Back to Journals » Clinical and Experimental Gastroenterology » Volume 12
Inflammation in gastrointestinal disorders: prevalent socioeconomic factors
Authors Ribaldone DG ![]() , Pellicano R
, Pellicano R ![]() , Actis GC
, Actis GC ![]()
Received 1 April 2019
Accepted for publication 24 June 2019
Published 19 July 2019 Volume 2019:12 Pages 321—329
DOI https://doi.org/10.2147/CEG.S210844
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Andreas M. Kaiser
Video abstract presented by Davide Giuseppe Ribaldone.
Views: 103
Davide Giuseppe Ribaldone,1 Rinaldo Pellicano,2 Giovanni Clemente Actis3
1Department of Surgical Sciences, University of Turin, Turin, Italy; 2Unit of Gastroenterology, Molinette-San Giovanni Antica Sede (SGAS) Hospital, Turin, Italy; 3The Medical Center Practice Office, Turin, Italy
Abstract: Western populations harbor a chronic inflammation pattern that lacks organ cardinal signs (edema, increased temperature, pain, and impaired function), releases increased levels of C-reactive protein, and often runs a creeping clinical course with generalized debilitating disease superimposed on system-specific involvement, mostly including nervous tissue (multiple sclerosis, Parkinson’s syndromes), joints (arthritis), and skin (psoriasis). A finalistic interpretation may apply to the consideration of the gut as the source of inflammation. In fact, these kind of local events as well as the remote manifestations named above, could be conditioned by the microbiome, the huge cell population indwelling the gut which is under growing scrutiny. The role of the gut as a barrier organ justifies lingering submucosal inflammation as a patrolling activity to maintain bodily integrity; the microbiome, launching inflammogenic signals in response to abrupt diet changes, confers to gut inflammation a socioeconomic vector calling for hitherto unrecognized multi-disciplinary interventions.
Keywords: colorectal cancer, inflammation, inflammatory bowel disease, irritable bowel syndrome, microbiome
Introduction
The last few years have witnessed a mounting interest for the phenomenon of inflammation, with all aspects of pathology, mechanisms, and clinics becoming reappraised closely. Rather than on classically known acute inflammation as an ancestral defense reaction, our attention, in accord with many investigators,1,2 has focused on its chronic manifestations. Roughly at the mid of the twentieth century, the inhabitants of the Western World began to present with an increasing incidence of (auto)immune disorders including arthritis, inflammatory bowel disease (IBD), and neurodegenerative phenomena, such as Parkinson’s disease, multiple sclerosis, amyotrophic lateral sclerosis, or Alzheimer’s disease. Suggesting a causative role of expanding civilization, these phenomena have manifested as a full-blown epidemic3 in some developing countries. Subjects attacked by this sort of creeping inflammation typically presented with weight loss, fatigue, and inability to sustain a productive life, deserving the fantasy label of “inflammacitizens”.4 Cutting-edge studies on laboratory markers have recently found5 that the inflammacitizens circulate abnormal levels of a marker named “GlycA”, which closely correlates with other inflammatory indicators and, interestingly, may herald reduction in life expectancy. The contemporary literature on chronic inflammation is abundant and covers variegated aspects of both internal medicine and specialties; we have arbitrarily chosen to succinctly expand on three publications dealing with both general and particular issues related to chronic inflammation.
Bennett et al,2 while covering and integrating the variegated medical, immunological, and psychosocial aspects of the individual’s interaction with the outer world, identified a few capital points listed below on a hypothetical anti-inflammatory policy, in close accordance with our recently published review:6
- Prioritize policies designed to reduce the stress impact on lifestyle, as stress poses lymphocytes into a pro-inflammatory economy
- Bear in mind the multi-systemic nature of pathobiological events, whereby failure of one anti-inflammatory barrier can easily trigger malfunction of other systems in a waterfall fashion
- Organized teams of specialists should treat a multisystem event such as inflammation
- Better food choices and physical activity should be prioritized
- Stress management skills to activate the para-sympathetic nerve system should be actively taught
- Sleep quality should be at the top of caretakers’ attention.
The multidisciplinary approaches mentioned above revealed7 that a sample student population undergoing stressful examination or job competition may develop a strong inflammation-like response, witnessed by increased blood levels of conventional markers, such as erythrocyte sedimentation rate and C-reactive protein. Such puzzling finding has recently received a tentative behavioral answer: in this context, inflammation could act as an ancestral response to the threat to disrupt an individual’s integrity, an equivalent to the ancestral fight between mankind and wilderness. Under these circumstances, inflammation could be considered as a novelty in neuroscience. In this context, in recent years the analysis of the gut-brain axis is focusing on the role of microbiota as its mediator. This axis would be bidirectional, being involved in the pathogenesis of both primitively intestinal diseases such as irritable bowel syndrome (IBS) and IBD, and of primitively neurological diseases such as Parkinson’s disease, Alzheimer’s disease, anxiety, depression, and autism. The common factor in these pathologies appears to be dysbiosis, that is an alteration of the number and function of the species of the microbiome. A reduction of bacterial butyrate-producing species results in a reduction of this short chain fatty acid (SCFA) which is a lipid with local and systemic anti-inflammatory characteristics.8
Finally, given the position of the gut as a barrier organ, inflammation cannot be ignored in the field of gastroenterology. Gastroenterologists repeatedly failed in their search for the causes of IBS. Threshold levels of inflammatory cells in the colonic biopsies have recently been found, suggesting that intestinal inflammation is a continuum from mild IBS to full-blown IBD. While this has been considered by some as a leap forward in taxonomy, others have protested again the total confusion that now seems to warrant immune suppression in an otherwise functional condition.9
The latter observation has advised us (as gastroenterologists) to further review data banks, and to test whether this point could enrich our concepts of inflammation.
Study concept
Henceforth, we shall: 1) dissect a few clinical conditions in which a dominant role of inflammation has been recently reiterated; 2) discuss the microbiome as the missing link between gene and inflammation; 3) discuss the theoretical and scientific consequences regarding disease classification; and 4) assess whether the progress described above can warrant a refinement of the current therapeutic options.
Articles published in English, on the inflammation as dominant causative factor in several fields of gastroenterology, were identified through MEDLINE, SCOPUS, ISI-Web of Knowledge, and EMBASE searches. The search of all studies published from January 1, 1988 to November 30, 2018 was conducted. The following medical subject headings were used: “inflammation” associated with one among “Irritable bowel syndrome”, colonic adenomas”, “colorectal cancer”, “gluten sensitivity”, “microbiome”, “microbiota”, “gut”, “bowel”, “IBD”, “inflammatory bowel disease”, “ulcerative colitis”, “Crohn’s disease”, “Helicobacter pylori”, “fecal microbiota transplantation”. Reference lists from published articles were also employed. Titles of the publications and their abstracts were scanned to eliminate duplicates and irrelevant articles. Two authors (G.C.A. and D.G.R.) independently reviewed the literature search results and selected relevant studies. The full-text studies were assessed by the two authors to determine whether the inclusion criteria were met.
Recent studies on inflammation in clinical entities
Irritable bowel syndrome
A recent paper (summarizing previous substantive evidence) has confirmed a significant role of an inflammatory-immunological etiopathogenesis of IBS.10 This is supported by two evidences: 1) an increased mast-cell density and activity in the IBS gut correlates with symptoms; 2) signs and symptoms of persisting IBS are known to often follow episodes of gastroenteritis.11,12 Indications to “eubiotic” treatments will be discussed briefly in “Classification and theoretic issues”).
Role of inflammatory transcripts in colonic adenomas and malignancy13
Evidence of the existence of inflammatory signals in colonic polyps and their resolution in response to NSAIDs have long been investigated by researchers. Recent studies have now pinpointed to the fact that arachidonic acid metabolism (cyclooxygenase-2 [COX-2] enzymes) link prostaglandin metabolism and cancer, promoting angiogenesis, cell growth, and invasion.14 Clinically, inhibition of the COX-2 pathways, and detection of prostaglandin signals in circulating cells may work as therapeutics and non-invasive diagnostic markers, respectively.
The increasing prevalence of gluten sensitivity has become a socio economic issue
The spectrum of gluten sensitivity disorders seems now to span from traditional celiac disease, to atypical sero-negative celiac allergy, up to ill-defined neurological and even autism-like manifestations.15 Typical elements of gut inflammatory onset (increased immune cell infiltrates and enhanced membrane permeability) seem to act as a continuum among the various forms of gluten reaction all the way to IBD.16,17
Linking the genetic message level with inflammation: the microbiome
Dysbiosis as a common etiologic factor of several diseases
Individuals live in a harmonious association with the gut microbiome, with people giving the ideal condition to the microbiota to thrive, and microscopic organisms consequently breaking down the nourishment sugars to short unsaturated fats, integrating certain nutrients and breaking down dietary oxalates.18
A condition of alteration of the gut microbiome is called “dysbiosis”, which in turn leads to an alteration of the immune system homeostasis.
This condition is frequent in metabolic disorders, obesity, IBS, IBD, allergy, asthma, cardiovascular diseases, and infections (Figure 1).19,20
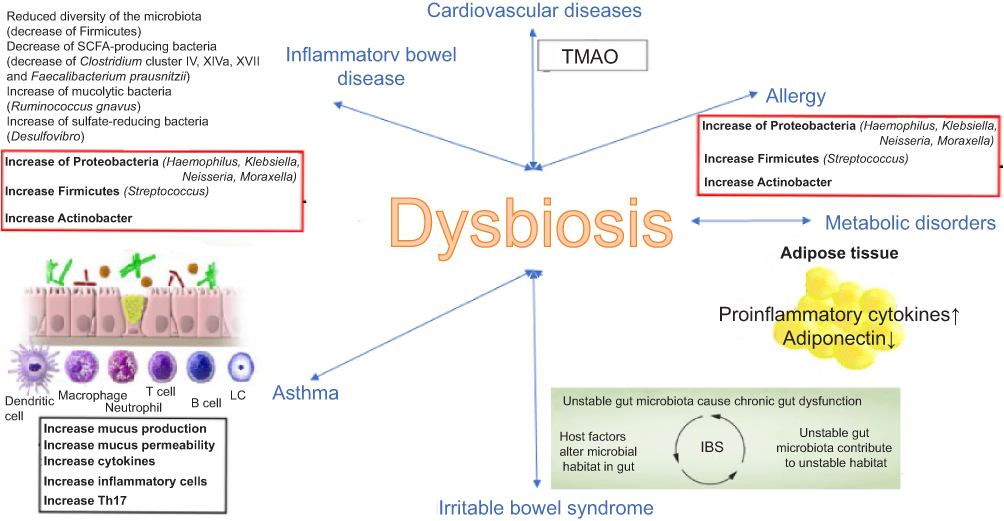 |
Figure 1 Mechanisms of association between dysbiosis and various diseases. |
There are gaps in knowledge concerning the interaction between the microbiome and the host as well as on the consequences of this interaction.
Recent technological advancements in metabolomics have vastly improved the sensitivity and accuracy at which metabolites can be detected and characterized, and metabolomic profiling might help answer these questions.21
The case of IBD
In the bowel are present most of the immune cells of the body, and only a single layer of columnar epithelial cells physically divides the numerous luminal antigens from the mucosal immune system. Microbial colonization importantly affects the immune system homeostasis. Anomalous feedback between gut microbiome and the immune system in the bowel mucosa has been recognized as the central alteration that prompts chronic phlogosis.22
IBD is believed to be incited by an unduly aggressive pattern of adaptive immune response mediated by T cells to a subset of commensal enteric bacteria in genetically susceptible hosts. Several studies have demonstrated that the activity phases of IBD are strongly associated with an overall increased prevalence of proteobacteria23 accompanied by a drop in intestinal richness involving the phylum Firmicutes,24 and the species Enterobacteriaceae, Bacteroidales, and Clostridiales25 of Clades IV and XIVa.25
A lower number of butyrate-producing bacteria, such as Clostridiales species, accounts for the reported reduction in unsaturated fats in the stools of IBD patients. Butyrate serves as a major source of energy for colonic epithelial cells and as an inhibitor of pro-inflammatory cytokine expression in the gut mucosa, through a mechanism that involves hyper-acetylation of histones and suppression of the nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) signaling.26 Butyrate reinforces mucosal barrier function by inducing production of mucin and antimicrobial peptides, and by increasing epithelial barrier integrity through the expression of tight junction proteins.27 Studies suggested a link between IBD and a decreased prevalence of butyrate-producing Faecalibacterium prausnitzii (firmicute), belonging to Clostridia cluster IV species.28
Even though dysbiosis is firmly thought to be linked with IBD, it is not clear whether this is a cause or a result of the inflammatory process. Hence, there is a need for prospective studies in healthy susceptible first-degree relatives of IBD patients, evaluating genetic, environmental and microbial factors, to clarify whether changes in the microbial communities precede or follow IBD onset.
The role of the diet
Differences in diet seem to be a factor that could impact synthesis of butyrate in the gut, impairing the composition of microbiome.
Compared with healthy controls, Crohn’s disease (CD) patients intake less vegetable and fruit, and more processed bread (white bread), and sugar foods. Patients with CD and a high number of butyrate-producing bacteria had a larger intake of nuts than those with a reduction in butyrate-producing bacteria. CD patients with a reduction in butyrate-producing bacteria intake less amount of certain foods containing fibers such as whole wheat, cereals, fruits, vegetables, and nuts, and increased intake of high non-whole wheat and sugar-rich foods. This proposes that microbiome modulation, either accomplished by probiotics or by prebiotics, may give increase more efficiently butyrate levels in the bowel respect to mere intake of butyrate itself.29
Prebiotics are non-edible sugars, for example, fiber and starch, which are broken down in the jejunum and the ileum by local bacteria. So, dietary fiber is a wellspring of microbiota available starches. Wellsprings of prebiotics incorporate inulins, soybeans, whole grain and wheat, crude oats, and non-edible oligosaccharides, for example, fructooligosaccharides (FOS), polydextrose, fructans, arabinooligosaccharides (AOS), xylooligosaccharides (XOS), and galactooligosaccharides (GOS).30 Non-absorbable sugar-rich diets (ie, whole wheat and grain) are connected to an expansion in gut Lactobacilli and Bifidobacteria.31 The positive impact of prebiotics on the bowel is thought to include a higher production of SCFAs.30
Polyphenols of the diet (ie, flavonols, catechins, proanthocyanidins, flavones, phenolic acids, and anthocyanins) are reported for their cancer prevention agent properties. Typical nourishments with rich polyphenol content incorporate natural products, vegetables, seeds, cocoa items, wine, and tea.30,32
In a recent meta-analysis curcumin, a biologically active phytochemical, with its unique anti-oxidant and anti-inflammatory activities and ability to modulate gut microbiota, resulted a potentially useful addition to our armamentarium of agents for IBS.33 It also appeared safe and well tolerated, with no adverse events reported in the available trials.
Cutting-edge research conducted in the last few months has placed a doubly committed attention on the role played by intestinal bacteria in causing or perpetuating IBD: bioremediation has been addressed by specifically investigating the effects of 5-aminosalicylic acid (5-ASA) compounds on gut microflora diversity and composition and in relation with the clinical response.34 It was found that 5-ASA exerted a beneficial effect on these parameters, favoring the establishment of a remission picture. Closer insights revealed that mucosal 5-ASA concentrations were positively associated with bacterial diversity; high mucosal drug concentrations correlated with a drop of harmful species concentrations (Proteobacteria) and increased abundance of favorable species such as Fecalibacterium.
Taken together, these data so far indicated that diet composition and use of drugs of the 5-ASA family can be effective to downregulate gastrointestinal inflammation in various disease contexts.
The gene-cytokine level
It is well known that genetic predisposition is involved in the onset of IBD. To date, over 230 genetic loci have been identified as associated with an increased risk of developing IBD. Most of the genes involved in the pathogenesis of IBD have a defense role against intestinal bacteria, including a protein related to autophagy 16–1 (ATG16L1), the IL-10 receptor and the receptor of the IL-23.35
The nucleotide-binding oligomerization domain-like receptors are a group of intracellular, highly conserved, receptors that recognize a signal of infections, through inflammasome cascades, NF-κB, and mitogen-activated protein kinase signaling.36,37 In patients with IBD there is a defect in recognition of infections.38 So, innate immune response would be involved in the pathogenesis of CD. Furthermore, a less level of tolerance to the normal bowel microbiome seems to play a pivotal role in IBD etiology.39 Autophagy is also involved, through ATG16L1 gene, favoring the entrance of the bacteria in the cells (Table 1).40
 |
Table 1 Main genes involved in inflammatory bowel diseases |
Classification and theoretic issues
The case of Helicobacter pylori (H. pylori)
The presence of an inflammatory adjunct to any pathogen can break the boundaries of the niche where it thrived, hence allowing trans-organ pathogenicity. This is the case of H. pylori. In addition to being unanimously recognized as the cause of gastritis and peptic ulcer (PU) disease,41 this bacterium, from its gastric niche, through the formation of inflammatory immune-toxin complexes has been shown to be involved in vasculitides affecting as disparate districts as the nervous system and the cardiac vessels.42 Recently, an updated review on H. pylori as one major factor triggering sporadic (non-genetic) Parkinson’s disease has been published.43 The extra-gastric manifestations of H. pylori may also be caused by a chronic, low-grade inflammation, induced by this bacterium, resulting in an increase in cytokines levels such as tumor necrosis factor (TNF)-α, IL-1, IL-6, IL-8, γ interferon and soluble molecules like intercellular adhesion molecule (ICAM-1) and vascular cell adhesion molecule (VCAM-1).44 In an interesting study,45 high-sensitivity assay C-reactive protein resulted higher in H. pylori-infected PU patients than in H. pylori-infected asymptomatic carriers or in uninfected subjects, likely through a cytokines-mediated mechanism (IL-1, IL-6, IL-8, TNF).46,47
Regarding the extra-gastric manifestation of H. pylori infection, for example, it is assumed that the gastric colonization with this bacterium is related with a higher probability of hyperemesis gravidarum, with a pooled OR of 1.3 (p<0.001).48 The changes of the woman body during pregnancy include a diminished cell-mediated immunity, subsequently making her progressively inclined to infections, such as a reactivation of a previous H. pylori infection.49 The gastric inflammation induced by H. pylori increases the typical nausea and vomiting in hyperemesis gravidarum.50 The finding of H. pylori in a woman suffering from hyperemesis gravidarum gives to this patient the possibility (treating the infection after the pregnancy) to prevent her symptoms in a future new pregnancy.
The epithelial barrier
With a wall formed by an epithelium and a lymphocyte-rich sub-epithelium, and a lumen indwelled by billions of germs (the microbiome), the gut is now classified as a barrier organ:51 the integrity of the latter function depends on the maintenance of the sealing status of the mucosa and the submucosa. The equilibrium of the system depends on the regulation of a subliminal lingering inflammation: infections, drugs, tobacco, psychological stress52 are but a few of the agents that can alter the sealing status mentioned above and let the luminal contents53 arouse the indwelling lymphocytes to tip the balance. It is not difficult to envisage that perturbation of this machinery makes a continuum of severity gradient ranking from (simple) IBS to the severest IBD, with consequent erosions of epithelial barrier being not always welcomed.9 The practical consequences of these etiopathogenetic mechanisms on therapeutic options will be dissected below.
Implications: treat-to-target strategies?
The concept of treat-to-target strategies was borrowed from rheumatologists,54 foreseeing that the natural history of rheumatic patients could be changed if they would be treated with strategies focused to identify pathological targets (for example, cartilage erosions) than toward generic clinical goals such as physical performance or well-being. The identification of pathological targets in IBD (for example, calprotectin under 250 µg/g or mucosal healing)55,56 favored the translation to gastroenterology of this rheumatologic frame of mind.
Several hints for a role of infectious pathogens in IBS warrant the pragmatic use of cycles of antibiotics followed by repopulation with “eubiota”; the evidence of inflammatory transcripts in gut polyps suggests the potential usefulness of the treatments with NSAIDs; suspicions of an enlarged pathogenic role of gluten proteins provide indications for tentative exclusion of gluten from the diets of otherwise seronegative individuals.
On the other hand, the search for inflammatory markers in colonic disorders can easily raise concerns regarding doctor–patient relationship. For instance, prescription of the anti-inflammatory aminosalycilic acid (5-ASA) can make an IBS patient believe that the diagnosis of full-blown IBD had been originally missed; moreover, UC patients, if offered to participate in a trial with the anti-STAT molecule tofacitinib,57 may suspect being offered a modern expensive version of a cortisone drug.
Finally, the discovery of the pivotal role of the microbiota in inflammatory diseases brought about an increasing number of studies regarding the efficacy of fecal microbiota transplantation (FMT) in intestinal and extra-intestinal diseases.58 In the randomized controlled trial conducted by Moayyedi et al,59 the authors found that early diagnosis of UC might result in better outcomes after FMT. This suggests a potential window of opportunity for FMT after diagnosis of UC. Accordingly, Paramsothy et al60 suggested that less severe grades of endoscopic inflammation may be a potential predictor of FMT response. Moreover, the increase of Clostridium clusters IV and XVIII and the high microbial diversity were identified as predictors of FMT efficacy. Conversely, the presence of Sutterella spp. and Fusobacterium spp. was associated with nonresponse to FMT.
Concluding remarks
In the environment of a “barrier organ” like the gut, inflammation can easily acquire specific significance. Potent scrutiny by modern techniques tends to blur the limits originally established between functional and organic disorders: mapping of the inflammatory patterns tends rather to delineate a disease continuum with an impact on therapeutic choices.61
Study highlights
What is current knowledge
The IBDs have been known for a century as inflammatory disorders hitting the gut at variable frequencies, and triggered by undefined factors, although an erratic response to antibiotics has at times refueled the quest for infectious origin. Prevalently positive results achieved by immune suppressive strategies have continued to fuel interest on the indications for steroids, then for immune suppression, culminated in the use of anti-cytokine monoclonals.
What is new here
The early decades of the twentieth century have witnessed an epidemic-like expansion of the IBDs hitting mostly developing countries and/or migrant populations from low-income to high-income areas of the world. Dissection of the elements in this apparently IBD-triggering Western life drew attention on factors such as diet (turned to a high elaborated protein and low fiber content); competitive work often carried out on night-shift, noise and artificial light pollution as hallmarks of a city life as opposed to often deserted countryside.62 The variegated etiologic array of IBD, wherein concrete factors such as genetics and diet mix up with the dynamics of psychology, namely working rhythms, noise, and artificial lights, is far from being understood in an organic way. The main points in the title (eg, the socio economic factors of IBD) begin here to shape up, heralding the final choral stage where the appearance on the scene of the microbiome (sub-title: what is new here) opens new perspectives in this field.
Inflammation was soon recognized as a protagonist, with at least a couple of novelties. It manifested as a creeping phenomenon lacking the known cardinal signs of the acute inflammatory bouts of the past century, and became the obviously leading pathology of hyperimmune diseases, ranging from rheumatoid arthritis to degenerative neuropathies.
IBD was easily positioned among this “modern” disease pattern, receiving a sort of “mark of the times” hitherto not clearly evaluated.
Yet, the specific variable that contributed to make IBD a disorder of the times was the microbiome. This huge bug colonic population is an example of mutual synergy. While the gut provides a uniquely suited environment, the microbiome components exert multiplex functions including inhibition of pathogenic bacteria, absorption of nutrients, regulation of the intestinal immune system, and control of cell renewal.
The key point is that the microbiome is a functional signal mediator, with a recorder end tuned to the environment, and an effector terminal capable to translate outside signals into modulating actions directed to the gut immune response. In this way, the immune gut cell environment is made to change accordingly and synchronously with the outside where diet components can be the primary drivers of change. Having set that the intestinal immune environment can preferentially respond to the diet through the “transponder” action of the microbiome, we can say that gut inflammation may chiefly respond to socio economic factors of which the diet is one of the primary probes. Connectivity may be the best definition for this novel network set forth by the microbiome.
This novel frame of mind offers therefore at least two materialized targets to work on (feeding habits and working rules), and calls on a huge number of social categories to lend their help and know-how toward a changing pathway. Thus, basic science knowledge and experience from real life are conducting us to merge with the holistic visions brought about by those interested in science forecast.63
Compliance with ethical standards
Informed consent was not necessary given the nature of the article.
Disclosure
The authors declare that they have no conflicts of interest.
References
1. Ribaldone DG, Pellicano R, Actis GC. Inflammation: a highly conserved Janus-like phenomenon – a gastroenterologist’ perspective. J Mol Med. 2018;96:861–871. doi:10.1007/s00109-018-1668-z
2. Bennett JM, Reeves G, Billman GE, Sturmberg JP. Inflammation–nature’s way to efficiently respond to all types of challenges: implications for understanding and managing “the epidemic” of chronic diseases. Front Med. 2018;5:316. doi:10.3389/fmed.2018.00316
3. Malekzadeh MM, Vahedi H, Gohari K, et al. Emerging epidemic of inflammatory bowel disease in a middle income country: a nation-wide study from Iran. Arch Iran Med. 2016;19:2–15. doi:10.161901/AIM.003
4. Aller MA, Arias N, Fuentes-Julian S, et al. Coupling inflammation with evo-devo. Med Hypotheses. 2012;78:721–731. doi:10.1016/j.mehy.2012.02.018
5. Gruppen EG, Connelly MA, Sluiter WJ, Bakker SJL, Dullaart RPF. Higher plasma GlycA, a novel pro-inflammatory glycoprotein biomarker, is associated with reduced life expectancy: the PREVEND study. Clin Chim Acta. 2018;488:7–12. doi:10.1016/j.cca.2018.10.029
6. Ribaldone DG, Pellicano R, Actis GC. Pathogenesis of IBD: basic science in the light of real-world epidemiology. Gastrointestinal Disord. 2019;1:129–146. doi:10.3390/gidisord1010010
7. Miller AH, Raison CL. The role of inflammation in depression: from evolutionary imperative to modern treatment target. Nat Rev Immunol. 2016;16:22–34. doi:10.1038/nri.2015.5
8. Russo R, Cristiano C, Avagliano C, et al. Gut-brain axis: role of lipids in the regulation of inflammation, pain and CNS diseases. Curr Med Chem. 2018;25:3930–3952. doi:10.2174/0929867324666170216113756
9. Quigley EM. Overlapping irritable bowel syndrome and IBD: less to this than meets the eye? Therap Adv Gastroenterol. 2016;9:199–212. doi:10.1177/1756283X15621230
10. Ng QX, Soh AYS, Loke W, Lim DY, Yeo W-S. The role of inflammation in irritable bowel syndrome (IBS). J Inflamm Res. 2018;11:345–349. doi:10.2147/JIR.S174982
11. O’Sullivan M, Clayton N, Breslin NP, et al. Increased mast cells in irritable bowel syndrome. Neurogastroenterol Motil. 2000;12:449–457.
12. Thabane M, Kottachchi DT, Marshall JK. Systematic review and meta-analysis: incidence and prognosis of post-infectious IBS. Aliment Pharmacol Ther. 2007;26:535–544. doi:10.1111/j.1365-2036.2007.03399.x
13. Alamro RA, Mustafa M, Al-Asmari A. Inflammatory gene mRNA expression in human peripheral blood and its association with colorectal cancer. J Inflamm Res. 2018;11:351–357. doi:10.2147/JIR.S155507
14. Wang D, Wang H, Brown J, et al. CXCL1 induced by prostaglandin E2 promotes angiogenesis in colorectal cancer. J Exp Med. 2006;203:941–951. doi:10.1084/jem.20052124
15. Alessandria C, Caviglia GP, Campion D, et al. HLA-DQ genotyping, duodenal histology, and response to exclusion diet in autistic children with gastrointestinal symptoms. J Pediatr Gastroenterol Nutr. 2019;69:39–44. [Epub ahead of print]. doi:10.1097/MPG.0000000000002293
16. Barmeyer C, Schumann M, Meyer T, Zielinski C. Long-term response to gluten-free diet as evidence for non-celiac wheat sensitivity in one third of patients with diarrhea dominant and mixed-type irritable bowel syndrome. Int J Colorectal Dis. 2017;32:29–39. doi:10.1007/s00384-016-2663-x
17. Pietzak M. Celiac disease, wheat allergy, and gluten sensitivity: when gluten free is not a fad. JPEN. 2012;36(Suppl 1):68S–75S. doi:10.1177/0148607111426276
18. Vernia P, Gnaedinger A, Hauck W, Breuer RI. Organic anions and the diarrhea of inflammatory bowel disease. Dig Dis Sci. 1988;33:1353–1358. doi:10.1007/BF01536987
19. Carding S, Verbeke K, Vipond DT, et al. Dysbiosis of the gut microbiota in disease. Microb Ecol Health Dis. 2015;26:26191.
20. Campion D, Ponzo P, Alessandria C, Saracco GM, Balzola F. The role of microbiota in autism spectrum disorders. Minerva Gastroenterol Dietol. 2018;64:333–350. doi:10.23736/S1121-421X.18.02493-5
21. Du C, Zhang B, He Y, et al. Biological effect of aqueous C60 aggregates on Scenedesmus obliquus revealed by transcriptomics and non-targeted metabolomics. J Hazard Mater. 2017;324:221–229. doi:10.1016/j.jhazmat.2016.10.052
22. Asquith M, Powrie F. An innately dangerous balancing act: intestinal homeostasis, inflammation, and colitis-associated cancer. J Exp Med. 2010;207:1573–1577. doi:10.1084/jem.20101330
23. Baumgart M, Dogan B, Rishniw M, et al. Culture independent analysis of ileal mucosa reveals a selective increase in invasive Escherichia coli of novel phylogeny relative to depletion of Clostridiales in Crohn’s disease involving the ileum. Isme J. 2007;1:403–418. doi:10.1038/ismej.2007.39
24. Frank DN, St Amand AL, Feldman RA, Boedeker EC, Harpaz N, Pace NR. Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc Natl Acad Sci USA. 2007;104:13780–13785. doi:10.1073/pnas.0706625104
25. Gevers D, Kugathasan S, Denson LA, et al. The treatment-naive microbiome in new-onset Crohn’s disease. Cell Host Microbe. 2014;15:382–392. doi:10.1016/j.chom.2014.02.005
26. Segain J, De Blétière DR, Bourreillea A, et al. Butyrate inhibits inflammatory responses through NFkappaB inhibition: implications for Crohn’s disease. Gut. 2000;47:397–403. doi:10.1136/gut.47.1.14
27. Vanhoutvin SA, Troost FJ, Hamer HM, et al. Butyrate-induced transcriptional changes in human colonic mucosa. PLoS One. 2009;4:e6759. doi:10.1371/journal.pone.0006759
28. Sokol H, Seksik P, Furet JP, et al. Low counts of faecalibacterium prausnitzii in colitis microbiota. Inflamm Bowel Dis. 2009;15:1183–1189. doi:10.1002/ibd.20903
29. Laserna-Mendieta EJ, Clooney AG, Carretero-Gomez JF. Determinants of reduced genetic capacity for butyrate synthesis by the gut microbiome in Crohn’s disease and ulcerative colitis. J Crohns Colitis. 2018;12:204–216. doi:10.1093/ecco-jcc/jjx137
30. Singh RK, Chang HW, Yan D, et al. Influence of diet on the gut microbiome and implications for human health. J Transl Med. 2017;15:73. doi:10.1186/s12967-017-1175-y
31. Carvalho-Wells AL, Helmolz K, Nodet C, et al. Determination of the in vivo prebiotic potential of a maize-based whole grain breakfast cereal: a human feeding study. Br J Nutr. 2010;104:1353–1356. doi:10.1017/S0007114510002084
32. Cuervo A, Valdés L, Salazar N, et al. Pilot study of diet and microbiota: interactive associations of fibers and polyphenols with human intestinal bacteria. J Agric Food Chem. 2014;62:5330–5336. doi:10.1021/jf501546a
33. Ng QX, Soh AY, Loke W, Venkatanarayanan N, Lim D, Yeo W-S. A meta-analysis of the clinical use of curcumin for irritable bowel syndrome (IBS). J Clin Med. 2018;7:298. doi:10.3390/jcm7100298
34. Olaisen M, Spigset O, Flatberg A, et al. Mucosal 5-aminosalicylic acid concentration, drug formulation and mucosal microbiome in patients with quiescent ulcerative colitis. Aliment Pharmacol Ther. 2019;49:1301–1313. doi:10.1111/apt.15227
35. Jostins L, Ripke S, Weersma RK, et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature. 2012;491:119–124. doi:10.1038/nature11582
36. Rubino SJ, Selvanantham T, Girardin SE, Philpott DJ. Nod-like receptors in the control of intestinal inflammation. Curr Opin Immunol. 2012;24:398–404. doi:10.1016/j.coi.2012.04.010
37. Girardin SE, Boneca IG, Viala J, et al. Nod2 is a general sensor of peptidoglycan through muramyl dipeptide (MDP) detection. J Biol Chem. 2003;278:8869–8872. doi:10.1074/jbc.C200651200
38. Lesage S, Zouali H, Cezard JP, et al. CARD15/NOD2 mutational analysis and genotype-phenotype correlation in 612 patients with inflammatory bowel disease. Am J Hum Genet. 2002;70:845–857. doi:10.1086/340449
39. Kobayashi KS, Chamaillard M, Ogura Y, et al. Nod2-dependent regulation of innate and adaptive immunity in the intestinal tract. Science. 2005;307:731–734. doi:10.1126/science.1104911
40. Travassos LH, Carneiro LA, Ramjeet M, et al. Nod1 and Nod2 direct autophagy by recruiting ATG16L1 to the plasma membrane at the site of bacterial entry. Nat Immunol. 2010;11:55–62. doi:10.1038/ni.1823
41. Pellicano R, Ribaldone DG, Fagoonee S, Astegiano M, Saracco GM, Mégraud F. A 2016 panorama of H Pylori infection: key messages for clinicians. Panminerva Med. 2016;58:304–317.
42. Lee M, Baek H, Park JS, et al. Current Helicobacter pylori infection is significantly associated with subclinical coronary ATHEROSCLEROSIS in healthy subjects: a cross-sectional study. PLoS One. 2018;13:e0193646. doi:10.1371/journal.pone.0193646
43. McGee DJ, Lu XK, Disbrow EA. Stomaching the possibility of a pathogenic role for H pylori in Parkinson’s disease. J Parkinson’s Dis. 2018;8:367–374. doi:10.3233/JPD-181327
44. Ribaldone DG, Fagoonee S, Hickman I, Altruda F, Saracco GM, Pellicano R. Helicobacter pylori infection and ischemic heart disease: could experimental data lead to clinical studies? Minerva Cardioangiol. 2016;64:686–696.
45. Jafarzadeh A, Hassanshahi GH, Nemati M. Serum levels of high-sensitivity C-reactive protein (hs-CRP) in Helicobacter pylori-infected peptic ulcer patients and its association with bacterial CagA virulence factor. Dig Dis Sci. 2009;54:2612–2616. doi:10.1007/s10620-008-0686-z
46. Vermeire S, Van Assche G, Rutgeerts P. C-reactive protein as a marker for inflammatory bowel disease. Inflamm Bowel Dis. 2004;10:661–665. doi:10.1097/00054725-200409000-00026
47. Mehmet N, Refik M, Harputluoglu M, et al. Serum and gastric fluid levels of cytokines and nitrates in gastric diseases infected with helicobacter pylori. New Microbiol. 2004;27:139–148.
48. Ng QX, Venkatanarayanan N, De Deyn MLZQ, et al. A meta-analysis of the association between Helicobacter pylori (H. pylori) infection and hyperemesis gravidarum. Helicobacter. 2018;23:e12455. doi:10.1111/hel.2018.23.issue-1
49. Chang J, Streitman D. Physiologic adaptations to pregnancy. Neurol Clin. 2012;30:
50. Mansour G, Nashaat E. Role of helicobacter pylori in the pathogenesis of hyperemesis gravidarum. Arch Gynecol Obstet. 2010;284:
51. Zhu TH, Zhu TR, Tran KA, Sivamani RK, Shi VY. Epithelial barrier dysfunction in atopic dermatitis: a skin-gut-lung model linking microbiome alteration and immune dysregulation. Br J Dermatol. 2018;179:570–581. doi:10.1111/bjd.16734
52. Actis GC. The changing face of IBD: etiology, physiopathology, epidemiology. Ann Colorectal Res. 2016;4:e32942.
53. Barko PC, McMichael MA, Swanson KS, Williams DA. The gastrointestinal microbiome: a review. J Vet Intern Med. 2018;32:9–25. doi:10.1111/jvim.14875
54. Allen PB, Olivera P, Emery P, et al. Review article: moving towards common therapeutic goals in Crohn’s disease and rheumatoid arthritis. Aliment Pharmacol Ther. 2017;45:1058–1072. doi:10.1111/apt.13995
55. Colombel JF, Panaccione R, Bossuyt P, et al. Effect of tight control management on Crohn’s disease (CALM): a multicentre, randomised, controlled phase 3 trial. Lancet. 2018;390:2779–2789. doi:10.1016/S0140-6736(17)32641-7
56. Caviglia GP, Ribaldone DG, Rosso C, Saracco GM, Astegiano M, Pellicano R. Fecal calprotectin: beyond intestinal organic diseases. Panminerva Med. 2018;60:29–34. doi:10.23736/S0031-0808.18.03405-5
57. Tsai HH. Editorial: tofacitinib and Biologics for moderate-to-severe ulcerative colitis – what is best in class? Aliment Pharmacol Ther. 2018;47:539–540. doi:10.1111/apt.14480
58. Bibbò S, Ianiro G, Gasbarrini A, Cammarota G. Fecal microbiota transplantation: past, present and future perspectives. Minerva Gastroenterol Dietol. 2017;63:420–430. doi:10.23736/S1121-421X.17.02374-1
59. Moayyedi P, Surette MG, Kim PT, et al. Fecal microbiota transplantation induces remission in patients with active ulcerative colitis in a randomized controlled trial. Gastroenterology. 2015;149:102–9.e6. doi:10.1053/j.gastro.2015.04.001
60. Paramsothy S, Kamm MA, Kaakoush NO, et al. Multidonor intensive faecal microbiota transplantation for active ulcerative colitis: a randomised placebo-controlled trial. Lancet. 2017;389:1218–1228. doi:10.1016/S0140-6736(17)30182-4
61. Actis GC. Inflammatory bowel disease 2018: consistency and controversy. Immunome Res. 2018;14:2. doi:10.4172/1745-7580
62. Ouyang JQ, Isaksson C, Schmidt C, Hutton P, Bonier F, Dominoni D. A new framework for urban ecology: an integration of proximate and ultimate responses to anthropogenic change. Integr Comp Biol. 2018;58:915–918. doi:10.1093/icb/icy110
63. Fiocchi C. Inflammatory bowel disease: complexity and variability need integration. Front Med (Lausanne). 2018;5:75. doi:10.3389/fmed.2018.00075
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.