Back to Journals » Breast Cancer: Targets and Therapy » Volume 12
Inflammation Mediated Hepcidin-Ferroportin Pathway and Its Therapeutic Window in Breast Cancer
Authors Shibabaw T ![]() , Teferi B, Molla MD
, Teferi B, Molla MD ![]() , Ayelign B
, Ayelign B ![]()
Received 10 August 2020
Accepted for publication 12 October 2020
Published 20 October 2020 Volume 2020:12 Pages 165—180
DOI https://doi.org/10.2147/BCTT.S276404
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Pranela Rameshwar
Tewodros Shibabaw,1 Banchamlak Teferi,2 Meseret Derbew Molla,1 Birhanu Ayelign3
1Department of Biochemistry, School of Medicine, College of Medicine and Health Sciences, University of Gondar, Gondar, Ethiopia; 2Department of Clinical Pharmacy, School of Pharmacy, College of Medicine and Health Sciences, University of Gondar, Gondar, Ethiopia; 3Department of Immunology and Molecular Biology, School of Biomedical and Laboratory Sciences, College of Medicine and Health Sciences, University of Gondar, Gondar, Ethiopia
Correspondence: Tewodros Shibabaw Tel +251 910162171
Email [email protected]
Abstract: Experimental and clinical data strongly support that iron is an essential element which plays a big role in cancer biology. Thus, hepcidin (Hp) and ferroportin (Fpn) are molecules that regulate and maintain the metabolism of iron. A peptide hormone hepcidin limits recycled and stored iron fluxes in macrophage and hepatic hepatocyte, respectively, to the blood stream by promoting degradation of the only iron exporter, Fpn, in the target cells. Moreover, the inflammatory microenvironment of breast cancer and altered hepcidin/ferroportin pathway is intimately linked. Breast cancer exhibits an iron seeking phenotype that is accomplished by tumor-associated macrophage (TAM). Because macrophages contribute to breast cancer growth and progression, this review will discuss TAM with an emphasis on describing how TAM (M2Ф phenotypic) interacts with their surrounding microenvironment and results in dysregulated Hp/Fpn and pathologic accumulation of iron as a hallmark of its malignant condition. Moreover, the underlying stroma or tumor microenvironment releases significant inflammatory cytokines like IL-6 and bone morphogenetic proteins like BMP-2 and 6 leading in aberrant Hp/Fpn pathways in breast cancer. Inflammation is primarily associated with the high intracellular iron levels, deregulated hepcidin/ferroportin pathway, and its upstream signaling in breast cancer. Subsequently, scholars have been reported that reducing iron level and manipulating the signaling molecules involved in iron metabolism can be used as a promising strategy of tumor chemotherapy. Here, we review the key molecular aspects of iron metabolism and its regulatory mechanisms of the hepcidin/ferroportin pathways and its current therapeutic strategies in breast cancer.
Keywords: breast cancer, hepcidin, ferroportin, inflammation, upstream signaling, bone morphogenetic proteins, tumor-associated macrophage
Introduction
Breast cancer (BC) is the most common term for a set of breast tumor subtypes with distinct molecular and cellular origins and clinical behavior. Most of them are the origin of ductal or lobular epithelial tumors. Globally, it is the most common malignant diseases of women’s diagnosed every year. It accounts for about 30% of the mortality of women ages of 40–49 years, followed by lung cancer, with an estimated 1.67 million cases worldwide each year worldwide.1 Approximately 12.5% of women in the United States have been affected by invasive breast cancer during their lifetime.2–4 It is also the second most common prevalent cancerous disease among women living in sub-Saharan Africa (SSA) with increasing over time1,5,6 and the survival rate of affected people are poor in this region.7 The incidence rates differ considerably across African countries; for example, 64.2 new cases and 18.8 mortality per 100 000 women per year in Mauritius compared with 41.8 cases and 23 mortality rate per 100 000 women per year in Ethiopia.8 There are many risk factors associated with the diagnosis of breast cancer some of them include BC susceptibility gene 1 (BRCA1) mutation, hormonal factor, obesity, alcohol intake, cigarettes smoking, infection, low-dose of irradiation and nutrition and dietary factor.2 Taking this step further, functional iron is an essential nutrient that is involved for many cellular processes, including electron transport chain (ETC), citric acid cycle, heme synthesis, the cofactor of DNA Polymerase, cell cycle phase transitioning (G1/S),9 and other macromolecule metabolisms.10,11 Extra-metabolic iron is stored in the form of ferritin and hemosiderin in the reticuloendothelial system mainly in the liver and macrophage of the spleen.12 In order to maintain sufficient and healthy iron level in the body, cells require the coordination of a wide range of genetic activities, which are tightly regulated by both intracellular (via iron response element (IRE)/iron regulatory protein (IRP) regulatory pathway) and systemic iron metabolism (Hp/FPN).11,13 However, reprogrammed iron metabolism has been identified as being one of the key metabolic hallmarks of cancer. It can potentially produce reactive oxygen species (ROS) and lipid peroxidation as well as mutagenic aldehyde product14,15 through Fenton or Haber–Weiss reaction (Figure 1).16,17 Therefore, an increased number of oxidative DNA lesion and an inflammatory condition that facilitates tumor formation.18 In addition, from numerous microenvironment components of the tumor, tumor-associated macrophage (TAM) takes 50% of the population. Tumor-associated macrophage is a major player in the connection between inflammation and dysregulated iron metabolism mainly Hp/FPN pathways of the cancer cell.19,20 It is Macrophage 2 (M2) phenotype oriented towards promoting tumor growth and angiogenesis, tissue remodelling and suppressing adaptive immunity.19,21 Therefore, this d-block transition metal consistently linked to carcinogenesis, either through persistent failure in the redox balance or due to its critical role in cellular proliferation.18 Several research reports elaborate that cellular, and systemic dysregulation in iron trafficking and storage, may lead to the development of to breast cancer.3,18,22 In this review article, we provided the current epidemiological, experimental and clinical finding regarding on the systemic iron trafficking, and its regulation through Hp/Fpn pathway in a normal cell, and dysregulated Hp/Fpn signaling secondary to inflammatory cytokine (IL-6) derived from exuberant malignancy, and bone morphogenic protein (BMP) mediated activation of JAK2-STAT3 and BMP-SMAD signaling, respectively, within the breast cancer cell. In addition, this review summarizes the current therapeutic approach for breast cancer by targeting the upstream signaling and Hp/Fpn pathway.
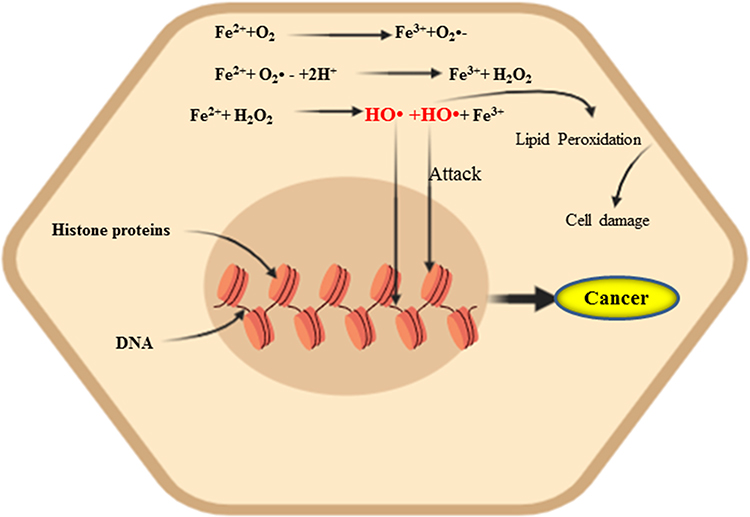 |
Figure 1 The Fenton/Haber–Weiss reaction. Iron is the most driving force for the generation of ROS and the malignant transformation of cells by directly damaging DNA, eventually leading to mutagenic transformation, resulting more aggressive tumor behavior. |
Systemic Iron Trafficking and Its Physiological Mechanism
Iron trafficking is controlled very carefully at the site of import and export.23 Iron is a double face molecule; in one side, it has great physiological importance for metabolism; on the other hand, it is potentially toxic when it becomes excess in the body and widely associated with the development and progression of several diseases, such as liver disease, heart failure, diabetes and cancers.24 Human iron metabolism is regulated through two different mechanisms.25 The first one is systemic balance iron via controlling at the site of dietary iron absorption (enterocyte), storage of excess iron (hepatocyte), and recycling of iron from senescent erythrocytes (reticuloendothelial macrophage) mediated by Hp/Fpn pathway (Figure 2). The second important mode of iron homeostasis is through the IRP/IRE system at the cellular level. Dietary iron is taken-up by the duodenal enterocyte and exported into the circulation through the iron exporting ferroportin (Fpn, Ireg1, MTP1, and SLC40A1). The released iron in the circulation is in charge of carrier protein transferrin (Tf). To be absorbed, in the duodenal lining of enterocyte iron in the non-heme dietary source must be changed into ferrous (Fe2+) form. In the brush border, ferric reductase enzyme or cytochrome B (Dcytb) reduces ferric Fe3+ to Fe2+ due to its associated with heme-containing b-type cytochrome.25,26 Divalent metal transporter 1 (DMT1 also called Nramp2, SLC11A2, DCT1) is the main transporter involved in cellular non-heme iron uptake. Divalent metal transporter 1 (568 amino acids) transports iron into and out of the duodenal cytosol and into the hepatocyte and macrophages.27 Divalent metal transporter 1 is active in a low-pH environment for efficient metal transport as H+/Fe2+ symporter.25 Similarly, dietary heme can also be transported across the apical membrane by a yet unknown mechanism or hypothetically using heme carrier protein 1 (HCP1) and subsequently metabolized in the enterocytes by heme oxygenase 1 (HO-1) to liberate (Fe2+).10,22,25,28 This process is more efficient than the absorption of inorganic iron and is independent of duodenal pH. It is thus not influenced by inhibitors such as phytate and polyphenols. Consequently, red meats high in hemoglobin are excellent nutrient sources of iron.29 Alternative to DMT1, iron can enter the cell directly via plasma membrane divalent cation (zinc, iron, and manganese) importers known as Zrt-Irt-like protein 14 (ZIP14 also known as SLC39A14), ZIP8 (also known as SLC39A8) or ZIP1130–32 Interestingly research study showed that in breast cancer tissue significantly have higher zinc levels than normal breast tissue.30 Basal breast cancer tumors expressed higher levels of ZIP4 and ZIP14 genes and lower levels of ZIP6, ZIP9 and ZIP11.30 The absorbed enterocyte iron can be either stored as ferritin33 or transported across the basolateral membrane of the enterocyte into the circulation.34 Besides to basolateral membranes of enterocyte, FPN is also expressed in various types of cells with a high level of expression in macrophages, liver kupffer cells, periportal hepatocytes, splenic red pulp macrophage, and placental syncytiotrophoblast. As far as our current understanding, it is the only known iron exporter in mammalian cells.34–36 Similar to DMT1, Ferroportin mediated efflux of Fe2+ is coupled with three multi-copper ferroxidase with distinct expression patterns such as ceruloplasmin (CP), hephaestin (HEPH), and zyklopen.37,38 Exported iron in the form of Fe2+ must be oxidized into Fe3+ for loading on top of transferrin (Tf) protein in the circulation.25,27,39 Under normal circumstances, 80-kD serum Tf has two high-affinity iron-binding sites and carries almost all serum iron, which solubilizes iron and dampens its reactivity.25 Transferrin bound iron is taken up by most cells, but it is especially important in erythroid precursors where it is the primary source of iron for heme synthesis. In erythroid cells, more than 90% of transferrin-derived iron from endosomes enters into mitochondria ferrochelatase till no clear. However, study on one hand stated mysteriously as its way into mitochondria through passive diffusion, other study hypothesis that the highly efficient transport of iron toward ferrochelatase in erythroid cells requires a direct and transit interaction between transferrin-endosomes and mitochondria (the“kiss-and-run” hypothesis). There are three key support of this hypothesis: 1) iron, delivered to mitochondria via the Tf-TfR pathway, is unavailable to cytoplasmic chelators 2) Tf-containing endosomes move to and contact mitochondria in erythroid cell, and 3) endosomal movement is required for iron delivery to mitochondria.40 Transferrin-Fe3+ binds with specific and 30-fold higher affinity dimeric Tf receptor (TfR1, CD71, 760 amino acids) than transferrin receptor 2 (TfR2) allows for iron uptake.10,41,42 The complex is clathrin-mediated endocytosed, and the acidic pH of the endosomal lumen induces a conformational change in Tf that accompanies iron release.43 Moreover, the acidic environment of the endosome cause the release Fe3+ from Tf- Fe3+ -TfR1 complex, the six-membrane epithelial antigen of the prostate 3 (STEAP3) then reduces Fe3+ to Fe2+ before it is released to the cytosol via DMT1.10,44,45 Iron taken up by cells enters a cytosolic pool called labile iron pool (LIP). The LIP is destined for storage in the form of ferritin, export via Fpn, or metabolic utilization such as cellular energy metabolism, DNA synthesis, and RBC formation (erythroblast to normoblast to reticulocyte to matured RBC) (Figure 2).38
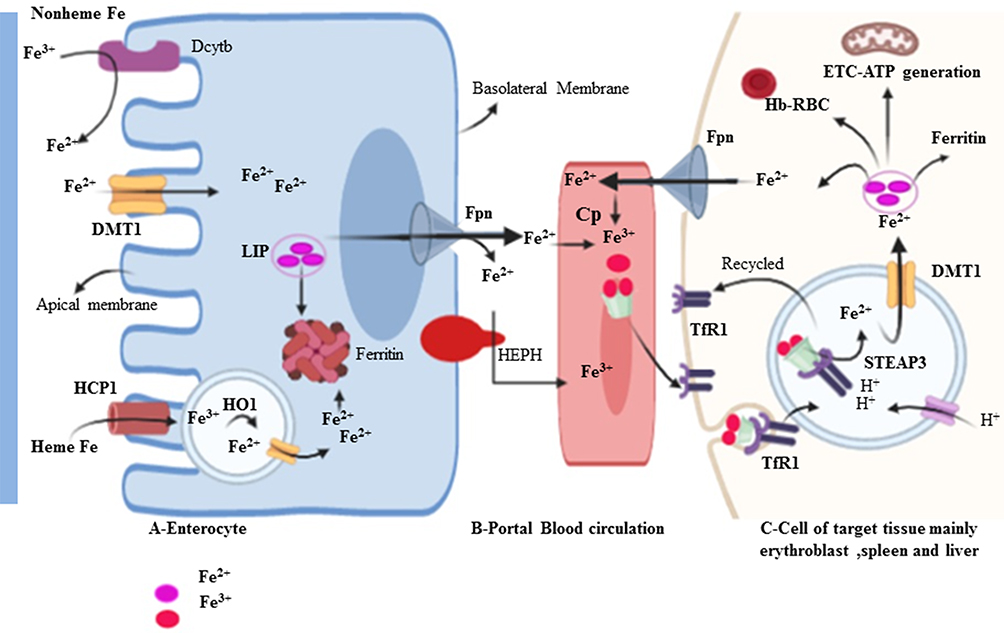 |
Figure 2 Intestinal iron uptake and its distribution to the reticuloendothelial system. Dietary iron can be either in the form of heme or non-heme. DMT1 is a ferrous ion (Fe2+) transporter; hence, Fe3+ is reduced to Fe2+ by apical membrane ferrireductases enzyme, Dcytb. IREG1 (Iron-regulated transporter-1) also known as Fpn is an iron exporter transporter to the circulation. |
Physiological Mechanism of Hepcidin/Ferroportin-Mediated Systemic Iron Homeostasis
Hepcidin (Hp) is the main regulatory molecule of plasma iron concentration. It was originally thought to be functioned solely as an antimicrobial peptide or Liver Expressed Antimicrobial Peptide (LEAP) such as it is up-regulated under inflammatory conditions and considered to be a type two acute-phase reactant due to its regulation via interleukin 6 (IL-6).46 Hepcidin with its gene located on chromosome 19q13.1 is normally synthesized by hepatocyte in a regulated manner for appropriate iron homeostasis.47,48 Thus, iron, inflammation, and erythropoiesis are the major factors for the regulation of Hp at the transcription level. Acute or chronic inflammation and iron increase Hp gene expression (positive regulators), whereas erythropoiesis and hypoxia suppress the expression of the gene (negative regulators).46,47,49,50 It is known that Hepcidin expression is predominantly regulated at the transcriptional level. Two families of cytokines are known to be major positive regulators of hepcidin: the IL-6–like family and the bone morphogenetic protein (BMP) family.51 Among more than 20 BMP related ligands, even though BMP2 and 6 are commonly recognized in Hp regulation but BMP 4, 5, 7 and 9 have been shown to induce hepcidin expression in isolated murine primary hepatocytes.52–54 High iron induces the production of BMP-6 in liver sinusoidal endothelial cells. Bone morphogenic protein 6 and 2 acts on the hepatocyte as paracrine fashion to BMP receptor with hemojuvelin (HJV) as a coreceptor.54,55 Following its binding, the cytoplasmic domain transduces the signaling via phosphorylation of S-mothers against decapentaplegic (SMAD) 1, 5, and 8 (also known as SMAD9 or R-SMAD)56 heterodimerize with common mediator called SMAD4. In turn, the R-SMAD-SMAD4 complex translocates to the nucleus and binds with the BMP response element (BMP-RE) and initiate Hp expression.39,55,56 Taking this step further, hepcidin promoter contains two BMP-responsive elements (BRE1 and BRE2) that are recognized by the SMADs (sometimes also abbreviated as BMP-RE1 and BMP-RE2).57 The wild type hepcidin promoter spans 3 kb upstream of the transcription start site and contains a proximal STAT-binding site (STATBS), a nearby BMP-responsive element located at positions −84/-79 (BRE1), and a distal BMP-responsive element located at positions −2,255/-2,250 (BRE2)57,58 of HAMP-1 gene are essential for both basal hepcidin mRNA expression and the hepcidin response to BMP-2 and BMP-6.58 Since iron is not only critical for life but toxic in excess because of iron-catalyzed formation of pro-oxidants that cause tissue damage in a range of disorders. By responding to toxic insults and controlling the expression of detoxification and antioxidant enzymes, the transcription factor nuclear factor erythroid 2-related factor 2 (Nrf2) maintains cellular health in the face of intracellular and environmental stresses.Nrf2 ultimately increasing the activity of antioxidant enzymes, including catalase, glutathione reductase, and glutathione peroxidase.59,60 As BMP signaling cascade, Hp gene expression is also enhanced by pro-inflammatory cytokine, in particular, interleukin 6 (IL-6) through Janus kinase 2 (JAK2)/signal transducer and activator of transcription 3 (STAT3) signaling pathway.39,49 In support of this, a study was done on mice with hepatocyte-specific deletion of IL-6 signal transducing gp130 receptor, with gp130 receptor lacking the essential region for the STAT-1 and −3 activation reported that Hp is expressed after IL-6 stimulation when gp130-STAT-3 signaling was intact both in vivo as well as in vitro.47,56 Therefore, IL-6/IL6R interaction activates STAT3 via phosphorylation. In turn, upon translocation of activated STAT3 to the nucleus, it binds with the STAT3 binding motif (STAT3-RE) located at position −64/-72 of the Hp promoter is also required for its transcriptional upregulation.39,61 In agreement, Hp is stimulated with lipopolysaccharide (LPS) or turpentine oil-injected mice and in vitro. Hepcidin inhibits iron absorption from the enterocyte, iron efflux from red pulp macrophage of the spleen, iron efflux from hepatocyte via its physical binding to hepcidin receptor domain of Fpn.62 Hp-Fpn complex activating intracellular Janus kinase 2 (Jak2), which results in the internalization and degradation of iron exporter IREG1 or FPN63,64 via ubiquitination proteasome and lysosomal proteolytic mechanism (Figure 3).50,62,65 Therefore, this loss of FPN from the cell membrane prevents cellular iron export and increase iron sequestration. In the contrary, Hp gene expressions are negatively regulated by the erythropoietic or hypoxic conditions. Iron is an essential functional unit of heme within the quaternary structure of haemoglobin and is required for the development of matured red blood cells from an erythropoietic stem cell.38,66 Under a decrease in circulating O2, erythropoietin (EPO) transcription is augmented in peritubular fibroblasts of the renal cortex by binding of heterodimeric (α/β) hypoxia-inducible transcription factors (predominantly HIF-2α) to hypoxia-responsive elements (HRE) of the EPO gene and in turn secret the hormone EPO.38,39,67 Erythropoietin is a glycoprotein growth factor that stimulates erythropoiesis by promoting the terminal differentiation of CFU-E into normoblasts and matures to erythrocytes. Therefore, it acts on the erythropoietin receptor (EPOR) causes JAK2/STAT5 phosphorylation leading to the production of erythroferrone (ERFE) in early erythroblasts.64,68 This signaling induces Hp negative regulator, ERFE, produced by erythroblast and bone marrow (in mice) and results from the differentiation and proliferation of erythroid cell.64 The mechanism action of ERFE on suppression of Hp production via suppressed SMAD1/5 phosphorylation in primary murine hepatocytes and Hep3B cell line of mouse model.47,56,64
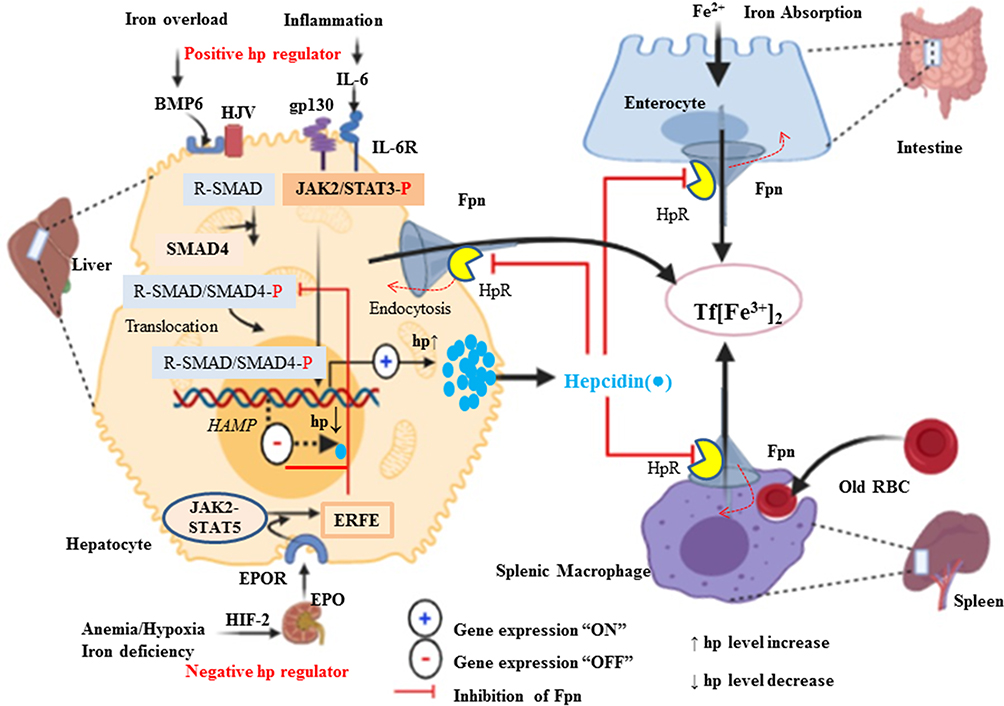 |
Figure 3 Positive and negative regulation of Hp (HAMP) transcription and systemic iron homeostasis through the Hp/Fpn signaling pathway. Hepatocyte production of Hp is affected by available iron from the store and demand. Under iron overload (sensed by liver sinusoidal endothelial cells), increased Hp in hepatocytes is release. Increased Hp binds to FPN and inhibits FPN-mediated iron export into the circulation to reduce Tf saturation. Conversely, when circulating iron is low and then Hp is low and increase iron release via Fpn into the circulation from liver, macrophage and intestine. Basal expression depends on regulation via BMPR and its downstream R-SMAD (SMAD1/5/8 signaling intermediates, which interact with common mediator SMAD (SMAD4) and translocate to the nucleus to activate HAMP transcription. Under inflammatory conditions, IL-6 is produced, which activates the STAT3 signaling pathway to promote transcription of Hp. Conversely, Hp expression is decrease under hypoxic or iron-deficient condition by stimulating kidney for expression of EPO through hypoxia-inducible factor (HIF1α/β) pathway as a transcription factor. Hp, which binds to ferroportin, causing the complex to be internalized and degraded, preventing iron export. |
During the developments of red blood cell, the iron absorption increases mobilization of stored iron and facilitate iron delivery to the marrow through decreasing blood Hp levels.66 This leads to increased Fpn expression and increased iron availability of the circulation for RBC maturation.68 Ferroportin plays a key role in the systemic iron homeostasis by delivering iron from the enterocyte to transferrin. In addition, it can also mediate the efflux of iron from red pulp macrophages of the spleen following catabolism of effete or old RBC and from hepatocyte ferritin (Figure 3).69 Moreover, in iron-deprived condition, liver transmembrane serine protease matriptase 2, encoded by transmembrane protease, serine 6 gene (TMPRSS6), down-regulate Hp expression via proteolytic cleavage of the BMP6 coreceptor hemojuvelin, and terminating BMP6-SMAD1/5/8 signaling pathway.55,64,70 Transmembrane protease, serine 6 encoding matriptase-2 (MTP-2), a transmembrane serine protease produced by the liver, identified to play a crucial role in regulating hepcidin expression, iron homeostasis and normal erythropoiesis.71,72 In both humans and mice, mutations in the TMPRSS6 gene lead to a strong increase in hepcidin expression, resulting in a dramatic decrease in ferroportin expression, and severe iron deficiency anemia.72 Indeed, TMPRSS6 mRNA expression has been demonstrated to be induced by erythropoietin (EPO), hypoxia and by acute iron deprivation.73 In vitro, treatment with BMP6 stimulates TMPRSS6 expression at the mRNA and protein levels and leads to an increase in matriptase-2 activity.72 Taken together TMPRSS6 is regulated by both iron deprivation, hypoxia or EPO and high iron or BMP6.
Aberration of Hepcidin/Ferroportin Signaling in Breast Cancer Cell
Dysregulated iron metabolism has a great role in breast cancer cells. An exuberant free radical generation can cause gene mutation which can accelerate tumor initiation.69 Taking this step further, Metabolic/oxidative stress generated by H2O2 promote the transition of low ROS quiescent breast cancer stem cells (BCSCs) to high ROS of proliferative epithelial-like (E) states.74 Secondly, iron is an essential element for its proliferation and DNA synthesis (iron-dependent ribonucleotide reductase) during S-phase of cell cycles.69,75 Accordingly, increased intracellular iron accumulation in BC cells is necessary to promote growth and its proliferation. Hepcidin is excessively detected in the serum of the patient presents with BC, which implies that it is not only originated from liver hepatocyte but also in the breast cancer cells.76,77 Clinically, the breast is often divided into four quadrants (upper-outer quadrant (UOQ), upper-inner quadrant (UIQ), lower-outer quadrant (LOQ), and lower-inner quadrant (LIQ)) based on horizontal and vertical lines crossing at the nipple. Breast cancers are most frequently arising from the UOQ of glandular tissues of the breast area.78 Moreover, breast cancer can also classified into four groups based on Immunohistochemistry (IHC) profile estrogen receptor (ER)/progesterone receptor (PR) and human epidermal growth factor receptor 2 (Her2/neu) expression, positive (+) and/or negative (-) such as ER/PR+,Her2+, ER/PR+,Her2-, ER/PR-,Her2+, and ER/PR-,Her2-.79 About more than 70% of BCs are ER-positive and tamoxifen is the most common and effective therapy for patients with ER-positive breast cancer.80 A retrospective study conducted in China revealed that serum Hp level has significantly correlated with the pro-inflammatory cytokine (IL-6) level in a patient present with breast cancer metastases to bone, and is considered as an independent risk factor for the breast cancer and its metastasis.81 Moreover, another similar study stated that Hp has a high diagnostic clinical value and potential prognostic marker than BMP6 and IL-6. Furthermore, Sun et al (2017) explored that, inflammatory cytokines such as IL-8, IL-6, and TNFα were higher with different stages of breast cancer and the highest is in stage IV.82 Recently, several research studies have reported on the hypothesis of “seed and soil”; the importance of tumor microenvironment (TME) as a (soil) for optimal growth and aggressive behavior of breast cancer as the (seed).83 From all cell types of TME, TAM is a major component of breast cancer.84 It is associated with poor prognosis of BC by promoting tumor survival, proliferation, invasion, and dissemination.85,86 In breast cancer, unlike other components of TME; TAM particularly invasive macrophage secrets matrix metalloproteinases (MMP-9), cysteine cathepsins and serine proteases (for degradation of the extracellular matrix and basement membranes), and angiogenic macrophages87 secrets proangiogenic factors such as VEGF (promoting tumor angiogenesis), IL-17, for developments of the new blood vessel and initiation of its metastasis.84,88,89 Recruitment of monocyte mediated with monocyte chemoattractant protein-1 (MCP-1) and certain breast cancer produces CSF-1, CCL2, STAT3 and STAT6 that promotes macrophage infiltration as well as polarizing TAMs towards the M2 phenotype.89 Interleukin-6 is also a multifunctional pro-inflammatory biomarker produced locally in the microenvironment of breast cancer such as lymphocytes, monocytes, endothelial cells, fibroblasts and many components of a breast cancer cell that can influence its proliferation90 and regulates erythropoiesis process.81 Among many activators, IL-10 switches the infiltrative monocytes into of M2-polarized and iron-donating phenotype in most types of malignancies and are contributing to their tumor-promoting properties.91 There are several stimuli that can induce further M2 polarization into M2a, M2b, M2c, and M2d. Of these, M2b and M2d mainly secrets anti or proinflammatory cytokines, such as IL1 and IL6.87 Similarly, a study done in Italy showed that high serum levels of IL-6 correlate with poor outcomes of patients with BC.92 Interestingly, a recent study explored that breast cancer-derived exosomes recognized as a means of communication between breast cancer and the immune cell of the microenvironment. Subsequently, it leads a capable of inducing IL-6 secretion and a pro-survival phenotype in macrophages, partially through gp130/STAT3 signaling.93 In vivo and in vitro, STAT3 is the key transcription factor responsible for IL-6 induced Hp (HAMP) gene expression in the liver hepatocyte.56,94 Therefore, IL-6 and Hp have a direct correlation with iron homeostasis via IL6/IL6R/JAK2/STAT3 signaling and likely contribute to the dysregulated Hp/Fpn signaling in breast cancer.90 IL-6 also increases the resistance of breast cancer cells to chemotherapeutic treatment95 and elevated serum IL-6 levels are a marker of poor prognosis in breast cancer.96 In addition, cancer cell and its inflammatory microenvironment manipulate hepcidin expression for their own metabolic needs.77 In breast cancer, BMPs are also linked with increased expression of hepcidin. In early breast cancer, there is no correlation of hepcidin with IL-6, erythropoietin and ERFE.97 Overexpressed BMP7 may be involved in hepcidin overexpression in early breast cancer and is linked with cancer metastasis.98 Understanding the differences in hepcidin regulation between non-cancerous and cancerous cells are important for our knowledge of tumor cell survival, proliferation and metastasis and can help us find new strategies to fight cancer. Collectively, a high level of Hp and followed post-translational regulation of Fpn with subsequent of its degradation are mainly due to malignancy-driven inflammation resulting in reduce iron export and increased retention in breast cancer.99 Consider unclear discrepancy, BMP7 is overexpressed in breast cancer tissue and is associated with cancer metastasis, while BMP7 administration has shown that it can recover Hp expression in BMP6 knockout mice.77 However, the liver is an important source of increased Hp levels in breast cancer, and this increase is related to BMP6 expression.63 Taking this step further, a recent study explored that BMP6 is very important mediators for Hp synthesis in BC cell derived from MCF-7 and MCF-10a cell lines studied in 2D and 3D cell culture system.4 Similar with prostate cancer and ovarian cancer, Fpn expression is reduced in breast cancer along with increased levels of the labile iron pool, TfR1, TfR2 and STEAP3.24,34,67 In contrast, Hp (a negative regulator of FPN) was found to be increased in the patients present with BC as compared with health control.67,100 Moreover, high Hp expressions are positively correlated with poor prognosis as observed in a patient with low breast cancer Fpn expression.24 Similarly, miRNAs are also involved in the dysregulation of iron metabolism in different cancer cells through gene silencing at the post-transcription level. For example, miR-20a and miR-485-3p overexpression can repress Fpn expression and give rise to elevated intracellular LIP content in lung cancer in particular non-small cell lung cancer (NSCLC).99,101
Further analysis of iron gene expression in breast cancer revealed that an iron import dyad TfR1 and an iron export dyad SLC40A1 (FPN) were complementary prognostic factors in predicting distant metastasis-free survival in a cohort of estrogen receptor-positive (ER+) patients treated with tamoxifen.102 Epigenetic modifications, such as DNA methylation, histone deacetylation (HDAC), and some transcriptional factors can control iron metabolism-related gene expression. For example, nuclear factor erythroid 2-like 2 (NRF2) and myeloid zinc finger-1 (MZF-1) could impact cancer cell growth by transcriptionally regulating Fpn and FTH/L expression in breast cancer.24,103 SLC40A1 transcription is inhabited by transcription factor BACH1 (Btb and Cnc Homology 1) and activated by NRF2. Deacetylase SIRT2 can deacetylate and repress NRF2 nuclear localization, reducing Fpn expression and iron export thus keeps an increased iron level of cancer cell.63,104 NAD-dependent Deacetylase sirtuin-3 (SIRT3) and TfR1 expression are negatively correlated in many human cancers such as breast cancer and pancreatic cancer. SIRT3 acts as a negative regulator of TfR1 expression via inhibiting IRP1 activity and prevent the growth of tumor cells (Figure 4). Research evidence showed that SIRT3 is significantly deleted in about 20% of all human cancer; in particular, its frequency of deletion increases by up to 40% in breast and ovarian cancer.105 SIRT3 loss increases ROS production, bringing about elevated IRP1 binding to IREs and increased TfR-1 expression as a consequence.105 However, SIRT3 may exhibit tumor-promoting capacity depending upon the type of cancer and probably, the context of intracellular signal pathways.106 This is exemplified by oesophageal cancer, in which high expression of SIRT3 is associated with a poor outcome; indeed, the level of SIRT3 expression is an independent predictor for cancer prognosis. High SIRT3 expression is also associated with poor prognosis in patients with grade 3 and tamoxifen or cisplatin-resistant breast cancer.106,107 In the other study, SIRT3 expression is markedly lower in breast cancer cells than in paired normal breast epithelium, and lower SIRT3 expression is associated with shorter locoregional relapse-free survival.108 A related study showed that the HEPH (Fe2+ to Fe3+) is also epigenetically repressed by G9a, a H3K9 methyltransferase, which forms a complex with transcription factor YY1 and HDAC1 leading to cellular LIP increase and promoting breast cancer proliferation.109 In turn, it is very clear that complete or partial deletion of SIRT3 results in the accumulation of iron in cancer cells due to the loss of the TFR1 expression. Poor prognosis was conferred by gene expression favoring increased cellular iron levels by upregulated import, TfR1 in 16/22 breast cancer patients69 and downregulated iron export, FPN (SLC40A1), or high HAMP, consistent with the role of TfR1 as a marker of poor response to tamoxifen and shortened breast cancer-specific survival.110 Conversely, research scholars explored that elevated Fpn and low Hp levels were associated with a more favorable prognosis.34
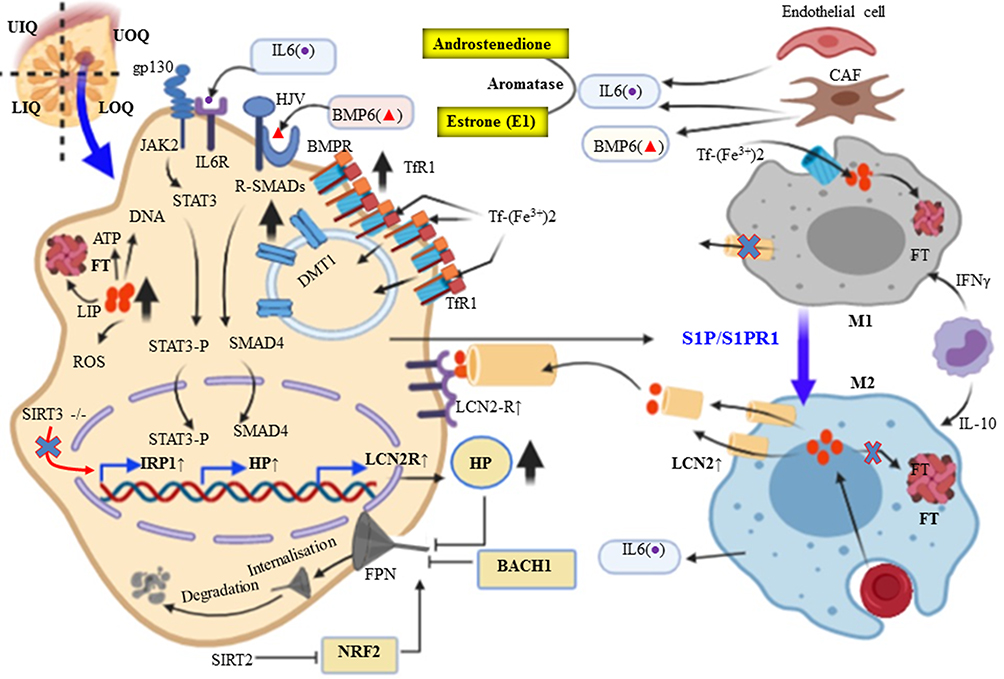 |
Figure 4 Altered iron homeostasis in breast Cancer Cells and the contribution of their microenvironment. Breast cancer cells usually have elevated expression of Tfr1, LCN2 and Hp, and low Fpn expression. Taken together, this breast cancer cell can decrease the level of iron efflux and increase the intracellular iron level to keep the highest demand for iron. |
Interestingly, recent research evidence meticulously showed that TAM express lipocalin 2 (LCN2) also known as Lipocalin 24p3, iron releasing phenotype and promotes tumor progression and metastasis (via activation of MMP9) of human cancer such as breast cancer, ovarian cancer, thyroid cancer, lung cancer, colon cancer, and pancreatic cancer.111–113 Numerous studies have indicated that LCN2 is also associated with high-grade malignancy, metastasis, and poor prognosis in breast cancer.111 The polarization of macrophage from iron sequestration (FThigh, FPN1low M1-like macrophages) to iron-donor phenotype (FT Low, FPN1 high M2-like subtype)99 is mediated by sphingosine-1-phosphate (S1P) released from dying MCF-7 breast cancer cell interacts with S1P receptor (S1PR1) that allows up-regulation of LCN2 in the membrane of macrophages.114,115 Iron exporting role of LCN2, not by direct binding with iron rather it depends on its association with mammalian siderophores. The iron-chelating biomolecule siderophores were first reported in bacteria as competitive with transferrin and lactoferrin. LCN2, an alternative form of iron exporting transport system in BC microenvironment correlates with increase invasiveness and poor prognosis.116 For example, LCN2 in non-invasive human MCF-7 BC cell promotes the activity of MMP9, a protease, and elicits tumor metastasis via binding with and forming MMP9-LCN2 complex protecting MMP9 auto-degradation,117,118 and cancer cell proliferation.116 In addition to this, overexpression of LCN2 can also regulate HIF-1 via signal-regulated kinase ERK or MAPK, and vascular endothelial growth factor (VEGF). This causes the induction of angiogenesis in the tumor microenvironment of MCF7 breast cancer cells.118 The iron storage protein ferritin (FT) facilitates increased iron storage while limiting increased ROS generation. Ferritin is upregulated in a number of cancers including breast cancer, glioblastoma, Hodgkin’s lymphoma, and pancreatic cancer.75 Taken together, due to the unregulated expression of BMP6 and IL6 by the microenvironment, breast cancer displayed exuberant Hp and TfR1 expression and downregulation of Fpn. In particular, iron release phenotype macrophage expresses an alternative iron exporter LCN2. Furthermore, systemic BMP6 is upregulated and acts in the liver hepatocyte contribute to the increased serum level of Hp. In turn, it contributes to an increase in iron uptake and reduced iron export, both of which play iron sequestration to maintain growth, proliferation, and metastasis of breast cancer (Figure 4).
Therapeutic Modulation of Iron Related Protein in Breast Cancer
As described earlier, the deregulation of iron metabolism may lead to abnormal elevation of cellular iron, which may in turn cause the progression of tumorigenesis. In support of this, studies have been reported that decreasing iron level and manipulating the proteins involved in iron metabolism can be used as efficient strategies in tumor chemotherapy.13,118 This is due to the fact that decreasing cellular iron import by blocking TF and increasing cellular iron export through Fpn overexpression reduces the growth of tumor.34 In agreement with this, another study explored that reducing the intracellular iron content (anti-import: TfR1 or pro-export: SLC40A1 (Fpn) (High)/HAMP (Low)) were associated with a more favorable prognosis (P<0.005).102 Moreover, targeting of Hp expression could be another option for control of tumorigenesis.119 In addition, BMP6/Smad4 as well as hemojuvelin (HJV), a BMP co-receptor is also essential for Hp expression.120 Collectively, Hp sequestration agents like Fpn stabilizer, TfR1, LCN2, BMP/SMAD and IL6/IL6R/STAT3 signaling inhibitors are considered as a promising therapeutic strategy against breast cancer.34,36,61
Iron Chelators and Anti-TfR1 as Therapeutics Option of Breast Cancer
The question would be: what is the Achilles’ heel behind the aggressive behavior of breast cancer? Cancer cells have an excessive demand for iron to retain their capacity to proliferate. Interfering of iron metabolism at the gene level or using chelators can be used as breast cancer therapy. Indeed, iron depletion by numerous natural and derived from bacterial siderophore iron chelators, such as desferrioxamine (DFO, Desferal), thiosemicarbazone Triapine,13 deferiprone (DFP, Ferriprox), deferasirox (DFX, Exjade) and tachpyridine were used as a preclinical or clinical inhibitory effect on tumor growth through starving it for iron and reducing its ability to proliferate.65,70 Taking this step further, DFO is the most common iron-chelating agent, relatively strong and non-toxic24,61,65,118 used as a therapeutic option for cancers, including breast cancer.61 In the circulation and tissues, DFO binds iron and the iron-bound forms are excreted efficiently in the urine and bile. As discussed earlier, breast cancer display TfR1 high, and Fpn low. Thus, available anti-cancer drugs like anti-TfR1 antibodies (HB21, 454A12, B3/25, OKT9, 7D3, 7579, and 42/6) were targeted TfR1.118
Therapeutic Options of Breast Cancer Through Hepcidin Modulation
Exacerbated expression and increments of Hp level due to malignancy-derived inflammation, and genetic mutation of the HAMP gene (gain function) are currently treated by a combination of erythropoiesis-stimulating agents (ESAs), such as epoetin-α and darbepoetin-α and RBC transfusions.49 However, their use has been become controversial due to their side effects linking with excessive thromboembolic events, inferior survival, and worse cancer outcomes. Hepcidin antagonist’s agents that either decrease Hp expression or prevent interaction with Fpn would be expected to relieve Hp/Fpn-mediated iron sequestration resulting release of more iron for erythropoiesis. Humanized monoclonal antibodies (mAb) (LY2787106 on Phase I clinical trial, 12B9m, and Ab2.7) have been developed that display a high affinity towards Hp leading to its premature degradation and neutralization of Hp.121–124 In addition, NOX-H94, an RNA-like oligonucleotides spiegelmers with L-stereochemistry also neutralizes human Hp.47,125 Thus, currently, NOX-H94 is under Phase II clinical trial to examine its efficacy in patient with anemia of cancer (AOC).13,121 To interior with Hp interaction with Fpn, because it depends on the extracellular loop of Fpn anti-Fpn mAb (LY2928057) was developed against this section of Fpn adjacent to the Hp receptor.126 Another study conducted by Ross et al stated that anti-Hp antibody (38G6 and 38C8) that pre-incubated with Hek-RExTMFPN-V5/BLA cells demonstrated a marginal ability to inhibit Hp induced internalization of Fpn.127 In addition to EPO and ESA, agents like prolyl hydroxylase inhibitors and HIF stabilizers may also reduce Hp expression by increasing the activity of HIF followed by synthesis of EPO and ERFE proposed as a negative regulator of Hp transcription.55,128
Suppressing Hepcidin by Anti-IL6/IL6R/STAT3 Strategy
Malignancy-derived inflammation induces the exuberant secretion of IL6 and results in activation of Hp via IL-6-mediated JAK2/STAT3. As discussed earlier, from several cancer cell types: TAM, T helper cell, myeloid-derived suppressor cells (MDSCs), and TAF are considered as the primary source of IL-6 in the TME.90 The responsiveness of breast cancer cells to IL-6 intimately depends on the expression of estrogen and progesterone receptors (ER and PR). Taking this step further, IL-6 can also increase levels of estrogen in the circulation and tumor sites by activating the enzymes that produce, including aromatase, estrone sulfatase, and 17β-hydroxysteroid dehydrogenase.129 They convert androstenedione (A) to estrone (E1), estrone sulfate (E1-S) to E1; and E1 to the biologically active estrogen, estradiol (E2), respectively. Thus, targeting against IL-6 signaling cascade has been investigated as therapeutic strategies to inhibit Hp as well as aromatase expression in breast cancer and could be a therapeutic option for the AOC.70,90 Various therapeutic agents, such as anti- IL-6/IL-6R or anti-SIL-6R mAb and specific inhibitors of STAT3 have been developed. The most commonly known anti-IL-6 mAb are siltuximab, sirukumab, olokizumab, clazakizumab, and MEDI5117. The anti-cancer effect of tocilizumab and sarilumab that target IL-6R has been demonstrated in breast cancer.36,130 Even though they are unlikely to be selective, microRNAs, such as miR-218 and miR-34a are also inhibiting IL6R expression. The second-generation STAT3 antisense oligonucleotide AZD9150 binds to and causes the degradation of STAT3 mRNA, thus decreasing its expression.131 STAT3 expression is inhibited by miR-17-5p, miR-20a, and miR-124, whereas Small-molecules of tyrosine kinase inhibitor (TKI) targets JAK such as tofacitinib (inhibits JAK1/3),132 ruxolitinib (inhibits JAK1/2)133,134 prevents phosphorylation and activation of STAT3. Taken together, STAT3-RE of Hp gene promoter free of activation and remained in a repressed state (Figure 5).
 |
Figure 5 Summary for Inhibitors of the upstream hepcidin signaling: IL-6/IL6R/JAK/STAT3 and BMP/SMAD pathway. MicroRNAs (miRNAs) are a class of small non-coding RNAs or untranslated RNA types that function as guide molecules in post-transcriptional RNA silencing. In miRNA 5′ ends are usually the site of interaction with target mRNA. |
Suppressing Hepcidin by Anti-BMP/SMAD Strategy
One of the most important positive regulators of Hp transcription is a BMP-SMAD signaling.49 Heparin, also known as unfractionated heparin (UFH) and low molecular weight heparin (LMWH) is used primarily to treat or prevent thromboembolic disease and naturally occurring glycosaminoglycan family of carbohydrate.70 Anticoagulant activity of Heparin is as a result of high-affinity binding to antithrombin. The Hp lowering effects of LMWH (enoxaparin and fondaparinux) were through sequestration of BMP and blocking SMAD phosphorylation, which then reduces Hp mRNA in HepG2 cell135 and in patients present with venous thrombosis.135 Taking this step further, the BMPs signaling interference capacity of heparins may be unrelated to anticoagulant activity, and with little or no anticoagulant activity. Glycol-split variants of heparins (RO-68 and RO-82) have both been shown to lack anti-thrombin binding while remaining potent Hp inhibitors or full suppression of Hp expression136 useful for the treatment of disorders with Hp excess. Antibodies (anti-BMP6 antibodies), miRNA (miR122), SMAD7 as well as small molecules (dorsomorphin and LDN-1913189) also target BMP/SMAD signaling or BMP co-receptor, HJV and leading to decrease Hp expression.38,70,120,121
Conclusion
The alteration and aberrantly hyperactivated Hp/Fpn pathways of iron metabolism are a key hallmark for breast cancer. The diversified contribution of the tumor microenvironment, particularly plasticity of TAM and TAF on the exuberant expression of Hp secondary to malignancy-derived inflammatory cytokine (IL-6) suggests the fundamental role of iron in breast cancer malignancy and therapeutic resistance. Taking this step further, TAM is frequently polarized and hijacked towards the iron-donor phenotype via expressing LCN2 as an alternative form of Fpn. These are evidence of its contribution to the aggressive subtypes of breast cancer and metastasis. Furthermore, activated IL6/JAK-STAT3 and BMP-SMAD pathways that target Hp gene transcription and possible therapeutic targets for many human cancers, including breast cancer. Thus, strategies targeting Hp/FPN, its upstream signaling cascade, and iron chelation are the promising choices for breast cancer therapy. Regardless of the current knowledge on iron modulating signaling pathways; meticulous understanding of the mechanism epigenetic modification and the cross-talk between tumor microenvironment and the cancer stem cell involved in iron metabolic reprogramming in breast cancer development needs further study.
Data Sharing Statement
All data used are included in the manuscript report.
Ethics Approval and Consent to Participate
Ethical approval is not applicable since the publication is based on the data that was previously published articles and not on its own investigations.
Consent for Publication
Not applicable.
Acknowledgments
We would like to forward our gratitude to the authors of the article, what we generate this review report.
Author Contributions
All authors made a significant contribution to the work reported, whether in conception, study design, execution, acquisition of data, analysis, interpretation, or all those areas; took part in drafting, revising, or critically reviewing the article; gave final approval to the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
For this review, we did not receive any grant from any funding agency.
Disclosure
The authors have no conflicts of interest to declare.
References
1. Collaborators GRF. Global, regional, and national comparative risk assessment of 79 behavioural, environmental and occupational, and metabolic risks or clusters of risks, 1990–2015: a systematic analysis for the Global Burden of Disease Study 2015. Lancet (London, England). 2016;388(10053):1659.
2. Siegel RL, Kimberly D, Miller M, et al. Cancer statistics, 2018. CA Cancer J Clin. 2018;68:7–30. doi:10.3322/caac.21442
3. Chekhun VF, Lukyanova NY, Burlaka АP, et al. Iron metabolism disturbances in the MCF-7 human breast cancer cells with acquired resistance to doxorubicin and cisplatin. Int J Oncol. 2013;43(5):1481–1486. doi:10.3892/ijo.2013.2063
4. Scimeca M, Bonanno E. New highlight in breast cancer development: the key role of hepcidin and iron metabolism. Ann Transl Med. 2018;6(Suppl):1.
5. Alarmo E-L, Kallioniemi A. Bone morphogenetic proteins in breast cancer: dual role in tumourigenesis? Endocr Relat Cancer. 2010;17(2):R123–R139. doi:10.1677/ERC-09-0273
6. Miguel F, Lopes LV, Ferreira E, et al. Breast cancer in Angola, molecular subtypes: a first glance. ecancermedicalscience. 2017;11.
7. Adeloye D, Sowunmi OY, Jacobs W, et al. Estimating the incidence of breast cancer in Africa: a systematic review and meta-analysis. J Glob Health. 2018;8:1. doi:10.7189/jogh.08.010419
8. Azubuike SO, Muirhead C, Hayes L, et al. Rising global burden of breast cancer: the case of sub-Saharan Africa (with emphasis on Nigeria) and implications for regional development: a review. World J Surg Oncol. 2018;16(1):1–13. doi:10.1186/s12957-018-1345-2
9. Yu Y, Kovacevic Z, Richardson DR. Tuning cell cycle regulation with an iron key. Cell Cycle. 2007;6(16):1982–1994. doi:10.4161/cc.6.16.4603
10. Anderson CP, Shen M, Eisenstein RS, et al. Mammalian iron metabolism and its control by iron regulatory proteins. Biochimica Et Biophysica Acta Mol Cell Res. 2012;1823(9):1468–1483. doi:10.1016/j.bbamcr.2012.05.010
11. Verma S, Cherayil BJ. Iron and inflammation–the gut reaction. Metallomics. 2017;9(2):101–111. doi:10.1039/C6MT00282J
12. Yiannikourides A, Latunde-Dada GO. A short review of iron metabolism and pathophysiology of iron disorders. Medicines. 2019;6(3):85.
13. Jung M, Mertens C, Tomat E, et al. Iron as a central player and promising target in cancer progression. Int J Mol Sci. 2019;20(2):273. doi:10.3390/ijms20020273
14. Chang VC, Cotterchio M, Khoo E. Iron intake, body iron status, and risk of breast cancer: a systematic review and meta-analysis. BMC Cancer. 2019;19(1):543. doi:10.1186/s12885-019-5642-0
15. Huang X. Does iron have a role in breast cancer? Lancet Oncol. 2008;9(8):803–807. doi:10.1016/S1470-2045(08)70200-6
16. Hecht F, Pessoa CF, Gentile LB, et al. The role of oxidative stress on breast cancer development and therapy. Tumor Biol. 2016;37(4):4281–4291. doi:10.1007/s13277-016-4873-9
17. Chifman J, Arat S, Deng Z, et al. Activated oncogenic pathway modifies Iron network in breast epithelial cells: a dynamic modeling perspective. PLoS Comput Biol. 2017;13(2):e1005352. doi:10.1371/journal.pcbi.1005352
18. Pantopoulos K, Porwal SK, Tartakoff A, et al. Mechanisms of mammalian iron homeostasis. Biochemistry. 2012;51(29):5705–5724. doi:10.1021/bi300752r
19. Soysal SD, Tzankov A, Muenst SE. Role of the tumor microenvironment in breast cancer. Pathobiology. 2015;82(3–4):142–152. doi:10.1159/000430499
20. Allen MD, Jones LJ. The role of inflammation in progression of breast cancer: friend or foe? Int J Oncol. 2015;47(3):797–805. doi:10.3892/ijo.2015.3075
21. Place AE, Huh SJ, Polyak K. The microenvironment in breast cancer progression: biology and implications for treatment. Breast Cancer Res. 2011;13(6):1–11. doi:10.1186/bcr2912
22. Manz DH, Blanchette NL, Paul BT, et al. Iron and cancer: recent insights. Ann N Y Acad Sci. 2016;1368(1):149. doi:10.1111/nyas.13008
23. Richardson DR, Lane DJ, Becker EM, et al. Mitochondrial iron trafficking and the integration of iron metabolism between the mitochondrion and cytosol. Proc Natl Acad Sci. 2010;107(24):10775–10782. doi:10.1073/pnas.0912925107
24. Zhou L, Zhao B, Zhang L, et al. Alterations in cellular iron metabolism provide more therapeutic opportunities for cancer. Int J Mol Sci. 2018;19(5):1545. doi:10.3390/ijms19051545
25. Andrews NC, Schmidt PJ. Iron homeostasis. Annu Rev Physiol. 2007;69:69–85. doi:10.1146/annurev.physiol.69.031905.164337
26. McKie AT, Barrow D, Latunde-Dada GO, et al. An iron-regulated ferric reductase associated with the absorption of dietary iron. Science. 2001;291(5509):1755–1759. doi:10.1126/science.1057206
27. Chen Y, Fan Z, Yang Y, et al. Iron metabolism and its contribution to cancer. Int J Oncol. 2019;54(4):1143–1154.
28. Wang J, Pantopoulos K. Regulation of cellular iron metabolism. Biochem J. 2011;434(3):365–381.
29. Abbaspour N, Hurrell R, Kelishadi R. Review on iron and its importance for human health. J Res Med Sci. 2014;19(2):164.
30. Bafaro E, Liu Y, Xu Y, et al. The emerging role of zinc transporters in cellular homeostasis and cancer. Signal Transduction Targeted Ther. 2017;2(1):1–12.
31. Lane D, Merlot A, Huang M-H, et al. Cellular iron uptake, trafficking and metabolism: key molecules and mechanisms and their roles in disease. Biochimica Et Biophysica Acta Mol Cell Res. 2015;1853(5):1130–1144. doi:10.1016/j.bbamcr.2015.01.021
32. Jenkitkasemwong S, Wang C-Y, Mackenzie B, et al. Physiologic implications of metal-ion transport by ZIP14 and ZIP8. Biometals. 2012;25(4):643–655. doi:10.1007/s10534-012-9526-x
33. Torti FM, Torti SV. Regulation of ferritin genes and protein. Blood J Am Soc Hematol. 2002;99(10):3505–3516.
34. Pinnix ZK, Miller LD, Wang W, et al. Ferroportin and iron regulation in breast cancer progression and prognosis. Sci Transl Med. 2010;2(43):43ra56–43ra56. doi:10.1126/scitranslmed.3001127
35. Torti SV, Torti FM. Ironing out cancer. Cancer Res. 2011;71(5):1511–1514. doi:10.1158/0008-5472.CAN-10-3614
36. Guo W, Zhang S, Chen Y, et al. An important role of the hepcidin–ferroportin signaling in affecting tumor growth and metastasis. Acta Biochim Biophys Sin (Shanghai). 2015;47(9):703–715. doi:10.1093/abbs/gmv063
37. Abboud S, Haile DJ. A novel mammalian iron-regulated protein involved in intracellular iron metabolism. J Biol Chem. 2000;275(26):19906–19912. doi:10.1074/jbc.M000713200
38. Muckenthaler MU, Rivella S, Hentze MW, et al. A red carpet for iron metabolism. Cell. 2017;168(3):344–361. doi:10.1016/j.cell.2016.12.034
39. Hentze MW, Muckenthaler MU, Galy B, et al. Two to tango: regulation of Mammalian iron metabolism. Cell. 2010;142(1):24–38. doi:10.1016/j.cell.2010.06.028
40. Hamdi A, Roshan TM, Kahawita TM, et al. Erythroid cell mitochondria receive endosomal iron by a “kiss-and-run” mechanism. Biochimica Et Biophysica Acta (BBA)-Molecular Cell Research. 2016;1863(12):2859–2867. doi:10.1016/j.bbamcr.2016.09.008
41. West AP, Bennett MJ, Sellers VM, et al. Comparison of the interactions of transferrin receptor and transferrin receptor 2 with transferrin and the hereditary hemochromatosis protein HFE. J Biol Chem. 2000;275(49):38135–38138. doi:10.1074/jbc.C000664200
42. Kawabata H, Germain RS, Vuong PT, et al. Transferrin receptor 2-α supports cell growth both in iron-chelated cultured cells and in vivo. J Biol Chem. 2000;275(22):16618–16625. doi:10.1074/jbc.M908846199
43. Cheng Y, Zak O, Aisen P, et al. Structure of the human transferrin receptor-transferrin complex. Cell. 2004;116(4):565–576. doi:10.1016/S0092-8674(04)00130-8
44. Gauss GH, Kleven MD, Sendamarai AK, et al. The crystal structure of six-transmembrane epithelial antigen of the prostate 4 (Steap4), a ferri/cuprireductase, suggests a novel interdomain flavin-binding site. J Biol Chem. 2013;288(28):20668–20682. doi:10.1074/jbc.M113.479154
45. Ohgami RS, Campagna DR, McDonald A, et al. The Steap proteins are metalloreductases. Blood. 2006;108(4):1388–1394. doi:10.1182/blood-2006-02-003681
46. Wrighting DM, Andrews NC. Interleukin-6 induces hepcidin expression through STAT3. Blood. 2006;108(9):3204–3209. doi:10.1182/blood-2006-06-027631
47. Zhao N, Zhang A-S, Enns CA. Iron regulation by hepcidin. J Clin Invest. 2013;123(6):2337–2343.
48. Nemeth E, Ganz T. Regulation of iron metabolism by hepcidin. Annu Rev Nutr. 2006;26:323–342. doi:10.1146/annurev.nutr.26.061505.111303
49. Ganz T, Nemeth E. The hepcidin-ferroportin system as a therapeutic target in anemias and iron overload disorders. Hematology Am Soc Hematol Educ Prog. 2011;2011(1):538–542.
50. Subha Palaneeswari M, Ganesh M, Karthikeyan T, et al. Hepcidin–Minireview. J Clin Diagnostic Res. 2013;7(8):1767.
51. Maes K, Nemeth E, Roodman GD, et al. In anemia of multiple myeloma, hepcidin is induced by increased bone morphogenetic protein 2. Blood J Am Soc Hematol. 2010;116(18):3635–3644.
52. Wu X, Yung L-M, Cheng W-H, et al. Hepcidin regulation by BMP signaling in macrophages is lipopolysaccharide dependent. PLoS One. 2012;7(9):e44622. doi:10.1371/journal.pone.0044622
53. Truksa J, Peng H, Lee P, et al. Bone morphogenetic proteins 2, 4, and 9 stimulate murine hepcidin 1 expression independently of Hfe, transferrin receptor 2 (Tfr2), and IL-6. Proc Natl Acad Sci. 2006;103(27):10289–10293. doi:10.1073/pnas.0603124103
54. Zhang A-S. Control of systemic iron homeostasis by the hemojuvelin-hepcidin axis. Advan Nutri. 2010;1(1):38–45. doi:10.3945/an.110.1009
55. Camaschella C, Nai A, Silvestri L. Iron metabolism and iron disorders revisited in the hepcidin era. haematologica. 2020;105(2):260–272. doi:10.3324/haematol.2019.232124
56. Wang C-Y, Core AB, Canali S, et al. Smad1/5 is required for erythropoietin-mediated suppression of hepcidin in mice. Blood J Am Soc Hematol. 2017;130(1):73–83.
57. Casanovas G, Banerji A, d’Alessio F, et al. A multi-scale model of hepcidin promoter regulation reveals factors controlling systemic iron homeostasis. PLoS Comput Biol. 2014;10(1):e1003421. doi:10.1371/journal.pcbi.1003421
58. Casanovas G, Mleczko-Sanecka K, Altamura S, et al. Bone morphogenetic protein (BMP)-responsive elements located in the proximal and distal hepcidin promoter are critical for its response to HJV/BMP/SMAD. J Mol Med. 2009;87(5):471–480. doi:10.1007/s00109-009-0447-2
59. Imam MU, Zhang S, Ma J, et al. Antioxidants mediate both iron homeostasis and oxidative stress. Nutrients. 2017;9(7):671. doi:10.3390/nu9070671
60. Fustinoni-Reis AM, Arruda SF, Dourado LP, et al. Tucum-do-cerrado (Bactris setosa Mart.) consumption modulates iron homeostasis and prevents iron-induced oxidative stress in the rat liver. Nutrients. 2016;8(2):38.
61. Fischer-Fodor E, Miklasova N, Berindan-Neagoe I, et al. Iron, inflammation and invasion of cancer cells. Clujul Med. 2015;88(3):272.
62. De Domenico I, Ward DM, Langelier C, et al. The molecular mechanism of hepcidin-mediated ferroportin down-regulation. Mol Biol Cell. 2007;18(7):2569–2578. doi:10.1091/mbc.e07-01-0060
63. Zhang S, Chen Y, Guo W, et al. Disordered hepcidin–ferroportin signaling promotes breast cancer growth. Cell Signal. 2014;26(11):2539–2550. doi:10.1016/j.cellsig.2014.07.029
64. Ganz T. Erythropoietic regulators of iron metabolism. Free Radic Biol Med. 2019;133:69–74. doi:10.1016/j.freeradbiomed.2018.07.003
65. Wang Y, Yu L, Ding J, et al. Iron metabolism in cancer. Int J Mol Sci. 2019;20(1):95. doi:10.3390/ijms20010095
66. Kautz L, Jung G, Valore EV, et al. Identification of erythroferrone as an erythroid regulator of iron metabolism. Nat Genet. 2014;46(7):678–684. doi:10.1038/ng.2996
67. Zhang J, Chen X. p53 tumor suppressor and iron homeostasis. FEBS J. 2019;286(4):620–629. doi:10.1111/febs.14638
68. Schmidt PJ. Regulation of iron metabolism by hepcidin under conditions of inflammation. J Biol Chem. 2015;290(31):18975–18983. doi:10.1074/jbc.R115.650150
69. Torti SV, Torti FM. Cellular iron metabolism in prognosis and therapy of breast cancer. Crit Rev Oncogenesis. 2013;18:5. doi:10.1615/CritRevOncog.2013007784
70. Hawula ZJ, Wallace DF, Subramaniam VN, et al. Therapeutic advances in regulating the hepcidin/ferroportin axis. Pharmaceuticals. 2019;12(4):170. doi:10.3390/ph12040170
71. Rausa M, Ghitti M, Pagani A, et al. Identification of TMPRSS 6 cleavage sites of hemojuvelin. J Cell Mol Med. 2015;19(4):879–888. doi:10.1111/jcmm.12462
72. Meynard D, Vaja V, Sun CC, et al. Regulation of TMPRSS6 by BMP6 and iron in human cells and mice. Blood J Am Soc Hematol. 2011;118(3):747–756.
73. Lakhal S, Schödel J, Townsend AR, et al. Regulation of Type II transmembrane serine proteinase TMPRSS6 by Hypoxia-inducible Factors new link Between Hypoxia Signaling And Iron Homeostasis. J Biol Chem. 2011;286(6):4090–4097. doi:10.1074/jbc.M110.173096
74. Luo M, Shang L, Brooks MD, et al. Targeting breast cancer stem cell state equilibrium through modulation of redox signaling. Cell Metab. 2018;28(1):69–86. e66. doi:10.1016/j.cmet.2018.06.006
75. Torti SV, Manz DH, Paul BT, et al. Iron and cancer. Annu Rev Nutr. 2018;38:97–125. doi:10.1146/annurev-nutr-082117-051732
76. Saneela S, Iqbal R, Raza A, et al. Hepcidin: a key regulator of iron. J Pak Med Assoc. 2019;69(8):1170–1175.
77. Vela D, Vela-Gaxha Z. Differential regulation of hepcidin in cancer and non-cancer tissues and its clinical implications. Exp Mol Med. 2018;50(2):e436–e436. doi:10.1038/emm.2017.273
78. Chan S, Chen J-H, Li S, et al. Evaluation of the association between quantitative mammographic density and breast cancer occurred in different quadrants. BMC Cancer. 2017;17(1):274. doi:10.1186/s12885-017-3270-0
79. Onitilo AA, Engel JM, Greenlee RT, et al. Breast cancer subtypes based on ER/PR and Her2 expression: comparison of clinicopathologic features and survival. Clin Med Res. 2009;7(1–2):4–13. doi:10.3121/cmr.2008.825
80. Xuan Q-J, Wang J-X, Nanding A, et al. Tumor-associated macrophages are correlated with tamoxifen resistance in the postmenopausal breast cancer patients. Pathol Oncol Res. 2014;20(3):619–624. doi:10.1007/s12253-013-9740-z
81. Shao X, Cao F, Tao M. The clinical value of hepcidin in breast cancer and its bone metastasis. Ann Clin Lab Sci. 2017;47(2):120–128.
82. Sun X-H, Cao Q, Xiao J-Y. Clinical significance of serum tumor markers and cytokines in the detection of breast cancer. J Hainan Med Univ. 2017;23(2):61–64.
83. Mao Y, Keller ET, Garfield DH, et al. Stromal cells in tumor microenvironment and breast cancer. Cancer Metastasis Rev. 2013;32(1–2):303–315. doi:10.1007/s10555-012-9415-3
84. Obeid E, Nanda R, Fu Y-X, et al. The role of tumor-associated macrophages in breast cancer progression. Int J Oncol. 2013;43(1):5–12. doi:10.3892/ijo.2013.1938
85. Jeong H, Hwang I, Kang SH, et al. Tumor-associated macrophages as potential prognostic biomarkers of invasive breast cancer. J Breast Cancer. 2019;22(1):38–51. doi:10.4048/jbc.2019.22.e5
86. Mahmoud S, Lee A, Paish E, et al. Tumour-infiltrating macrophages and clinical outcome in breast cancer. J Clin Pathol. 2012;65(2):159–163. doi:10.1136/jclinpath-2011-200355
87. Poh AR, Ernst M. Targeting macrophages in cancer: from bench to bedside. Front Oncol. 2018;8:49. doi:10.3389/fonc.2018.00049
88. Qiu S-Q, Waaijer SJ, Zwager MC, et al. Tumor-associated macrophages in breast cancer: innocent bystander or important player? Cancer Treat Rev. 2018;70:178–189. doi:10.1016/j.ctrv.2018.08.010
89. Mukhtar RA, Nseyo O, Campbell MJ, et al. Tumor-associated macrophages in breast cancer as potential biomarkers for new treatments and diagnostics. Expert Rev Mol Diagn. 2011;11(1):91–100. doi:10.1586/erm.10.97
90. Masjedi A, Hashemi V, Hojjat-Farsangi M, et al. The significant role of interleukin-6 and its signaling pathway in the immunopathogenesis and treatment of breast cancer. Biomed Pharmacother. 2018;108:1415–1424. doi:10.1016/j.biopha.2018.09.177
91. Cairo G, Recalcati S, Mantovani A, et al. Iron trafficking and metabolism in macrophages: contribution to the polarized phenotype. Trends Immunol. 2011;32(6):241–247. doi:10.1016/j.it.2011.03.007
92. Sanguinetti A, Santini D, Bonafè M, et al. Interleukin-6 and pro inflammatory status in the breast tumor microenvironment. World J Surg Oncol. 2015;13(1):129. doi:10.1186/s12957-015-0529-2
93. Ham S, Lima LG, Chai EPZ, et al. Breast cancer-derived exosomes alter macrophage polarization via gp130/STAT3 signaling. Front Immunol. 2018;9:871. doi:10.3389/fimmu.2018.00871
94. Verga Falzacappa MV, Vujic Spasic M, Kessler R, et al. STAT3 mediates hepatic hepcidin expression and its inflammatory stimulation. Blood. 2007;109(1):353–358. doi:10.1182/blood-2006-07-033969
95. Conze D, Weiss L, Regen PS, et al. Autocrine production of interleukin 6 causes multidrug resistance in breast cancer cells. Cancer Res. 2001;61(24):8851–8858.
96. Knüpfer H, Preiß R. Significance of interleukin-6 (IL-6) in breast cancer. Breast Cancer Res Treat. 2007;102(2):129–135. doi:10.1007/s10549-006-9328-3
97. Ciniselli CM, De Bortoli M, Taverna E, et al. Plasma hepcidin in early-stage breast cancer patients: no relationship with interleukin-6, erythropoietin and erythroferrone. Expert Rev Proteomics. 2015;12(6):695–701. doi:10.1586/14789450.2015.1099436
98. Pauk M, Grgurevic L, Brkljacic J, et al. Exogenous BMP7 corrects plasma iron overload and bone loss in Bmp6-/-mice. Int Orthop. 2015;39(1):161–172. doi:10.1007/s00264-014-2550-4
99. Pfeifhofer-Obermair C, Tymoszuk P, Petzer V, et al. Iron in the tumor microenvironment—connecting the dots. Front Oncol. 2018;8:549.
100. Orlandi R, De Bortoli M, Ciniselli C, et al. Hepcidin and ferritin blood level as noninvasive tools for predicting breast cancer. Ann Oncol. 2014;25(2):352–357. doi:10.1093/annonc/mdt490
101. Sangokoya C, Doss JF, Chi J-T. Iron-responsive miR-485-3p regulates cellular iron homeostasis by targeting ferroportin. PLoS Genet. 2013;9(4):e1003408. doi:10.1371/journal.pgen.1003408
102. Miller LD, Coffman LG, Chou JW, et al. An iron regulatory gene signature predicts outcome in breast cancer. Cancer Res. 2011;71(21):6728–6737. doi:10.1158/0008-5472.CAN-11-1870
103. Chen Y, Zhang S, Wang X, et al. Disordered signaling governing ferroportin transcription favors breast cancer growth. Cell Signal. 2015;27(1):168–176. doi:10.1016/j.cellsig.2014.11.002
104. Yang X, Park S-H, Chang H-C, et al. Sirtuin 2 regulates cellular iron homeostasis via deacetylation of transcription factor NRF2. J Clin Invest. 2017;127(4):1505–1516. doi:10.1172/JCI88574
105. Jeong SM, Lee J, Finley L, et al. SIRT3 regulates cellular iron metabolism and cancer growth by repressing iron regulatory protein 1. Oncogene. 2015;34(16):2115–2124. doi:10.1038/onc.2014.124
106. Xiong Y, Wang M, Zhao J, et al. Sirtuin 3: A Janus face in cancer. Int J Oncol. 2016;49(6):2227–2235. doi:10.3892/ijo.2016.3767
107. Torrens‐Mas M, Pons DG, Sastre‐Serra J, et al. SIRT3 silencing sensitizes breast cancer cells to cytotoxic treatments through an increment in ROS production. J Cell Biochem. 2017;118(2):397–406. doi:10.1002/jcb.25653
108. Desouki MM, Doubinskaia I, Gius D, et al. Decreased mitochondrial SIRT3 expression is a potential molecular biomarker associated with poor outcome in breast cancer. Hum Pathol. 2014;45(5):1071–1077. doi:10.1016/j.humpath.2014.01.004
109. Wang Y-F, Zhang J, Su Y, et al. G9a regulates breast cancer growth by modulating iron homeostasis through the repression of ferroxidase hephaestin. Nat Commun. 2017;8(1):1–14.
110. Habashy HO, Powe DG, Staka CM, et al. Transferrin receptor (CD71) is a marker of poor prognosis in breast cancer and can predict response to tamoxifen. Breast Cancer Res Treat. 2010;119(2):283. doi:10.1007/s10549-009-0345-x
111. Hu C, Yang K, Li M, et al. Lipocalin 2: a potential therapeutic target for breast cancer metastasis. Onco Targets Ther. 2018;11:8099. doi:10.2147/OTT.S181223
112. Jung M, Mertens C, Bauer R, et al. Lipocalin-2 and iron trafficking in the tumor microenvironment. Pharmacol Res. 2017;120:146–156. doi:10.1016/j.phrs.2017.03.018
113. Mertens C, Mora J, Ören B, et al. Macrophage-derived lipocalin-2 transports iron in the tumor microenvironment. Oncoimmunology. 2018;7(3):e1408751. doi:10.1080/2162402X.2017.1408751
114. Sola A, Weigert A, Jung M, et al. Sphingosine‐1‐phosphate signalling induces the production of Lcn‐2 by macrophages to promote kidney regeneration. J Pathol. 2011;225(4):597–608. doi:10.1002/path.2982
115. Jung M, Ören B, Mora J, et al. Lipocalin 2 from macrophages stimulated by tumor cell–derived sphingosine 1-phosphate promotes lymphangiogenesis and tumor metastasis. Sci Signal. 2016;9(434):ra64–ra64. doi:10.1126/scisignal.aaf3241
116. Yang J, Bielenberg DR, Rodig SJ, et al. Lipocalin 2 promotes breast cancer progression. Proc Natl Acad Sci. 2009;106(10):3913–3918. doi:10.1073/pnas.0810617106
117. Yan L, Borregaard N, Kjeldsen L, et al. The high molecular weight urinary matrix metalloproteinase (MMP) activity is a complex of gelatinase B/MMP-9 and neutrophil gelatinase-associated lipocalin (NGAL) modulation of MMP-9 activity by NGAL. J Biol Chem. 2001;276(40):37258–37265. doi:10.1074/jbc.M106089200
118. Zhang C, Zhang F. Iron homeostasis and tumorigenesis: molecular mechanisms and therapeutic opportunities. Protein Cell. 2015;6(2):88–100. doi:10.1007/s13238-014-0119-z
119. Kautz L, Meynard D, Monnier A, et al. Iron regulates phosphorylation of Smad1/5/8 and gene expression of Bmp6, Smad7, Id1, and Atoh8 in the mouse liver. Blood. 2008;112(4):1503–1509. doi:10.1182/blood-2008-03-143354
120. Ruchala P, Nemeth E. The pathophysiology and pharmacology of hepcidin. Trends Pharmacol Sci. 2014;35(3):155–161. doi:10.1016/j.tips.2014.01.004
121. Langer AL, Ginzburg YZ. Role of hepcidin‐ferroportin axis in the pathophysiology, diagnosis, and treatment of anemia of chronic inflammation. Hemodialysis Int. 2017;21:S37–S46. doi:10.1111/hdi.12543
122. Cooke KS, Hinkle B, Salimi-Moosavi H, et al. A fully human anti-hepcidin antibody modulates iron metabolism in both mice and nonhuman primates. Blood J Am Soc Hematol. 2013;122(17):3054–3061.
123. Vadhan-Raj S, Abonour R, Goldman JW, et al. A first-in-human Phase 1 study of a hepcidin monoclonal antibody, LY2787106, in cancer-associated anemia. J Hematol Oncol. 2017;10(1):73. doi:10.1186/s13045-017-0427-x
124. Sasu BJ, Cooke KS, Arvedson TL, et al. Antihepcidin antibody treatment modulates iron metabolism and is effective in a mouse model of inflammation-induced anemia. Blood. 2010;115(17):3616–3624. doi:10.1182/blood-2009-09-245977
125. White J. The regulation and expression of hepcidin in carcinogenesis as a result of iron overload disorders: an extended literature review. J Cancer Prev Curr Res. 2015;3(2):00077.
126. Katsarou A, Pantopoulos K. Hepcidin therapeutics. Pharmaceuticals. 2018;11(4):127. doi:10.3390/ph11040127
127. Ross SL, Biswas K, Rottman J, et al. Identification of antibody and small molecule antagonists of ferroportin-hepcidin interaction. Front Pharmacol. 2017;8:838. doi:10.3389/fphar.2017.00838
128. Peyssonnaux C, Zinkernagel AS, Schuepbach RA, et al. Regulation of iron homeostasis by the hypoxia-inducible transcription factors (HIFs). J Clin Invest. 2007;117(7):1926–1932. doi:10.1172/JCI31370
129. Purohit A, Newman SP, Reed MJ. The role of cytokines in regulating estrogen synthesis: implications for the etiology of breast cancer. Breast Cancer Res. 2002;4(2):1–5. doi:10.1186/bcr425
130. Jiang X-P, Yang DC, Elliott RL, et al. Down-regulation of expression of interleukin-6 and its receptor results in growth inhibition of MCF-7 breast cancer cells. Anticancer Res. 2011;31(9):2899–2906.
131. Hong D, Kurzrock R, Kim Y, et al. AZD9150, a next-generation antisense oligonucleotide inhibitor of STAT3 with early evidence of clinical activity in lymphoma and lung cancer. Sci Transl Med. 2015;7(314):314ra185–314ra185. doi:10.1126/scitranslmed.aac5272
132. Meyer DM, Jesson MI, Li X, et al. Anti-inflammatory activity and neutrophil reductions mediated by the JAK1/JAK3 inhibitor, CP-690,550, in rat adjuvant-induced arthritis. J Inflamm. 2010;7(1):1–12. doi:10.1186/1476-9255-7-41
133. Pemmaraju N, Kantarjian H, Kadia T, et al. A phase I/II study of the Janus kinase (JAK) 1 and 2 inhibitor ruxolitinib in patients with relapsed or refractory acute myeloid leukemia. Clin Lymphoma Myeloma Leuk. 2015;15(3):171–176. doi:10.1016/j.clml.2014.08.003
134. Bose P, Verstovsek S. JAK2 inhibitors for myeloproliferative neoplasms: what is next? Blood J Am Soc Hematol. 2017;130(2):115–125.
135. Poli M, Girelli D, Campostrini N, et al. Heparin: a potent inhibitor of hepcidin expression in vitro and in vivo. Blood J Am Soc Hematol. 2011;117(3):997–1004.
136. Poli M, Asperti M, Naggi A, et al. Glycol-split nonanticoagulant heparins are inhibitors of hepcidin expression in vitro and in vivo. Blood J Am Soc Hematol. 2014;123(10):1564–1573.
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.