Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 11 » Issue 1
Inhaled corticosteroid normalizes some but not all airway vascular remodeling in COPD
Authors Soltani A, Walters EH, Reid D, Dhar Shukla S ![]() , Nowrin K, Ward C
, Nowrin K, Ward C ![]() , Muller HK, Singh Sohal S
, Muller HK, Singh Sohal S
Received 19 May 2016
Accepted for publication 7 July 2016
Published 22 September 2016 Volume 2016:11(1) Pages 2359—2367
DOI https://doi.org/10.2147/COPD.S113176
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Richard Russell
Amir Soltani,1 Eugene Haydn Walters,1,* David W Reid,1,2 Shakti Dhar Shukla,1 Kaosia Nowrin,1 Chris Ward,3 H Konrad Muller,1 Sukhwinder Singh Sohal1,4,*
1NHMRC Center of Research Excellence for Chronic Respiratory Disease, School of Medicine, University of Tasmania, Hobart, TAS, Australia; 2Iron Metabolism Laboratory, Queensland Institute of Medical Research, Brisbane, QLD, Australia; 3Institute of Cellular Medicine, Newcastle University, Newcastle upon Tyne, Tyne and Wear, UK; 4School of Health Sciences, University of Tasmania, Launceston, TAS, Australia
*These authors contributed equally to this work
Background: This study assessed the effects of inhaled corticosteroid (ICS) on airway vascular remodeling in chronic obstructive pulmonary disease (COPD).
Methods: Thirty-four subjects with mild-to-moderate COPD were randomly allocated 2:1 to ICS or placebo treatment in a double-blinded clinical trial over 6 months. Available tissue was compared before and after treatment for vessel density, and expression of VEGF, TGF-β1, and TGF-β1-related phosphorylated transcription factors p-SMAD 2/3. This clinical trial has been registered and allocated with the Australian New Zealand Clinical Trials Registry (ANZCTR) on 17/10/2012 with reference number ACTRN12612001111864.
Results: There were no significant baseline differences between treatment groups. With ICS, vessels and angiogenic factors did not change in hypervascular reticular basement membrane, but in the hypovascular lamina propria (LP), vessels increased and this had a proportionate effect on lung air trapping. There was modest evidence for a reduction in LP vessels staining for VEGF with ICS treatment, but a marked and significant reduction in p-SMAD 2/3 expression.
Conclusion: Six-month high-dose ICS treatment had little effect on hypervascularity or angiogenic growth factors in the reticular basement membrane in COPD, but normalized hypovascularity in the LP, and this was physiologically relevant, though accompanied by a paradoxical reduction in growth factor expression.
Keywords: airway remodeling, bronchial biopsy, COPD, inhaled corticosteroid, vascular remodeling
Introduction
Smoking and resulting chronic obstructive pulmonary disease (COPD) are major worldwide health problems.1 Previous insights into the details of airway remodeling in the airway wall in COPD have been quite limited, and our knowledge about the effects of inhaled corticosteroid (ICS) on such airway remodeling is even scantier.
We have reported some novel characteristics of airway remodeling using bronchial biopsies (BB) in COPD. For example, we found that the subepithelial reticular basement membrane (Rbm) was markedly fragmented and hypervascular (Figure 1), in association with increased vessel expression of angiogenic factors VEGF and TGF-β1.2–7 Hiroshima et al8 have reported vessel growth up into the epithelium in COPD. These changes in combination are reminiscent of epithelial–mesenchymal transition (EMT) type-3, which is thought of as a procancerous condition.4,6,9
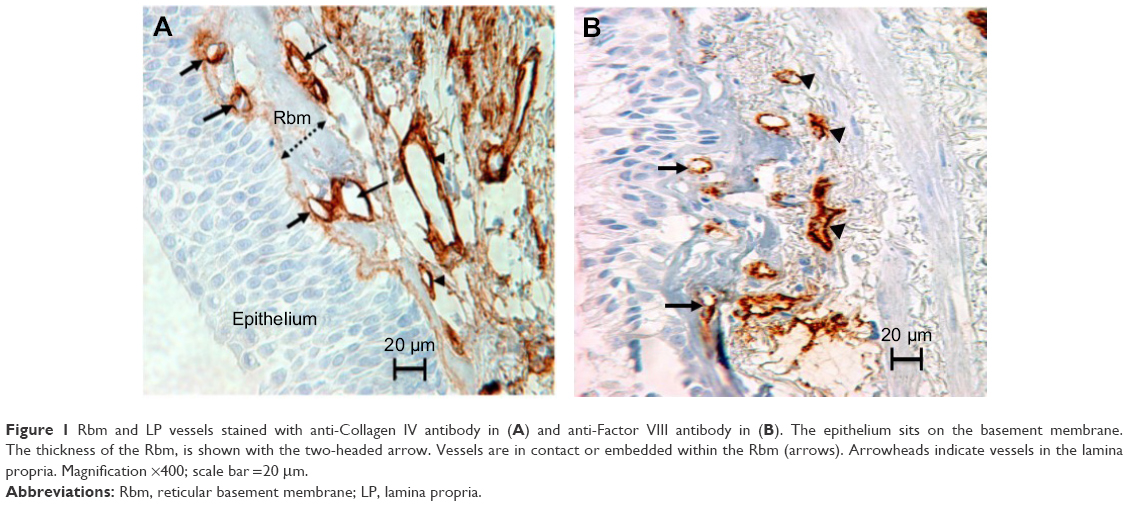 | Figure 1 Rbm and LP vessels stained with anti-Collagen IV antibody in (A) and anti-Factor VIII antibody in (B). The epithelium sits on the basement membrane. The thickness of the Rbm, is shown with the two-headed arrow. Vessels are in contact or embedded within the Rbm (arrows). Arrowheads indicate vessels in the lamina propria. Magnification ×400; scale bar =20 μm. |
In contrast to the Rbm and epithelium, we described the subepithelial/sub-Rbm lamina propria (LP) as being hypovascular in current smokers with COPD.4,7
ICS has become standard treatment in more severe COPD, on the basis of empirical results from large multicenter studies.1,10,11 Studies have shown some limited clinical improvement, anti-inflammatory effects, and also changes in extracellular matrix in airways with ICS treatment.11–16 Some positive effects on mortality and a protective effect of ICS on lung cancer development have also been reported in COPD.11,17–20 The mechanisms of any lung (airway) cancer protection with ICS are not clear,20 although we have demonstrated, using airway biopsies, that ICS therapy improves EMT-related changes.9,21
Our group has previously shown that ICS therapy has antiangiogenic effects in asthma.22 Therefore, we hypothesized that ICS may have similar antiangiogenic activity in COPD airways, as another potential cancer-protective effect.
Methods
Our human airway tissue material originated from a double-blinded, randomized, and placebo-controlled clinical trial performed in 2000–2003 and involving 34 COPD subjects (Figure 2).9 After a 2-week run-in period, baseline assessments of spirometry and fiberoptic bronchoscopy with BB were undertaken. Then, using a computer-generated random-numbers table, the participants were randomized 2:1 to receive fluticasone propionate (Accuhaler; Glaxo Wellcome, Middlesex, UK; 500 μg twice daily) or placebo for 6 months via an identical dry placebo inhaler. At the end of the treatment period, lung function and BB were obtained again. The details of study design and data on inflammatory cell profiles have been published previously by our group.12 The study was approved by the Alfred and Royal Hobart Hospital Ethics Committees. Unfortunately, because these tissues have already been depleted by previous analyses, the paired samples available for this study was rather limited (26 matching biopsy sets), but it still turned out to be highly informative.
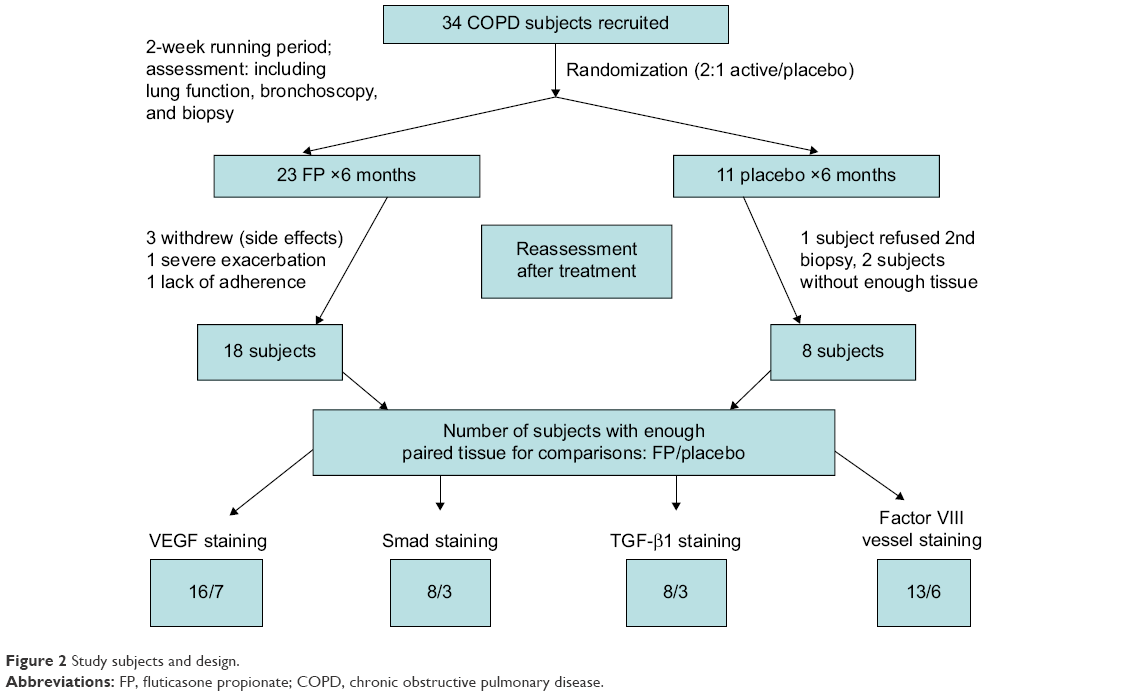 | Figure 2 Study subjects and design. |
Subjects
All subjects gave written informed consent prior to participation. We included those who were older than 45 years with more than a 15 pack-year history of smoking. Subjects with a history suggestive of asthma, those who had used any steroids (oral or inhaled), or who had experienced an exacerbation of COPD within 12 weeks before recruitment were excluded. Other exclusion criteria included significant uncontrolled comorbidities such as diabetes, angina, or cardiac failure, and other coexisting respiratory disorders, including pulmonary fibrosis, lung cancer, and bronchiectasis.12,23 The diagnosis of COPD met Global initiative for chronic Obstructive Lung Disease physiological criteria.24
Lung function
Spirometry, diffusing capacity of the lung for carbon monoxide, and lung volumes by body plethysmography were performed according to American Thoracic Society/European Respiratory Society Task Force guidelines.25
Fiberoptic bronchoscopies
Bronchoscopies and biopsies were performed as previously described.4
Tissue processing and immunostaining
We used anti-Factor VIII antibody for staining of vessels in our samples because, as we reported previously, it is best at demonstrating newer vessels;7 we also undertook a preliminary evaluation of the effects of ICS on selected vascular growth factors in our biopsy tissues.
Details of tissue processing are available in our previously published paper.26 Briefly, following removal of paraffin and subsequent hydration, immunostaining for von Willebrand factor (Factor VIII-related antigen) (Dacocytomation, Copenhagen, Denmark; Dako M06160 1:150 for 90 minutes at room temperature, following heat retrieval using Dako S1699 for 20 minutes in a pressure cooker), VEGF (Fitzgerald, Concord, MA, USA; Catalog number 10R – V101ax: 1/500 dilution, overnight at room temperature), anti-TGF-β1 (Abcam, Cambridge, UK; abcam ab 27969 clone TB1 at 1/16,000–6.25×10−5 mg/mL – overnight at room temperature after blocking with Dako serum block X0909; Dacocytomation), and p-Smad 2/3 (Santa Cruz SRC11769R incubated at 1/100 60 minutes at room temperature following heat retrieval using Dako PT link low PH – [K8005] for 30 minutes at 97°C) was performed on two 3.5 μm sections that were separated by 50 μm.
The primary antibody was elaborated using either anti-mouse or anti-rabbit horseradish peroxidase conjugated DAKO Envision + reagent (K4001 or K4003) for secondary antibody binding and color resolution using Dako DAB+ (K3468). Nuclei were counterstained using Mayer’s Hematoxylin and sections dehydrated by using ascending grades of ethanol, cleared in xylene, and mounted in per mount. In each case, a nonimmune IgG1 negative control (X0931 clone DAK-GO1; Dacocytomation) was performed to eliminate false-positive staining, and endogenous peroxidase in tissue was removed by incubation in a 3% hydrogen peroxide solution for 15 minutes prior to incubation with the primary antibody. A known lung tissue for positive tissue control was included with each staining.
Quantification of end points
Quantification of end points was performed before and after treatments with ICS or placebo using a computer-assisted image analysis (Leica DM 2500 microscope; Leica Microsystems, Wetzlar, Germany), a Spot insight 12 digital camera (Diagnostic Instruments, Inc., Sterling Heights, MI, USA), and Image Pro V5.1 (Media Cybernetics, Inc., Rockville, MD, USA) software. The details of measurements have been explained previously.4,6,7,26 All slides were coded and randomized by an independent person, and then counted by experienced observers (AS and SSS) blinded to subject, diagnosis, therapy, or sequence, with quality assurance on randomly selected slides provided by an academic clinical pathologist (HK).
Number of vessels in the Rbm were normalized by dividing by the length of Rbm (Figure 1). The number of vessels within the LP (Figure 1) was counted to a depth of 150 μm, and vascular density was calculated two dimensionally. Vessels stained with VEGF and TGF-β1 in the Rbm were also quantified. As we reported previously, TGF-β1 staining was heavy and diffused in the LP in most COPD samples and could not be adequately quantitated by this immunohistological system,6,27 and so we substituted p-Smad 2/3 expressions as an indicator of activity of the canonical intracellular TGF-β1 pathway.28
Statistical analyses
At the time of developing this study, there were no data on which to base power calculation, and so we relied on previous precedent in similar asthma research, eg, Richmond et al’s29 study which suggested numbers needed of around 12–15 per group, but also what was feasible in terms of volunteer recruitment. In retrospect, using representative data from our control group, we can say that we did have 90% power at the 5% level to detect at least a 50% change in all our outcome measures at least within the active treatment group, but not between groups. The effect of interventions within groups was measured by comparing each outcome before and after treatment using the Wilcoxon’s two related-samples test, as the variables were non-normally distributed. Groups were compared using the Mann–Whitney test. Two-tailed P-values of 0.05 or less were considered as significant. For nominal variables, a χ2 test was used. Correlations were tested with Spearman’s correlation analyses. All the statistical analyses were done using SPSS 16.0 (IBM Corporation, Armonk, NY, USA).
Results
The demographics of the study groups are presented in Table 1, and for contextual contrast, some comparisons are made with the normal control group from a comparable previous study.7 There were no significant differences between the two treatment COPD groups in demographics, or in lung function, or in airway pathological indices of interest before intervention (Tables 1 and 2) (see Figure 1 and Figures S1 and S2). Thirty-four COPD subjects initially participated, but not all could be represented in these analyses. Figure 2 summarizes the final number of subjects with adequate paired tissue for each immune stain; as might be expected, the demographics and other baseline data for this specifically included group of volunteers were very similar on average to the whole group.
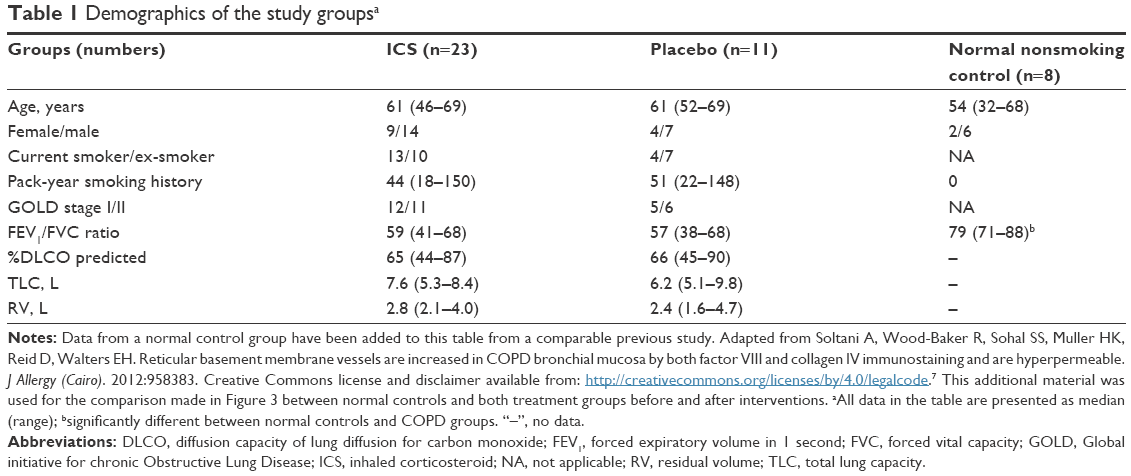 | Table 1 Demographics of the study groupsa |
 | Table 2 Comparison of baseline tissue vascular parameters between groupsa |
Vessels in the Rbm
Vessel number in the Rbm did not change significantly with ICS (Table 3). This also applied to current smokers who showed this signal most markedly.
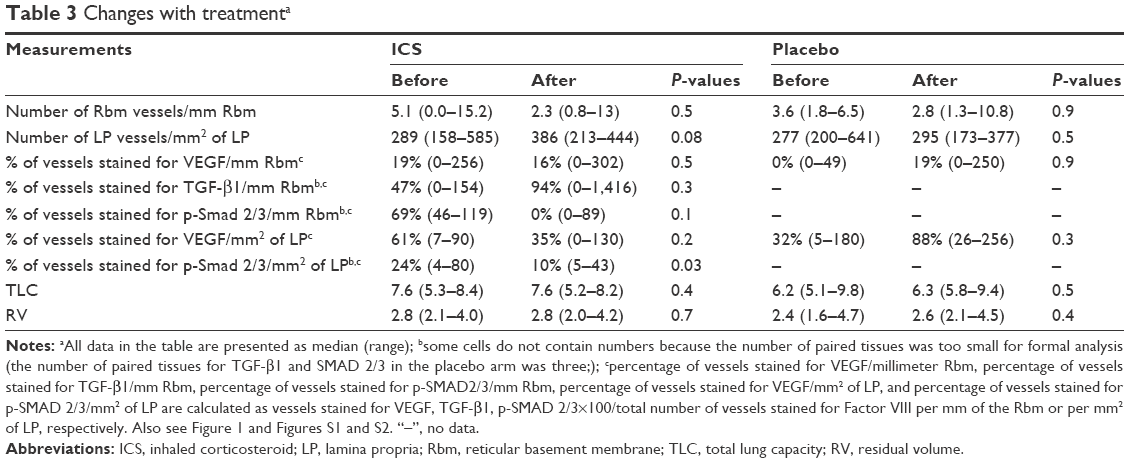 | Table 3 Changes with treatmenta |
Vessels in the LP
Overall, there was moderate evidence for an overall increase in vascular density with ICS (Table 3 and Figure 3). Furthermore, in the current smoking COPD subgroup, where the LP was most markedly hypovascular at baseline, there was even stronger evidence for a normalization of vessels (median [range] 219 [158–437] before vs 356 [213–413] after ICS, P=0.05). This was also well illustrated when we performed a regression of change in vessel number with baseline vessel number in the LP (Figure 4): there was a negative correlation between the baseline number of vessels in the LP and the changes with treatment in the whole COPD group (r=−0.7, P<0.01) and also in the current smoking subgroup alone (r=−0.7, P=0.05). Thus, vessels increased in density with ICS mainly in those subjects most hypovascular, and especially in those below the median in the nonsmoking controls (ie, 408/mm2 of the LP, Figure 3). Above this cutoff, there was essentially no change in vessel number with ICS, suggesting that there was not merely a regression toward the mean across the range.
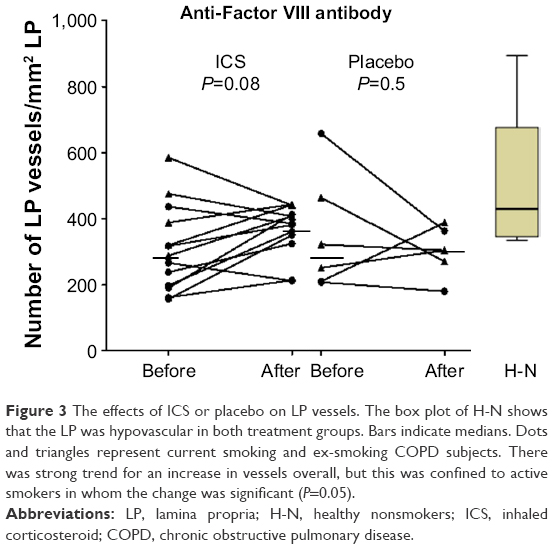 | Figure 3 The effects of ICS or placebo on LP vessels. The box plot of H-N shows that the LP was hypovascular in both treatment groups. Bars indicate medians. Dots and triangles represent current smoking and ex-smoking COPD subjects. There was strong trend for an increase in vessels overall, but this was confined to active smokers in whom the change was significant (P=0.05). |
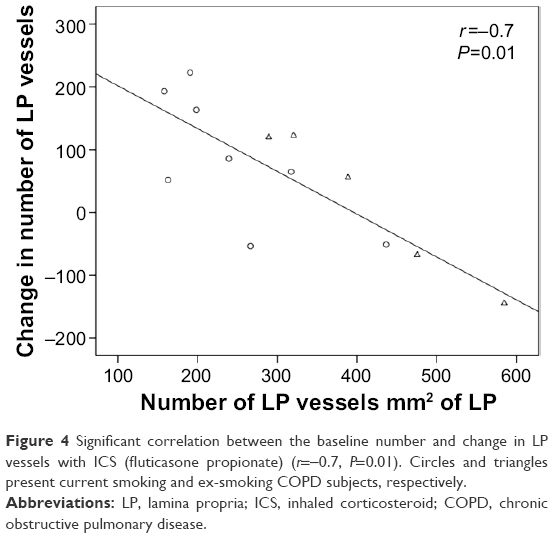 | Figure 4 Significant correlation between the baseline number and change in LP vessels with ICS (fluticasone propionate) (r=−0.7, P=0.01). Circles and triangles present current smoking and ex-smoking COPD subjects, respectively. |
VEGF, TGF-β1, and p-SMAD 2/3 in the Rbm
The percentage of vessels stained for VEGF in the Rbm did not change with either treatment (Table 3), nor was there any effect of ICS treatment for percentage of vessels stained for TGF-β1. There was little suggestion for any change in the placebo group (median 93% before vs 79% after intervention; P=1.00), though the number of pairs of tissues was too small for formal analysis (Figure 2). There was modest evidence (given the numbers involved) for a decrease in percentage of vessels stained for p-SMAD 2/3 in the Rbm with ICS. The number of data points in the placebo arm (n=3 paired slides) was too small for formal analysis, but there was little suggestion of any change.
VEGF and p-SMAD 2/3 in the LP
There was little convincing evidence for a real change in percentage of VEGF-stained vessels with ICS although the numbers fell (Table 3). However, there was a more definite decrease in the percentage of p-SMAD 2/3-positive vessels with ICS treatment. Again, there was little suggestion of change in the placebo arm, but numbers available with both stains per individual were very small.
Relation with lung function
There were strong negative correlations between changes of LP vessels and changes in residual volume (r=−0.8, P<0.05) and between changes in total lung capacity (r=−0.9, P<0.01) and ICS treatment in the current smoking COPD subgroup.
Discussion
This study, to our knowledge, is the first double-blinded placebo-controlled trial examining the effects of ICS on vascular remodeling in COPD airways. We made some potentially important observations that were reasonably robust:
- ICS had no effect on vessel numbers, nor on angiogenic factor expression in the Rbm, though there was some suggestive evidence that p-SMAD 2/3 expression was attenuated.
- ICS normalized the low number of vessels in the LP, and this was associated with, rather paradoxically perhaps, some evidence for a decrease in VEGF and more definite evidence for decreased p-SMAD 2/3 expressions.
- Physiological indices of air trapping showed negative correlations with increased vessel numbers, ie, more vessels, less air trapping.
A previous report demonstrated the effectiveness of ICS in decreasing Rbm fragmentation and markers of EMT.9,21,27 This current study has evaluated the effects of ICS on airway vascular remodeling in COPD and revealed that, in contrast to our hypothesis, hypervascularity of the Rbm did not respond to ICS treatment.
In contrast, this study found that, especially in the current smoking COPD group, ICS increased the density of LP vessels back to normal. This finding was not seen in the ex-smoking COPD subgroup, but vessel numbers here at baseline were essentially normal.4,7 These findings, accompanied by little change or indeed reduction in growth factor activity, may suggest that ICS reduced vessel destruction rather than promotion of new vessels. However, this does not easily fit with our previous observation in COPD airways of a switch from older to newer vessels.7 An alternative explanation could be that there was up-modulation of growth factor receptors on endothelial cells, as occurs in asthmatics on ICS,30 ie, making them more sensitive to growth factors.
Increase of LP vessels may have physiological significance perhaps through increasing airway stiffness, thus reducing expiratory dynamic airway compression and consequent air trapping.
A previously published “cross-sectional” study by Zanini et al13 compared bronchial tissues from ex-smokers with COPD who were treated with ICS to those who were not. Paradoxically, this study found increased vascularity in the COPD group that was not on ICS, suggesting an antiangiogenic effect of ICS. Why their conclusions are different to ours is not easy to tell, but it is notable that we have not found vessel changes to be as marked in ex-smokers with COPD as in current smokers.4,7 Furthermore, a cross-sectional study is not as robust as a longitudinal one.
Limitations
One important “limitation” of our study was its quite selective choice of the angiogenic factors studied. This is potentially a hugely complex area with very many pro- and antiangiogenic factors that could be in play, including collagen breakdown products such as tumstatin, which is an angiogenic inhibitor said to be absent in asthma airways.31,32 But a comprehensive survey would be logistically very difficult, and one at least needs to start with the likely main players gleaned from the literature. In previous studies, we assessed the associations of VEGF and TGF-β1 with vessel changes in the airway sub-epithelial Rbm in smokers and COPD subjects and showed increased activity of both angiogenic factors in hypervascular Rbm, a relationship between VEGF and lung function, and a correlation between TGF-β1 and total number of vessels in this compartment.4,6,7 VEGF is the most potent vascular growth factor, and its level is increased in chronic inflammation, including in airway inflammation in asthma and smokers.8,30,33,34 TGF-β1 is a multifunction cytokine with angiogenic activity, the level of which is increased in airways in smokers and COPD.6,34 In addition, TGF-β1 receptors play an important role in the pathogenesis of COPD through their regulation of Smad pathways.35 A second major problem was limitation of paired biopsies from before and after treatment, mainly in the placebo treatment arm. This was partly due to the original 2:1 treatment allocation and because this study was dependent on “remnants” of tissues not already used in a number of previous cellular and remodeling analyses. We always intended looking at vessel changes, and indeed, the signals that we have picked up as significant seem important and quite striking, in spite of these limitations.
Conclusion
In summary, this longitudinal, repeat bronchoscopy study, for the first time has attempted an evaluation of the effects of ICS on airway vessel remodeling in COPD. LP, but not Rbm vessels, seem responsive to treatment, with an increase in younger vessels stained with anti-Factor VIII antibody. Our physiological data suggest that increases in LP vessels decrease air trapping, perhaps because of resulting stiffer airways that can better resist expiratory dynamic airway compression.
Acknowledgments
This research was funded by NHMRC Australia, GSK Pharmaceutical Company, and Clifford Craig Medical Research Trust.
Disclosure
The authors report no conflicts of interest in this work.
References
Barnes PJ, Shapiro SD, Pauwels RA. Chronic obstructive pulmonary disease: molecular and cellular mechanisms. Eur Respir J. 2003;22(4):672–688. | ||
Sohal SS, Reid D, Soltani A, et al. Reticular basement membrane fragmentation and potential epithelial mesenchymal transition is exaggerated in the airways of smokers with chronic obstructive pulmonary disease. Respirology. 2010;15(6):930–938. | ||
Sohal S, Reid D, Soltani A, et al. Evaluation of epithelial mesenchymal transition in patients with chronic obstructive pulmonary disease. Respir Res. 2011;12(1):130. | ||
Soltani A, Reid D, Sohal S, et al. Basement membrane and vascular remodelling in smokers and chronic obstructive pulmonary disease: a cross-sectional study. Respir Res. 2010;11(1):105. | ||
Soltani A, Muller HK, Sohal SS, et al. Distinctive characteristics of bronchial reticular basement membrane and vessel remodelling in chronic obstructive pulmonary disease (COPD) and in asthma: they are not the same disease. Histopathology. 2012;60(6):964–970. | ||
Soltani A, Sohal SS, Reid D, et al. Vessel-associated transforming growth factor-beta1 (TGF-β1) is increased in the bronchial reticular basement membrane in COPD and normal smokers. PLoS One. 2012;7(6):e39736. | ||
Soltani A, Wood-Baker R, Sohal SS, Muller HK, Reid D, Walters EH. Reticular basement membrane vessels are increased in COPD bronchial mucosa by both factor VIII and collagen IV immunostaining and are hyperpermeable. J Allergy (Cairo). 2012:958383. | ||
Hiroshima K, Iyoda A, Shibuya K, et al. Evidence of neoangiogenesis and an increase in the number of proliferating cells within the bronchial epithelium of smokers. Cancer. 2002;95(7):1539–1545. | ||
Sohal SS, Soltani A, Reid D, et al. A randomized controlled trial of inhaled corticosteroids (ICS) on markers of epithelial-mesenchymal transition (EMT) in large airway samples in COPD: an exploratory proof of concept study. Int J Chron Obstruct Pulmon Dis. 2014;9:533–542. | ||
Magnussen H, Disse B, Rodriguez-Roisin R, et al. Withdrawal of inhaled glucocorticoids and exacerbations of COPD. N Engl J Med. 2014;371(14):1285–1294. | ||
Yang IA, Fong KM, Sim EH, Black PN, Lasserson TJ. Inhaled corticosteroids for stable chronic obstructive pulmonary disease. Cochrane Database Syst Rev. 2012;7:CD002991. | ||
Reid DW, Wen Y, Johns DP, Williams TJ, Ward C, Walters EH. Bronchodilator reversibility, airway eosinophilia and anti-inflammatory effects of inhaled fluticasone in COPD are not related. Respirology. 2008;13(6):799–809. | ||
Zanini A, Chetta A, Saetta M, et al. Bronchial vascular remodelling in patients with COPD and its relationship with inhaled steroid treatment. Thorax. 2009;64(12):1019–1024. | ||
Lapperre TS, Snoeck-Stroband JB, Gosman MM, et al. Effect of fluticasone with and without salmeterol on pulmonary outcomes in chronic obstructive pulmonary disease: a randomized trial. Ann Intern Med. 2009;151(8):517–527. | ||
Hoonhorst SJ, ten Hacken NH, Vonk JM, et al. Steroid resistance in COPD? Overlap and differential anti-inflammatory effects in smokers and ex-smokers. PLoS One. 2014;9(2):e87443. | ||
Kunz LI, Strebus J, Budulac SE, et al. Inhaled steroids modulate extracellular matrix composition in bronchial biopsies of COPD patients: a randomized, controlled trial. PLoS One. 2013;8(5):e63430. | ||
Parimon T, Chien JW, Bryson CL, McDonell MB, Udris EM, Au DH. Inhaled corticosteroids and risk of lung cancer among patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2007;175(7):712–719. | ||
Lam S, leRiche JC, McWilliams A, et al. A randomized phase IIb trial of pulmicort turbuhaler (budesonide) in people with dysplasia of the bronchial epithelium. Clin Cancer Res. 2004;10(19):6502–6511. | ||
Veronesi G, Szabo E, DeCensi A, et al. Randomized phase ii trial of inhaled budesonide versus placebo in high-risk individuals with CT screen–detected lung nodules. Cancer Prev Res (Phila). 2011;4(1):34–42. | ||
Kiri VA, Fabbri LM, Davis KJ, Soriano JB. Inhaled corticosteroids and risk of lung cancer among COPD patients who quit smoking. Respir Med. 2009;103(1):85–90. | ||
Soltani A, Sohal SS, Reid DW, et al. Airway angiogenesis and basement membrane remodeling in smokers and chronic obstructive pulmonary disease: cross-sectional and longitudinal studies [Abstract]. Am J Respir Crit Care Med. 2010;181:A3874. | ||
Feltis BN, Wignarajah D, Reid DW, Ward C, Harding R, Walters EH. Effects of inhaled fluticasone on angiogenesis and vascular endothelial growth factor in asthma. Thorax. 2007;62(4):314–319. | ||
Sohal SS, Reid D, Soltani A, et al. Changes in airway histone deacetylase2 in smokers and COPD with inhaled corticosteroids: a randomized controlled trial. PLoS One. 2013;8(5):e64833. | ||
Andersson CK, Mori M, Bjermer L, Löfdahl CG, Erjefält JS. Alterations in lung mast cell populations in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2010;181(3):206–217. | ||
Miller MR, Hankinson J, Brusasco V. Standardisation of spirometry. Eur Respir J. 2005;26(2):319–338. | ||
Soltani A, Ewe YP, Lim ZS, et al. Mast cells in COPD airways: relationship to bronchodilator responsiveness and angiogenesis. Eur Respir J. 2012;39(6):1361–1367. | ||
Sohal SS, Reid DW, Soltani A, et al. Fluticasone propionate attenuates epithelial mesenchymal transition (EMT) in COPD. Respirology. 2012;17(Suppl 1):25. | ||
Postma DS, Timens W. Remodeling in asthma and chronic obstructive pulmonary disease. Proc Am Thorac Soc. 2006;3(5):434–439. | ||
Richmond I, Booth H, Ward C, Walters EH. Intrasubject variability in airway inflammation in biopsies in mild to moderate stable asthma. Am J Respir Crit Care Med. 1996;153(3):899–903. | ||
Feltis BN, Wignarajah D, Zheng L, et al. Increased vascular endothelial growth factor and receptors: relationship to angiogenesis in asthma. Am J Respir Crit Care Med. 2006;173(11):1201–1207. | ||
Verhamme FM, Bracke KR, Joos GF, Brusselle GG. TGF-beta superfamily in obstructive lung diseases: more suspects than TGF-beta alone. Am J Respir Cell Mol Biol. 2015;52(6):653–662. | ||
Burgess JK, Boustany S, Moir LM, et al. Reduction of tumstatin in asthmatic airways contributes to angiogenesis, inflammation, and hyperresponsiveness. Am J Respir Crit Care Med. 2010;181(2):106–115. | ||
Jackson JR, Seed MP, Kircher CH, Willoughby DA, Winkler JD. The codependence of angiogenesis and chronic inflammation. FASEB J. 1997;11(6):457–465. | ||
Knox AJ, Stocks J, Sutcliffe A. Angiogenesis and vascular endothelial growth factor in COPD. Thorax. 2005;60(2):88–89. | ||
Hogg JC, Timens W. The pathology of chronic obstructive pulmonary disease. Annu Rev Pathol. 2009;4:435–459. |
Supplementary materials
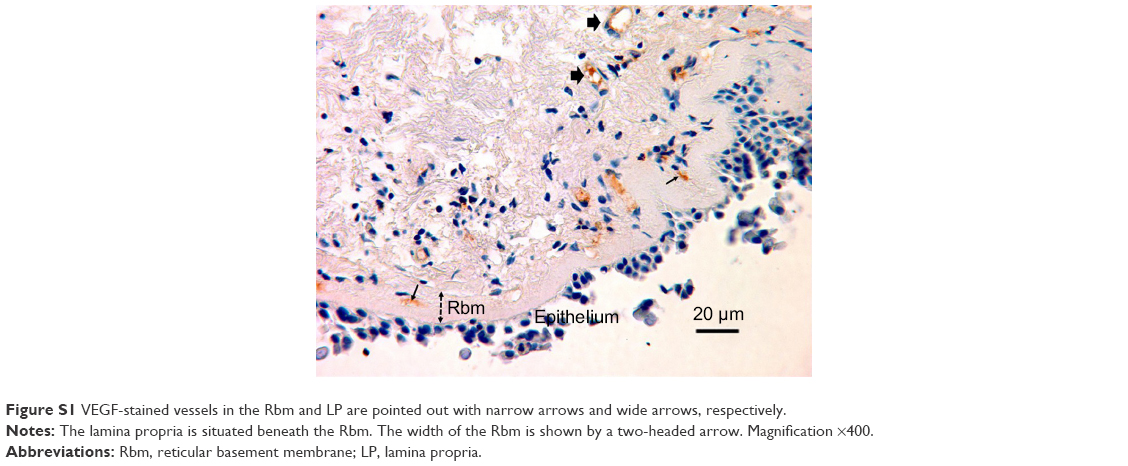 | Figure S1 VEGF-stained vessels in the Rbm and LP are pointed out with narrow arrows and wide arrows, respectively. |
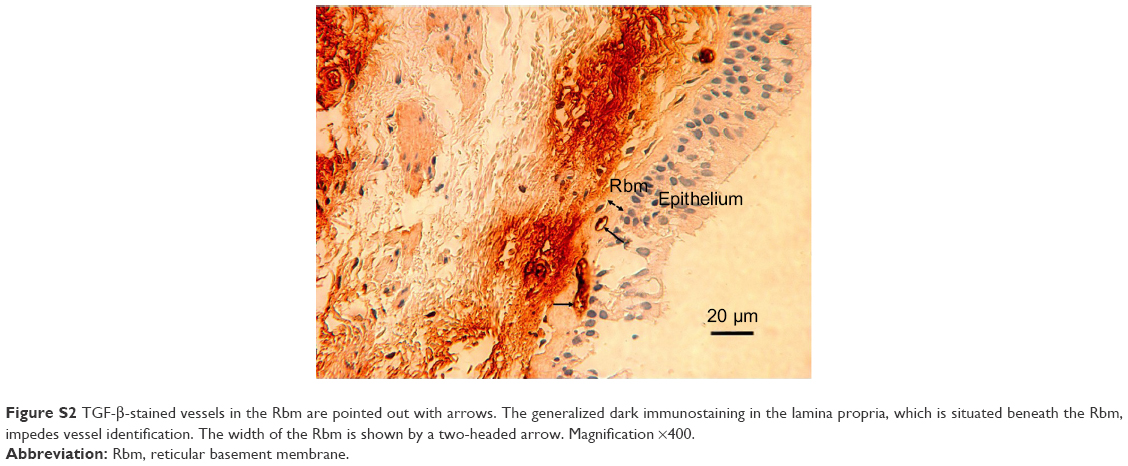 | Figure S2 TGF-b-stained vessels in the Rbm are pointed out with arrows. The generalized dark immunostaining in the lamina propria, which is situated beneath the Rbm, impedes vessel identification. The width of the Rbm is shown by a two-headed arrow. Magnification ×400. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.