Back to Journals » Journal of Inflammation Research » Volume 13
Inflammatory Cytokine: IL-17A Signaling Pathway in Patients Present with COVID-19 and Current Treatment Strategy
Authors Shibabaw T ![]()
Received 22 August 2020
Accepted for publication 15 September 2020
Published 6 October 2020 Volume 2020:13 Pages 673—680
DOI https://doi.org/10.2147/JIR.S278335
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Ning Quan
Tewodros Shibabaw
Department of Biochemistry, School of Medicine, College of Medicine and Health Sciences, University of Gondar, Gondar, Ethiopia
Correspondence: Tewodros Shibabaw
Department of Biochemistry, School of Medicine, University of Gondar, P.O. Box 196, Gondar, Ethiopia
Tel +251910162171
Email [email protected]
Abstract: Coronavirus disease 2019 (COVID-19) is a globally communicable public health disease caused by severe acute respiratory syndrome coronavirus 2 (SARS‐CoV-2). Eradication of COVID-19 appears practically impossible but, therefore, more effective pharmacotherapy is needed. The deteriorated clinical presentation of patients with COVID-19 is mainly associated with hypercytokinemia due to notoriously elevated pro-inflammatory cytokines such as interleukin (IL)-1B, IL-6, IL-8, IL-17, granulocyte-macrophage colony-stimulating factor (GM-CSF), granulocyte colony-stimulating factor (G-CSF), interferon-γ-inducible protein (IP10), monocyte chemoattractant protein (MCP1), and tumor necrosis factor-α (TNFα), and is usually responsible for cytokine release syndrome. In the cytokine storm, up-regulation of T-helper 17 cell cytokine IL-17A, and maybe also IL-17F, is mostly responsible for the immunopathology of COVID-19 and acute respiratory distress syndrome. Herein, I meticulously review the exuberant polarization mechanism of naïve CD4+ T cells toward Th17 cells in response to SARS-CoV-2 infection and its associated immunopathological sequelae. I also, propose, for clinical benefit, targeting IL-17A signaling and the synergic inflammatory cytokine IL-6 to manage COVID-19 patients, particularly those presenting with cytokine storm syndrome.
Keywords: IL-17A, inflammation, immunopathology, COVID-19, cytokine storm, Th17, IL-6, ARDS
Introduction
Coronavirus disease 2019 (COVID-19) is a current pandemic infectious disease caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), which first appeared in Wuhan, China, in December 2019, and has since spread globally.1–3 To date, official figures released by the World Health Organization (WHO) indicate over 21 million confirmed cases of COVID-19 worldwide, with 761,018 deaths.4 This 2019-nCoV is the third and the most lethal pathogenic human positive sense RNA coronavirus identified following the outbreak of zoonotic transmission of CoV that has been recognized as SARS-CoV (in 2003) and MERS-CoV (in 2012), affecting birds and a wide range of animals including humans.5–7 The hallmark of each of these infections is viral pneumonia accompanied by host inflammation, leading to pulmonary edema and a syndrome that resembles acute respiratory distress syndrome (ARDS).8
Following SARS-CoV-2 infection, patients present with exuberant activation of T cells, and Th17 cell infiltration leads to an elevation of inflammatory cytokines such as interleukin-17A (IL-17A) (also known as IL-17) and related families such as IL-17B, IL-17C, IL-17D, IL-17E (also known as IL-25), and IL-17F. In turn, inefficient production of type 1 interferons and an impaired antiviral response are seen, while increasing activation of NF-κB contributes to the cytokine storm.9–12 Secondary to a large amount of inflammatory cell infiltration and the cytokine storm, the alveolar–capillary membrane becomes congested, damaged, and leaky, allowing increased movement of water and proteins from the intravascular space to the interstitial space.13 In turn, pulmonary edema, lung failure, and death ensue.9 Taking this a step further, severely infected patients with ARDS have elevated serum levels of IL-1B, IL1RA, IL-6, IL-7, IL-8, IL-17, IL-9, IL-10, fibroblast growth factor (FGF), granulocyte–macrophage colony-stimulating factor (GM-CSF), interferon-γ (IFNγ), granulocyte colony-stimulating factor (G-CSF), interferon-γ-inducible protein (IP10), monocyte chemoattractant protein (MCP1), macrophage inflammatory protein-1α (MIP1α), platelet-derived growth factor (PDGF), tumor necrosis factor (TNFα), and vascular endothelial growth factor (VEGF) (Table 1).9,14–17
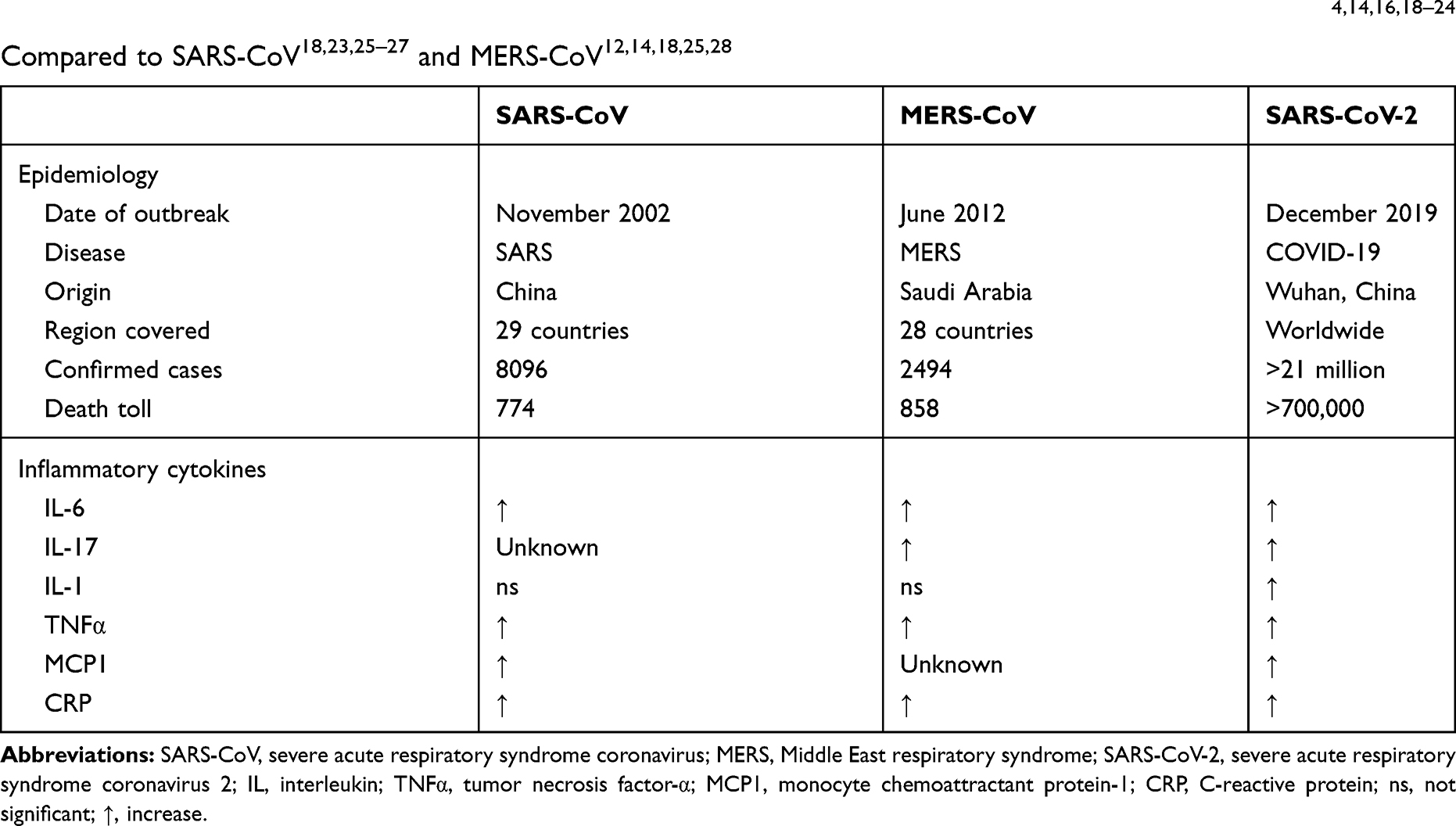 |
Table 1 Epidemiology and Common Inflammatory Cytokines in Patients Presenting with SARS-CoV-2,4,14,16,18–24 Compared to SARS-CoV18,23,25–27 and MERS-CoV12,14,18,25,28 |
Because there is no specific antiviral therapy for COVID-19, understanding of the cytokine storm mechanisms in this disease could help to reveal possible therapeutic interventions. Therefore, among several inflammatory cells and cytokines, T-helper (Th)-17 cells are a unique subset of a cluster of differentiation 4 (CD4+) T-helper cells characterized by the production of pro-inflammatory cytokines such as IL-17A, IL-17F, and IL-22. Differentiation of naïve CD4+ T cells to Th-17 cells is mediated by the activation of T-helper cells in the presence of a combination of TGF-β, IL-6, IL-1β, and IL-23.10,11 In turn, IL-17A acts by activating inducible nitric oxide synthase (iNOS) and inducing the expression of macrophages, IL-1β, IL-6, IL-8, TNFα, and several chemokines, which collaborate to potentiate the inflammatory process of ARDS.13 Elevated Th17 (as well as Th1) responses or enhanced IL-17-related pathways are also observed in MERS-CoV and SARS-CoV patients.14,18 ARDS is not commonly due to the viral load but due to an exuberant immune response, and results in cytokine release syndrome (CRS). Thus, there is an urgent need for anti-inflammatory drugs for COVID-19 patients presenting with CRS. This article provides the proposed immunopathological mechanism of IL-17A/F and drugs against IL-17–IL-17R signaling, including the synergistic IL-6–IL-6R axis, as a potential therapeutic option.
Th-17 Cell Response to SARS-CoV-2
Th17 cells are pro-inflammatory and were introduced in 2005 as a third subset of the CD4+ T-cell lineage.30,31 Functionally, Th17 cells play a role in host defense against extracellular pathogens by virtue of their production of IL-17 and IL-17F, and by mediating the recruitment of neutrophils and macrophages to infected tissues. Moreover, it has become evident that aberrant regulation of Th17 cells may play a significant role in the pathogenesis of multiple inflammatory and autoimmune disorders.31 Th-17 cells are promoted by antigen-presenting cells (APCs) through transforming growth factor-beta (TGF-β) in mice, and by IL-6, IL-21, and IL-23 in both mice and humans.11,14,31–33 Most people seem to be affected less severely and either remain asymptomatic or develop only mild symptoms during COVID-19.34 There is limited evidence regarding Th-17 cells and the related inflammatory cell profile in asymptomatic people; rather, elevated IL-17 serves as a biomarker of disease severity.35 Moreover, in SARS-CoV-2 infection, APCs such as alveolar macrophages release IL-6, IL-23, and many more cytokines. Taking this a step further, following the binding of IL-6 and IL-23 with their respective receptors,36 key factors in the polarization of naïve CD4+ T cells toward differentiation, as well as the maturation of Th17 cells, are signal transducer and activator of transcription-3 (STAT3) and retinoic acid receptor-related orphan receptors gamma (RORγ) and alpha (RORα).14,36,37 In the lung alveoli, IL-17A, IL-17F, IL-21, and IL-22 are produced as the signature cytokines by Th17 cells in response to polarizing cytokines secondary to presentation of viral infection (Figure 1).10,36,38,39 Furthermore, Xu et al showed in a patient with severe COVID-19 that the peripheral blood had a strikingly high number of CCR6+ Th17 cells,14,40 further supporting a Th17-type cytokine storm in this disease. Elevated Th17 responses or enhanced IL-17-related pathways are also observed in MERS-CoV and SARS-CoV patients.18,41 Inflammatory cytokines continue to be disordered, perhaps leading to lymphocyte apoptosis. Basic research has confirmed that TNFα, IL-6, and other pro-inflammatory cytokines could induce lymphocyte deficiency.42,43 Collectively, the Th17-type response contributes to the cytokine storm in pulmonary viral infection, including SARS-CoV-2, which results in tissue damage and likely promotes pulmonary edema; therefore, targeting the Th17 pathway may benefit patients with a Th17-dominant immune profile.44
 |
Figure 1 Th-17 cell polarization from naïve CD4+ T cells and activation. In response to SARS-CoV-2, antigen-presenting cells (APCs) such as dendritic cells as well as macrophages can present the fragments of the antigen to the naïve CD4+ T cells, and upon activation APCs release IL-6, TGF-β, and IL-23 polarizing cytokines. In turn, IL-6 binds with its receptor and through JAK-STAT3 causes polarization, maturation, and expansion of CD4+ T cells to Th17 cells with the expression of RORγt. In turn, the activated Th-17 cells produce inflammatory cytokines such as IL-17A, IL-17F, IL-21, and IL-22. |
IL-17A Signaling and Pathological Effects During COVID-19
In SARS-CoV, it is immune dysregulation, rather than viral load, which results in aberrant pro-inflammatory cytokine secretion by alveolar macrophages. As a new type of highly contagious disease in humans, the pathophysiology of the unusually high pathogenicity of COVID-19 is not yet completely understood,45 but most likely ARDS in the case of SARS-CoV-2 infections is a result of exuberant infiltration of inflammatory cells responding to the SARS-CoV-2 viral infection, and the subsequent synthesis and release of inflammatory cytokines.46 Among the many cytokines involved in the storm, IL-17 is a notable and predominant mediator of pulmonary inflammation. The pro-inflammatory properties of IL-17 also make it crucial to mediators of inflammation and immunopathology.47,48 IL-17 activates many signaling pathways, which in turn leads to the production of many other cytokines (such as IL-6, IL-1β, TNFα, G-CSF, GM-CSF, and TGF-β) and chemokines (including IL-8 and MCP1) from many alveolar cell types (endothelial cells, epithelial cells, and macrophages).44,49 Among the chemokines, profoundly elevated G-CSF and IL-8 (CXCL8) lead to the recruitment of immune cells such as neutrophils to the inflamed area of the alveoli, and hence to dysregulated activation of immune cells, which results in a cytokine storm (Figure 2).44,50,51 During IL-17 signaling, IL-17 family cytokines (IL-17A, Il-17F, and others) bind with both IL-17RA and IL-17RC subunits to transduce signaling at the target cells and generate effector molecules.52 Both receptors are engaged in Act1 as an adaptor molecule to recruit TRAF6. In turn, upon recruitment of TRAF6 and its ubiquitination (Act1 may indeed function as an E3 ubiquitin ligase), a cascade of molecular interactions is turned on, leading to the phosphorylation and consequent proteasomal degradation of IκB, ultimately allowing the nuclear translocation of NFκB and the activation of NFκB targeted inflammatory cytokine and chemokine encoding genes.52,53 In turn, the gene products of inflammatory cytokines and chemokines are responsible for the cytokine storm.49 Moreover, clinical symptoms of patients presenting with COVID-19 result from a consequent significant inflammatory cell infiltrate and release of pro-inflammatory cytokines.54 In particular, IL-6, IL-17, and IL-8 are synergistically responsible for pulmonary fibrosis (promoting collagen deposition) secondary to aberrant fibroblast and epithelial cell function or pleural effusion of the lung, and viral persistence results in dyspnea and provokes SARS (Figure 2).55 I suggest that the presence of these cells may be a primary driver of the signature pathology observed in COVID-19 patients. Taken together, IL-17-induced dysregulated and exuberant immune responses have been shown to potentially cause stage 3 (characterized by a hyperinflammatory phase, cytokine storm) COVID-19 disease, likely through increased pulmonary pathology or lung damage and diminished survival.25,56
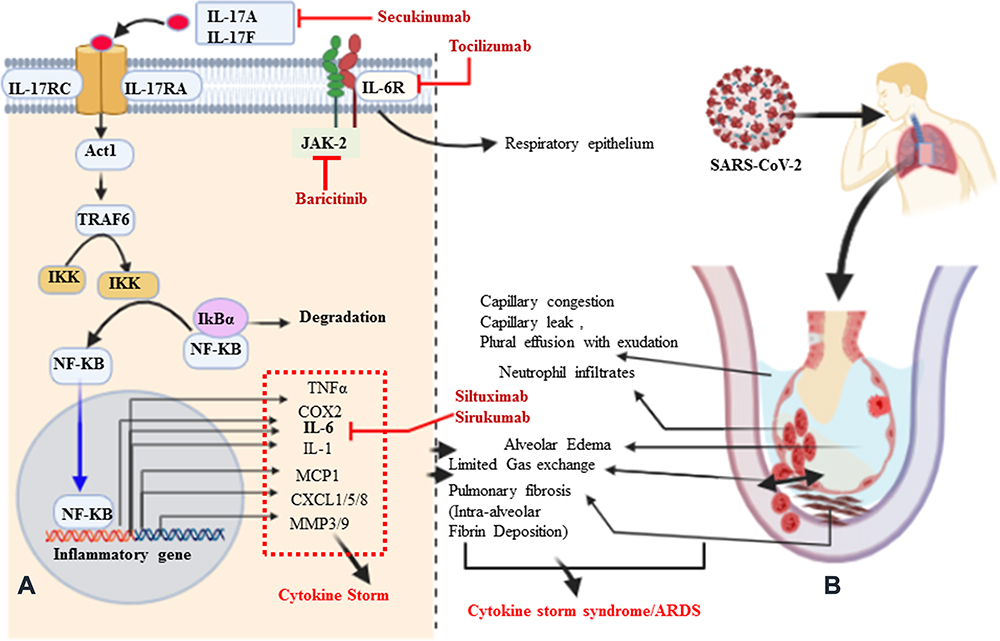 |
Figure 2 Signaling pathways of IL-17A, pathological effect, and potential drug targets. (A) Signaling transduction of IL-17A at type I epithelial cells of the alveoli of the lung. IL-17A and IL-17F are secreted predominantly by Th-17 cells; they are structurally very similar, bind the IL-17RA–IL-17RC receptor combination, and can form heterodimers together, signaling via the adaptor protein nuclear factor (NF)-κ activator (Act1). Many IL-17 target genes contain a promoter region that binds with NF-κB. IL-17 is not a potent inducer of inflammation by itself. Its strong effects during inflammation are derived from its ability to recruit immune cells via chemokine expression such as CXCL1, CXCL5, and MCP1, as well as from its synergistic action with other cytokines such as IL-6, IL-1, and TNFα. Thus, IL-17, acting in synergy with IL-6 and TNF, is a powerful inflammatory signal that results in the rapid recruitment and sustained presence of neutrophils and leads to a cytokine storm. (B) Schematic representation of IL-17A-mediated immunopathological effect of ARDS during SARS-CoV-2 infection. Pulmonary fibrosis is one of the pathological changes due to the activation of fibroblasts mediated by IL-6 and results in abnormal deposition of collagen. Moreover, the stimulation of fibroblasts can produce IL-8 and leads to the attraction of neutrophils to the site of injury. MMP3/9 (which causes tissue destruction) and PGE2 (increases capillary permeability) are also responsible for neutrophil infiltration, alveolar edema, and protein-positive (exudative) pleural effusion. Altogether, the proposed pathological mechanism suggests that IL-17 can mediate numerous immunopathological effects in CRS secondary to SARS-CoV-2 infection. |
IL-17A Targeting as a Treatment Strategy Against COVID-19
COVID-19 is a global public health problem.4 Currently, many different treatment approaches are under investigation and clinical trials are ongoing. There are currently no known specific treatment measures except for meticulous supportive care such as mechanical ventilation when indicated.57 Taking the pathogenesis of this novel coronavirus from the experience of the first SARS-CoV, it is clear that the exuberant immune response and inflammatory cell infiltration are the pathological mechanisms most responsible for the catastrophic death toll.58 Among the anti-cytokines, monoclonal antibodies are a potential and possible approach to the prevention of SARS, although none is yet available for clinical purposes. Anti-IL-17A humanized monoclonal antibodies such as secukinumab (AIN457) (approved fully human IL-17-specific IgG1k monoclonal antibody) and Ixekizumab (LY2439821) (under a phase III clinical trial for psoriasis) are well-known and recognized treatment options for psoriasis.44,59–61 These anti-IL-17A monoclonal antibodies are also utilized in different inflammatory diseases, including ARDS,13 rheumatism,61 and pulmonary fibrosis.62 Moreover, patients presenting with COVID-19 show elevated serum IL-6, which correlates with respiratory failure.63 As discussed earlier, IL-6 plays a key role not only in the polarization of Th17 cell from naïve CD4+ T cells but also in promoting pulmonary inflammation in synergy with IL-17A. Indirectly, tocilizumab is now approved by the US Food and Drug Administration (FDA) for the treatment of chimeric antigen receptor T cell (also known as CAR T cell)-induced CRS, with confirmed efficacy and minimal side effects in hundreds of patients.63 Taking this a step further, tocilizumab against IL-6 has been shown to be efficient for patients presenting with inflammatory rheumatism (Figure 2).61,64,65 In addition, IL-23 and IL-6 are involved in Th17 differentiation, and they act through the JAK-STAT3 signaling pathway. This implies that STAT3 could be a potential target, at the convergence point of different upstream activators. On the other hand, the anti-JAK tofacitinib56,65 (oral inhibitor, selective for JAK1 and JAK3), is under a phase III clinical trial for the treatment of inflammatory rheumatic disorders as well as COVID-19.66 JAK inhibitors interfere with phosphorylation of signal transducer and activator of transcription (STAT) proteins and the production of downstream inflammatory molecules. Thus, anti-IL-17 (secukinumab) combined with blockade of IL-6 (sirukumab and siltuximab), IL-6R (tocilizumab), or JAK (baricitinib, selective for JAK1 and JAK2) may be beneficial in controlling the cytokine storm while boosting antiviral IFN-I responses during SARS-CoV-2 infection (Table 2).57,66
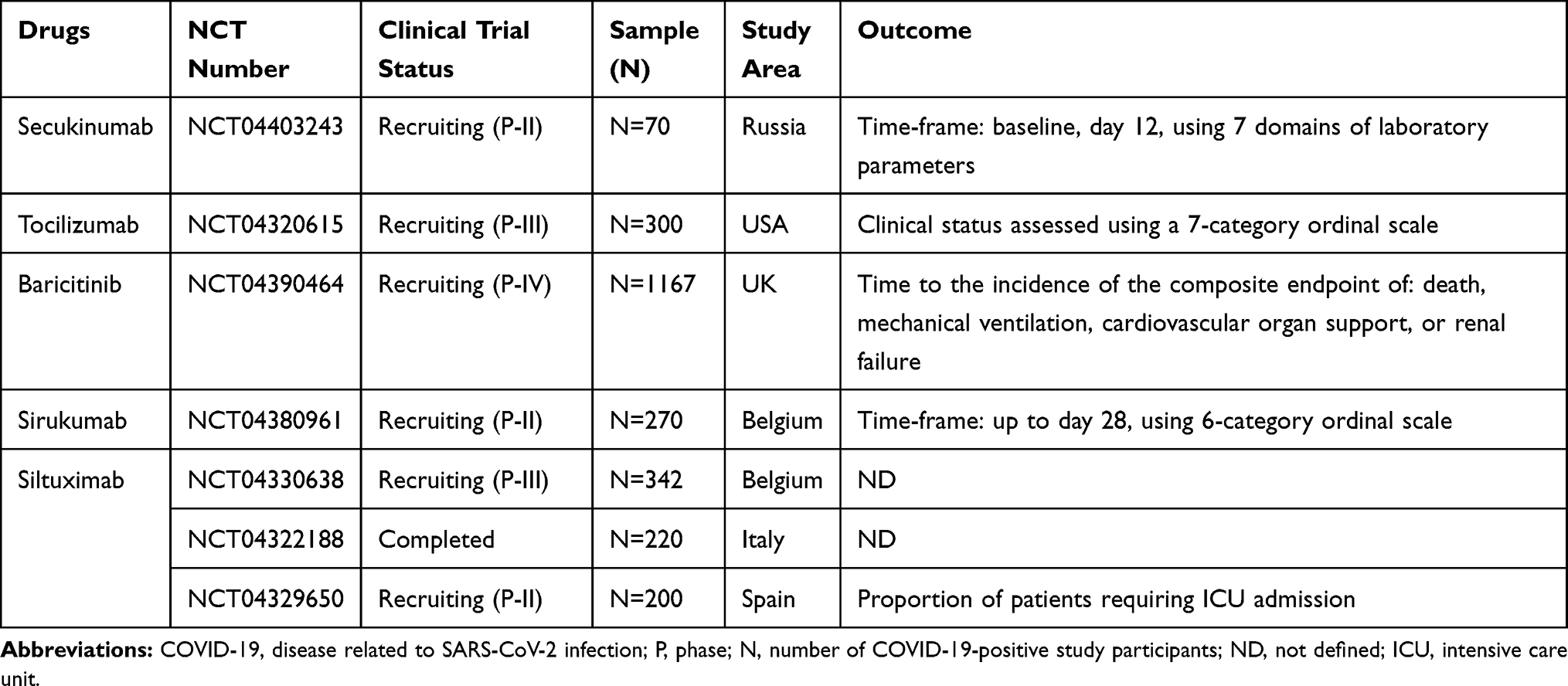 |
Table 2 Summary of Clinical Trials Investigating the Implications of Anti-Inflammatory Agents in COVID-19 Patients, Registered Under Clinicaltrials.gov, WHO Trial Registry Network, and NIH |
Conclusion
In this review, I have discussed the current understanding of IL-17A signaling and the underlying immunopathological role as a pro-inflammatory cytokine during ARDS secondary to SARS-CoV-2 infection. Profound elevation of IL-17A and downstream synergetic effector molecules such as cytokines (IL-6, IL-1β, TNFα) and chemoattractants (IL-8 and MCP1) are indicative of both pathological progression and the severity of COVID-19. The synergistic interaction of IL-17A and IL-6 (required during polarization of Th17 cell) is the central player in the development of pulmonary fibrosis and an impaired respiratory system. Regarding therapeutic strategies, specific therapies for SARS-CoV-2 infections and related complications remain challenging. Although there is limited research evidence, here it is proposed that anti-IL-17A (secukinumab), anti-IL-6, or anti-IL-6R antibody (tocilizumab), or an anti-JAK-2-STAT3 drug (baricitinib) as a promising therapeutic option to terminate pulmonary inflammation, could decrease dangerous cytokines and limit lung tissue pathology, and help to save life if used judiciously in appropriate COVID-19 cases.
Abbreviations
ARDS, acute respiratory distress syndrome; COVID-19, coronavirus disease 2019; CRS, cytokine release syndrome; IL-17, interleukin-17; JAK, janus kinase; RORγ/α, retinoic acid receptor-related orphan receptors gamma and alpha; SARS, severe acute respiratory syndrome; STAT3, signal transducer and activator of transcription 3.
Acknowledgments
I would like to forward my gratitude to the authors of the articles from which this review was generated.
Author Contributions
The author made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Disclosure
The author reports no conflicts of interest in this work.
References
1. Zhu N, Zhang D, Wang W, et al. A novel coronavirus from patients with pneumonia in China, 2019. N Engl J Med. 2020;382(8):727–733. doi:10.1056/NEJMoa2001017
2. Xu C, Luo X, Yu C, Cao S-J. The 2019-nCoV epidemic control strategies and future challenges of building healthy smart cities. Indoor and Built Environment. 2020;29(5):639–644. doi:10.1177/1420326X20910408
3. Rothe C, Schunk M, Sothmann P, et al. Transmission of 2019-nCoV infection from an asymptomatic contact in Germany. N Engl J Med. 2020;382(10):970–971. doi:10.1056/NEJMc2001468
4. World Health Organization. Coronavirus disease (COVID-19): situation report. 2019.
5. Chen Y, Liu Q, Guo D. Emerging coronaviruses: genome structure, replication, and pathogenesis. J Med Virol. 2020. doi:10.1002/jmv.26234
6. Chen J. Pathogenicity and transmissibility of 2019-nCoV—A quick overview and comparison with other emerging viruses. Microbes Infect. 2020;22(2):69–71. doi:10.1016/j.micinf.2020.01.004
7. Benvenuto D, Giovannetti M, Ciccozzi A, Spoto S, Angeletti S, Ciccozzi M. The 2019‐new coronavirus epidemic: evidence for virus evolution. J Med Virol. 2020;92(4):455–459. doi:10.1002/jmv.25688
8. Hotez PJ, Bottazzi ME, Corry DB. The Potential Role of TH17 Immune Responses in Coronavirus Immunopathology and Vaccine-Induced Immune Enhancement. Elsevier; 2020.
9. Zhang W, Zhao Y, Zhang F, et al. The use of anti-inflammatory drugs in the treatment of people with severe coronavirus disease 2019 (COVID-19): the experience of clinical immunologists from China. Clin Immunol. 2020;214:108393. doi:10.1016/j.clim.2020.108393
10. Jin W, Dong C. IL-17 cytokines in immunity and inflammation. Emerg Microbes Infect. 2013;2(1):1–5. doi:10.1038/emi.2013.58
11. Bulat V, Situm M, Azdajic MD, Likic R. Potential role of IL‐17 blocking agents in the treatment of severe COVID‐19? Br J Clin Pharmacol. 2020. doi:10.1111/bcp.14437
12. Mahallawi WH, Khabour OF, Zhang Q, Makhdoum HM, Suliman BA. MERS-CoV infection in humans is associated with a pro-inflammatory Th1 and Th17 cytokine profile. Cytokine. 2018;104:8–13. doi:10.1016/j.cyto.2018.01.025
13. Righetti RF, Santos TMD, Camargo LDN, et al. Protective effects of anti-IL17 on acute lung injury induced by LPS in mice. Front Pharmacol. 2018;9:1021. doi:10.3389/fphar.2018.01021
14. Wu D, Yang XO. TH17 responses in cytokine storm of COVID-19: an emerging target of JAK2 inhibitor Fedratinib. J Microbiol Immunol Infect. 2020;53(3):368–370. doi:10.1016/j.jmii.2020.03.005
15. Du F, Liu B, Zhang S. COVID-19: the role of excessive cytokine release and potential ACE2 down-regulation in promoting hypercoagulable state associated with severe illness. J Thromb Thrombolysis. 2020;1–17.
16. Huang C, Wang Y, Li X, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. 2020;395(10223):497–506. doi:10.1016/S0140-6736(20)30183-5
17. Girija A, Shankar EM, Larsson M. Could SARS-CoV-2-induced hyperinflammation magnify the severity of coronavirus disease (CoViD-19) leading to acute respiratory distress syndrome? Front Immunol. 2020;11:1206. doi:10.3389/fimmu.2020.01206
18. Faure E, Poissy J, Goffard A, et al. Distinct immune response in two MERS-CoV-infected patients: can we go from bench to bedside? PLoS One. 2014;9(2):e88716. doi:10.1371/journal.pone.0088716
19. Wang C, Horby PW, Hayden FG, Gao GF. A novel coronavirus outbreak of global health concern. Lancet. 2020;395(10223):470–473. doi:10.1016/S0140-6736(20)30185-9
20. Zhou F, Yu T, Du R, et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet. 2020.
21. Jose RJ, Manuel A. COVID-19 cytokine storm: the interplay between inflammation and coagulation. Lancet Respir Med. 2020;8(6):e46–e47. doi:10.1016/S2213-2600(20)30216-2
22. Wang Z, Yang B, Li Q, Wen L, Zhang R. Clinical features of 69 cases with coronavirus disease 2019 in Wuhan, China. Clin Infect Dis. 2020. doi:10.1093/cid/ciaa538
23. Fang Y, Zhang H, Xu Y, Xie J, Pang P, Ji W. CT manifestations of two cases of 2019 novel coronavirus (2019-nCoV) pneumonia. Radiology. 2020;295(1):208–209. doi:10.1148/radiol.2020200280
24. Xu J, Zhao S, Teng T, et al. Systematic comparison of two animal-to-human transmitted human coronaviruses: SARS-CoV-2 and SARS-CoV. Viruses. 2020;12(2):244. doi:10.3390/v12020244
25. Diao B, Wang C, Tan Y, et al. Reduction and functional exhaustion of T cells in patients with coronavirus disease 2019 (COVID-19). Front Immunol. 2020;11:827. doi:10.3389/fimmu.2020.00827
26. Su S, Wong G, Shi W, et al. Epidemiology, genetic recombination, and pathogenesis of coronaviruses. Trends Microbiol. 2016;24(6):490–502. doi:10.1016/j.tim.2016.03.003
27. Zhang Y, Li J, Zhan Y, et al. Analysis of serum cytokines in patients with severe acute respiratory syndrome. Infect Immun. 2004;72(8):4410–4415. doi:10.1128/IAI.72.8.4410-4415.2004
28. Jiang Y, Xu J, Zhou C, et al. Characterization of cytokine/chemokine profiles of severe acute respiratory syndrome. Am J Respir Crit Care Med. 2005;171(8):850–857. doi:10.1164/rccm.200407-857OC
29. Quirch M, Lee J, Rehman S. Hazards of the cytokine storm and cytokine-targeted therapy in patients with COVID-19. J Med Internet Res. 2020;22(8):e20193. doi:10.2196/20193
30. Llosa NJ, Geis AL, Orberg ET, Housseau F. Interleukin-17 and type 17 helper T cells in cancer management and research. ImmunoTargets Ther. 2014;3:39.
31. Tesmer LA, Lundy SK, Sarkar S, Fox DA. Th17 cells in human disease. Immunol Rev. 2008;223(1):87–113.
32. McGeachy MJ, Cua DJ. Th17 cell differentiation: the long and winding road. Immunity. 2008;28(4):445–453. doi:10.1016/j.immuni.2008.03.001
33. Sekine T, Perez-Potti A, Rivera-Ballesteros O, et al. Robust T cell immunity in convalescent individuals with asymptomatic or mild COVID-19. Cell. 2020. doi:10.1016/j.cell.2020.08.017
34. Pacha O, Sallman MA, Evans SE. COVID-19: a case for inhibiting IL-17? Nat Rev Immunol. 2020;20(6):345–346. doi:10.1038/s41577-020-0328-z
35. Ghilardi N, Ouyang W Targeting the development and effector functions of TH17 cells. Paper presented at: Seminars in immunology. 2007; 19(6):383–393. Academic Press.
36. Ivanov II, McKenzie BS, Zhou L, et al. The orphan nuclear receptor RORγt directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell. 2006;126(6):1121–1133. doi:10.1016/j.cell.2006.07.035
37. Raucci F, Mansour AA, Casillo GM, et al. Interleukin-17A (IL-17A), a key molecule of innate and adaptive immunity, and its potential involvement in COVID-19-related thrombotic and vascular mechanisms. Autoimmun Rev. 2020;19(7):102572. doi:10.1016/j.autrev.2020.102572
38. Bunte K, Beikler T. Th17 cells and the IL-23/IL-17 axis in the pathogenesis of periodontitis and immune-mediated inflammatory diseases. Int J Mol Sci. 2019;20(14):3394. doi:10.3390/ijms20143394
39. Xu Z, Shi L, Wang Y, et al. Pathological findings of COVID-19 associated with acute respiratory distress syndrome. Lancet Respir Med. 2020;8(4):420–422. doi:10.1016/S2213-2600(20)30076-X
40. Josset L, Menachery VD, Gralinski LE, et al. Cell host response to infection with novel human coronavirus EMC predicts potential antivirals and important differences with SARS coronavirus. MBio. 2013;4:3. doi:10.1128/mBio.00165-13
41. Tan L, Wang Q, Zhang D, et al. Lymphopenia predicts disease severity of COVID-19: a descriptive and predictive study. Signal Transduct Target Ther. 2020;5(1):1–3.
42. Bermejo-Martin JF, Almansa R, Menéndez R, Mendez R, Kelvin DJ, Torres A. Lymphopenic community acquired pneumonia as signature of severe COVID-19 infection. J Infect. 2020;80(5):e23. doi:10.1016/j.jinf.2020.02.029
43. Cafarotti S. Severe acute respiratory syndrome–Coronavirus-2 Infection and patients with lung cancer: the potential role of interleukin-17 target therapy. J Thorac Oncol. 2020;15(7):e101–e103. doi:10.1016/j.jtho.2020.04.015
44. Qin C, Zhou L, Hu Z, et al. Dysregulation of immune response in patients with COVID-19 in Wuhan, China. Clin Infect Dis. 2020.
45. Yajing F, Cheng Y, Wu Y. Understanding SARS-CoV-2-mediated inflammatory responses: from mechanisms to potential therapeutic tools. Virol Sin. 2020;6.
46. Ma W-T, Yao X-T, Peng Q, Chen D-K. The protective and pathogenic roles of IL-17 in viral infections: friend or foe? Open Biol. 2019;9(7):190109. doi:10.1098/rsob.190109
47. Veldhoen M. Interleukin 17 is a chief orchestrator of immunity. Nat Immunol. 2017;18(6):612. doi:10.1038/ni.3742
48. Badawi A. Hypercytokinemia and pathogen–host interaction in COVID-19. J Inflamm Res. 2020;13:255. doi:10.2147/JIR.S259096
49. Ryzhakov G, Lai CC-K, Blazek K, To K-W, Hussell T, Udalova I. IL-17 boosts proinflammatory outcome of antiviral response in human cells. J Immunol. 2011;187(10):5357–5362. doi:10.4049/jimmunol.1100917
50. Guglani L, Khader SA. Th17 cytokines in mucosal immunity and inflammation. Curr Opin HIV AIDS. 2010;5(2):120. doi:10.1097/COH.0b013e328335c2f6
51. Onishi RM, Gaffen SL. Interleukin‐17 and its target genes: mechanisms of interleukin‐17 function in disease. Immunology. 2010;129(3):311–321.
52. Brembilla NC, Senra L, Boehncke W-H. The IL-17 family of cytokines in psoriasis: IL-17A and beyond. Front Immunol. 2018;9:1682. doi:10.3389/fimmu.2018.01682
53. Frieman M, Heise M, Baric R. SARS coronavirus and innate immunity. Virus Res. 2008;133(1):101–112. doi:10.1016/j.virusres.2007.03.015
54. Satija N, Lal SK. The molecular biology of SARS coronavirus. Ann N Y Acad Sci. 2007;1102(1):26–38. doi:10.1196/annals.1408.002
55. Calabrese LH. Cytokine storm and the prospects for immunotherapy with COVID-19. Cleve Clin J Med. 2020.
56. Organization WH. Clinical Management of Severe Acute Respiratory Infection (SARI) When COVID-19 Disease is Suspected: Interim Guidance, 13 March 2020. World Health Organization; 2020.
57. Quartuccio L, Semerano L, Benucci M, Boissier M-C, De Vita S. Urgent avenues in the treatment of COVID-19: targeting downstream inflammation to prevent catastrophic syndrome. Joint Bone Spine. 2020;87(3):191. doi:10.1016/j.jbspin.2020.03.011
58. Ritchlin CT, Krueger JG. New therapies for psoriasis and psoriatic arthritis. Curr Opin Rheumatol. 2016;28(3):204. doi:10.1097/BOR.0000000000000274
59. Fletcher JM, Moran B, Petrasca A, Smith CM. IL‐17 in inflammatory skin diseases psoriasis and hidradenitis suppurativa. Clin Exp Immunol. 2020;201(2):121–134. doi:10.1111/cei.13449
60. Truchetet M-E, Mossalayi MD, Boniface K. IL-17 in the rheumatologist’s line of sight. Biomed Res Int. 2013;2013:1–18. doi:10.1155/2013/295132
61. Robak E, Gerlicz-Kowalczuk Z, Dziankowska-Bartkowiak B, Wozniacka A, Bogaczewicz J. Serum concentrations of IL-17A, IL-17B, IL-17E and IL-17F in patients with systemic sclerosis. Arch Med Sci. 2019;15(3):706. doi:10.5114/aoms.2019.84738
62. Moore JB, June CH. Cytokine release syndrome in severe COVID-19. Science. 2020;368(6490):473–474. doi:10.1126/science.abb8925
63. Georgiev T. Coronavirus disease 2019 (COVID-19) and anti-rheumatic drugs. Rheumatol Int. 2020;1.
64. Abdin SM, Elgendy SM, Alyammahi SK, Alhamad DW, Omar HA. Tackling the cytokine storm in COVID-19, challenges, and hopes. Life Sci. 2020;257:118054. doi:10.1016/j.lfs.2020.118054
65. Favalli EG, Biggioggero M, Maioli G, Caporali R. Baricitinib for COVID-19: a suitable treatment? Lancet Infect Dis. 2020;20(9):1012–1013. doi:10.1016/S1473-3099(20)30262-0
66. Zhong J, Tang J, Ye C, Dong L. The immunology of COVID-19: is immune modulation an option for treatment? Lancet Rheumatol. 2020;2(7):e428–e436. doi:10.1016/S2665-9913(20)30120-X
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.