")
Back to Journals » International Journal of Nanomedicine » Volume 10 » Supplement 1 Challenges in biomaterials research
Influence of needle-like morphology on the bioactivity of nanocrystalline wollastonite – an in vitro study
Authors Lakshmi R, Sasikumar S
Received 7 January 2015
Accepted for publication 5 March 2015
Published 1 October 2015 Volume 2015:10(Supplement 1 Challenges in biomaterials research) Pages 129—136
DOI https://doi.org/10.2147/IJN.S79986
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 6
Editor who approved publication: Dr Thomas Webster
R Lakshmi, S Sasikumar
Materials Chemistry Division, School of Advanced Sciences, VIT University, Vellore, Tamil Nadu, India
Abstract: In the past 2 decades, wollastonite has been studied thoroughly for its application as a bone implant material due to its biocompatibility, high mechanical strength, and excellent bioactivity when compared to calcium phosphates bioceramics. Wollastonite was prepared through the low-temperature sol-gel combustion method using urea as the fuel, nitrate ions and nitric acid as the oxidizer. Calcium nitrate and tetraethyl orthosilicate were taken as the source of calcium and silica. The synthesized wollastonite were characterized by Fourier transform infrared spectroscopy for the identification of characteristic functional group and powder X-ray diffraction for the phase identification. Employing urea as a fuel resulted in needle-like morphology of the particles, which was confirmed by scanning electron microscopy and transmission electron microscopy. It was observed that the needle-like morphology enhances the mechanical properties such as elasticity and compressive strength and also increases the surface area of the material, which could help in a rapid deposition of hydroxyapatite layer. These properties of wollastonite warrant its application as a new artificial bone material in the field of hard tissue engineering.
Keywords: sol-gel combustion synthesis, bioceramics, hydroxyapatite, compressive strength, morphology
Introduction
Bioactive ceramics actively interact with the biological environment and are able to chemically integrate and form a direct chemical bond with the surrounding bone tissues.1–4 Hydroxyapatite and calcium phosphates have received considerable attention for biomedical applications for many years due to their excellent biocompatible properties.5–7 But they have poor mechanical properties like low fracture toughness, which creates space to find alternative bioceramics.8,9 Thus, wollastonite is considered an ideal candidate for artificial bone implants in hard tissue regeneration and bone tissue engineering10,11 and is also widely used in drug delivery applications.12 The formation rate of hydroxyapatite on the surface of the wollastonite proved to be faster than that of any other biocompatible glasses, glass ceramics, and calcium phosphates due to the presence of silicate group which actively takes part in the metabolic processes during bone formation.13–17
Bones are made up of 70% inorganic constituent hydroxyapatite having rod-like-shaped morphology or needle-like morphology with diameter ranges between 25±5 nm. An artificial implant material with the same morphology and a particle size similar to that of natural bone can be a bone regenerative material and is expected to possess more bone-bonding capability than other morphological bioceramics with different morphology.18
Silicon deficiency leads to abnormal bone formation and, during the bone formation, osteoblastic proliferation is enhanced due to the presence of silicon ions. Hence, silicon is found to be an essential trace element for the bone cell activity. Pseudowollastonite was reported to possess good osteoblastic formation. Si ion bonded with calcium ion makes the ionic dissolution easier, thus provides the bond formation between the implant and surrounding bone tissue. Wollastonite is expected to induce osteogenesis by satisfying all these criteria, hence it can be used as an artificial bone implant material.19,20
The various synthetic methods employed for the preparation of wollastonite are solid-state,21 hydrothermal,22 coprecipitation,23 and solution combustion synthesis.24 The most widely used method for the preparation of wollastonite is the sol-gel method, as the synthesized particles are homogeneous and the particles were found to be in the nano-regime.25 In the existing literature, the synthesis of single phase wollastonite at 1,400°C–1,000°C for 24 hours has been reported.26,27 The synthesis of wollastonite below 1,000°C was not achieved to date. The current study reports that wollastonite was prepared using urea as the fuel at 900°C for 5 hours.
Conventional methods of synthesizing ceramic material, such as the solid-state method, need very high calcination temperature and result in excessive grain growth, leading to the poor mechanical property of the material. Sol-gel combustion synthesis of wollastonite using urea as the complexing agent is a new approach in the preparation of wollastonite. The calcination temperature is reduced to a great extent and hence the present method needs less energy and time. Routine sol-gel method is carried out first and the resultant gel is subjected to a combustion process in the furnace, which results in the formation of the precursor. The precursor is calcined to form the final product, which has a porous structure.28,29
In the present work, a modified sol-gel method assisted by combustion process is proposed in order to prepare wollastonite nanoparticles with different morphology which will improve the biocompatibility when placed in simulated body fluid (SBF).
Materials and methods
Sol-gel combustion method
Stoichiometric ratios of 1 M aqueous solution of calcium nitrate tetrahydrate (SD fine 98%) and tetraethyl orthosilicate (98%; Acros Organics) were taken and mixed with 1 M solution of urea (99%; HiMedia Laboratories), which acts as a complexing agent at the time of gel formation and as fuel during combustion. Nitric acid is used as a catalyst for the hydrolysis of tetraethyl orthosilicate and acts as an oxidizing agent, hence the solution’s pH was maintained at 1 by adding concentrated HNO3. The above solution was thoroughly mixed by using a magnetic stirrer, and the mixture was heated at 60°C. The pH and temperature favor the formation of a thick, transparent gel. The gel kept aging for 24 hours. The obtained gel was dried at 150°C for 2 hours. The dried gel was decomposed at 400°C, which resulted in a white color precursor. The precursor was calcined at 900°C for 5 hours, which led to the formation of single-phasic bioactive nanocrystalline wollastonite. The acellular SBF was prepared to examine the bioactive behavior of the synthesized wollastonite. The SBF was prepared according to the procedure proposed by Kokobo et al.30
Characterization
The phase purity of wollastonite and the nucleation of hydroxyapatite were carried out by X-ray diffraction (XRD) analysis by using the Bruker D8 Advance powder X-ray diffractometer with copper Kα radiations (λ=1.5406) over the 2θ range from 10°–80°. The functional groups were identified in the spectral range of 4,000–400 cm–1 by the KBr pellet method by using a Shimadzu (TR Affinity 1) Fourier transform infrared (FT-IR) spectrophotometer. A morphological analysis was carried out using an FEI Quanta FEG 200 high-resolution scanning electron microscope. The particle size of wollastonite was analyzed with a JEOL JEM 2100 high-resolution transmission electron microscope (TEM).
Mechanical properties
The compression strength was measured according to the ASTM standard D695-02a with a universal testing machine (Instron, USA). The dimensions of the cylindrical specimen were 13 mm diameter and 2 mm thickness of the sample was measured. The compression test was carried out at a loading rate of 1 mm/min, until a maximum reduction in sample height of 80% was obtained.
In vitro bioactivity studies of the wollastonite
To analyze the bioactive behavior of synthesized wollastonite, it was pressed into a pellet of 13 mm diameter and 2 mm thickness.31 The pellet was immersed in the SBF for 21 days to check the in vitro bioactivity process (which is the deposition of hydroxyapatite layer). The pellet was soaked in 30 mL of SBF, being replaced every 24 hours to avoid changes in the cationic concentrations. The deposition of the hydroxyapatite layer on the surface of the wollastonite was examined with a powder X-ray diffractometer at an interval of 7 days. Analysis of the pellet surface by powder XRD for its bioactivity was carried out after washing the pellet with deionized water and air-drying the pellet in a hot-air oven at 60°C.
Dissolution studies
The quantitative analysis of Ca, Si, and P ion concentrations released from the wollastonite scaffold in the SBF medium was determined with an inductively coupled plasma optical emission spectrometer (PerkinElmer OPTIMA DV 5300 ICP-OES). The samples were analyzed after immersing the pellet for various durations. Three samples were measured for each data point and the results were obtained in triplicate from three separate samples for each test.
Results and discussion
Urea was used as a fuel and complexing agent and it formed calcium ion complexes during the combustion process. Initially urea forms complex with calcium ions and simultaneously the polycondensation of silanol results in the formation of thick gel. The polycondensation process is catalyzed by nitric acid followed by the decomposition process, during which the dried gel was decomposed at 400°C, which initiated the combustion reaction:
Ca(NO3)2·4H2O + CO(NH2)2 + Si–OH | (1) |
 CaCO3 + SiO2 + CO2↑ + N2↑ + H2O
CaCO3 + SiO2 + CO2↑ + N2↑ + H2OThe exothermic reaction took place very rapidly and liberated a large amount of gases and favored the formation of calcium carbonate and silica after the combustion process.32
When the sample was calcined at 900°C for 5 hours, the calcium carbonate and silica formed the calcium silicate:
CaCO3 + SiO2 → CaSiO3 | (2) |
FT-IR spectra
The FT-IR spectra (Figure 1) of the wollastonite shows a broad band in the region 3,500 cm−1 and 1,639 cm−1 by the stretching and the bending vibration of O–H groups due to the moisture content of the sample. The FT-IR spectra (Figure 1b) of the precursor shows a band at 1,381 cm−1 due to the C=O stretching vibration of the calcium carbonate phase. The peak at 868 cm−1 was due to the CO32− ions of CaCO3. The broad band at 1,092 cm−1 was observed due to the formation of amorphous silica. This indicates the formation of a calcium carbonate phase and amorphous silica after the combustion process. The spectrum did not display the bands at 868 cm−1 after calcination and 1,381 cm−1, which shows that calcium carbonate and amorphous silica got converted into single-phasic wollastonite. The bands occurred in the region of 1,067 cm−1, which was because of the Si–O–Si stretching vibration, and 892 cm−1, which was due to Si–O–Ca groups. The band near 515 cm−1 was due to the rocking vibration of the Si–O bond. The strong peaks of Figure 1a indicates the single-phasic wollastonite without any secondary phases.33,34
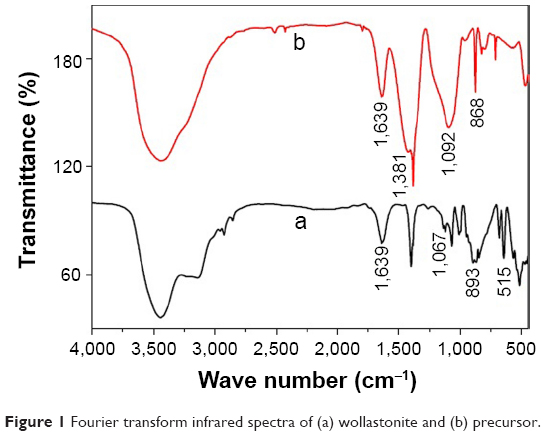 | Figure 1 Fourier transform infrared spectra of (a) wollastonite and (b) precursor. |
XRD analysis
The powder XRD pattern (Figure 2) of the calcined wollastonite powder indicates pure single-phase wollastonite. The XRD pattern is indexed according to the standard JCPDS card no 00-043-1460. The average crystallite size of wollastonite was calculated by using the Scherrer formula, and the particle size was found to be in the range of 30–40 nm. The lattice parameter values calculated by using the XRD pattern were found to be a=15.4089 Å, b=7.3258 Å, and c=7.0685 Å.
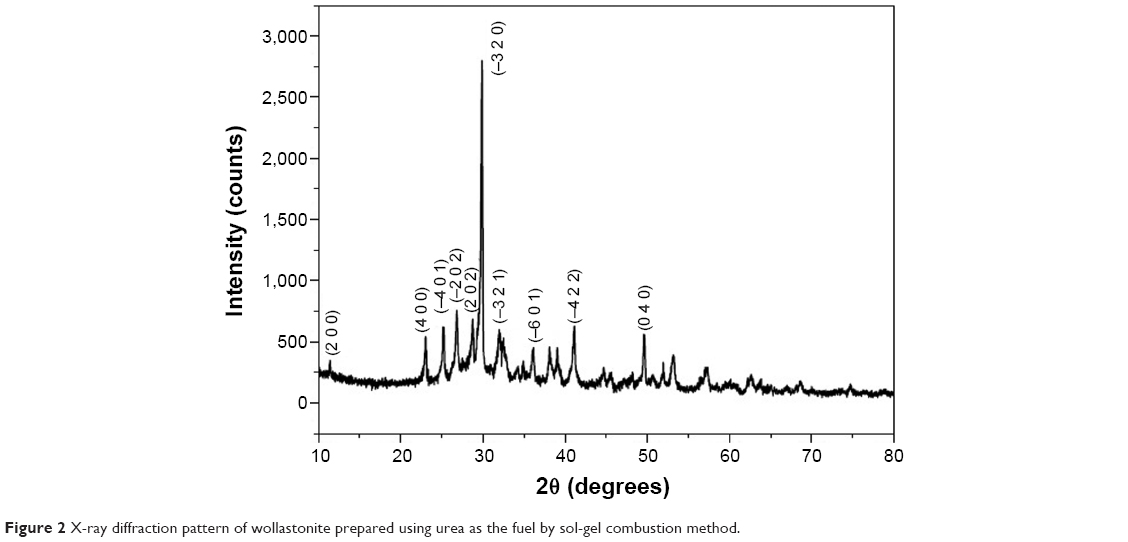 | Figure 2 X-ray diffraction pattern of wollastonite prepared using urea as the fuel by sol-gel combustion method. |
Scanning electron microscope observation
The scanning electron microscope images (Figure 3) of the wollastonite show the particles were heterogeneous in nature. At lower magnification (Figure 3A), the particle size was found to be in the range of 50–100 μm and was irregular in shape. At higher magnification (Figure 3B), sample morphology was observed as a needle-like structure.
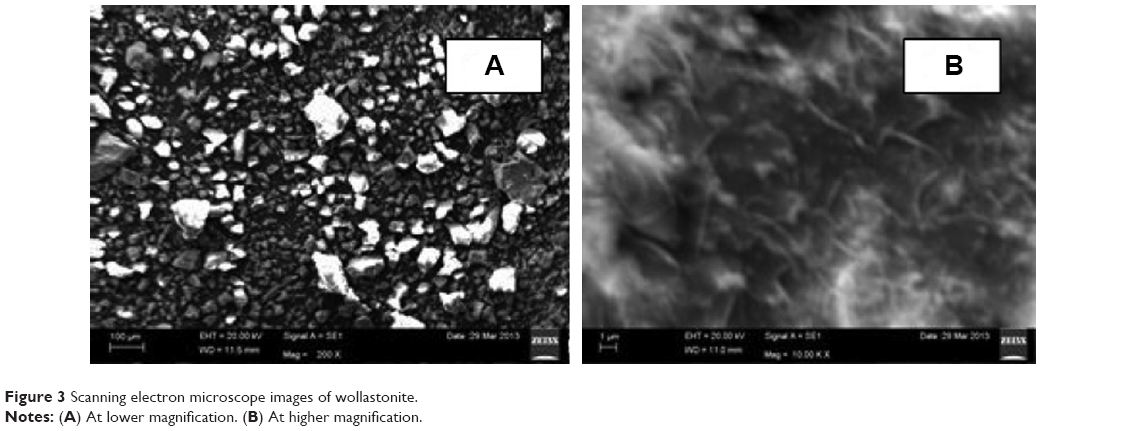 | Figure 3 Scanning electron microscope images of wollastonite. |
TEM observation
TEM micrographs (Figure 4A) and high-resolution TEM micrographs (Figure 4B) of synthesized wollastonite sample confirm the needle-like morphology with a mean particle size of 25–28 nm in diameter. The average length of the particles fell in the range of 0.2–0.5 μm. The interplanar distance taken from the zone axis along the direction (−2 0 2) was measured to be 3.319 Å. The needle-like morphology provides a high surface area, hence the synthesized compound may possess higher bioactivity.35
 | Figure 4 TEM (A) and high-resolution TEM (B) images of wollastonite. |
XRD analysis of wollastonite after immersion in SBF
The ceramic material was expected to be bioactive, and this was proved by immersing it in the SBF and analyzing its surface periodically for the biomineralization process. The temperature of the human body has to be maintained throughout the in vitro bioactivity studies, hence the temperature was maintained at 37°C. The primary factor required for the bioactive surface is that, when it is immersed in SBF to nucleate the hydroxyapatite on its surface, it should have the chemical composition similar to that of human bone’s inorganic constituent. If the ceramic compound possesses good in vitro bioactivity, then it is assumed to have a good bone-bonding capability during implantation.
The sintered pellet was soaked in SBF for 21 days and the surface of the pellet was characterized with powder XRD to analyze its bioactivity in terms of deposition of hydroxyapatite. After 21 days, it was found that the XRD pattern (Figure 5) of the wollastonite pellet showed the intensity of the diffraction peaks corresponding to wollastonite becoming less intense, and there was the appearance of a small hydroxyapatite peak after 7 days’ immersion in SBF, which is shown in Figure 5a. The hydroxyapatite peak is indexed according to the JCPDS file no 01-072-1243. The XRD pattern (Figure 5b) of the surface taken after soaking in SBF for 14 days shows that the intensity of hydroxyapatite was more prominent than the wollastonite peaks. After 21 days of immersion, almost the entire surface of the wollastonite pellet was covered by the crystalline hydroxyapatite layer, and the intensity of the wollastonite peaks appeared to be very low and the intensity of hydroxyapatite peaks was very high, as shown in Figure 5c.
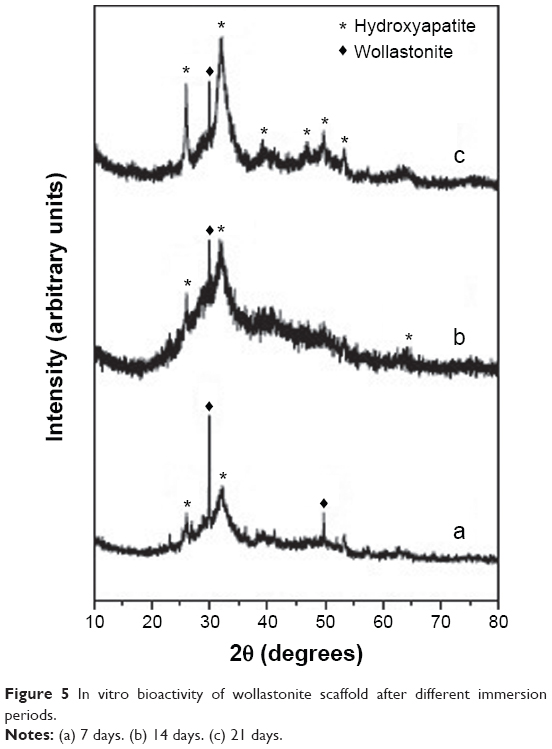 | Figure 5 In vitro bioactivity of wollastonite scaffold after different immersion periods. |
The kinetics of hydroxyapatite nucleation of the synthesized sample depends on the scale factor of the compound. Scale factor plays a crucial role in fast deposition of hydroxyapatite layer. Nanocrystalline needle-shaped wollastonite provides the maximum number of nucleation sites for the deposition of hydroxyapatite crystals (shown in Figure 6). This is due to high surface energy of the grain boundary, leading to the rapid deposition of hydroxyapatite crystals.36,37
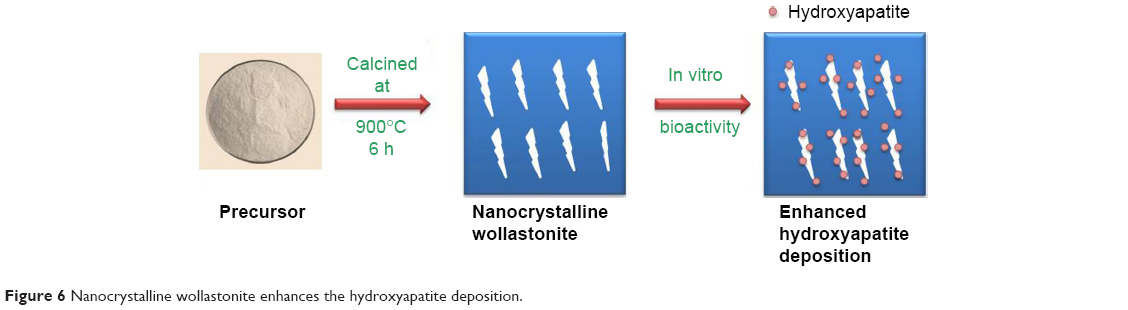 | Figure 6 Nanocrystalline wollastonite enhances the hydroxyapatite deposition. |
Figure 7A and B shows scanning electron microscope images of a cross-section of the scaffold after hydroxyapatite nucleation. The change in surface morphology of the scaffold was observed after 21 days due to the nucleation of hydroxyapatite on the surface of the scaffold. The upper surface of the scaffold was completely covered by the continuous and dense layer of hydroxyapatite. The morphology of the nucleated hydroxyapatite was irregular agglomerated particles.38 Figure 7C is the energy dispersive X-ray spectroscopy pattern of wollastonite scaffold after immersion in SBF for 21 days. It was found that, besides Si and Ca peaks, the pattern for phosphorous appeared in the spectrum, indicating the formation of hydroxyapatite on the top surface of the scaffold.39
 | Figure 7 Normal view and cross sectional view of wollastonite scaffold after hydroxyapatite. |
Release of ions in SBF
Figure 8 shows the variations of Ca, Si, and P ion concentrations after soaking in SBF for different time periods. After 7 days’ immersion, the Ca ion concentration in the SBF decreased, which reveals that the Ca ion in the SBF was consumed for the hydroxyapatite deposition, and it decreased gradually after the 14th and 21st days. As for the P ion concentration of SBF, it decreased after 1 day of immersion. The bioactivity of silicate-based materials indicates that the presence of PO43- ions in the composition is an essential requirement for the development of an hydroxyapatite layer, which consumes calcium and phosphate ions. Si concentrations increase with the increase in the duration of the incubation.40
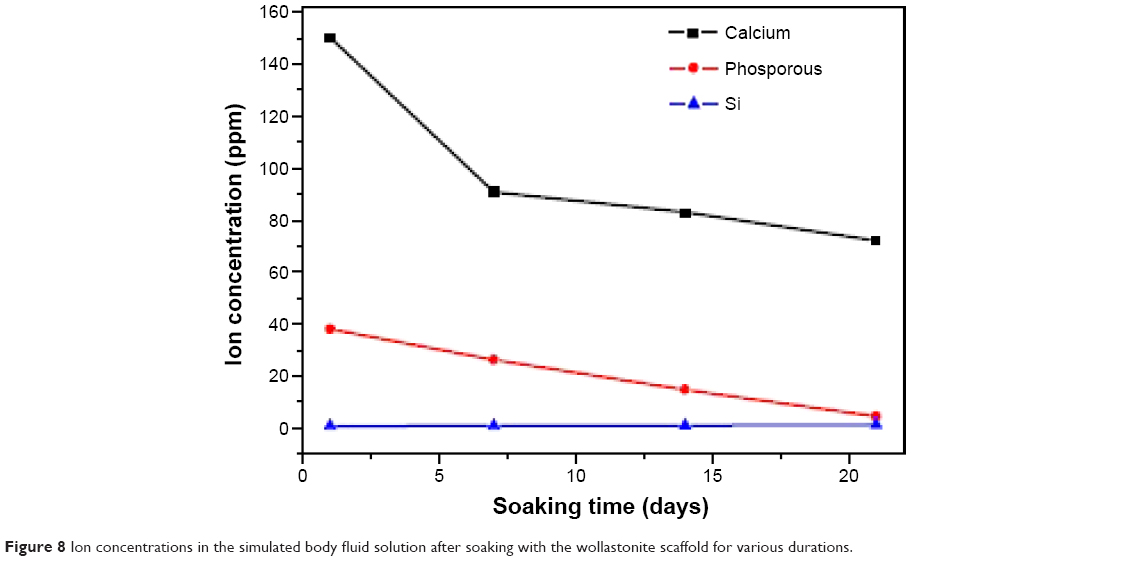 | Figure 8 Ion concentrations in the simulated body fluid solution after soaking with the wollastonite scaffold for various durations. |
Compressive strength
The compressive strength was determined from stress–strain curves by applying load until the scaffold was crushed. A typical compressive stress–strain curve of the ceramic samples is provided in Figure 9. The mechanical response shows an early abrupt descent, with stress values increasing until a maximum was reached. It is evident that, as the test progressed, the sample started to be crushed. Eventually, the sample densification occurred and, due to this phenomenon, some of the tested samples did not even break. The wollostonite appears with needle-like morphology and it provides higher compressive strength.41 The maximum compressive strength for the prepared scaffold was determined as 343 MPa.42
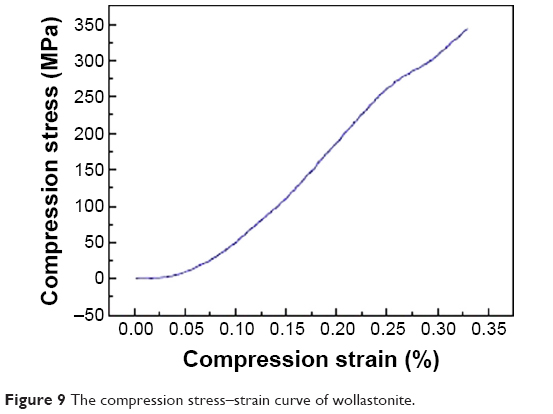 | Figure 9 The compression stress–strain curve of wollastonite. |
Conclusion
The method adopted for preparing wollastonite required less processing time and the calcination temperature was reduced from 1,200°C to 900°C. Wollastonite was prepared by using urea as the fuel for the first time and the calcined product was found to be highly single-phasic without any secondary phases. The morphology of wollastonite crystals were found to be needle-like in structure and the diameter of the crystal fell in the range of 25–28 nm, which mimics the natural human bone mineral hydroxyapatite. Needle-like morphology plays a significant role in the in vitro bioactivity process.
Acknowledgments
This research was financially supported by the Vellore Institute of Technology Research Grants for Engineering, Management and Science (VITRGEMS) and the authors thank DST-FIST for the XRD facility. The authors express their sincere gratitude to Johnson & Johnson for sponsoring the article publication.
Disclosure
The authors report no conflicts of interest in this work.
References
Manzano M, Vallet-Regi M. Revisiting bioceramics: bone regenerative and local drug delivery systems. Progress in Solid State Chemistry. 2012;40:17–30. | ||
Salgado AJ, Coutinho OP, Reis RL. Bone tissue engineering: state of the art and future trends. Macromol Biosci. 2004;4:743–765. | ||
Kawahara H. Bioceramics for hard tissue replacements. Clin Mater. 1987;2:181–206. | ||
Thamaraiselvi TV, Rajeswari S. Biological evaluation of bioceramic materials – a review. Trends Biomater Artif Organs. 2004;18:9–17. | ||
Zhou H, Lee J. Nanoscale hydroxyapatite particles for bone tissue engineering. Acta Biomater. 2011;7:2769–2781. | ||
Zhao H, Jin H, Cai J. Preparation and characterization of nano-hydroxyapatite/chitosan composite with enhanced compressive strength by urease-catalyzed method. Mater Lett. 2014;116:293–295. | ||
Felekori MN, Mesgar ASM, Mohammadi Z. Development of composite scaffolds in the system of gelatin-calcium phosphate whiskers/fibrous spherulites for bone tissue engineering. Ceram Int. 2015;41: 6013–6019. | ||
Jyoti MA, Thai VV, Min YK, Lee BT, Song HY. In vitro bioactivity and biocompatibility of calcium phosphate cements using Hydroxy-propyl-methyl-Cellulose (HPMC). Appl Surf Sci. 2010;257:1533–1539. | ||
Milovac D, Ferrer GG, Ivankovic M, Ivankovic H. PCL-coated hydroxyapatite scaffold derived from cuttlefish bone: Morphology, mechanical properties and bioactivity. Mater Sci Eng C. 2014;34:437–445. | ||
Xiong K, Shi H, Liu J, Shen Z, Li H, Ye J. Control of the dissolution of Ca and Si ions from CaSiO3 bioceramic via tailoring its surface structure and chemical composition. J Am Ceram Soc. 2013;96:691–696. | ||
Raquez JM, Barone DT, Luklinska Z, et al. Osteoconductive and bioresorbable composites based on poly-(L,L-lactide) and pseudowollastonite: from synthesis and interfacial compatibilization to in vitro bioactivity and in vivo osseointegration studies. Biomacromolecules. 2011;12:692–700. | ||
Li H, Chang J. Preparation, characterization and in vitro release of gentamicin from PHBV/wollastonite composite microspheres. J Control Release. 2005;107:463–473. | ||
Liu X, Ding C, Chu PK. Mechanism of apatite formation on wollastonite coatings in simulated body fluids. Biomaterials. 2004;25:1755–1761. | ||
Magallanes-Perdomo M, Luklinska ZB, De Aza AH, Carrodeguas RG, De Aza S, Pena P. Bone-like forming ability of apatite–wollastonite glass ceramic. J Eur Ceram Soc. 2011;31:1549–1561. | ||
Padmanabhann SK, Gervaso F, Carrozzo M, Scalera F, Sannino A, Licciulli A. Wollastonite/hydroxyapatite scaffolds with improved mechanical, bioactive and biodegradable properties for bone tissue engineering. Ceram Int. 2013;39:619–627. | ||
Magallanes-Perdomo M, De Aza AH, Mateus AY, et al. In vitro study of the proliferation and growth of human bone marrow cells on apatite-wollastonite-2M glass ceramics. Acta Biomater. 2010;6:2254–2263. | ||
Liu X, Zhao M, Lu J, Ma J, Wei J, Wei S. Cell responses to two kinds of nanohydroxyapatite with different sizes and crystallinities. Int J Nanomedicine. 2012;7:1239–1250. | ||
Hing KA, Revell PA, Smith N, Buckland T. Effect of silicon level on rate, quality and progression of bone healing within silicate-substituted porous hydroxyapatite scaffolds. Biomaterials. 2006;27:5014–5026. | ||
Ha SW, Sikorski JA, Weitzmann MN, Beck GR Jr. Bio-active engineered 50 nm silica nanoparticles with bone anabolic activity: therapeutic index, effective concentration, and cytotoxicity profile in vitro. Toxicol In Vitro. 2014;28:354–364. | ||
Sprio S, Ruffini A, Valentini F, et al. Biomimesis and biomorphic transformations: new concepts applied to bone regeneration. J Biotechnol. 2010;156:347–355. | ||
Sainz MA, Pena P, Serena S, Caballero A. Influence of design on bioactivity of novel CaSiO3-CaMg(SiO3)2 bioceramics: in vitro simulated body fluid test and thermodynamic simulation. Acta Biomater. 2010;6:2797–2807. | ||
Lin K, Chang J, Lu J. Synthesis of wollastonite nanowires via hydrothermal microemulsion methods. Mater Lett. 2006;60:3007–3010. | ||
Hazar ABY. Preparation and in vitro bioactivity of CaSiO3 powders. Ceram Int. 2007;33:687–692. | ||
Chakradhar RPS, Nagabhushana BM, Chandrappa GT, Ramesh KP, Rao JL. Solution combustion derived nanocrystalline macroporous wollastonite ceramics. Mater Chem Phys. 2006;95:169–175. | ||
Meiszterics A, Sinkó K. Sol–gel derived calcium silicate ceramics. Colloids Surf A Physicochem Eng Asp. 2008;319:143–148. | ||
Iimori Y, Kameshima Y, Okada K, Hayashi S. Comparative study of apatite formation on CaSiO3 ceramics in simulated body fluids with different carbonate concentrations. J Mater Sci Mater Med. 2005;16:73–79. | ||
Zhou L, Yan B. Sol–gel synthesis and photoluminescence of CaSiO3:Eu3+ nanophosphors using novel silicate sources. J Phys Chem Solids. 2008;69:2877–2882. | ||
Mali A, Petric A. Synthesis of sodium β”-alumina powder by sol–gel combustion. J Eur Ceram Soc. 2012;32:1229–1234. | ||
Lakshmi R, Velmurugan V, Sasikumar S. Preparation and phase evolution of wollastonite by sol-gel combustion method using sucrose as the fuel. Combustion Science and Technology. 2013;185:1777–1785. | ||
Kokubo T, Kushitani H, Sakka S, Kitsugi T, Yamamuro T. Solutions able to reproduce in vivo surface-structure changes in bioactive glass–ceramic A-W. J Biomed Mater Res. 1990;24:721–734. | ||
Ragel CV, Vallet-Regí M, Rodríguez-Lorenzo LM. Preparation and in vitro bioactivity of hydroxyapatite/solgel glass biphasic material. Biomaterials. 2002;23:1865–1872. | ||
Biamino S, Badini C. Combustion synthesis of lanthanum chromite starting from water solutions: investigation of process mechanism by DTA–TGA–MS. J Eur Ceram Soc. 2004;24:3021–3034. | ||
Saravanapavan P, Hench LL. Mesoporous calcium silicate glasses. I. Synthesis. J Non Cryst Solids. 2003;318:1–13. | ||
Tangboriboon N, Khongnakhon T, Kittikul S, Kunanuruksapong R, Sirivat A. An innovative CaSiO3 dielectric material from eggshells by sol–gel process. J Solgel Sci Technol. 2011;58:33–41. | ||
Bakan F, Laçin O, Sarac H. A novel low temperature sol–gel synthesis process for thermally stable nano crystalline hydroxyapatite. Powder Technol. 2013;233:295–302. | ||
Surmenev RA, Surmeneva MA, Ivanova AA. Significance of calcium phosphate coatings for the enhancement of new bone osteogenesis – a review. Acta Biomater. 2014;10:557–579. | ||
Ergun C, Evis Z, Webster TJ, Sahin FC. Synthesis and microstructural characterization of nano-size calcium phosphates with different stoichiometry. Ceram Int. 2011;37:971–977. | ||
Plazas Bonilla CE, Trujillo S, Demirdögen B, Perilla JE, Murat Elcin Y, Gómez Ribelles JL. New porous polycaprolactone-silica composites for bone regeneration. Mater Sci Eng C Mater Biol Appl. 2014;40:418–426. | ||
Lu J, Wei J, Gan Q, et al. Preparation, bioactivity, degradability and primary cell responses to an ordered mesoporous magnesium–calcium silicate. Microporous Mesoporous Mater. 2012;163:221–228. | ||
Kao CT, Huang TH, Chen YJ, Hung CJ, Lin CC, Shie MY. Using calcium silicate to regulate the physicochemical and biological properties when using β-tricalcium phosphate as bone cement. Mater Sci Eng C Mater Biol Appl. 2014;43:126–134. | ||
Jie W, Yubao L. Tissue engineering scaffold material of nano-apatite crystals and polyamide composite. Eur Polym J. 2004;40:509–515. | ||
Tripathi G, Basu B. A porous hydroxyapatite scaffold for bone tissue engineering: physico-mechanical and biological evaluations. Ceram Int. 2012;38:341–349. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.