")
Back to Journals » International Journal of Nanomedicine » Volume 10 » Issue 1
Influence of cationic lipid concentration on properties of lipid–polymer hybrid nanospheres for gene delivery
Authors Bose R , Arai Y, Ahn JC, Park H , Lee S
Received 22 April 2015
Accepted for publication 23 June 2015
Published 2 September 2015 Volume 2015:10(1) Pages 5367—5382
DOI https://doi.org/10.2147/IJN.S87120
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Thomas Webster
Rajendran JC Bose,1,2 Yoshie Arai,1 Jong Chan Ahn,1 Hansoo Park,2 Soo-Hong Lee1
1Department of Biomedical Science, College of Life Science, CHA University, Seongnam, 2Department of Integrative Engineering, Chung-Ang University, Seoul, South Korea
Abstract: Nanoparticles have been widely used for nonviral gene delivery. Recently, cationic hybrid nanoparticles consisting of two different materials were suggested as a promising delivery vehicle. In this study, nanospheres with a poly(D,l-lactic-co-glycolic acid) (PLGA) core and cationic lipid shell were prepared, and the effect of cationic lipid concentrations on the properties of lipid polymer hybrid nanocarriers investigated. Lipid–polymer hybrid nanospheres (LPHNSs) were fabricated by the emulsion-solvent evaporation method using different concentrations of cationic lipids and characterized for size, surface charge, stability, plasmid DNA-binding capacity, cytotoxicity, and transfection efficiency. All LPHNSs had narrow size distribution with positive surface charges (ζ-potential 52–60 mV), and showed excellent plasmid DNA-binding capacity. In vitro cytotoxicity measurements with HEK293T, HeLa, HaCaT, and HepG2 cells also showed that LPHNSs exhibited less cytotoxicity than conventional transfection agents, such as Lipofectamine and polyethyleneimine–PLGA. As cationic lipid concentrations increased, the particle size of LPHNSs decreased while their ζ-potential increased. In addition, the in vitro transfection efficiency of LPHNSs increased as lipid concentration increased.
Keywords: core–shell hybrid nanospheres, lipid concentration, surface modification, low cytotoxicity, transfection efficiency
Introduction
Gene therapy has emerged as a potent therapeutic approach for numerous diseases, including cancer.1 Accordingly, gene therapy using traditional Chinese medicine, gene-delivery systems, and genetic engineering have been widely studied. However, the clinical success of gene therapy is still uncertain.2–4 In particular, designing efficient gene-delivery vectors with low cytotoxicity is considered a major challenge among scientists.5,6 In clinical trials, viral vectors that have been widely used as gene-delivery carriers have exhibited severe toxicity and caused side effects.1,2,7 Therefore, there is an increasing demand for the development of nonviral delivery vectors with the ability to overcome physiological barriers.8
Among the various nonviral gene vectors, cationic lipids are ideal gene carriers because of their high transfection efficiency, excellent gene-incorporation ability, and ease of preparation.1,9 However, their clinical uses are limited, because of their instability and poor reproducibility caused by high batch-to-batch variation.10 Alternatively, biodegradable polymeric nanoparticles (NPs) have been investigated, due to their small particle size, ease of surface functionalization, excellent physiological stability, and sustained-release profiles.11 However, the low transfection efficiency of polymeric NPs has hindered their clinical success.
Therefore, there is an increasing demand for a hybrid vector to overcome the barriers associated with conventional gene carriers.12 Recently, lipid–polymer hybrid nanospheres (LPHNSs) were designed to achieve complementary benefits of liposomes and polymeric NSs.13–15 They were designed to have a biodegradable polymer core and lipid layers.14,16 Depending on their application, specific fabrication methods have been reported.17 In the past, LPHNSs were prepared by two-step methods that required introducing lipid vesicles into preformed polymeric NSs.18 However, this method is inefficient, because of practical difficulties and process complexity.12 Alternatively, a single-step fabrication method was developed, which combined the fabrication process of emulsion-solvent evaporation with the self-assembly of lipids.15 Although successful outcomes have been achieved using the single-step method, a better understanding of the parameters involved in the fabrication process is required in order to significantly improve the design and fabrication of LPHNSs.19,20 Specifically, cationic lipids forming outer shells could allow for superior gene-delivery capabilities. However, it is still not clear how lipid concentration affects the formation of LPHNSs. Furthermore, it is important to balance the amount of lipids, because despite being a key factor for DNA delivery, a high concentration of cationic lipids could result in cytotoxicity. Therefore, in order to optimize their performance, it is necessary to understand the influence of cationic lipid concentration on various properties of LPHNSs.21
The present study describes a simple and efficient method for fabrication of LPHNSs, and examined the influence of cationic lipid concentration on the structural and functional characteristics of LPHNSs. We rationally designed LPHNS formulations with four different ratios of cationic lipids to polymer during the fabrication step. Then, the LPHNSs were compared with various controls, including poly(D,L-lactic-co-glycolic acid) (PLGA), liposomes, and polyethyleneimine (PEI)–PLGA NSs.
Materials and methods
Materials
PLGA (50:50) (Resomer® RG 502 H, molecular weight 7,000–17,000) was purchased from Sigma-Aldrich Co (St Louis, MO, USA). Cationic lipid 1,2-di-(9Z-octadecenoyl)-3-trimethylammonium-propane (chloride salt) and fluorescent lipid 1-oleoyl-2-[12-[(7-nitro-2-1,3-benzoxadiazol-4-yl)amino]dodecanoyl]-sn-glycero-3-phosphocholine] (NBD-PC) were purchased from Avanti Polar Lipids (Alabaster, AL, USA). Lipofectamine 2000 was obtained from Life Technologies Korea (Seoul, South Korea). TE buffer (10 mM Tris/HCl, pH 8.0, 1 mM ethylenediaminetetraacetic acid) was purchased from Cosmo Genetech (Seoul, South Korea). Plasmid EGFP (pEGFP) was obtained from Clontech (Palo Alto, CA, USA), and the plasmids were amplified in Escherichia coli and purified using a Qiagen Plasmid Giga Kit (Qiagen NV, Venlo, the Netherlands). Polyvinyl alcohol (PVA; molecular weight 13–23 kDa), Protamine sulfate, rhodamine (Rho), and all other chemicals were obtained from Sigma-Aldrich.
Lipid–polymer hybrid nanosphere preparation and characterization
The four sets of LPHNSs were prepared as described previously by the modified double-emulsion solvent-evaporation method with self-assembly.20 Briefly, protamine sulfate (10 μg/mL) was dissolved in aqueous solution. The cationic lipid (1,2-di-(9Z-octadecenoyl)-3-trimethylammonium-propane (chloride salt) [DOTAP]) and PLGA (3% w/v) were dissolved in dichloromethane. The water-in-oil emulsion was formed by the addition of an aqueous phase into an organic phase with sonication (ultrasonic probe; Sonics & Materials Inc, Newtown, CT, USA). The primary emulsion was transferred to 1% w/v PVA aqueous solution and sonicated in an ice bath. The resultant secondary (water in oil in water) emulsion was stirred overnight at room temperature until evaporation of dichloromethane was complete. The final formulated LPHNSs were collected by ultracentrifugation at 20,000 rpm for 30 minutes (Optima L-100 XP Ultra Centrifuge; Beckman Coulter, Brea, CA, USA). Then, they were washed three times by a repeating centrifugation step and freeze-dried at −20°C for 48 hours. At least three batches were prepared for each formulation. To investigate the influence of cationic lipid concentrations on size, charge, and in vitro performance, we prepared four formulation groups of LPHNSs with different concentrations of cationic lipid (DOTAP) to polymer ratio, as shown in Table 1. All other parameters were kept constant. PLGA NSs were also fabricated as described and used to prepare the PEI–PLGA NSs for comparison.22
 | Table 1 Physical characterizations of LPHNSs |
Preparation of fluorescent lipid–polymer hybrid nanospheres
Fluorescent (NBD-PC lipid and Rho–PLGA) NSs were prepared using the same modified solvent evaporation with self-assembly procedure with few changes.23 Briefly, 200 μL of a 1 mg/mL of Rho B solution was added to the polymer–solvent mixture (3% w/v of polymer) with 0.5mg of NBD-PC and DOTAP (24% w/w to polymer). The resultant water-in-oil emulsion was processed in the same way as the aforementioned procedure.
Characterization of hybrid nanospheres
The mean particle diameter (z-average) and size distribution (polydispersity index) of LPHNSs and LPHNS–plasmid DNA (pDNA) complexes were determined by dynamic light scattering (DLS) using a Zetasizer Nano ZS (Malvern Instruments, Malvern, UK), and the surface charge of the LPHNSs and LPHNS–pDNA complexes was determined by analysis of the ζ-potential as described previously.24,25 Both types of measurements were performed at 25°C using deionized water and repeated three times for each sample (50 μg/L). The shape and surface morphology of the LPHNSs were initially investigated by field-emission scanning electron microscopy (FESEM; JSM-6700F; JEOL, Tokyo, Japan) at an accelerating voltage of 5 kV. For the sample preparation, one drop of the NS dispersion was drop casted on a carbon tape supported by the stub, and the water was evaporated under reduced pressure. Thin layers of dried particle were sputter coated with platinum by an Auto Fine Coater (JEOL) for 30 seconds at 30 mA. The core–shell structures of LPHNSs and LPHNS–pDNA complexes were further confirmed by energy-filtered transmission electron microscopy (EFTEM).26 EFTEM experiments were carried out with a Libra 120 microscope (Carl Zeiss Meditec AG, Jena, Germany), and samples were prepared by depositing 20 μL of LPHNS suspension (0.5 mg/mL) onto a 200-mesh carbon-coated copper grid. Samples were blotted away after 30 minutes’ incubation, and grids were negatively stained with freshly prepared and sterile-filtered 2% (w/v) uranyl acetate aqueous solution. The grids were then washed twice with distilled water and air-dried prior to imaging. The core–shell hybrid structure of LPHNSs was further confirmed by a confocal laser scanning microscopy (CLSM; Carl Zeiss LSM 510 Meta) with fluorescent LPHNSs.
Formation of LPHNS–pDNA complexes and gel retardation assay
LPHNS–pDNA complexes were prepared using 1 μg of pDNA mixed with the resuspended freeze-dried LPHNS group (A, B, C, and D) at different ratios of NSs to pDNA (LPHNS:pDNA 15:1, 30:1, 60:1, and 90:1 w/w, respectively) in nuclease-free deionized water. The mixture was incubated at room temperature for 30 minutes to allow for LPHNS–pDNA complex formation. After incubation, the LPHNS–pDNA complexes were diluted in serum-free medium (Dulbecco’s Modified Eagle’s Medium [DMEM]). The incorporation efficiency of each LPHNS group (A, B, C, and D) was verified by gel retardation assay.27 For the assay, we prepared LPHNS–pDNA complexes from each experimental group (A, B, C, and D) by mixing 15, 30, 60, and 90 μg of LPHNSs with 1 μg of pDNA and incubating them for 30 minutes at room temperature. After 30 minutes, 5 μL of each group of LPHNS–pDNA complexes was mixed with 1 μL of loading buffer (0.25% w/w bromophenol blue in TE buffer) and applied to 1% agarose gel. Electrophoresis was carried out at 100 V for 20 minutes at room temperature in 0.5× TAE buffer. The gels were stained with ethidium bromide solution (10 mg/mL) for DNA visualization. Images were captured by a gel documentation system (GDS-200 D; Korea Lab Tech, Seongnam, Korea). Similarly, to investigate the supportive role of protamine, we fabricated LPHNSs (DOTAP 24% w/w) without protamine for comparison.
Cell cultures and transfection
HEK293, HeLa, HepG2, and HaCaT cells were obtained from the American Type Culture Collection (Manassas, VA, USA) and cultured at 37°C and 5% CO2 in DMEM supplemented with 10% fetal bovine serum (FBS) and 1% penicillin (5,000 U/mL)–streptomycin (5,000 U/mL). For LPHNS transfections, HEK293, HeLa, HaCaT, and HepG2 cells were seeded in 24-well plates at a density of 6×104 cells per well in 0.5 mL DMEM (GE Healthcare Bio-Sciences AB, Uppsala, Sweden) supplemented with 10% FBS for 24 hours to reach approximately 60%–80% confluence before transfection. For the transfection experiment, the cells was cultured with serum-free medium (0.5 mL) and incubated with each group for 4 hours. Then, the medium was replaced with serum containing medium and incubated for 48 hours. After 48 hours of incubation, transfected cells were examined by fluorescence microscopy (Leica Microsystems, Wetzlar, Germany), and the fluorescence intensity was quantified by C6 flow cytometry (Accuri™ C6; BD Biosciences, San Jose, CA, USA) and analyzed under excitation and emission wavelengths of 488 and 520 nm, respectively. The autofluorescence of untreated cells was used as an internal control. Forward and side light-scatter gates were set to exclude dead cells, debris, and cell aggregates. At least 10,000 events were acquired and analyzed per sample. To calculate the relative transfection efficiency of LPHNSs, all experiments were designed to compare LPHNS groups with Lipofectamine 2000. PEI–PLGA NSs were used as a positive control, and pDNA alone was used as the negative control. Lipofectamine–pDNA complexes were prepared according to manufacturer’s instructions and equalized to our experimental conditions by optimizing the concentration (1–2.5 μL with 1 μg of DNA). Similarly, PEI-PLGA–pDNA complexes were prepared by a mixing (90:1 w/w) ratio. The cell experiment in this study was done in the CHA University. All the immortalized human cell lines were purchased from ATCC and have been subcultured with the approval of the Ethics Committee at CHA University.
Cellular uptake and intracellular release study
The cellular uptake and intracellular release behaviors of the LPHNS groups (A, B, C, and D) were investigated as described previously.28 HeLa cells were seeded in 24-well plates at a density of 6×104 cells per well in 0.5 mL DMEM supplemented with 10% FBS to reach approximately 60%–80% confluence. After 24 h, fluorescently labeled Rho LPHNSs (90 μg) with different cationic lipid-concentration groups (A, B, C, and D) were added into the culture medium. Following various incubation times of 0.5, 1, 2, 4, 6, 12, and 24 hours, the cells were washed twice with phosphate-buffered saline (PBS), trypsinized, and quantified by flow cytometry. Initially, a threshold of fluorescence was generated using HeLa cells without exposure to the LPHNSs as a control sample. The autofluorescence of untreated cells was used as an internal control. Forward and side light-scatter gates were set to exclude dead cells, debris, and cell aggregates. At least 10,000 events were acquired and analyzed per sample. All events corresponding to the control sample were located at intensities below this threshold. The number of cells carrying Rho LPHNSs was found from the area matching the events located at higher intensities than the threshold. The cellular uptake ratio was calculated as follows:
|
|

Further, the cellular uptake and intracellular behaviors of LPHNSs with different surface coatings were studied in HeLa cells with CLSM (LSM 510) using fluorescently labeled Rho LPHNSs. The PEI-modified PLGA NSs were used as controls.28 In brief, HeLa cells were plated on coverslips in 24-well plates (cell density 6×104 cells/well) and maintained in DMEM supplemented with 10% FBS at 37°C in a humidified atmosphere containing 5% CO2 for 1 day. Then, fluorescent Rho LPHNSs (90 μg) with different cationic lipid-concentration surface coatings were added to the cells and incubated for 12 hours at 37°C. The cells were washed with PBS and fixed with 4% paraformaldehyde solution in PBS and washed three times, and the nuclei were stained with 5 μL of DAPI (4′,6-diamidino-2-phenylindole; 1 μg/mL) for 1–2 minutes at room temperature and washed. Finally, the cells were mounted and observed using CLSM.
Cytotoxicity assay
The cytotoxicity of LPHNS–pDNA complexes were evaluated using HEK293, HeLa, HaCaT, and HepG2 cells by CCK-8 assay.29 Cells were seeded in a 24-well plate at a density of 6×104 cells/mL. After incubation for 24 hours, the medium was exchanged with 0.5 mL of culture medium containing 90 μg of LPHNS–pDNA complexes (90:1 w/w) from each group. Lipofectamine 2000 and PEI–PLGA NSs were used as control groups. After incubation for 48 hours, the medium was replaced with fresh medium. CCK-8 solution (10 μL) was added to each well, followed by incubation for 1 hour at 37°C. Then, the absorbance at 450 nm was measured by a VersaMax ELISA Microplate Reader (Molecular Devices LLC, Sunnyvale, CA, USA). Cell viability was expressed as a percentage based on control (untreated) cells. Additionally, CCK-8 assays were performed with HEK293 cells for groups with different concentrations (15, 30, 60, and 90 μg).
Short-term stability testing
Stability is a crucial factor affecting the practicality of hybrid NS formulations. For stability testing, three batches of each LPHNS group (A, B, C, and D) and bare PLGA were stored in transparent glass vials at 4°C. The particle sizes of the experimental groups were used to determine the stability by DLS (Nano ZS), and measurements were taken at selected time intervals.
Statistical analysis
At least three independent sets of experiments for each condition were performed in triplicate. Data were pooled, and are statistically expressed in terms of means and standard deviation. Analysis of variance was used for analysis of quantitative values, and the Bonferroni post hoc test was used for comparisons among groups. Differences were considered significant at P<0.05. The Prism software package (version 5.02; GraphPad Software Inc, La Jolla, CA, USA) was used to perform the statistical tests.
Results and discussion
In the past few decades, numerous nanocarriers have been developed for safe and efficient gene delivery.30 We aimed to develop a simple and efficient hybrid nanocarrier as a nonviral gene-delivery vehicle, which has the complementary features of both liposome and polymeric NSs. We synthesized relatively monodisperse core–shell LPHNSs (Figure 1), consisting of a positively charged cationic lipid (DOTAP) with a protamine layer forming the outer shell and an inner spherical PLGA core. The LP HNSs were fabricated by modifying the double-emulsion solvent-evaporation process by allowing lipids and protamine to self-assemble on the surface of a polymer core.14,20 Protamine was used as the condensing agent.31 In this study, four different concentrations of lipids with constant protamine were used to construct LPHNSs, and their properties and transfection efficiencies were compared.
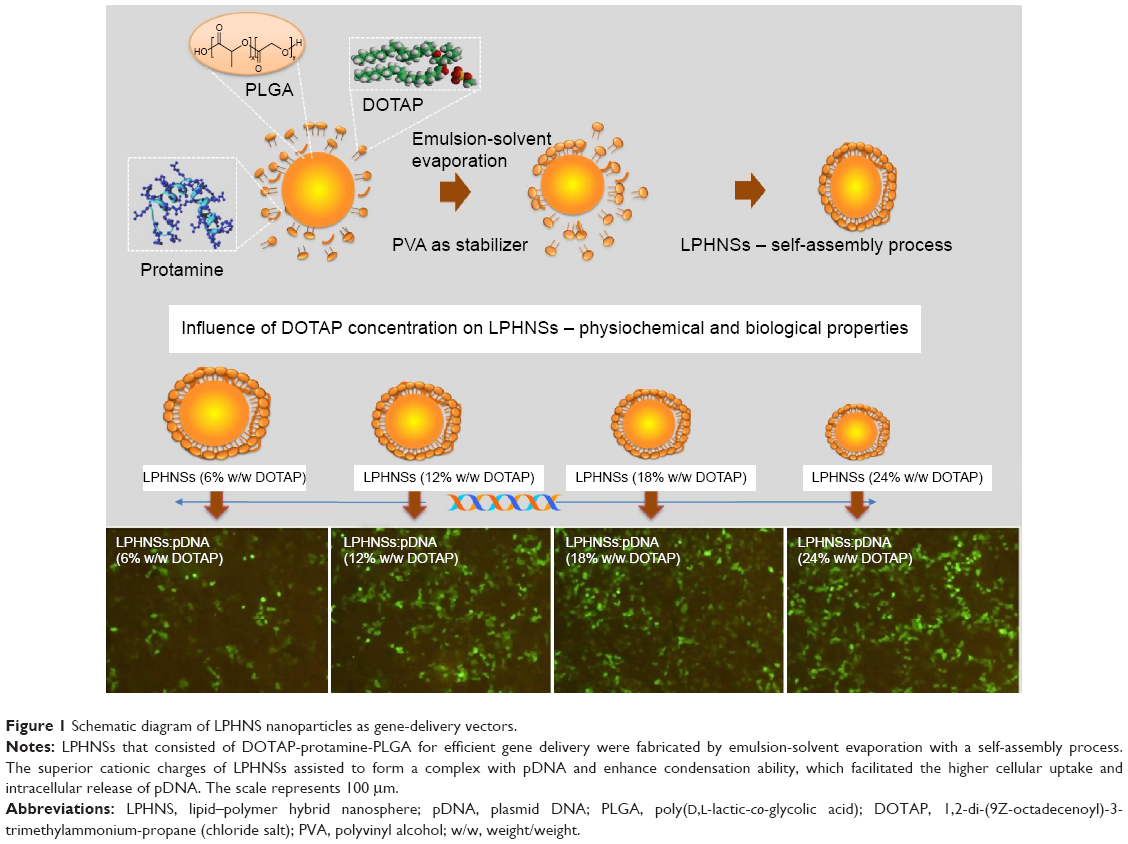 | Figure 1 Schematic diagram of LPHNS nanoparticles as gene-delivery vectors. |
Influence of cationic lipid concentration on size and size distribution of LPHNSs
In order to study the influence of cationic lipid concentration on LPHNS properties, LPHNSs with different concentrations of lipids were prepared using different ratios of cationic lipid to polymer in the organic phase, with a constant amount of protamine (10 μg/mL) in the aqueous phase. As shown in Table 1, it was observed that the mean diameters (z-average sizes) of LPHNSs with cationic lipid concentrations of 6% w/w (group A), 12% w/w (group B), 18% w/w (group C), and 24% w/w (group D) were 209±10.4, 186±7.3, 163±5.6, and 154±5.2 nm, respectively. DLS analysis revealed that there was a significant size reduction (Figures 2A, S1) compared to bare PLGA NSs (247±12.8 nm), which could be explained by the fact that the processing of those particles in a single step was stabilized by the function of the lipids with PVA.15,20 It was also found that there was a decrease in particle size (209–154 nm) as the lipid concentrations increased in the organic phase from 6% to 24% w/w (lipid-to-polymer weight ratio). The concentration of cationic lipids could play a significant role in controlling the size of LPHNSs, possibly reducing the coalescence of particles.32 Furthermore, it was found that the higher lipid concentration (24% w/w DOTAP) with constant protamine considerably decreased LPHNS–pDNA complex size (z-average) than the less lipid group (6% w/w DOTAP). The synergistic effect of cationic lipid and protamine could have been the possible reason, as observed in earlier studies.33
 | Figure 2 Characterizations of LPHNSs. |
Influence of cationic lipid concentration on surface charge of LPHNSs
Surface charge is an important indication of the stability of a colloidal system in a particular medium.34 We investigated the influence of cationic lipid concentrations on LPHNS surface charge (Figures 2B, S1). As expected, the inclusion of cationic lipids changed the surface charges of the particles. All LPHNSs had positive charges compared with the negative charge of bare PLGA NSs (−24±4 mV). As shown in Table 1, the ζ-potentials of LPHNSs with cationic lipid concentrations of 6% w/w (group A), 12% w/w (group B), 18% w/w (group C), and 24% w/w (group D) were 36±3, 50±1.8, 57±2.2, and 64±2 mV, respectively. However, the addition of negatively charged pDNA to the LPHNSs led to a slight charge reduction, as shown in Figure 3C. Considering that the surface area of LPHNSs decreased with increasing concentration, which might lead to a decrease in the incorporation of cationic lipids, an increase in the surface charges with increasing lipid concentration was unexpected.35 We believe that this finding will help with the optimization of LPHNSs for the purpose of transfection and delivery of multiple bioactive molecules in the future.
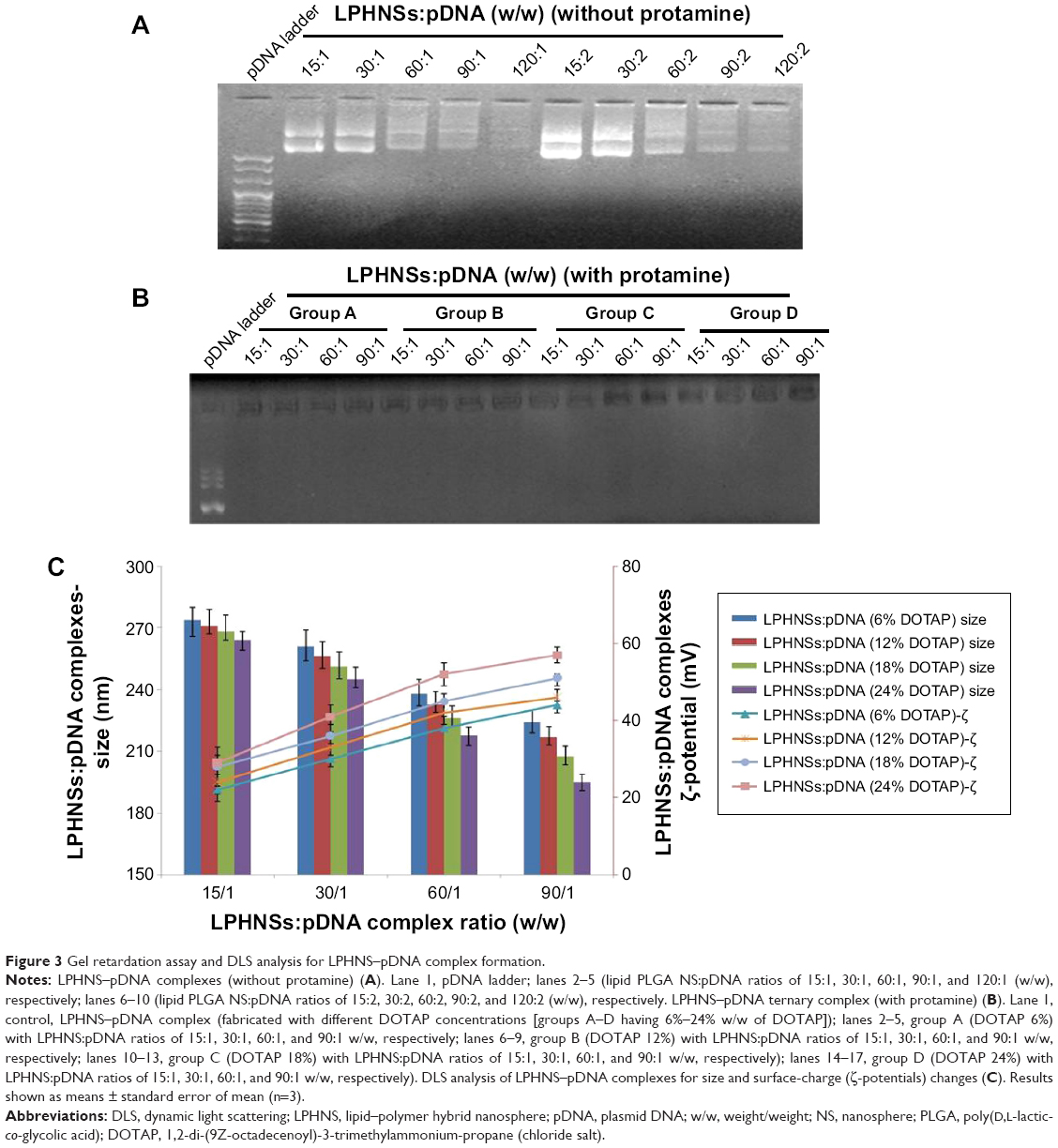 | Figure 3 Gel retardation assay and DLS analysis for LPHNS–pDNA complex formation. |
Influence of cationic lipid concentration on LPHNS morphology
The overall structure of the NSs was examined to ensure that they were hybrid particles of lipid and a polymeric core, rather than a random combination of liposomes and unprotected PLGA NPs. As shown in Figure 2C and D, FESEM and EFTEM showed that all formulation groups (A, B, C, and D) exhibited the perfect HNS size (~150–250 nm). The particle size observed from the EFTEM image was in agreement with that determined by DLS (Figures 2A, S1). Further, EFTEM confirmed the formation of an HNS consisting of a PLGA core covered by a thin lipid monolayer. The negative staining further confirmed the core–shell structure of LPHNSs. Additionally, EFTEM confirmed the particle-size reduction (~209–154 nm) when the lipid concentration was increased from 6% to 24% w/w. It appeared that 6% DOTAP was unable to control the NS aggregation, whereas 24% w/w DOTAP effectively controlled the aggregation. We speculated that the DOTAP played a synergistic role as a stabilizer of PVA more efficiently at higher concentrations than at lower concentrations. To visualize the core–shell structure and lipid assembly further, two different fluorescent dyes – NBD-PC (0.5 mg/mL) with DOTAP and Rhodamine (1 mg/mL) with PLGA – were used to synthesize fluorescent LPHNSs and examined using confocal microscopy. The fluorescence image in Figure 2E clearly illustrates the self-assembly process of the lipid–polymer core–shell structure by forming a uniform green fluorescence on a red fluorescence from the surface of the inner core. Furthermore, EFTEM clearly shows the difference between LPHNSs and LPHNS–pDNA complexes (Figure S2).
Influence of cationic lipid concentration on LPHNS–DNA complexes and transfection efficiency
The binding ability of the cationic LPHNSs to the polyanionic pDNA (pEGFP) was studied using electrophoresis, DLS, and microscopic analysis.36 It has been proven that the condensation of DNA is important for the protection of DNA from enzymatic degradation, as well for allowing easy entry into the nucleus. The LPHNS–pDNA complexes were prepared as described in the Materials and methods section. As shown in Figure 3A, with increasing cationic lipid (DOTAP) concentration in the LPHNS formulation, DNA-incorporation efficiency increased. When the DOTAP lipid concentration was increased from 6% to 24% w/w, the pDNA-incorporation ability improved significantly (results not shown). However, the NS–pDNA ratio played a crucial role in DNA incorporation, and it was observed that at higher LPHNS particle concentrations (>130 μg), 1 μg of pDNA was retained. However, at such a high NS concentration, cytotoxicity increased and cell viability decreased below 80% (results not shown). On the other hand, less than 100 μg of LPHNS particle concentration (<100 μg) was unable to compact the pDNA completely, as shown in Figure 3A. Therefore, we included protamine in the LPHNSs during the fabrication process (Figure 1). Incorporation of protamine in the LPHNS formulation resulted in strong pDNA binding and retention in all LPHNS-formulation groups (A, B, C, and D) at various NS concentrations (15, 30, 60, and 90 μg) (Figure 3B and C). Furthermore, the synergistic effect of protamine and lipid resulted in a higher degree of complexation with pDNA, explaining the role of protamine as a DNA-condensing agent (Figures 3C, S2).37
Influence of cationic lipid concentration on cell viability
Cytotoxicity is a crucial problem associated with most nonviral vectors, limiting their clinical use.38 The cytotoxicity of LPHNSs–pDNA complexes as well as control groups (PEI–PLGA and Lipofectamine 2000–pDNA complexes) was investigated with HEK293, HeLa, HaCaT, and HepG2 cells by CCK-8 assays. It is well known that lipoplexes and polyplexes exhibit high cytotoxicity.8 The experimental results confirmed similar cytotoxicity for Lipofectamine and PEI-PLGA–pDNA complexes, as shown in Figure 4. Notably, all LPHNS–pDNA complexes exhibited low cytotoxicity (cell viability >70%), even at higher NS concentrations (90 μg) for 293T, HeLa, HaCaT, and HepG2 cells. In addition, groups with higher lipid concentrations, such as LPHNS group C with 18% w/w DOTAP and group D with 24% w/w DOTAP, did not exhibit severe cytotoxicity (>70% cell viability in all tested cells). Furthermore, we investigated the effect of concentration of LPHNS–pDNA complexes on cell viability of HEK293 cells by using 15, 30, 60, and 90 μg of LPHNSs with 1 μg of pDNA. The experimental results confirmed that all tested NS–pDNA complexes exhibited comparatively lower cytotoxicity than PEI–PLGA NS–pDNA complexes (90:1 w/w) (Figure S3).
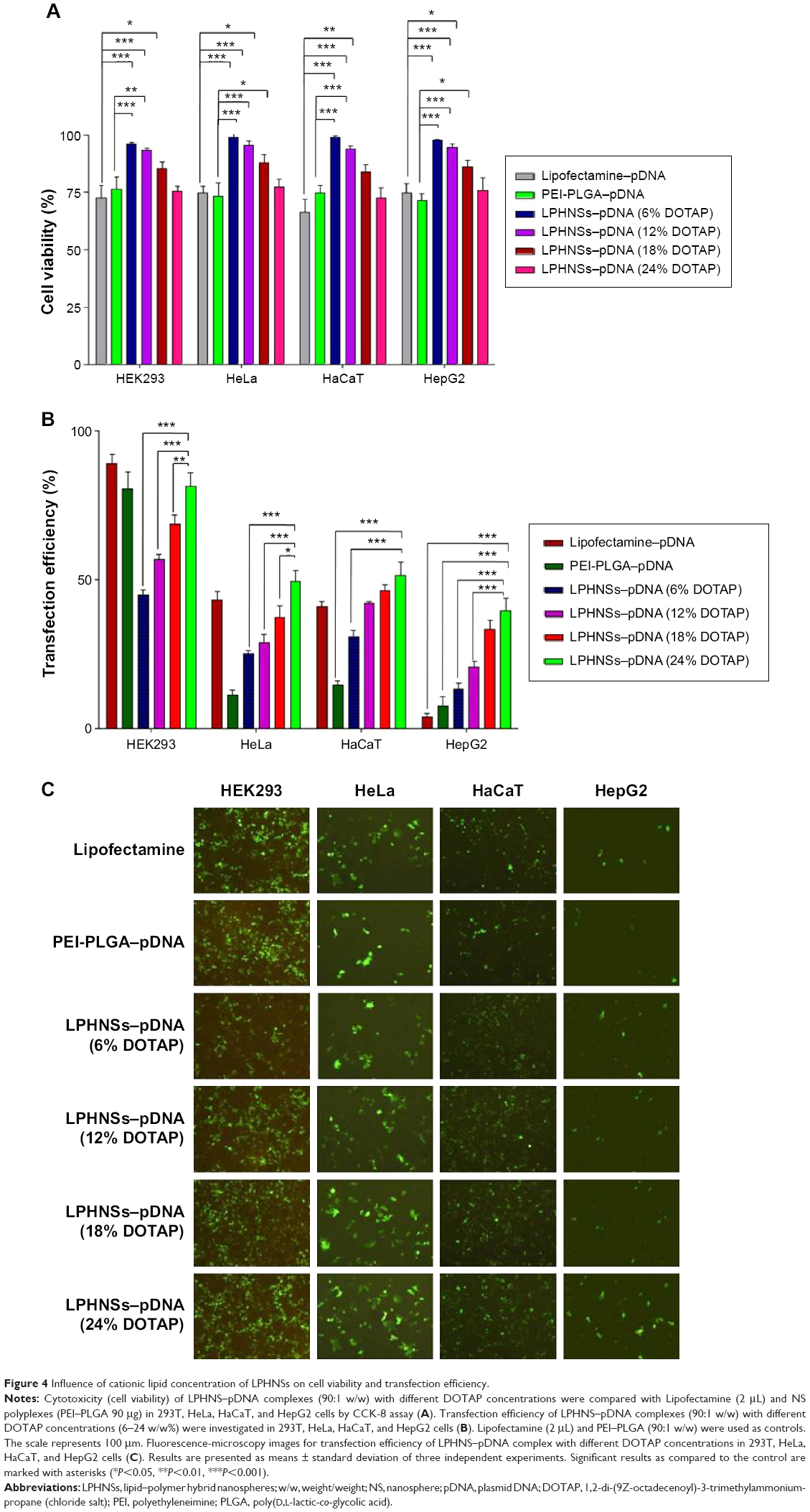 | Figure 4 Influence of cationic lipid concentration of LPHNSs on cell viability and transfection efficiency. |
It was also observed that there was a slight increase in cytotoxicity for both PEI–PLGA and LPHNSs. It has been confirmed in various tissues that cationic liposome-based lipoplexes show dose-dependent toxicity.8,39 The size, charge ratio, and amount of free lipid present in the cationic liposome may be the reason for the increased cytotoxicity of cationic lipoplexes.40 However, our LPHNS system’s nanosize (~150–200 nm) and biodegradable lipid–polymer hybrid could be the major contributors to the low cytotoxicity. Furthermore, the spherical shape and less aggregation of LPHNSs could be the reason for low cytotoxicity, since cytotoxicity of NSs depends on the nature of the aggregates formed and morphology of lipoplexes (Figure 3).41 Ample evidence exists showing that polymeric NPs or liposomes alone cause cytotoxicity. Therefore, to overcome this problem, lipid–polymer hybrid architectures have been designed, which combine the mechanical (stability) advantages of biodegradable polymeric NPs and the biomimetic advantages of liposomes.8,17
Influence of cationic lipid concentration on transfection efficiency of LPHNSs
The in vitro transfection activity of LPHNSs with protamine at different concentrations of cationic lipid were studied in HEK293, HeLa, HaCaT, and HepG2 cells using pEGFP as a reporter gene at a 90:1 w/w (NS:pDNA) ratio.
Transfection efficiency exhibited a strong correlation with cationic lipid concentration and NS concentration (results not shown) of the LPHNS gene carriers. Transfection efficiency in all four cell lines was slightly different, as shown in Figure 4B. When the cationic lipid concentration increased from 6% to 24% w/w at a specific NS concentration (90 μg), the transgene-expression level increased from 48% to 85% in HEK293 cells. At the same NS concentration, the transgene-expression level increased from 27% to 50% in the HeLa cells. Similar trends were observed in human hepatocellular carcinoma cells, where transfection efficiency increased from 13% to 40%, and in immortalized human keratinocytes, where transgene-expression level increased from 30% to 53%. Interestingly, all formulation groups of LPHNSs showed markedly enhanced transfection efficiencies in HEK293 cells compared with other tested cells, as shown in Figure 4C. This could be attributed to the biochemical machinery of HEK293 cells, capable of carrying out most of the posttranslational folding and processing required to generate functional and mature protein from a wide spectrum of both mammalian and nonmammalian nucleic acids.42
Lipid concentration-dependent transfection efficiency was observed in all four tested cell lines (Figure 4C). The transfection efficiencies of LPHNSs with protamine increased significantly as expected, but was slightly different in different cell types. For instance, in HEK293 cells, LPHNS group D exhibited enhanced gene expression (85%) compared to group A (48%), group B (61%), and group C (72%), while transgene expression in other cell types was not as different as in HEK293 cells. However, we observed that the type of cells marginally influenced transgene expression (Figure 4C). It was evident in the cervical cancer (HeLa) cells that experimental group D had a higher transgene-expression level (50%) compared to group A (27%), group B (33%), and group C (45%). Particularly, the transgene-expression capability of LPHNSs and the effect of cell type on transgene expression among cell lines were investigated in additional cells. (Figure 4C) A similar trend of lipid concentration-dependent transfection was also observed in HepG2 cells, where group D had a higher gene-expression level (40%) compared to group A (17%), group B (24%), and group C (32%). In immortalized human keratinocytes, the transgene expression of group D showed superior efficiency (54%) compared to the lower lipid-concentration counterparts, as observed in group A (30%), group B (42%), and group C (46%), demonstrating that the transfection activity of the cationic lipid–protamine polymer hybrid nanocarrier was significantly dependent on their cationic lipid concentration.
The transgene-expression level induced by the LPHNSs at the optimal NS:pDNA ratio in HEK cells was lower than that mediated by Lipofectamine, whereas it was at the same order of magnitude as that mediated by PEI–PLGA in HEK293 cells. However, groups D and C exhibited 30% more transfection efficiency than that induced by PEI–PLGA in HeLa cells and 25% higher transfection in HepG2 cells. Specifically, group D demonstrated 2.6-fold higher transfection activity in HepG2 than Lipofectamine. One possible explanation for the good transfection activity of LPHNSs may be the cationic lipid–protamine ternary complex having a hybrid structure with optimal cationic charge and uniform size.
Influence of cationic lipid concentration of LPHNSs on cellular uptake and intracellular processing
The cellular uptake of NSs can take place through multiple pathways, depending on the hybrid NS characteristics and specific cell type.28 High cellular uptake of LPHNSs is an essential qualification for gene transfection.43 The fluorescently labeled Rho LPHNSs taken up by HeLa cells were quantified as a function of incubation time. The cellular uptake of LPHNSs in all tested groups (A, B, C, and D) was higher than PEI-modified PLGA NSs for the same incubation time. As shown in Figure 5A, group D (DOTAP 24% w/w) had a higher cellular uptake, reaching more than 85% after 24 hours of incubation, while group A had ~60% uptake of LPHNSs. Based on these results, the particle-uptake rate and the final amount of HNSs inside the cells were strongly dependent on the cationic lipid concentrations. The results from flow cytometry were further evaluated with CLSM, as shown in Figure 5B. Interestingly, LPHNSs were found in the cytoplasm of cells, and were in close proximity to the nucleus. With increasing concentrations of DOTAP in the LPHNS formulation, the HNSs were formed as aggregates in the proximity of the nucleus. The possible explanation for the formation of an aggregated vesicle-like structure may be that DOTAP-mediated gene-delivery carriers are internalized by cells via clathrin-mediated endocytosis and efficiently released from the acidic endosomal compartment prior to endosome/lysosomal degradation. Therefore, the DNA is accumulated near the perinuclear region of the cells; our results demonstrated that the size of the aggregates may be indirectly proportional to the DOTAP concentration. Alternately, polyplexes (PEI–PLGA) probably follow caveolae-mediated uptake, leading to scattered fluorescence in the cytoplasm.44
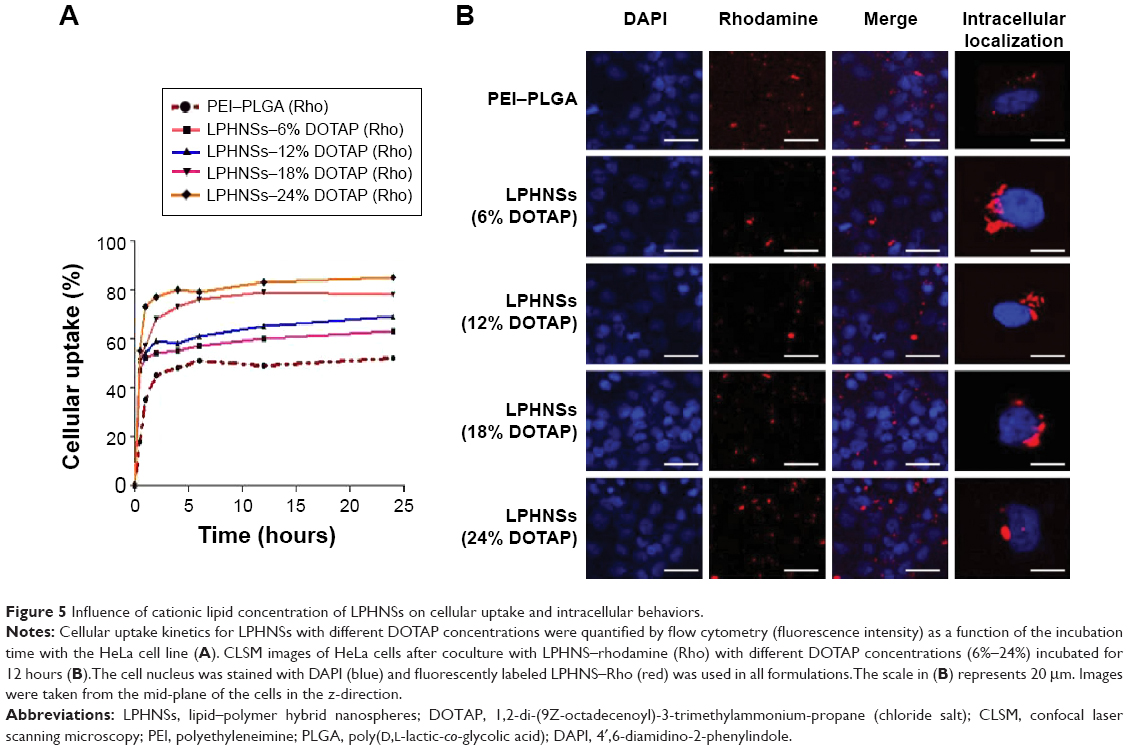 | Figure 5 Influence of cationic lipid concentration of LPHNSs on cellular uptake and intracellular behaviors. |
The group with the lowest concentration of cationic lipid (group A [6% w/w of DOTAP]) formed large, scattered aggregates close to the nucleus. When the concentration of cationic lipid in the LPHNS formulation was increased from 6% to 24% w/w (Figure 5B), the ζ-potentials (positive charge) of the hybrid nanocarriers also increased significantly, probably leading to vesicle-like aggregate formation.45 The propensity to form aggregates in the cytoplasm, particularly close to the nucleus, is a special feature of lipid-based nanocarriers.46 On the contrary, PEI–PLGA NSs do not form vesicle-like structures, and become scattered in the cytoplasm.28 Analysis of LPHNSs with different concentrations of DOTAP lipid (6%, 12%, 18%, and 24% w/w) demonstrated that cationic lipid concentration played a crucial role in cellular uptake and transfection efficiency of LPHNSs.28 The data were consistent with the major role of clathrin-mediated endocytosis in the internalization of DOTAP lipoplexes.44
Stability and aggregation of LPHNSs during storage
Nanocarrier stability is a critical factor for long-term storage and practical use. To test the shelf life of LPHNSs, three batches of each LPHNS formulation from groups A, B, C, and D were prepared by modified solvent evaporation with self-assembly and stored in transparent glass vials at 4°C. The average intensity-weighted diameters (z-average) were used to study LPHNS stability by DLS measurements at 5-day intervals.19 As shown in Figure S4, LPHNSs and control NSs stored at 4°C remained stable during the entire observed period of 15 days, maintaining a z-average slightly higher than that on the day of production and showing no visible signs of instability, such as sedimentation or aggregation.
Conclusion
Herein, we have demonstrated the potential role of cationic lipid concentration in the physical and biological performances of LPHNSs. Cationic lipids potentially reduce the size of LPHNSs, and significantly increase the surface charge toward the positive side. We postulate that the transfection efficiency of our LPHNSs could be greatly enhanced by optimizing the cationic lipid concentration and controlling process. We believe that these LPHNS vectors will allow for the optimization of LPHNSs for safe and efficient nonviral gene transfection, and have great promise as a systematic delivery system for multiple bioactive molecules, such as small interfering RNA and small-molecule drugs for the treatment of various diseases.
Acknowledgments
This research was supported by the National Research Foundation of Korea (NRF), funded by the Ministry of Science, ICT and Future Planning (NRF-2013R1A2A1A09013980). This research was also supported by the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health and Welfare, Republic of Korea (HI14C3484).
Disclosure
The authors report no conflicts of interest in this work.
References
Ginn SL, Alexander IE, Edelstein ML, Abedi MR, Wixon J. Gene therapy clinical trials worldwide to 2012 – an update. J Gene Med. 2013;15(2):65–77. | ||
Hacein-Bey-Abina S, Garrigue A, Wang GP, et al. Insertional oncogenesis in 4 patients after retrovirus-mediated gene therapy of SCID-X1. J Clin Invest. 2008;118(9):3132–3142. | ||
Check E. A tragic setback. Nature. 2002;420(6912):116–118. | ||
Thomas CE, Ehrhardt A, Kay MA. Progress and problems with the use of viral vectors for gene therapy. Nat Rev Genet. 2003;4(5):346–358. | ||
Pack DW, Hoffman AS, Pun S, Stayton PS. Design and development of polymers for gene delivery. Nat Rev Drug Discov. 2005;4(7):581–593. | ||
Yang H, Li Y, Li T, et al. Multifunctional core/shell nanoparticles cross-linked polyetherimide-folic acid as efficient Notch-1 siRNA carrier for targeted killing of breast cancer. Sci Rep. 2014;4:70–72. | ||
Raper SE, Chirmule N, Lee FS, et al. Fatal systemic inflammatory response syndrome in a ornithine transcarbamylase deficient patient following adenoviral gene transfer. Mol Genet Metab. 2003;80(1):148–158. | ||
Zhong Q, Chinta D, Pamujula S, et al. Optimization of DNA delivery by three classes of hybrid nanoparticle/DNA complexes. J Nanobiotechnology. 2010;8:6. | ||
Wasungu L, Hoekstra D. Cationic lipids, lipoplexes and intracellular delivery of genes. J Control Release. 2006;116(2):255–264. | ||
Wang Y, Miao L, Satterlee A, Huang L. Delivery of oligonucleotides with lipid nanoparticles. Adv Drug Deliv Rev. Epub 2015 Feb 27. | ||
Petros RA, DeSimone JM. Strategies in the design of nanoparticles for therapeutic applications. Nat Rev Drug Discov. 2010;9(8):615–627. | ||
Yi Y, Li Y, Wu H, et al. Single-step assembly of polymer-lipid hybrid nanoparticles for mitomycin C delivery. Nanoscale Res Lett. 2014;9(1):560. | ||
Yang XZ, Dou S, Wang YC, et al. Single-step assembly of cationic lipid-polymer hybrid nanoparticles for systemic delivery of siRNA. ACS Nano. 2012;6(6):4955–4965. | ||
Zhang L, Chan JM, Gu FX, et al. Self-assembled lipid-polymer hybrid nanoparticles: a robust drug delivery platform. ACS Nano. 2008;2(8):1696–1702. | ||
Moon JJ, Suh H, Polhemus ME, Ockenhouse CF, Yadava A, Irvine DJ. Antigen-displaying lipid-enveloped PLGA nanoparticles as delivery agents for a Plasmodium vivax malaria vaccine. PLoS One. 2012;7(2):e31472. | ||
Krishnamurthy S, Vaiyapuri R, Zhang L, Chan JM. Lipid-coated polymeric nanoparticles for cancer drug delivery. Biomater Sci. 2015;3(7):923–936. | ||
Mandal B, Bhattacharjee H, Mittal N, et al. Core-shell-type lipid-polymer hybrid nanoparticles as a drug delivery platform. Nanomedicine. 2013;9(4):474–491. | ||
Hadinoto K, Sundaresan A, Cheow WS. Lipid-polymer hybrid nanoparticles as a new generation therapeutic delivery platform: a review. Eur J Pharm Biopharm. 2013;85(3 Pt A):427–443. | ||
Fang RH, Chen KN, Aryal S, Hu CM, Zhang K, Zhang L. Large-scale synthesis of lipid-polymer hybrid nanoparticles using a multi-inlet vortex reactor. Langmuir. 2012;28(39):13824–13829. | ||
Bershteyn A, Hanson MC, Crespo MP, et al. Robust IgG responses to nanograms of antigen using a biomimetic lipid-coated particle vaccine. J Control Release. 2012;157(3):354–365. | ||
Basha G, Novobrantseva TI, Rosin N, et al. Influence of cationic lipid composition on gene silencing properties of lipid nanoparticle formulations of siRNA in antigen-presenting cells. Mol Ther. 2011;19(12):2186–2200. | ||
Krishnakumar D, Kalaiyarasi D, Bose J, Jaganathan K. Evaluation of mucoadhesive nanoparticle based nasal vaccine. J Pharm Investig. 2012;42(6):315–326. | ||
Hu Y, Ehrich M, Fuhrman K, Zhang C. In vitro performance of lipid-PLGA hybrid nanoparticles as an antigen delivery system: lipid composition matters. Nanoscale Res Lett. 2014;9(1):434. | ||
Liu Y, Pan J, Feng SS. Nanoparticles of lipid monolayer shell and biodegradable polymer core for controlled release of paclitaxel: effects of surfactants on particles size, characteristics and in vitro performance. Int J Pharm. 2010;395(1):243–250. | ||
Yuan H, Zhang W, Du YZ, Hu FQ. Ternary nanoparticles of anionic lipid nanoparticles/protamine/DNA for gene delivery. Int J Pharm. 2010;392(1):224–231. | ||
Langlois CT, Oikawa T, Bayle-Guillemaud P, Ricolleau C. Energy-filtered electron microscopy for imaging core-shell nanostructures. J Nanopart Res. 2008;10(6):997–1007. | ||
Vighi E, Ruozi B, Montanari M, Battini R, Leo E. pDNA condensation capacity and in vitro gene delivery properties of cationic solid lipid nanoparticles. Int J Pharm. 2010;389(1):254–261. | ||
Romero G, Sanz DJ, Qiu Y, et al. Lipid layer engineering of poly(lactide-co-glycolide) nanoparticles to control their uptake and intracellular co-localisation. J Mater Chem B Mater Biol Med. 2013;1(17):2252–2259. | ||
Han SB, Shin YJ, Hyon JY, Wee WR. Cytotoxicity of voriconazole on cultured human corneal endothelial cells. Antimicrob Agents Chemother. 2011;55(10):4519–4523. | ||
Li J, He YZ, Li W, Shen YZ, Li YR, Wang YF. A novel polymer-lipid hybrid nanoparticle for efficient nonviral gene delivery. Acta Pharmacol Sin. 2010;31(4):509–514. | ||
Brgles M, Šantak M, Halassy B, Forcic D, Tomašić J. Influence of charge ratio of liposome/DNA complexes on their size after extrusion and transfection efficiency. Int J Nanomedicine. 2012;7:393–401. | ||
Mainardes RM, Evangelista RC. PLGA nanoparticles containing praziquantel: effect of formulation variables on size distribution. Int J Pharm. 2005;290(1–2):137–144. | ||
Li S, Rizzo M, Bhattacharya S, Huang L. Characterization of cationic lipid-protamine-DNA (LPD) complexes for intravenous gene delivery. Gene Ther. 1998;5(7):930–937. | ||
Valencia PM, Basto PA, Zhang L, et al. Single-step assembly of homogenous lipid-polymeric and lipid-quantum dot nanoparticles enabled by microfluidic rapid mixing. ACS Nano. 2010;4(3):1671–1679. | ||
Harush-Frenkel O, Rozentur E, Benita S, Altschuler Y. Surface charge of nanoparticles determines their endocytic and transcytotic pathway in polarized MDCK cells. Biomacromolecules. 2008;9(2):435–443. | ||
Liao ZX, Peng SF, Chiu YL, et al. Enhancement of efficiency of chitosan-based complexes for gene transfection with poly(γ-glutamic acid) by augmenting their cellular uptake and intracellular unpackage. J Control Release. 2014;193:304–315. | ||
Chen J, Yu Z, Chen H, Gao J, Liang W. Transfection efficiency and intracellular fate of polycation liposomes combined with protamine. Biomaterials. 2011;32(5):1412–1418. | ||
Dinçer S, Türk M, Pişkin E. Intelligent polymers as nonviral vectors. Gene Ther. 2005;12 Suppl 1:S139–S145. | ||
Armeanu S, Pelisek J, Krausz E, et al. Optimization of nonviral gene transfer of vascular smooth muscle cells in vitro and in vivo. Mol Ther. 2000;1(4):366–375. | ||
Lv H, Zhang S, Wang B, Cui S, Yan J. Toxicity of cationic lipids and cationic polymers in gene delivery. J Control Release. 2006;114(1):100–109. | ||
Ma B, Zhang S, Jiang H, Zhao B, Lv H. Lipoplex morphologies and their influences on transfection efficiency in gene delivery. J Control Release. 2007;123(3):184–194. | ||
Thomas P, Smart TG. HEK293 cell line: a vehicle for the expression of recombinant proteins. J Pharmacol Toxicol Methods. 2005;51(3):187–200. | ||
Xue HY, Liu S, Wong HL. Nanotoxicity: a key obstacle to clinical translation of siRNA-based nanomedicine. Nanomedicine. 2014;9(2):295–312. | ||
Rejman J, Bragonzi A, Conese M. Role of clathrin-and caveolae-mediated endocytosis in gene transfer mediated by lipo- and polyplexes. Mol Ther. 2005;12(3):468–474. | ||
Harush-Frenkel O, Debotton N, Benita S, Altschuler Y. Targeting of nanoparticles to the clathrin-mediated endocytic pathway. Biochem Biophys Res Commun. 2007;353(1):26–32. | ||
Rivolta I, Panariti A, Lettiero B, et al. Cellular uptake of coumarin-6 as a model drug loaded in solid lipid nanoparticles. J Physiol Pharmacol. 2011;62(1):45–53. |
Supplementary materials
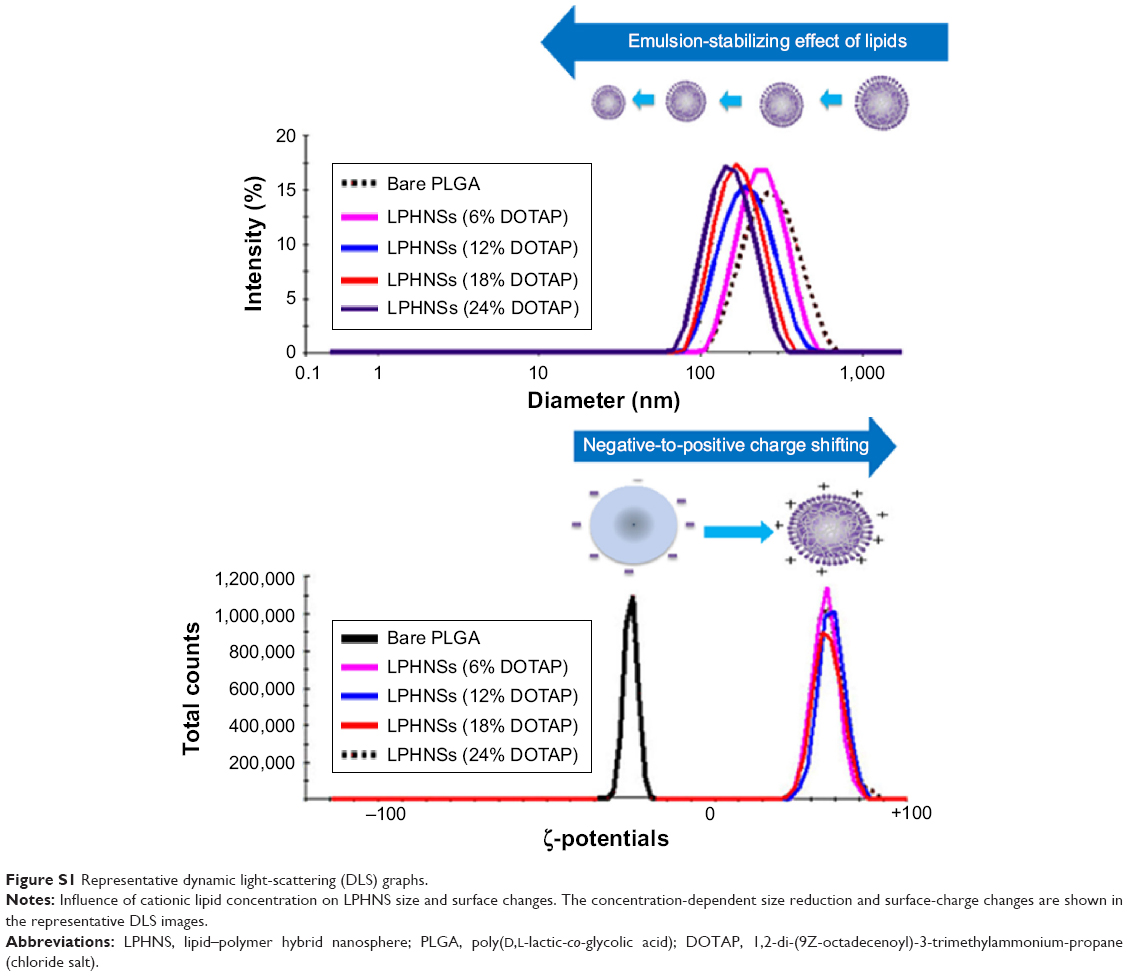 | Figure S1 Representative dynamic light-scattering (DLS) graphs. |
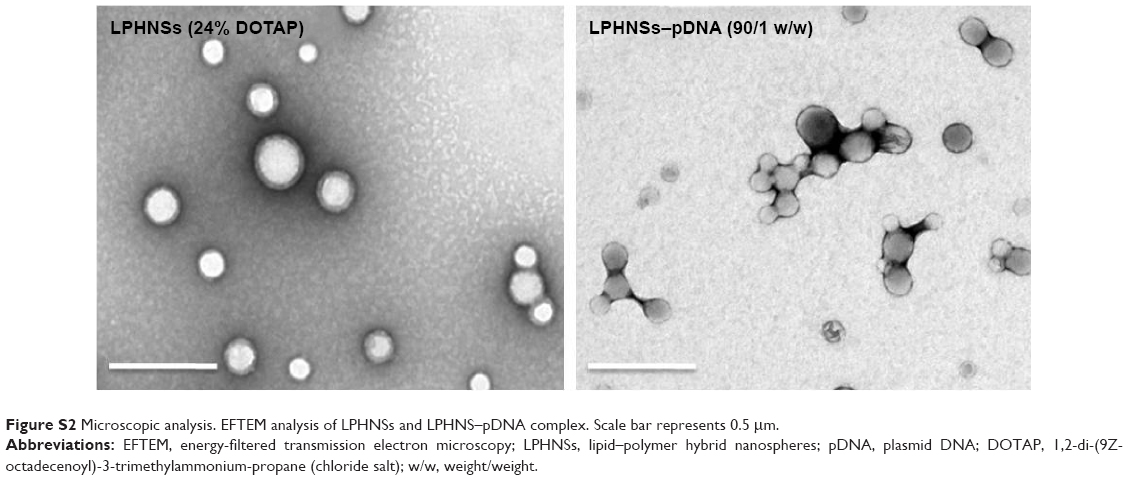 | Figure S2 Microscopic analysis. EFTEM analysis of LPHNSs and LPHNS–pDNA complex. Scale bar represents 0.5 μm. |
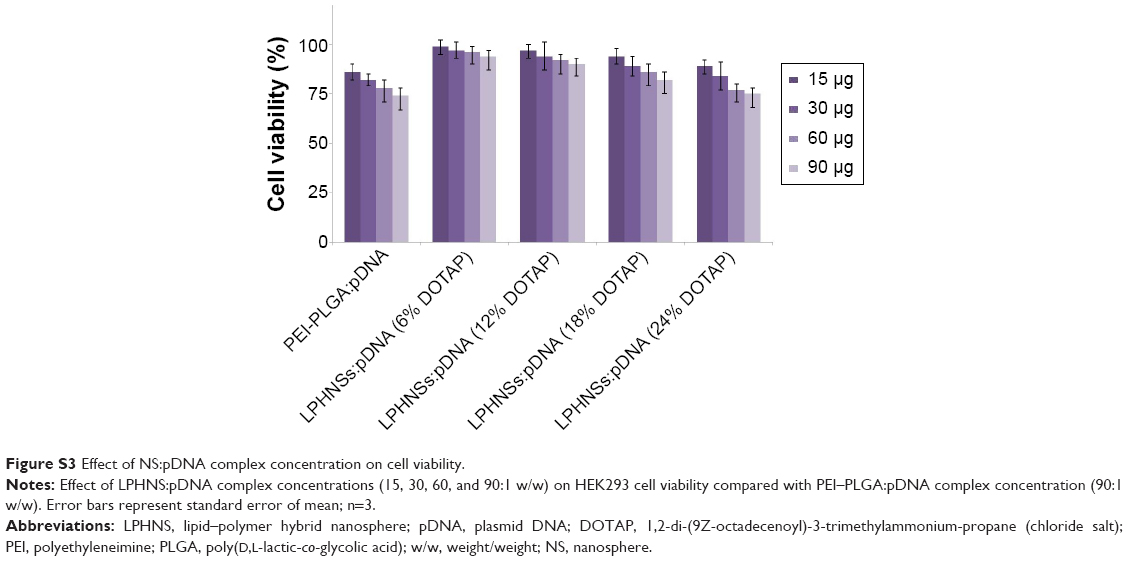 | Figure S3 Effect of NS:pDNA complex concentration on cell viability. |
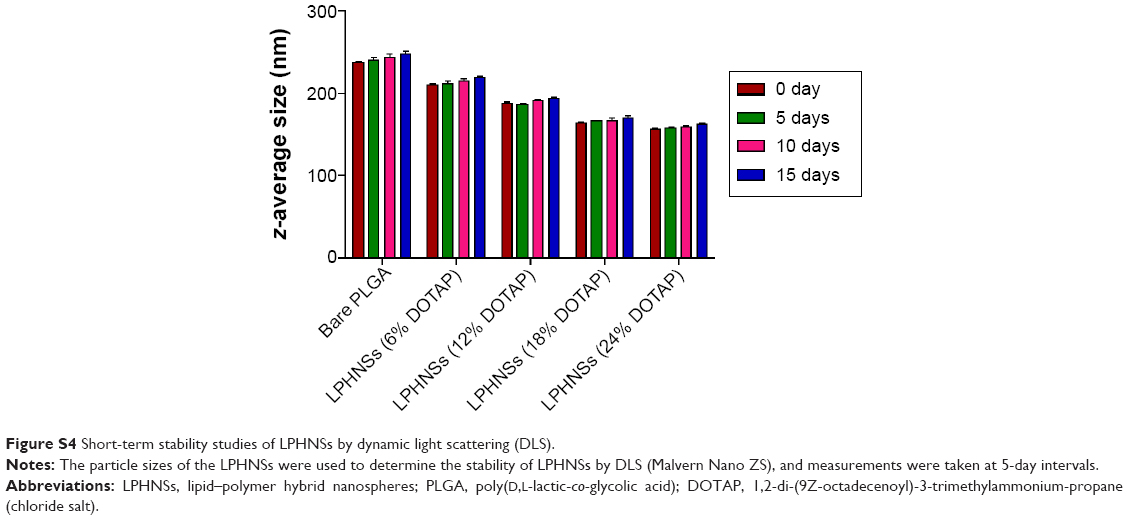 | Figure S4 Short-term stability studies of LPHNSs by dynamic light scattering (DLS). |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.