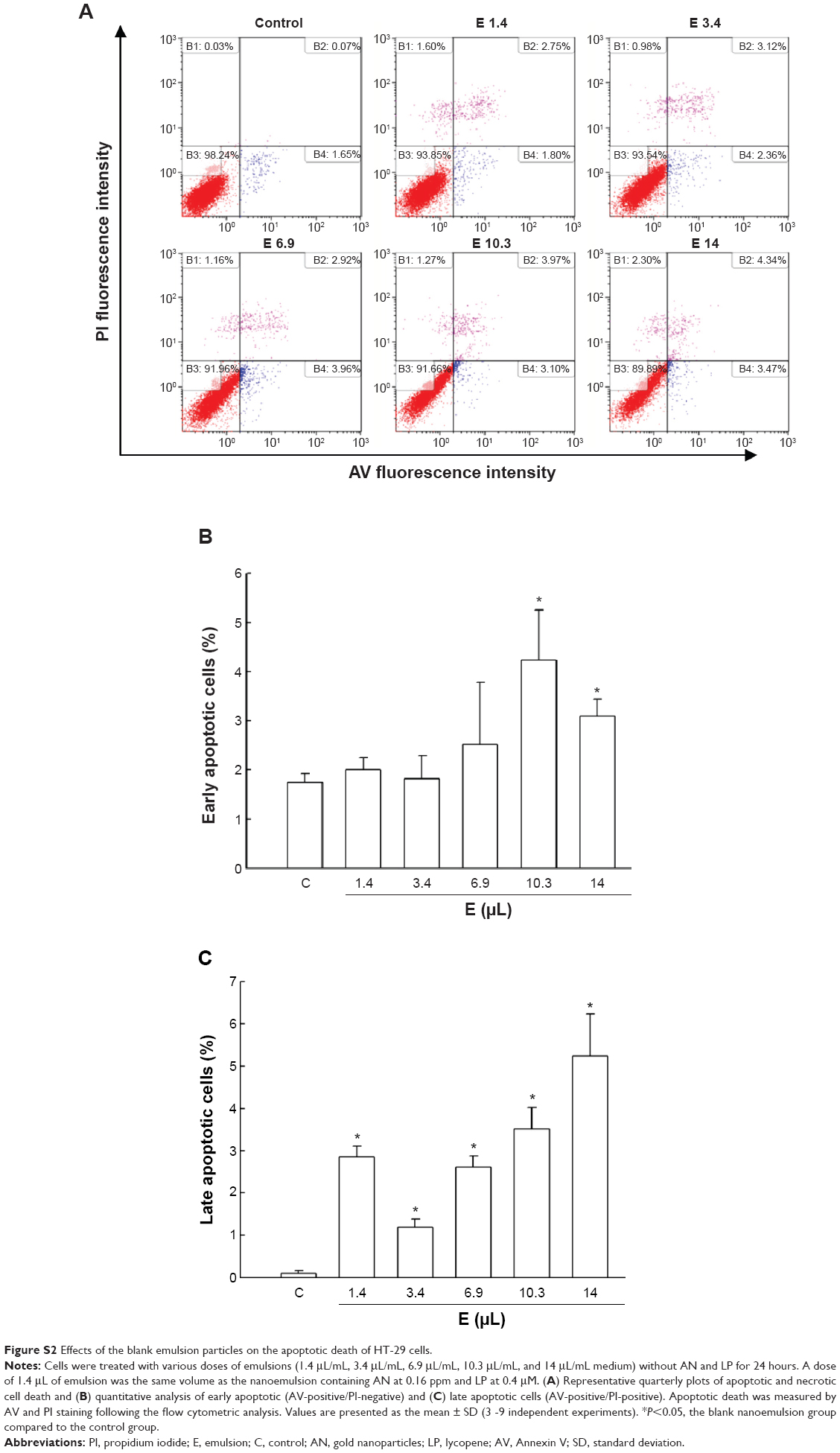 | Figure S2 Effects of the blank emulsion particles on the apoptotic death of HT-29 cells. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.