Back to Journals » The Application of Clinical Genetics » Volume 7
Influence of oncogenic transcription factors on chromatin conformation and implications in prostate cancer
Received 21 March 2014
Accepted for publication 3 April 2014
Published 12 May 2014 Volume 2014:7 Pages 81—91
DOI https://doi.org/10.2147/TACG.S35598
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Yeqing Angela Yang,1 Jung Kim,1 Jindan Yu1,2
1Division of Hematology/Oncology, Department of Medicine, 2Robert H Lurie Comprehensive Cancer Center, Northwestern University, Feinberg School of Medicine, Chicago, IL, USA
Abstract: In recent years, facilitated by rapid technological advances, we are becoming more adept at probing the molecular processes, which take place in the nucleus, that are crucial for the hierarchical regulation and organization of chromatin architecture. With an unprecedented level of resolution, a detailed atlas of chromosomal structures (histone displacement, variants, modifications, chromosome territories, and DNA looping) and mechanisms underlying their establishment, provides invaluable insight into physiological as well as pathological phenomena. In this review, we will focus on prostate cancer, a prevalent malignancy in men worldwide, and for which a curative treatment strategy is yet to be attained. We aim to catalog the most frequently observed oncogenic alterations associated with chromatin conformation, while emphasizing the TMPRSS2-ERG fusion, which is found in more than one-half of prostate cancer patients and its functions in compromising the chromatin landscape in prostate cancer.
Keywords: chromatin conformation, ERG, prostate cancer
Introduction to chromatin organization and three-dimensional topology
The nucleus is a fascinating organelle within a cellular entity, not only due to the fact that it contains the entire genetic blueprint required for a cell to survive and propagate, but – more importantly – how it is capable of organizing this vast sea of information in an efficient and effective manner. It has been known that the human genome consists of more than 3 billion base pairs, and in fact the total deoxyribonucleic acid (DNA) in a diploid human cell would sum up to approximately 2 m in length when completely stretched.1 Moreover, the extent of compaction for metaphase chromosome is estimated to be between 10,000- and 20,000-fold.2
To achieve this high level of proficiency and accuracy, the nucleus employs multiple levels of packaging methods to generate what is known as the higher-order structure of chromatin, which is composed of a combination of DNA and proteins that intertwine together to separate genes into regulatory hubs and to form a three-dimensional (3D) topology best suited for a cell’s functions.
About 40 years ago, the use of electron microscopy enabled identification of the classical beads-on-a-string type of structure of DNA, which has been generally accepted as the basic level of chromatin organization.3–6 Further demonstrated by biochemical and X-ray diffraction studies,5,7 the chromatin has been described to be formed by repeating units of nucleosomes, octameric structures consisting of four different histone proteins (two each of H2A, H2B, H3, and H4), which are wound by an estimated number of 147 base pairs of DNA, giving rise to 1.7 superhelical turns.7,8
The advent of fluorescence in situ hybridization (FISH) technology has provided evidence for the nonrandom spatial organization of the genome, allowing visualization of position and interaction of chromosomes, chromatin domains, as well as individual genes. It was revealed that gene density is one of the indicators of nuclear positional organization, which is present generally in a radial pattern where gene-dense chromosomal regions prefer to congregate in the nuclear interior, while gene-poor regions are located around the nuclear periphery.9 It has been observed that chromosomes are segregated into subnuclear compartments, known as chromosome territories,10 where the edge of the nucleus is host to mainly repressed genes packed into heterochromatin form,11,12 and the nuclear interior is concentrated in early replicating DNA and frequently transcribed genes.13,14 In addition, FISH experiments have also demonstrated that during differentiation, specific loci can reposition either toward or away from the nuclear periphery, which is concordant with repression or activation of those nearby genes.15 More recently, a series of chromosome conformation capture (3C)-based approaches, which can achieve a high-resolution interrogation of the chromatin landscape, further confirmed that intrachromosomal associations in metazoan genome can serve to concentrate and segregate active gene-rich and gene-poor domains.16
It is known that – indeed – there are topologically associated domains (TADs) that are pervasive throughout the genome and function to compartmentalize the genome into local and distinct regions, therefore modulating gene expression.17
Role of chromatin organization in gene transcription
The structure of chromatin has been well-known to associate with the status of gene transcription. As early as the 1980s, scientists were able to demonstrate that the mere presence of nucleosomes can inhibit initiation by ribonucleic acid polymerase II (RNAPII) and thus stall transcription.18 The mechanisms for regulation of the chromatin structure with respect to gene transcription are diverse, and may involve histone displacement, histone variant incorporation, posttranslational modifications, chromosome territories, and DNA looping (Figure 1).19 Each of these mechanisms has its unique influence on chromatin conformation, which in turn dictates gene transcription status.
 | Figure 1 Different chromatin remodeling regulates gene transcription. |
While packaging of the DNA into nucleosomes can inhibit transcription in vitro, this stereochemical constraint may be relieved by structural changes in nucleosomes.18 Histones have been observed to exhibit high turnover properties from the core nucleosome. It is reported that histone dimers of H2A and H2B are relatively more susceptible to displacement when compared to H3 and H4.20 Results from biochemical and genetic studies consistently reinforce the notion that histone eviction from the nucleosome typically occurs at promoters during gene activation, and such process may be mediated by events including but not limited to adenosine triphosphate (ATP)-dependent chromatin remodeling, as well as histone chaperones.21
For instance, chromatin remodeling complexes, such as switch/sucrose nonfermentable (SWI/SNF)22–24 and chromatin structure remodeling (RSC) complex,25,26 and additionally active RNAP II27 can all take part in evicting H2A and H2B to assist nucleosome unraveling. Thus, in a stepwise manner, these chromatin remodeling complexes can mediate repositioning28 or ejection29 of nucleosomes at promoters to initiate transcription activation. Moreover, histone chaperone proteins (Asf1,30,31 Nap1,32 and nucleophosmin33), which act by sequestering the evicted histones to prevent their reincorporation into the nucleosome, are also an indispensable component for proper histone displacement and ultimately gene transcription.
In addition to the physical exchange of histone proteins, the incorporation of variant histones can also lead to modifications in chromatin structure and transcriptional regulation. Unlike canonical core histones, generally these unconventional histone proteins are distinguished by the fact that they are expressed outside of the S phase and their deposition into the nucleosome is deemed DNA replication-independent.34
As a result of changes in their amino acid sequence, variant forms of histones could acquire divergent biophysical properties predisposing them to localize in specific regions of the genome. One prominent histone variant is H2A.Z, which is an alternative form of H2A, and differs from its counterpart in that its N-terminal tail sequence and several key internal residues, which can effectively alter its ability to interact with H2B as well as the H3/H4 tetramer that eventually manifests in reduced nucleosome stability.35,36 The deposition of H2A.Z is reportedly carried out by ATP-dependent histone exchange reactions through SWR1,37 or by the aforementioned chaperone protein Nap1.38 Another well-studied histone variant is H3.3, and in spite of the fact that it only differs in four amino acids from its canonical form H3, H3.3 has its distinct deposition pattern where it is preferentially enriched in transcriptionally active chromatin and regulatory sites.39,40
On the other hand, certain variants, such as macroH2A (mH2A), have the ability to repress gene transcription by remodeling the chromatin to impede RNAPII binding. The name mH2A is derived from the structural feature of this histone variant, which contains a large nonhistone region (NHR), known as the macro domain, on its N-terminus.41 As a consequence, the NHR of mH2A alters nucleosome structure and interferes with the transcription machinery.42
Furthermore, a significant category of mechanisms contributing to chromatin organization is posttranslational modifications (PTMs) on histone proteins. There has been extensive research conducted to compile and characterize existing histone modifications, depicting a close relationship between histone PTMs and chromatin structure. Some of the most widely studied histone PTMs include acetylation, methylation, phosphorylation, ubiquitination, and sumoylation. They covalently modify the N-terminal and/or the C-terminal histone tails, while affecting the globular domains at a lesser extent.43
These various forms of histone marks generate a code that can be interpreted by specialized proteins to regulate gene expression or to mediate DNA repair.44 Modifications that reflect in active transcription have been elucidated and include acetylation of H3 and H4, and di- or trimethylation of H3 at lysine position 4 (H3K4me2 or me3). In contrast, modifications that instigate inactivation of transcription include methylation at H3K9 and H3K27.19
In eukaryotes, individual chromosomes can occupy spatially defined territories in the interphase nucleus, and repositioning of these genomic regions has an impact on the regulation of gene expression. FISH analysis has shown that chromosome territories adjoin at their borders to create boundaries between chromatin domains. More recently, it is demonstrated that TADs are enclosed by sharp boundaries enriched for the insulator-binding protein CTCF, as well as the heterochromatin mark H3K9me3.17 Since boundaries of these topological domains display properties of classical insulator and barrier features, it is therefore suggested that TADs may be linked to transcriptional control.
Concordantly, another study reported that the positions of TADs align with repressive epigenetic marks, as well as lamina-associated domains, and disrupting a TAD boundary can lead to the long-range deregulation in gene expression during X-chromosome inactivation.45 Therefore, the evidence is convincing that TADs indeed play a role in shaping transcriptional landscapes by clearly defining which sequences belong to the same regulatory network.
Last, as DNA is packaged inside the nucleus, long-range chromatin interactions inevitably occur and – as a result – form loop structures, a majority of which take place between cis-regulatory elements and promoters. It is reported that the dynamic alterations of chromatin looping can either activate or suppress gene expression by facilitating the interactions between enhancers or silencers and their target genes.
One study revealed that only approximately 7% of looping is bridging its nearest gene, reflecting that this chromatin structure is not restrained by genomic proximity and is capable of engaging promoters with distal sites to form complex networks.46 At the same time, these long-range interactions are not inhibited by CTCF and cohesin occupancy,46 which argues against previous notions that CTCF’s binding to insulator sequences may prevent promoter-enhancer interactions.
Moreover, evidence suggests that the enhancer-promoter loop interactions are formed, in a cell type-specific manner, prior to the binding of transcription factors, indicating their critical role in laying the groundwork for transcriptional control during lineage specification.47 Furthermore, in terms of thermodynamic properties of DNA looping, it is understood that this mechanism of bringing together multiple components into one functional unit serves to simultaneously increase specificity and affinity and reduce transcriptional noise.48
Role of chromatin conformation in cancer
Due to the crucial role chromatin structure has on determining gene transcription, it is intuitive that chromatin conformation could be manipulated during oncogenic transformation of cancerous cells. It has been demonstrated that under the employment of tumor cells, these chromatin organization machineries become deregulated, disrupting the 3D architecture and undermining the genomic integrity. One of the most recurring phenomena that is associated with cancer development is chromosomal translocations.49 In the past several decades, a copious number of translocation events have been identified to play pivotal roles in development of a wide range of hematological malignancies as well as solid tumors, which have in turn been utilized as valuable diagnostic and prognostic markers.
Aside from chromosomal translocations, a myriad of events have been implicated in cancer, most of which are deviations from the physiological occurrences of chromatin organization discussed previously. Here, we will catalog the most significant aberrations pertinent to chromatin topology that contribute to cancer development, with a particular emphasis on prostate cancer (Figure 2).
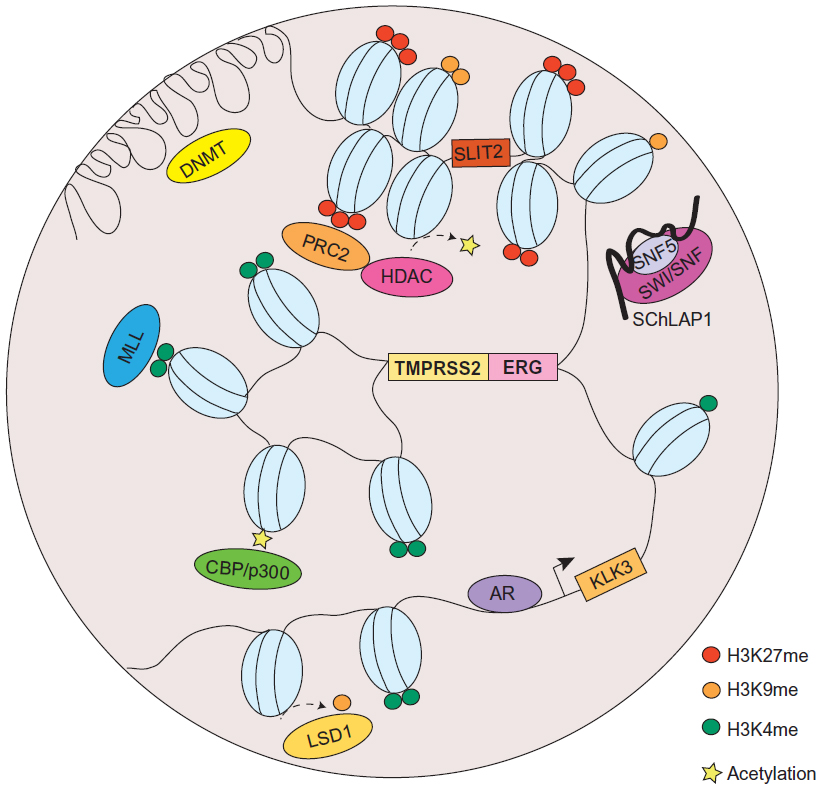 | Figure 2 Chromatin organization aberrations in prostate cancer. |
The Philadelphia chromosome is recognized as one the most prominent cancer-associated cytogenetic abnormality that was first reported by Nowell and Hungerford in 1960.50 It is a highly frequent oncogenic event found in more than 90% of chronic myelogenous leukemia. The translocation is characterized by a reciprocal interchange between chromosome 9 and chromosome 22, which inopportunely generates a BCR-ABL tyrosine kinase gene fusion product.51 As a result of juxtaposing the breakpoint cluster region (BCR) promoter with the coding region of the ABL gene, the hyperactive BCR-ABL fusion protein confers myeloproliferative properties and leads to leukemogenesis.52 Clinical successes obtained through pharmacological therapies directly inhibiting the activity of BCR-ABL (eg, imatinib mesylate) have provided a promising paradigm in which chromosomal organization could be a critical target for cancer development and, certainly, cancer treatment.
It was not until recently, however, that chromosomal translocations have been identified in solid tumors. In 2005, Tomlins et al made the breakthrough discovery of the fusion of the TMPRSS2 and ERG genes in prostate cancer.53 According to their study, a striking proportion of 50% of prostate cancers were found to contain a merged product of the 5′ untranslated region of TMPRSS2(21q22), an androgen-regulated gene, and the protein-coding sequences of ERG (21q22), an erythroblast transformation-specific (ETS) transcription factor (Figure 2). The TMPRSS2-ERG rearrangement has been confirmed to be present in 36%–78% of prostate cancers.54 In addition, other members of the ETS family, including ETV1 (7p21), ETV4 (17q21), and ETV5 (3q28), were also uncovered as fusion partners with TMPRSS2 in prostate cancer, but they were detected in lower frequency.55 Unlike the BCR-ABL translocation, the fusion between TMPRSS2 and ETS genes does not generate a chimeric protein, but instead it promotes the overexpression of oncogenic factors directed by a corrupted promoter element. While solely TMPRSS2 has been identified as a fusion partner of ERG, other 5′ partners of ETS genes have also been observed. These include androgen-induced genes SLC45A3, KLK2, CANT1, and NDRG1, and an endogenous retroviral element HERV-K_22q11.23, which are functionally comparable to TMPRSS2, as well as androgen-repressed gene C15orf21.56–58
It was also reported that rearrangements in the rapidly accelerated fibrosarcoma (RAF) pathway also occur in advanced prostate cancer (SLC45A3-BRAF, ESRP1-RAF1), which can be targeted by RAF kinase inhibitors.59 Moreover, a recent study was able to identify a median of 90 rearrangements in seven prostate cancer tumor samples.60 Examples of disrupted genes due to rearrangement include CADM2, which is a cell adhesion molecule, and phosphatase and tensin homolog (PTEN), a well-established tumor suppressor, as well as a PTEN-interacting protein, MAGI2. These findings depict a convoluted network of genomic rearrangements and chromatin conformation, which synergistically confer deregulated gene expressions and contribute to tumorigenesis.
In addition to chromosomal translocations, modifications to histone could also place a huge impact on the 3D structure of chromatin and has been widely implicated in cancer. In prostate cancer, H3K4 methylation and H3K27 methylation are among the most extensively investigated histone PTMs; while the former is generally associated with activation of proto-oncogenes, the latter is associated with silencing of tumor suppressors. The repressive epigenetic PTM, H3K27 trimethylation (H3K27me3), has been found to be significantly enriched in promoters of numerous tumor suppressor genes (eg, ADRB2,61 SLIT2,62 DAB2IP,63,64 etc), in metastatic prostate cancer. Meanwhile, H3K9me1 and me2, generally accompanied by heterochromatin assembly,65 are also implicated in prostate cancer. Demethylation of H3K9 has been reported to reflect in derepression of AR-regulated genes.66
H3K4 mono- and dimethylation (H3K4me1, H3K4me2) have been thought of as markers for enhancer sites in directing the androgen receptor (AR) transcriptional program, by facilitating AR binding directly or indirectly through the recruitment of coactivators, such as FOXA1, GATA2, and MED1.67 Moreover, an endeavor combining high-resolution nucleosome positioning with histone marks mapping showed strong evidence that H3K4me2-containing nucleosomes spaced 250–450 bp (base pair) apart can flank binding sites of AR prior to its ligand-mediated activation, while the binding site is occluded by a well-positioned nucleosome. Following AR activation, nucleosomes with altered H3K4me2 marks become destabilized at AR binding sites and are comparably more stable at the two flanking loci.68
In addition, the study revealed that the labile H2A.Z variant was more likely to be present in the central nucleosome relative to the flanking nucleosomes, which further contributes to reduced stability of the nucleosome occupied at the AR binding site. Also, it has been shown that androgen treatment can increase the level of H2A.Z and that the incorporation of H2A.Z in enhancer and proximal promoter sites of the AR-induced gene prostate-specific antigen (PSA; or KLK3) can poise the gene for activation by AR.69
Established and maintained by protein–protein interaction between transcription factors bound at enhancers and at promoters,70 DNA looping and chromatin compartmentalization are essential processes governing gene transcription; hence, they are a frequent target for disruption during cancer development. In the case of prostate cancer, AR-mediated chromatin looping has been a longtime research interest in the field, and extensive efforts have been devoted to elucidate the process of how AR signaling may lead to changes in chromatin conformation during prostate tumorigenesis. Studies using chromatin immunoprecipitation (ChIP) techniques showed a striking feature of AR genome-wide binding pattern that, approximately 86%–95% of AR localization occurs in nonpromoter regions.67,71
This evidence strongly indicates that AR, as a transcription factor, is able to direct its specific transcriptional program from a distance – sometimes, even hundreds of kilobases –away from its target gene. Therefore, it is plausible to presume that a looping model is the mechanism by which AR can regulate its targets from afar. In fact, this model has been proven to be true through 3C-based assays, which demonstrated that distal AR enhancer regions form long-range physical contacts with transcription start sites of AR-regulated genes, such as PSA and TMPRSS2,72,73 as well as UBE2C, which is a critical enzyme involved in promoting growth of castration-resistant prostate cancer.67,74
Oncogene-mediated alterations in chromatin conformation
Oncogenes have long been implicated in cancer through chromatin alterations, and one route they take is histone modification. It was discovered in 2002 that EZH2, the enzymatic component of the polycomb repressive complex 2 (PRC2), was among the most upregulated genes in prostate cancer.75 The tumorigenic role of EZH2 has been well-documented, and it involves epigenetic silencing of tumor suppressors and developmental regulators to maintain a dedifferentiated state for cancer cells.76 EZH2 catalyzes trimethylation of H3K27, creating repressive chromatin structures over long genomic distances.77,78 It also recruits several other players, such as PRC1, DNA methyltransferase (DNMT), and histone deacetylase (HDAC) (Figure 2), which are concordantly upregulated in prostate cancer.79–81 It has been revealed that 50% of hypermethylated genes in prostate cancer display preestablished EZH2-mediated H3K27me3 marks, which then leads to de novo DNA methylation.82 Therefore, EZH2 acts in concert with additional epigenetic enzymes to implement chromatin compaction in a cooperative manner.
Furthermore, methylation at H3K9 has also shown to be deregulated in prostate cancer, through perturbed activities of lysine-specific demethylase 1 (LSD1) (Figure 2).66 However, the functions of LSD1 in prostate cancer appear to multifaceted, since it is capable to demethylate not only H3K9, but also H3K4. Since H3K4me1 and me2 are essential marks on AR enhancer sites, erasing these modifications consequently result in gene repression.83 Moreover, an exome-sequencing study recently revealed that several members of the mixed-lineage leukemia (MLL) complex (MLL, MLL2, and ASH2L), which acts as H3K4-specific methyltransferase, can physically interact with AR and are significantly mutated in prostate cancer84 (Figure 2). From a translational standpoint, pharmacological targeting of these histone-modifying enzymes has been envisaged and shown clinical triumph.85
While histone modifications are carried out by specific enzymes, the molecular process underlying the formation of chromatin looping may be effected through a network of coregulators (eg, MED12, SRC-1, p300/CBP, BRG1, etc) that are collectively responsible for sustaining the loop structure.1 Additionally, the GATA, OCT, PAX, NKX, and LEF family proteins have been observed to have sequence motifs near nuclear hormone receptors, including AR and estrogen receptor (ER).86 Disruption of chromosomal structures, therefore, can significantly impair proper gene transcription. A central protein in AR/ER signaling, the pioneer factor forkhead box A1 (FOXA1), has been regarded as a key mediator of AR/ER transcription regulation through chromatin remodeling and recruitment of AR/ER to target sites.87
The fact that FOXA1 is overexpressed and mutated in hormone-dependent cancers, prostate cancer, and breast cancer, is in concordance with its predominant role in directing AR/ER signaling to drive cancer development.88,89 In addition, knowledge about the multiprotein Mediator complex, which is well-known for its role in bridging enhancer and promoter into close proximity,90 has also contributed to our understanding about chromosome looping involving AR, wherein the silencing of a Mediator subunit MED1 can significantly impair AR transactivation.91
Moreover, in the past decade, the AR signaling pathway has also been shown to play essential roles in altering chromatin conformation, primarily due to its involvement in a majority of chromosomal translocations identified in prostate cancer. Through FISH analyses, it was discovered that androgen stimulation can induce the spatial proximity between TMPRSS2 and ERG, thus highly augmenting the probability of forming a fusion product when under the stress of DNA double-strand breaks.92 Further evidence demonstrated that AR binding at specific intronic loci near break sites in TMPRSS2, ERG, and ETV1 could result in rapid formation of intra- and interchromosomal interactions that in turn generate enough spatial proximity to predispose the genes for translocation.
In addition, the ensuing modifications of chromatin architecture sensitize these regions to genotoxic stress, making the translocation loci particularly susceptible to double-stranded breaks. The liganded AR, upon binding to DNA, can recruit enzymes – including activation-induced cytidine deaminase (AID) and LINE-1 repeat-encoded ORF2 endonuclease, as well as topoisomerase II beta (TOP2B) to create double-stranded breaks at break sites, which then become ligated through nonhomologous end joining.93,94
Aside from proteins playing an oncogenic function, there has been emerging evidence that long noncoding RNAs (lncRNAs; >200 nt)95 may also adversely affect chromatin structures. For instance, it is recently reported that HOTAIR, a 2.2 kb lncRNA residing in the HOXC locus, serves as a crucial interface between DNA and the chromatin-modifying complex PRC2. As a result, in breast cancer, an overexpression of HOTAIR is causally linked to alterations in the chromatin state reimposed by PRC2 occupancy, consequently permitting a gene expression program that is conducive to cell motility and invasion by silencing key metastasis suppressor genes.96
Another prominent member of the lncRNAs that is recently implicated in cancer is SChLAP1, which was identified as an overexpressed gene in prostate tumor samples.97 Similar to HOTAIR, SChLAP1, in context of prostate cancer, can promote cancer invasion and metastasis. The molecular mechanisms underlying SChLAP1’s oncogenic function were also connected to a chromatin modifying complex, namely SWI/SNF (Figure 2). Through antagonizing the genomic binding of SWI/SNF, SChLAP1 significantly impairs the transcriptional program directed by SWI/SNF that, as alluded to previously, is a complex that utilizes ATP to mobilize nucleosomes and remodel chromatin.97
Numerous links have been established between SWI/SNF and carcinogenesis;98 however, the latest discovery of SChLAP1, in addition to HOTAIR, sheds new light on the mechanistic basis of how deregulation of lncRNAs may result in defective chromatin organization, which ultimately contributes to oncogenesis. Furthermore, a recent study reports that two lncRNAs overexpressed in prostate cancer, PCGEM1 and PRNCR1 (PCAT8), can bind with AR and facilitate the enhancer-promoter loop formation required for AR transcriptional regulation. It was demonstrated that these lncRNAs can promote AR activation in a hormone-independent environment, providing novel mechanistic insight into the pathogenesis of castration-resistant prostate cancer.99
Therefore, taken together, these lines of encouraging evidence keep propelling scientists forward to continuously uncover novel mechanisms associated with deregulation of chromosomal organization – to provide beneficial insight into strategies for diagnosing as well as treating cancer.
ERG overexpression and chromatin conformation
ERG is overexpressed in prostate cancer due to AR-mediated changes in chromosome rearrangement. As a consequence, ERG overexpression, in turn, can also lead to chromatin structure alterations, which further contribute to prostate cancer development in a feed-forward vicious cycle. The function of ERG involves physical interaction with a number of cofactors as well as transcription factors, including AR – ultimately leading to a transcriptional program favoring the dedifferentiation, invasion, and neoplastic transformation of prostate epithelial cells.100
To characterize the molecular crosstalk between ERG and AR, studies showed that ERG can effectively attenuate AR signaling by the direct transcriptional repression of AR, and additionally ERG occupancy at AR target genes correlates with negative regulation, which is potentiated by ERG-induced EZH2 activity.76,101 It is becoming increasingly clear that ERG complexes with other molecules to coordinately organize chromatin structure. Several interacting partners of ERG have been identified in recent years, including EZH271,102 and HDAC1,103 which – along with ERG – form a repressor coregulatory network that is important for mediating androgen response in prostate cancer.101 This notion is further supported by the fact that ERG overexpression also dictates changes in the genomewide DNA methylation landscape,104 reflecting a complex regulatory program directed by ERG to impose structural alterations in the overall 3D chromatin topology.
Employing a combination of advanced technologies, including Hi-C, ChIP-seq, and RNA-seq, and integrative bioinformatic analyses, Rickman et al100,105 showed that an overexpression of ERG could induce dramatic changes in 3D chromatin topology, corresponding to the changes in the expression of a group of genes implicated in aggressive prostate cancer. Since ERG binding strongly associated with hotspots of differential chromatin interactions, an upregulation of ERG when fused to TMPRSS2 upon androgen stimulation consequently leads to altered regulation of transcription events.
Among these ERG-regulated genes are genes associated with invasion and migration (eg, FYN, PLAU, MMP3, MMP9, LEF1, and miR200c) and urogenital development (eg, HOXA, B, C gene cluster members, PYGO1, and NKX3.1).100,106–108
Concluding remarks and therapeutic implications
Over the past several decades, we have witnessed a plethora of pioneering studies that established the essential role of chromatin conformation during normal biological processes and oncogenic cellular transformations.
Through investigations of molecular mechanisms governing the alterations in chromatin architecture, researchers have been able to strategically design therapeutic agents which, by abolishing the enzymatic activity of certain chromatin-modifying proteins, to achieve the correct 3D chromatin topology. Several drugs were recently approved by the FDA due to their improved efficacy in prolonging survival and reduced toxicity compared to conventional chemotherapy. Some prominent examples include DNA methylation inhibitor azacitidine (Vidaza®) and decitabine (Dacogen®) and HDAC inhibitors vorinostat (Zolinza®) and romidepsin (Istodax®), which were FDA-approved successively in the last 10 years, for the treatment of myelodysplastic syndrome and cutaneous T-cell lymphoma, respectively.109
Currently, clinical trials are being conducted to examine the pharmacological efficacy of DNMT and HDAC inhibitors in prostate cancer, as adjuvant therapies to complement androgen deprivation.110 In addition, a wide range of chemical inhibitors targeting enzymes, such as EZH2 (eg, DZNep111 and GSK126112) and LSD1 (eg, TCP113 and ORY-100185), have demonstrated promising potential in various in vitro and in vivo studies for multiple cancer types.85
These molecules hold hopeful prospective for treatment of prostate cancer, in which oncogenic contributors to chromosomal abnormalities are abundant. It is anticipated that future pharmaceutical therapies aimed to restore the physiological activity level of key chromatin modulators would provide desirable curative effects.
Acknowledgments
This work was supported in part by: the National Institutes of Health/National Cancer Institute training grant T32CA09560 (to YAY); National Institutes of Health R01CA172384 (to JY); the US Army Medical Research and Materiel Command grant W81XWH-13-1-0319 (to JY); and the Research Scholar Award RSG-12-085-01 (to JY) from the American Cancer Society.
Disclosure
The authors report no conflicts of interest in this work.
References
Wu D, Zhang C, Shen Y, Nephew KP, Wang Q. Androgen receptor-driven chromatin looping in prostate cancer. Trends Endocrinol Metab. 2011;22(12):474–480. | |
Woodcock CL, Ghosh RP. Chromatin higher-order structure and dynamics. Cold Spring Harb Perspect Biol. 2010;2(5):a000596. | |
Oudet P, Gross-Bellard M, Chambon P. Electron microscopic and biochemical evidence that chromatin structure is a repeating unit. Cell. 1975;4(4):281–300. | |
Hewish DR, Burgoyne LA. Chromatin sub-structure. The digestion of chromatin DNA at regularly spaced sites by a nuclear deoxyribonuclease. Biochem Biophys Res Commun. 1973;52(2):504–510. | |
Kornberg RD. Chromatin structure: a repeating unit of histones and DNA. Science. 1974;184(4139):868–871. | |
Olins AL, Olins DE. Spheroid chromatin units (v bodies). Science. 1974;183(4122):330–332. | |
Finch JT, Lutter LC, Rhodes D, et al. Structure of nucleosome core particles of chromatin. Nature. 1977;269(5623):29–36. | |
Luger K, Mäder AW, Richmond RK, Sargent DF, Richmond TJ. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature. 1997;389(6648):251–260. | |
Bickmore WA. The spatial organization of the human genome. Annu Rev Genomics Hum Genet. 2013;14:67–84. | |
Cremer T, Cremer C. Chromosome territories, nuclear architecture and gene regulation in mammalian cells. Nat Rev Genet. 2001;2(4):292–301. | |
Croft JA, Bridger JM, Boyle S, Perry P, Teague P, Bickmore WA. Differences in the localization and morphology of chromosomes in the human nucleus. J Cell Biol. 1999;145(6):1119–1131. | |
Bridger JM, Bickmore WA. Putting the genome on the map. Trends Genet. 1998;14(10):403–409. | |
Xing Y, Johnson CV, Moen PT Jr, McNeil JA, Lawrence J. Nonrandom gene organization: structural arrangements of specific pre-mRNA transcription and splicing with SC-35 domains. J Cell Biol. 1995;131(6 Pt 2):1635–1647. | |
Carter KC, Bowman D, Carrington W, et al. A three-dimensional view of precursor messenger RNA metabolism within the mammalian nucleus. Science. 1993;259(5099):1330–1335. | |
Kosak ST, Skok JA, Medina KL, et al. Subnuclear compartmentalization of immunoglobulin loci during lymphocyte development. Science. 2002;296(5565):158–162. | |
Simonis M, Klous P, Splinter E, et al. Nuclear organization of active and inactive chromatin domains uncovered by chromosome conformation capture-on-chip (4C). Nat Genet. 2006;38(11):1348–1354. | |
Dixon JR, Selvaraj S, Yue F, et al. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature. 2012;485(7398):376–380. | |
Knezetic JA, Luse DS. The presence of nucleosomes on a DNA template prevents initiation by RNA polymerase II in vitro. Cell. 1986;45(1):95–104. | |
Li B, Carey M, Workman JL. The role of chromatin during transcription. Cell. 2007;128(4):707–719. | |
Kimura H, Cook PR. Kinetics of core histones in living human cells: little exchange of H3 and H4 and some rapid exchange of H2B. J Cell Biol. 2001;153(7):1341–1353. | |
Workman JL. Nucleosome displacement in transcription. Genes Dev. 2006;20(15):2009–2017. | |
Owen-Hughes T, Utley RT, Côté J, Peterson CL, Workman JL. Persistent site-specific remodeling of a nucleosome array by transient action of the SWI/SNF complex. Science. 1996;273(5274):513–516. | |
Bruno M, Flaus A, Stockdale C, Rencurel C, Ferreira H, Owen-Hughes T. Histone H2A/H2B dimer exchange by ATP-dependent chromatin remodeling activities. Mol Cell. 2003;12(6):1599–1606. | |
Phelan ML, Schnitzler GR, Kingston RE. Octamer transfer and creation of stably remodeled nucleosomes by human SWI-SNF and its isolated ATPases. Mol Cell Biol. 2000;20(17):6380–6389. | |
Lorch Y, Zhang M, Kornberg RD. RSC unravels the nucleosome. Mol Cell. 2001;7(1):89–95. | |
Lorch Y, Maier-Davis B, Kornberg RD. Chromatin remodeling by nucleosome disassembly in vitro. Proc Natl Acad Sci U S A. 2006;103(9):3090–3093. | |
Kireeva ML, Walter W, Tchernajenko V, Bondarenko V, Kashlev M, Studitsky VM. Nucleosome remodeling induced by RNA polymerase II: loss of the H2A/H2B dimer during transcription. Mol Cell. 2002;9(3):541–552. | |
Whitehouse I, Flaus A, Cairns BR, White MF, Workman JL, Owen-Hughes T. Nucleosome mobilization catalysed by the yeast SWI/SNF complex. Nature. 1999;400(6746):784–787. | |
Lorch Y, Zhang M, Kornberg RD. Histone octamer transfer by a chromatin-remodeling complex. Cell. 1999;96(3):389–392. | |
Adkins MW, Howar SR, Tyler JK. Chromatin disassembly mediated by the histone chaperone Asf1 is essential for transcriptional activation of the yeast PHO5 and PHO8 genes. Mol Cell. 2004;14(5):657–666. | |
Schwabish MA, Struhl K. Asf1 mediates histone eviction and deposition during elongation by RNA polymerase II. Mol Cell. 2006;22(3):415–422. | |
Walter PP, Owen-Hughes TA, Côté J, Workman JL. Stimulation of transcription factor binding and histone displacement by nucleosome assembly protein 1 and nucleoplasmin requires disruption of the histone octamer. Mol Cell Biol. 1995;15(11):6178–6187. | |
Swaminathan V, Kishore AH, Febitha KK, Kundu TK. Human histone chaperone nucleophosmin enhances acetylation-dependent chromatin transcription. Mol Cell Biol. 2005;25(17):7534–7545. | |
Kamakaka RT, Biggins S. Histone variants: deviants? Genes Dev. 2005;19(3):295–310. | |
Zhang H, Roberts DN, Cairns BR. Genome-wide dynamics of Htz1, a histone H2A variant that poises repressed/basal promoters for activation through histone loss. Cell. 2005;123(2):219–231. | |
Raisner RM, Hartley PD, Meneghini MD, et al. Histone variant H2A.Z marks the 5′ ends of both active and inactive genes in euchromatin. Cell. 2005;123(2):233–248. | |
Mizuguchi G, Shen X, Landry J, Wu WH, Sen S, Wu C. ATP-driven exchange of histone H2AZ variant catalyzed by SWR1 chromatin remodeling complex. Science. 2004;303(5656):343–348. | |
Park YJ, Chodaparambil JV, Bao Y, McBryant SJ, Luger K. Nucleosome assembly protein 1 exchanges histone H2A-H2B dimers and assists nucleosome sliding. J Biol Chem. 2005;280(3):1817–1825. | |
Henikoff S. Labile H3.3+H2A.Z nucleosomes mark ‘nucleosome-free regions’. Nat Genet. 2009;41(8):865–866. | |
Yuen BT, Knoepfler PS. Histone H3.3 mutations: a variant path to cancer. Cancer Cell. 2013;24(5):567–574. | |
Pehrson JR, Fried VA. MacroH2A, a core histone containing a large nonhistone region. Science. 1992;257(5075):1398–1400. | |
Doyen CM, An W, Angelov D, et al. Mechanism of polymerase II transcription repression by the histone variant macroH2A. Mol Cell Biol. 2006;26(3):1156–1164. | |
Berger SL. The complex language of chromatin regulation during transcription. Nature. 2007;447(7143):407–412. | |
Kouzarides T. Chromatin modifications and their function. Cell. 2007;128(4):693–705. | |
Nora EP, Lajoie BR, Schulz EG, et al. Spatial partitioning of the regulatory landscape of the X-inactivation centre. Nature. 2012;485(7398):381–385. | |
Sanyal A, Lajoie BR, Jain G, Dekker J. The long-range interaction landscape of gene promoters. Nature. 2012;489(7414):109–113. | |
Jin F, Li Y, Dixon JR, et al. A high-resolution map of the three-dimensional chromatin interactome in human cells. Nature. 2013;503(7475):290–294. | |
Vilar JM, Saiz L. DNA looping in gene regulation: from the assembly of macromolecular complexes to the control of transcriptional noise. Curr Opin Genet Dev. 2005;15(2):136–144. | |
Göndör A. Dynamic chromatin loops bridge health and disease in the nuclear landscape. Semin Cancer Biol. 2013;23(2):90–98. | |
Nowell PC, Hungerford DA. Chromosome studies on normal and leukemic human leukocytes. J Natl Cancer Inst. 1960;25:85–109. | |
Rowley JD. Letter: A new consistent chromosomal abnormality in chronic myelogenous leukaemia identified by quinacrine fluorescence and Giemsa staining. Nature. 1973;243(5405):290–293. | |
Lugo TG, Pendergast AM, Muller AJ, Witte ON. Tyrosine kinase activity and transformation potency of bcr-abl oncogene products. Science. 1990;247(4946):1079–1082. | |
Tomlins SA, Rhodes DR, Perner S, et al. Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science. 2005;310(5748):644–648. | |
Demichelis F, Fall K, Perner S, et al. TMPRSS2:ERG gene fusion associated with lethal prostate cancer in a watchful waiting cohort. Oncogene. 2007;26(31):4596–4599. | |
Clark JP, Cooper CS. ETS gene fusions in prostate cancer. Nat Rev Urol. 2009;6(8):429–439. | |
Tomlins SA, Laxman B, Dhanasekaran SM, et al. Distinct classes of chromosomal rearrangements create oncogenic ETS gene fusions in prostate cancer. Nature. 2007;448(7153):595–599. | |
Hermans KG, Bressers AA, van der Korput HA, Dits NF, Jenster G, Trapman J. Two unique novel prostate-specific and androgen-regulated fusion partners of ETV4 in prostate cancer. Cancer Res. 2008;68(9):3094–3098. | |
Pflueger D, Rickman DS, Sboner A, et al. N-myc downstream regulated gene 1 (NDRG1) is fused to ERG in prostate cancer. Neoplasia. 2009;11(8):804–811. | |
Palanisamy N, Ateeq B, Kalyana-Sundaram S, et al. Rearrangements of the RAF kinase pathway in prostate cancer, gastric cancer and melanoma. Nat Med. 2010;16(7):793–798. | |
Berger MF, Lawrence MS, Demichelis F, et al. The genomic complexity of primary human prostate cancer. Nature. 2011;470(7333):214–220. | |
Yu J, Cao Q, Mehra R, et al. Integrative genomics analysis reveals silencing of beta-adrenergic signaling by polycomb in prostate cancer. Cancer Cell. 2007;12(5):419–431. | |
Yu J, Cao Q, Yu J, et al. The neuronal repellent SLIT2 is a target for repression by EZH2 in prostate cancer. Oncogene. 2010;29(39):5370–5380. | |
Chen H, Tu SW, Hsieh JT. Down-regulation of human DAB2IP gene expression mediated by polycomb Ezh2 complex and histone deacetylase in prostate cancer. J Biol Chem. 2005;280(23):22437–22444. | |
Min J, Zaslavsky A, Fedele G, et al. An oncogene-tumor suppressor cascade drives metastatic prostate cancer by coordinately activating Ras and nuclear factor-kappaB. Nat Med. 2010;16(3):286–294. | |
Nakayama J, Rice JC, Strahl BD, Allis CD, Grewal SI. Role of histone H3 lysine 9 methylation in epigenetic control of heterochromatin assembly. Science. 2001;292(5514):110–113. | |
Metzger E, Wissmann M, Yin N, et al. LSD1 demethylates repressive histone marks to promote androgen-receptor-dependent transcription. Nature. 2005;437(7057):436–439. | |
Wang Q, Li W, Zhang Y, et al. Androgen receptor regulates a distinct transcription program in androgen-independent prostate cancer. Cell. 2009;138(2):245–256. | |
He HH, Meyer CA, Shin H, et al. Nucleosome dynamics define transcriptional enhancers. Nature Genet. 2010;42(4):343–347. | |
Dryhurst D, McMullen B, Fazli L, Rennie PS, Ausió J. Histone H2A.Z prepares the prostate specific antigen (PSA) gene for androgen receptor-mediated transcription and is upregulated in a model of prostate cancer progression. Cancer Lett. 2012;315(1):38–47. | |
Bulger M, Groudine M. Functional and mechanistic diversity of distal transcription enhancers. Cell. 2011;144(3):327–339. | |
Yu J, Yu J, Mani RS, et al. An integrated network of androgen receptor, polycomb, and TMPRSS2-ERG gene fusions in prostate cancer progression. Cancer Cell. 2010;17(5):443–454. | |
Wang Q, Carroll JS, Brown M. Spatial and temporal recruitment of androgen receptor and its coactivators involves chromosomal looping and polymerase tracking. Mol Cell. 2005;19(5):631–642. | |
Wang Q, Li W, Liu XS, et al. A hierarchical network of transcription factors governs androgen receptor-dependent prostate cancer growth. Mol Cell. 2007;27(3):380–392. | |
Chen Z, Zhang C, Wu D, et al. Phospho-MED1-enhanced UBE2C locus looping drives castration-resistant prostate cancer growth. EMBO J. 2011;30(12):2405–2419. | |
Varambally S, Dhanasekaran SM, Zhou M, et al. The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature. 2002;419(6907):624–629. | |
Yu J, Yu J, Rhodes DR, et al. A polycomb repression signature in metastatic prostate cancer predicts cancer outcome. Cancer Res. 2007;67(22):10657–10663. | |
Francis NJ, Kingston RE. Mechanisms of transcriptional memory. Nat Rev Mol Cell Biol. 2001;2(6):409–421. | |
Zhao JC, Yu J, Runkle C, et al. Cooperation between Polycomb and androgen receptor during oncogenic transformation. Genome Res. 2012;22(2):322–331. | |
Cao Q, Mani RS, Ateeq B, et al. Coordinated regulation of polycomb group complexes through microRNAs in cancer. Cancer Cell. 2011;20(2):187–199. | |
Weichert W, Röske A, Gekeler V, et al. Histone deacetylases 1, 2 and 3 are highly expressed in prostate cancer and HDAC2 expression is associated with shorter PSA relapse time after radical prostatectomy. Br J Cancer. 2008;98(3):604–610. | |
Nelson WG, Yegnasubramanian S, Agoston AT, et al. Abnormal DNA methylation, epigenetics, and prostate cancer. Front Biosci. 2007;12: 4254–4266. | |
Schlesinger Y, Straussman R, Keshet I, et al. Polycomb-mediated methylation on Lys27 of histone H3 pre-marks genes for de novo methylation in cancer. Nat Genet. 2007;39(2):232–236. | |
Cai C, He HH, Chen S, et al. Androgen receptor gene expression in prostate cancer is directly suppressed by the androgen receptor through recruitment of lysine-specific demethylase 1. Cancer Cell. 2011;20(4):457–471. | |
Grasso CS, Wu YM, Robinson DR, et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature. 2012;487(7406):239–243. | |
Helin K, Dhanak D. Chromatin proteins and modifications as drug targets. Nature. 2013;502(7472):480–488. | |
Lupien M, Brown M. Cistromics of hormone-dependent cancer. Endocr Relat Cancer. 2009;16(2):381–389. | |
Lupien M, Eeckhoute J, Meyer CA, et al. FoxA1 translates epigenetic signatures into enhancer-driven lineage-specific transcription. Cell. 2008;132(6):958–970. | |
Robinson JL, Holmes KA, Carroll JS. FOXA1 mutations in hormone-dependent cancers. Front Oncol. 2013;3:20. | |
Jin HJ, Zhao JC, Ogden I, Bergan RC, Yu J. Androgen receptor-independent function of FoxA1 in prostate cancer metastasis. Cancer Res. 2013;73(12):3725–3736. | |
Kagey MH, Newman JJ, Bilodeau S, et al. Mediator and cohesin connect gene expression and chromatin architecture. Nature. 2010;467(7314):430–435. | |
Wang Q, Sharma D, Ren Y, Fondell JD. A coregulatory role for the TRAP-mediator complex in androgen receptor-mediated gene expression. J Biol Chem. 2002;277(45):42852–42858. | |
Mani RS, Tomlins SA, Callahan K, et al. Induced chromosomal proximity and gene fusions in prostate cancer. Science. 2009; 326(5957):1230. | |
Lin C, Yang L, Tanasa B, et al. Nuclear receptor-induced chromosomal proximity and DNA breaks underlie specific translocations in cancer. Cell. 2009;139(6):1069–1083. | |
Haffner MC, Aryee MJ, Toubaji A, et al. Androgen-induced TOP2B-mediated double-strand breaks and prostate cancer gene rearrangements. Nat Genet. 2010;42(8):668–675. | |
Djebali S, Davis CA, Merkel A, et al. Landscape of transcription in human cells. Nature. 2012;489(7414):101–108. | |
Gupta RA, Shah N, Wang KC, et al. Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature. 2010;464(7291):1071–1076. | |
Prensner JR, Iyer MK, Sahu A, et al. The long noncoding RNA SChLAP1 promotes aggressive prostate cancer and antagonizes the SWI/SNF complex. Nat Genet. 2013;45(11):1392–1398. | |
Roberts CW, Orkin SH. The SWI/SNF complex – chromatin and cancer. Nat Rev Cancer. 2004;4(2):133–142. | |
Yang L, Lin C, Jin C, et al. lncRNA-dependent mechanisms of androgen-receptor-regulated gene activation programs. Nature. 2013;500(7464):598–602. | |
Rickman DS, Soong TD, Moss B, et al. Oncogene-mediated alterations in chromatin conformation. Proc Natl Acad Sci U S A. 2012;109(23):9083–9088. | |
Chng KR, Chang CW, Tan SK, et al. A transcriptional repressor co-regulatory network governing androgen response in prostate cancers. EMBO J. 2012;31(12):2810–2823. | |
Kunderfranco P, Mello-Grand M, Cangemi R, et al. ETS transcription factors control transcription of EZH2 and epigenetic silencing of the tumor suppressor gene Nkx3.1 in prostate cancer. PLoS One. 2010;5(5):e10547. | |
Iljin K, Wolf M, Edgren H, et al. TMPRSS2 fusions with oncogenic ETS factors in prostate cancer involve unbalanced genomic rearrangements and are associated with HDAC1 and epigenetic reprogramming. Cancer Res. 2006;66(21):10242–10246. | |
Kron K, Trudel D, Pethe V, et al. Altered DNA methylation landscapes of polycomb-repressed loci are associated with prostate cancer progression and ERG oncogene expression in prostate cancer. Clin Cancer Res. 2013;19(13):3450–3461. | |
Elemento O, Rubin MA, Rickman DS. Oncogenic transcription factors as master regulators of chromatin topology: a new role for ERG in prostate cancer. Cell Cycle. 2012;11(18):3380–3383. | |
Tomlins SA, Laxman B, Varambally S, et al. Role of the TMPRSS2-ERG gene fusion in prostate cancer. Neoplasia. 2008;10(2):177–188. | |
Kim J, Wu L, Zhao JC, Jin HJ, Yu J. TMPRSS2-ERG gene fusions induce prostate tumorigenesis by modulating microRNA miR-200c. Oncogene. 2013. | |
Wu L, Zhao JC, Kim J, Jin HJ, Wang CY, Yu J. ERG is a critical regulator of Wnt/LEF1 signaling in prostate cancer. Cancer Res. 2013;73(19):6068–6079. | |
Boumber Y, Issa JP. Epigenetics in cancer: what’s the future? Oncology (Williston Park). 2011;25(3):220–226, 228. | |
Lin J, Wang C, Kelly WK. Targeting epigenetics for the treatment of prostate cancer: recent progress and future directions. Semin Oncol. 2013;40(3):393–401. | |
Tan J, Yang X, Zhuang L, et al. Pharmacologic disruption of Polycomb-repressive complex 2-mediated gene repression selectively induces apoptosis in cancer cells. Genes Dev. 2007;21(9):1050–1063. | |
McCabe MT, Ott HM, Ganji G, et al. EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature. 2012;492(7427):108–112. | |
Schenk T, Chen WC, Göllner S, et al. Inhibition of the LSD1 (KDM1A) demethylase reactivates the all-trans-retinoic acid differentiation pathway in acute myeloid leukemia. Nat Med. 2012;18(4):605–611. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.