Back to Journals » Journal of Inflammation Research » Volume 11
Inflammasomes: Pandora’s box for sepsis
Authors Kumar V ![]()
Received 23 June 2018
Accepted for publication 11 July 2018
Published 11 December 2018 Volume 2018:11 Pages 477—502
DOI https://doi.org/10.2147/JIR.S178084
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Ning Quan
Video abstract presented by Vijay Kumar.
Views: 1796
Vijay Kumar1,2
1Children’s Health Queensland Clinical Unit, School of Clinical Medicine, Faculty of Medicine, Mater Research, University of Queensland, Brisbane, Australia; 2School of Biomedical Sciences, Faculty of Medicine, University of Queensland, Brisbane, Australia
Abstract: Sepsis was known to ancient Greeks since the time of great physician Hippocrates (460–377 BC) without exact information regarding its pathogenesis. With time and medical advances, it is now considered as a condition associated with organ dysfunction occurring in the presence of systemic infection as a result of dysregulation of the immune response. Still with this advancement, we are struggling for the development of target-based therapeutic approach for the management of sepsis. The advancement in understanding the immune system and its working has led to novel discoveries in the last 50 years, including different pattern recognition receptors. Inflammasomes are also part of these novel discoveries in the field of immunology which are <20 years old in terms of their first identification. They serve as important cytosolic pattern recognition receptors required for recognizing cytosolic pathogens, and their pathogen-associated molecular patterns play an important role in the pathogenesis of sepsis. The activation of both canonical and non-canonical inflammasome signaling pathways is involved in mounting a proinflammatory immune response via regulating the generation of IL-1β, IL-18, IL-33 cytokines and pyroptosis. In addition to pathogens and their pathogen-associated molecular patterns, death/damage-associated molecular patterns and other proinflammatory molecules involved in the pathogenesis of sepsis affect inflammasomes and vice versa. Thus, the present review is mainly focused on the inflammasomes, their role in the regulation of immune response associated with sepsis, and their targeting as a novel therapeutic approach.
Keywords: inflammasomes, sepsis, cytokines, inflammation, pathogens
Introduction
The word sepsis originated from the Greek word “σηψις” (pronounced as “sipsis”) that is originally used in Greek language for describing deterioration of the animal or vegetable organic matter due to the presence of microbes.1 However, the word sepsis was used in medical literature for the first time by Homer in his poems almost 2,700 years ago and the word was directly derived from the word “sepo” (σηπω) meaning “I rot”.2 The term sepsis was also used by the Greek physician Hippocrates in 430 BC to describe deterioration of the organic matter.1 He is well known for his work in ancient Greek medicine. He introduced the concept of impairment in the body humors or fluids including blood, bile (yellow and black bile), and phlegm as a major cause of the disease. Thereafter, Ibna Sina (also known as Avicenna, AD 980–1037), a Persian philosopher and the father of modern medicine, described sepsis/septicemia as putrefaction of blood and tissues with fever.3 The emergence of classic or modern concept of sepsis happened in the 19th century. Ignaz Semmelweis (a Vienna, Austria-based obstetrician, 1818–1865) is considered as the first physician to provide the modern view of sepsis while studying the cause of death in infants getting puerperal fever (also called childbed fever). He concluded that the childbed fever was caused by the decomposed animal matter that entered their blood system. He is also considered an early pioneer in developing antiseptic techniques.
It was the beginning toward understanding sepsis. In 1914, a German physician called Hugo Schottmüller established for the first time that the presence of infection is the basic requirement for the development of sepsis.3,4 In 1989, a USA-based intensive care unit specialist Roger C Bone (1941–1997) defined sepsis as the invasion of microorganisms and/or their toxins into the bloodstream of patients and induction of the host’s immune response against the microbial invasion called systemic inflammatory response syndrome (SIRS).5,6 This definition of sepsis remained valid for more than two decades. However, in the year 2016, the third consensus on sepsis, which is also called sepsis 3, defined sepsis as a condition of life-threatening organ dysfunction generated due to the dysregulation of the host’s immune response during infection.7 Sepsis 3 also led to the development of quick Sequential Organ Failure Assessment (qSOFA) scoring system for the early identification of simultaneous organ dysfunction in sepsis and challenged the SIRS criteria for sepsis definition because sepsis involves an early activation of both pro- and anti-inflammatory immune responses.8–11 Hence, sepsis 3 emphasized on early diagnosis and quick initiation of treatment during the ongoing process of sepsis to prevent mortality. Sepsis accompanied with the incidence of hypoperfusion and hypotension along with organ dysfunction is considered as severe sepsis,12,13 while septic shock is defined as severe sepsis with profound hypotension that persists during volume resuscitation and needs the administration of vasopressors including epinephrine, norepinephrine, isoproterenol, dobutamine, and so on.10,13,14 Septic shock predisposes the patient towards an increased risk of mortality as compared to sepsis/severe sepsis alone.
Thus, sepsis is a condition of severely dysregulated immune response against a pathogen, where both pronounced and overwhelming activation of innate immunity along with the suppression of conventional T-cell–mediated immune response and an upregulation of regulatory T cells (Tregs) causing sepsis-associated immunosuppression are observed.10,15–22 It is now well established that sepsis is a condition of an aberrant immune response where an initial innate immune activation fails to clear the source of the infection or pathogen, causing the generation of an overwhelming proinflammatory reaction responsible for multiorgan damage or failure (Figure 1). Furthermore, patients surviving the episode of sepsis suffer from long-term immunosuppression, making them prone to develop secondary infections.8,9,23,24
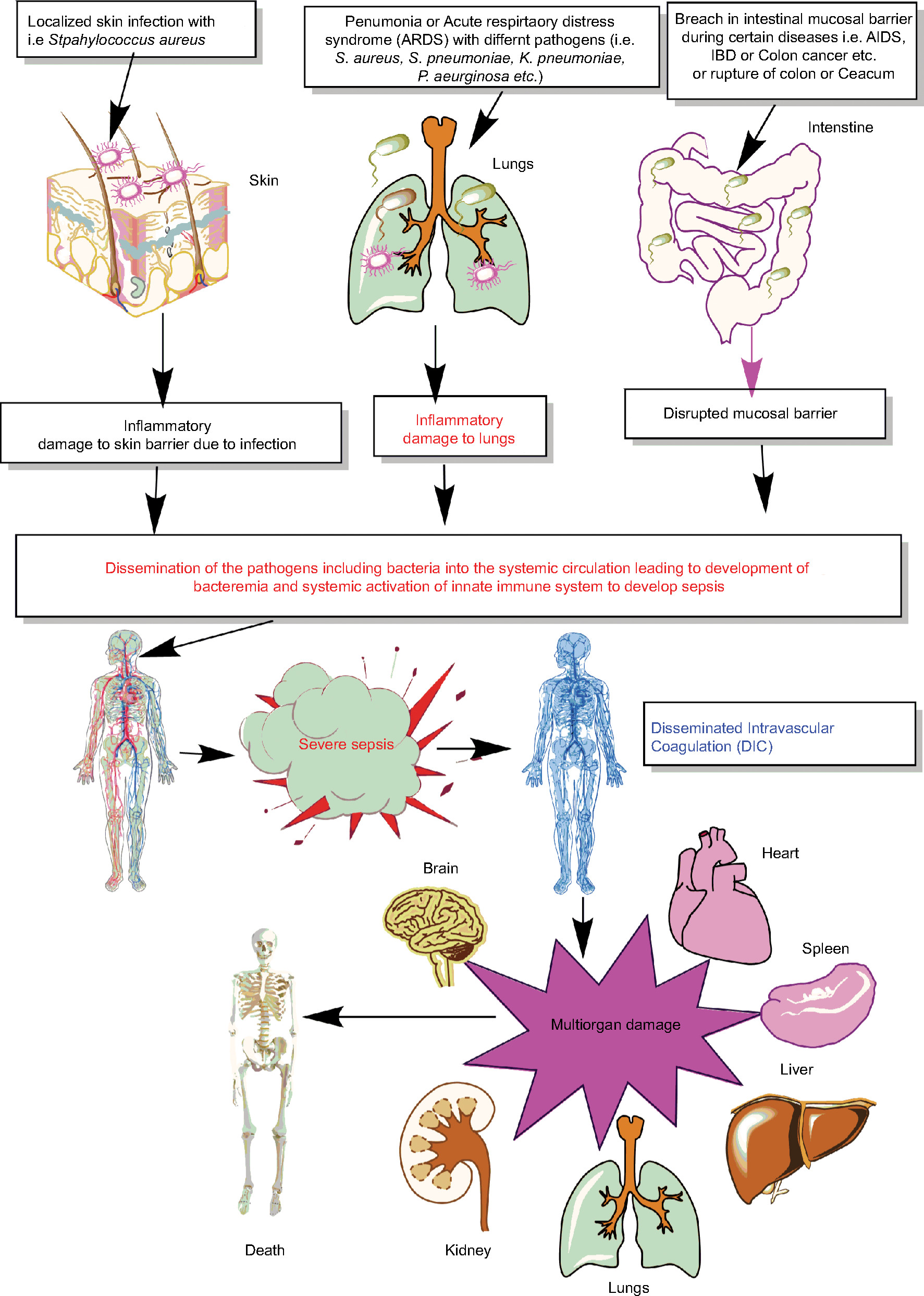 | Figure 1 Schematic representation of breach in host innate immunity due to different bacterial infections and development of bacteremia and sepsis. Notes: Once the bacteremia is developed in the host and the initial control of systemic infection fails, it leads to increased activation of the innate immune response systemically. This exaggerated innate immune response causes MOD or MODS. The development of MODS leads to the ultimate death of the patients. Abbreviations: ARDS, acute respiratory distress syndrome; DIC, disseminated intravascular coagulation; IBD, inflammatory bowel disease; MOD, multiorgan damage; MODS, multiorgan dysfunction syndrome. |
Inflammasomes are the essential components of host’s immune defense mechanisms that regulate the generation of proinflammatory immune response along with the recognition of intracellular pathogens.25–29 However, activation of NLRP3 inflammasome is also shown to exhibit an inhibitory action on protective gastrointestinal immune response against helminthic infections.30 The activation of both the classical/canonical and non-canonical/non-classical inflammasome signaling pathways under diverse inflammatory conditions is shown in Figure 2 and the detailed mechanisms are described elsewhere in detail.31–38 The primary aim of the present review is to highlight the role of inflammasomes in sepsis-associated proinflammatory immune response and its targeting.
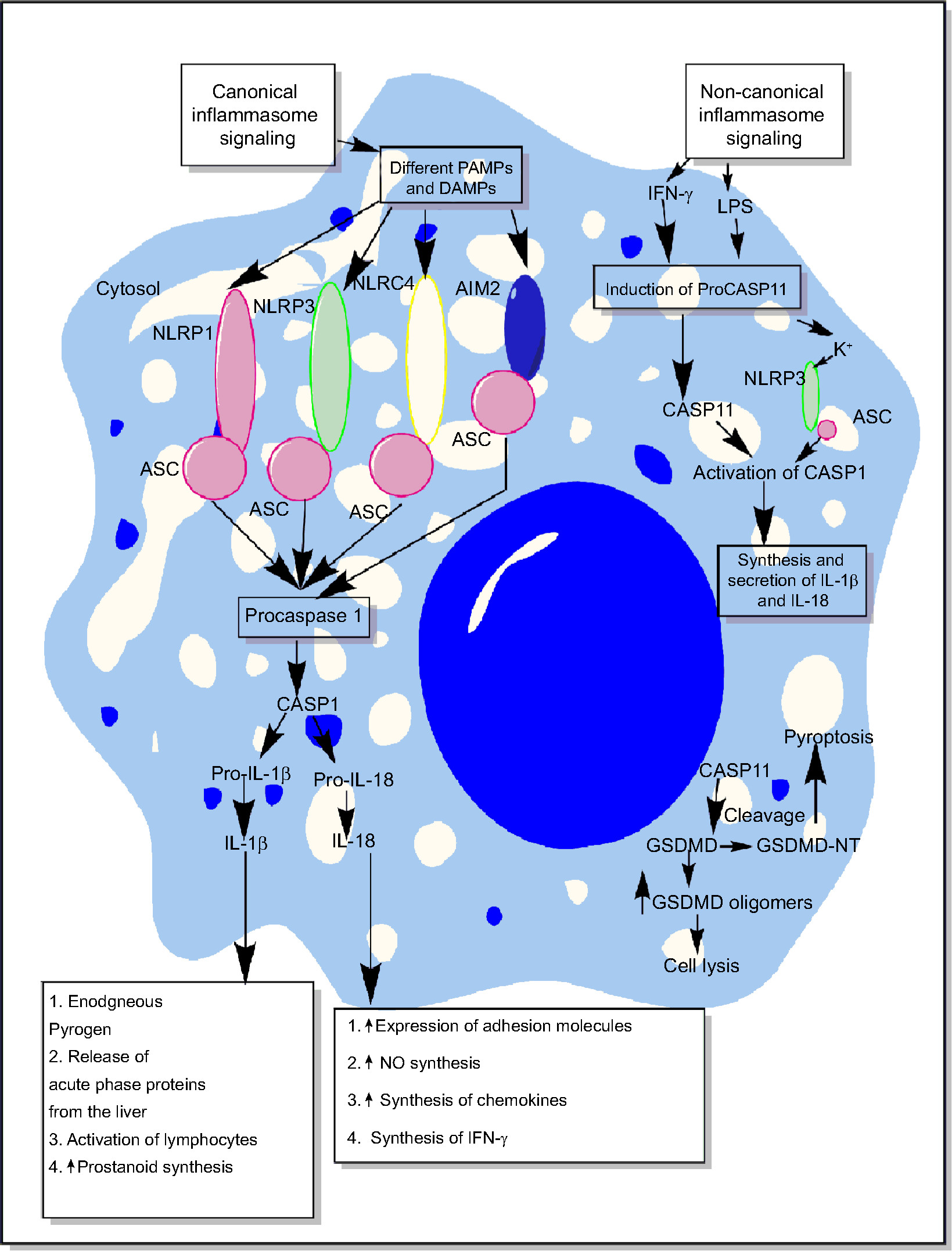 | Figure 2 Schematic representation of canonical and non-canonical inflammasome signaling pathways. Notes: The figure describes both the canonical and non-canonical signaling pathways involved in the activation of different inflammasome components including NLRP1, NLRP3, NLRC4, and AIM2. The canonical inflammasome signaling pathway causes activation of procaspase 1 into mature CASP1 that cleaves immature proIL-1β and proIL-18 into mature IL-1β and IL-18 involved in the generation of inflammatory immune response. The intracellular LPS and type-1 IFNs cause induction of non-canonical signaling in macrophages and DCs via the induction of procaspase 11 that further activates NLRP3 inflammasome by inducing a decrease in cytosolic K+ through different mechanisms described in the text. Additionally, CASP11 cleaves GSDMD into N- and C-terminal components that, upon their cytosolic accumulation, form GSDMD oligomer causing cell death, while GSDMD-NT causes pyroptosis, but it does not cause the death of adjacent cells upon its extracellular release. Detailed mechanism of inflammasome signaling pathway is mentioned elsewhere in the text. Abbreviations: DAMP, death/damage-associated molecular pattern; DCs, dendritic cells; GSDMD, gasdermin D; IFN, interferon; LPS, lipopolysaccharide; PAMP, pathogen-associated molecular pattern. |
Inflammasomes as a regulator of immune homeostasis during infection and inflammation
Inflammasomes were first discovered as a caspase-activating complex comprising mainly caspase-1 (CASP1), caspase-5 (CASP5), Pycard/Asc, and NALP1, a pyrin domain-containing protein sharing structural homology with NODs, in the year 2002 by the research team led by Jurg Tschopp.39 After 16 years of their discovery, they have been studied extensively and are still the topic of research targeting infectious and inflammatory diseases including cancer and neurodegeneration.40–47 Thus, inflammasomes might be playing an important role in the homeostasis of the immune system to maintain a healthy stage within the host. For example, inflammasome-deficient mice harbor an aberrant microbial community that can be dominantly transmissible to healthy animals.48 This alteration in the microbial community due to the deficiency of inflammasomes causes a profound defect in intestinal immunity along with that in distant organs systemically.48 In addition, the AIM2 and NLRP3-deficient New Zealand Black mice exhibit autoimmune phenotype responsible for the development of anti-erythrocyte antibodies and autoimmune hemolytic anemia.49,50 These mice also develop anti-nuclear antibodies typical of systemic lupus erythematosus.49
The recognition of PAMPs or DAMPs involves specific germline-encoded receptors called pattern recognition receptors (PRRs) expressed either on the outer surface of the plasma membrane or intracellular cytoplasmic vesicles including endosomes, lysosome, or endolysosomes.51–53 In comparison to a class of PRRs called Toll-like receptors (TLRs) that are present both on the cell surface and intracellular environment, certain receptors called Nod-like receptors (NLRs), AIM-2 like receptors (ALRs), pyrin and HIN domain-containing family proteins, and cytosolic nucleoside sensors are present only intracellularly and guard against potential pathogens or inflammogens.25,54 These NLRs and ALRs are distinct from other cytosolic PRRs mentioned, as activation of these receptors forms a specific complex via the assembly and activation of a complex called inflammasomes to activate CASP1 and CASP11 (also known as caspase-4).31,39 Only five PRRs including NLRP1, NLRP3, NLRC4, pyrin, and AIM2 are able to form inflammasomes.55,56 Under resting conditions, the inflammasomes are considered to exist in an inhibitory state.57,58 Once activated, they cause conversion of proinflammatory procaspases into their mature form. This inflammasome-mediated activation of CASP1 and CASP11 causes the release of proinflammatory cytokines called IL-1β and IL-18.55,59 Thus, the activation of inflammasomes under diverse conditions causes proinflammatory cell death via several mechanisms including pyroptosis (inflammatory cell death) and necroptosis (a form of lytic inflammatory cell death or a physiological suicide initiated by RIPK3-mediated phosphorylation of MLKL).57,60–63 Necroptosis is involved in the development, inflammation, and disease pathogenesis.64 Hence, via inducing necroptosis or regulating the release of IL-1β and IL-18, inflammasomes play an active role in the process of generation of inflammatory immune response and in the disease pathogenesis, depending on the condition/stimulus.65,66
Inflammasome-mediated release of IL-1β and IL-18 and the maintenance of immune homeostasis
IL-1β and IL-18 are the very first proinflammatory cytokines released by the macrophages and dendritic cells (DCs) upon an encounter with different pathogens.59,67 IL-1β acts as an endogenous pyrogen and is responsible for the induction of fever, release of hepatic acute-phase proteins, activation of lymphocytes (ie, release of IL-2 form T-cells), and an upregulation of prostanoid synthesis.67–69 However, IL-18 does not produce fever or induce prostaglandins (PGs) or prostanoids due to its inability to induce cyclooxygenase-2 (COX-2), but induces inflammation via other mechanisms.70–72 The inability of IL-18 to induce COX-2 activation and, thus, fever can be attributed to the fact that it acts by stimulating p38 mitogen-activated protein (MAP) kinase pathway, not via NF-κB activation.71 In addition, IL-18 can attenuate IL-1beta (an endogenous pyrogen) induced fever.73 Instead, it induces the overexpression of adhesion molecules required for neutrophil infiltration at the site of infection or inflammation and increases nitric oxide (NO) synthesis and chemokines required for neutrophil and T-cell chemotaxis.74 IL-18 was found to induce the synthesis of interferon (IFN)-γ, and thus plays an important role in the polarization of T-cells toward proinflammatory Th1 cells.67,75 IL-18 controls energy homeostasis by suppressing appetite and feed efficiency in mice, as IL-18-/- mice exhibit hyperphagia leading to the development of obesity, and insulin resistance causing an induction of metabolic syndrome phenotype due to a defective STAT3 phosphorylation.76,77 Thus, inflammasome-mediated CASP1 and CASP11 activation controls several immunological mechanisms via mediating the release of these cytokines.
Additionally, inflammasomes have also been implicated in the maintenance of normal and healthy intestinal microbiota.78,79 For example, NLRP6 inflammasome affects the intestinal microbiota in mice via recognizing various low-molecular-weight metabolites including taurine, pinitol, sebacate, and undecanedioate derived from colonic bacteria.37,80,81 Thus, any disruption in the function of inflammasomes in the intestinal immune environment has a potential to induce a dysfunctional immune response that may lead to the development of an environment supportive to the dysbiosis of the gut bacteria into peripheral circulation, leading to the development of bacteremia and sepsis.82–84 Thus, inflammasomes are one of the essential components of vertebrate immunity playing an important role in the defense against pathogens via regulating the generation of inflammatory immune response and other components of immunity required for the host defense.
Inflammasomes in the immunopathogenesis of sepsis
The induction of profound and dysregulated immune response during uncontrolled infections of different origins including bacterial, viral, parasitic, and fungal infections may lead to the development of sepsis. However, sepsis in most cases seems to be clinically associated with the occurrence of an uncontrolled bacterial infection caused either by gram-negative or gram-positive bacteria or both (polymicrobial sepsis). The induction of an effective innate immune response to clear the pathogen requires the identification of pathogens via different PRRs that recognize different PAMPs.85 PAMPs are evolutionarily conserved molecular structures expressed by different pathogens, but are not synthesized or expressed by the host cells.86 These PRRs can be classified into phagocytic PRRs and sensor PRRs. The phagocytic PRRs directly bind to the pathogens and prepare them for the process of phagocytosis via opsonization.87,88 On the other hand, sensor PRRs include different TLRs (TLR1–TLR13) that are present both extracellularly on the plasma membrane or intracellularly in endosomes or lysosomes, NLRs, and ALRs.89–98 Thus, any defect in any of these PRRs and associated signaling pathway during the recognition of the potential pathogen and their PAMPs proves detrimental to the host by supporting the development of sepsis.
The pathogen is recognized by the various innate immune cells (including neutrophils recruited at the site of infection) located in the vicinity of their route of exposure, that is, respiratory tract, gastrointestinal tract, genitourinary tract, and skin.99–101 Thus, the initial recognition of the pathogen by PRRs (ie, TLRs including TLR4 [present both on the cell membrane and intracellularly in endosomes and phagosomes], TLR2, TLR1, TLR6, and TLR5) located on the surface of immune cells initiates the signaling cascade responsible for the generation of proinflammatory immune response via releasing various proinflammatory molecules including various cytokines and chemokines (ie, tumor necrosis factor [TNF]-α, IL-1α, IL-6, IL-8 [CXCL8], monocyte chemoattractant protein 1 [MCP1 or CCL2], etc) along with the generation of type 1 IFNs.102–106 The intact pathogens or PAMPs that get into the cytosol are also recognized by intracellular PRRs. Inflammasomes act as a major class of intracellular PRRs, along with TLR3, TLR7, TLR8, and TLR9. However, inflammasomes are freely circulating in the cytosol, while intracellular TLRs are present in cellular organelles including endosomes, lysosomes, and phagosomes. Thus, the initial recognition of the pathogens and PAMPs by TLRs strengthens the inflammasome signaling required for the release of potent proinflammatory mediators and generates several other molecules that act as endogenous activators of inflammasomes. Both the canonical and non-canonical inflammasomes are activated under different infectious conditions, which may lead to the development of sepsis if not regulated/controlled efficiently.31,32,34,107–111 All these factors and events are described subsequently under various subheadings.
Adhesion molecules, sepsis, and inflammasomes
A higher level of circulating adhesion molecules called intracellular adhesion molecule 1 (ICAM-1) and vascular cell adhesion molecule 1 (VCAM-1) are seen in the clinical cases of sepsis in patients of all age groups112–114 (Figure 3A). The increased circulating levels of ICAM-1 and VCAM-1 are well correlated with the higher incidence of sepsis-associated multiorgan failure (MOF) and mortality in patients admitted to the hospitals.115,116 The elevated serum level of VCAM-1 is associated with septic encephalopathy.117 Thus, an increase in the circulating adhesion molecules is used as a prognostic marker for sepsis and septic shock.113,116 The expression of adhesion molecules on endothelial cells is required for the migration of immune cells (neutrophils, monocytes, and T-cells) at the site of infection to clear the pathogens.118 However, overexpression of these adhesion molecules during sepsis leads to the increased infiltration of proinflammatory immune cells at the site of infection, which also causes tissue and organ damage.118–121 One of the factors contributing to the increased serum levels of adhesion molecules and MOF during sepsis is endothelial dysfunction and their death due to apoptosis or inflammasome-mediated pyroptosis122,123 (Figure 3A).
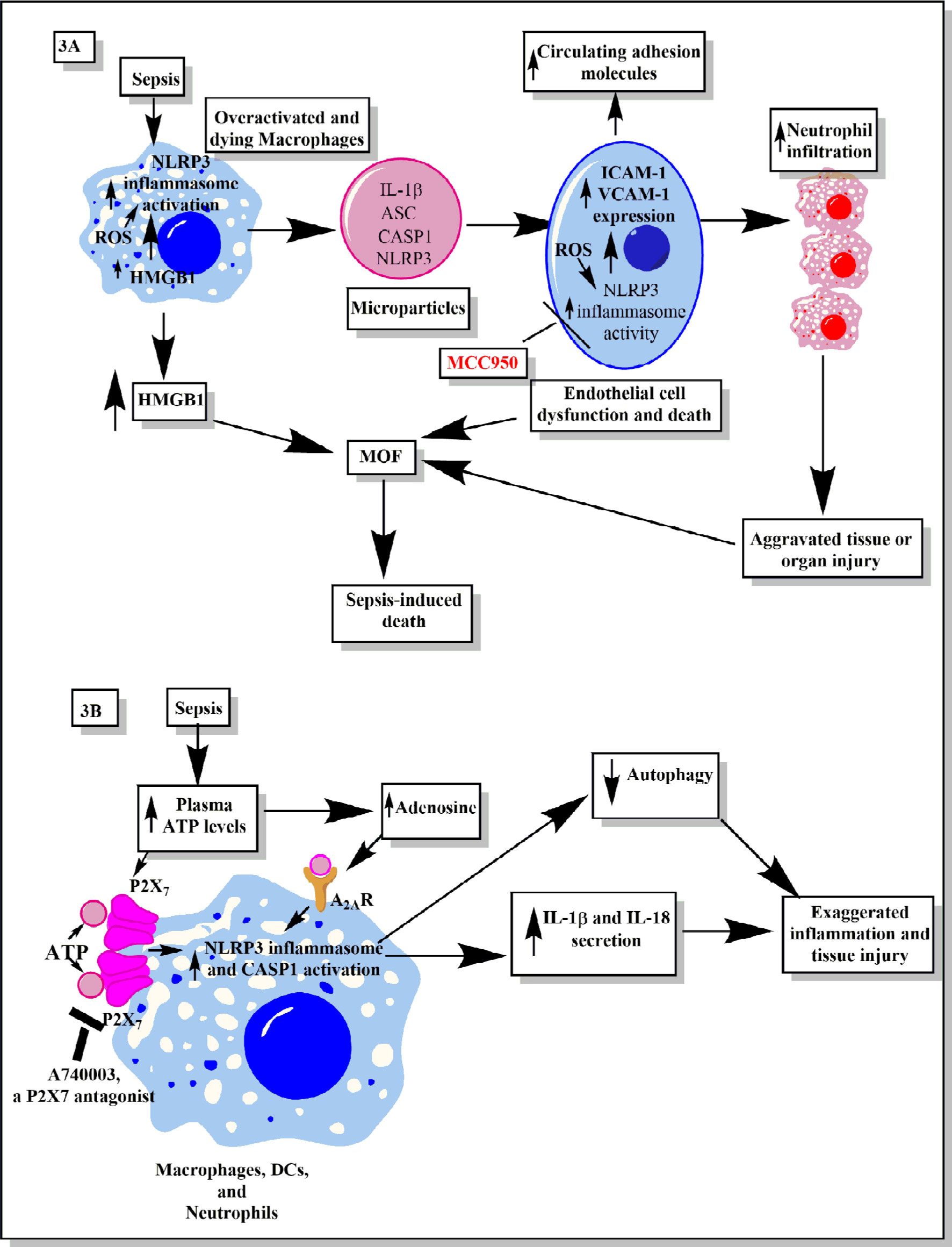 | Figure 3 Schematic representation of the role of inflammasomes and their regulation via different molecules or factors elevated during sepsis. Notes: (A) This part figure shows the induction of NLRP3 inflammasomes during sepsis in macrophages, causing their death, release of high-mobility group box 1 protein (HMGB1), microparticles (consisting of IL-1β, NLRP3, ASC, and CASP1) binding to endothelial cells and stimulating the induction of higher expression of adhesion molecules (ICAM-1 and VCAM-1), causing profound infiltration of neutrophils at the site of infection, which leads to increased tissue or organ damage and death of endothelial cells, in turn leading to increased circulating levels of ICAM-1 and VCAM-1, MOF, and finally death of the septic patient. MCC950, an NLRP3 inflammasome inhibitor, caused inhibition of increased expression of ICAM-1 and VCAM-1 on the endothelial cells and prevented MOF. (B) This part figure shows increased circulating levels of ATP and its metabolite called adenosine act via different receptors called P2X7 and A2ARs expressed on the neutrophils, DCs, and macrophages. The binding then activates the NLRP3 inflammasomes and CASP1 to release IL-1β and IL-18. These inflammatory cytokines cause increased neutrophil infiltration and tissue/organ damage during sepsis. The P2X7 receptor antagonist is shown to inhibit the activation of NLRP3 inflammasome and, thus, the inflammatory tissue damage. Furthermore, activation of NLRP3 inflammasomes decreases the process of autophagy that contains the inflammation, as restoring autophagy has been used to target sepsis. See text for full details. Abbreviations: ATP, adenosine 5’-triphosphate; DC, dendritic cell; ICAM, intracellular adhesion molecule; MOF, multiorgan failure; VCAM, vascular cell adhesion molecule. |
NLRP3 inflammasome activation also increases the expression of chemotactic molecules (ie, α4β1 integrin, CCL7, CCL8, and CXCL16).124 The overactivated and dying macrophages during sepsis also release microparticles (MPs) in the systemic circulation that consist of IL-1β and components of the inflammasome, including ASC, CASP-1, and NLRP3125 (Figure 3A). These MPs bind to and are internalized by human endothelial cells, causing induction of phosphorylation of extracellular signal-related kinase (ERK)1/2, NF-κB activation, and expression of ICAM-1, VCAM-1, and E-selectin.125 However, macrophages lacking NLRP3 inflammasome exhibit reduced production and activity of MPs.125 The endothelial cells lacking IL-1 receptor (IL-1R) showed decreased expression of adhesion molecules in the presence of these MPs.125 Thus, NLRP3 inflammasome-mediated release of MPs containing IL-1β is essential to upregulate the expression of adhesion molecules on endothelial cells. The expression of different adhesion molecules on endothelial cells plays an important role in neutrophil chemotaxis and their infiltration at the site of infection to clear the infection initially. However, in the case of sepsis, persistent infection along with overactivation of the immune response may cause increased activation of the NLRP3 inflammasome. This causes increased release of MPs from the dying macrophages, which leads to higher expression of various adhesion molecules on the endothelial cells required for neutrophil/monocyte adhesion and migration to the distant organs. This increased infiltration of neutrophils/monocytes along with clearing the pathogen also exerts collateral damage and, if not controlled, may cause severe organ damage, including lungs, kidneys, liver, and so on.126,127 On the other hand, this overactivation of endothelial cells also leads to their destruction and release of MPs which further enhances the pathogenic effect of endothelial cells.127 Thus, a prolonged interaction between the neutrophils/monocytes and endothelial cells, where NLRP3 inflammasome activation induces increased expression of adhesion molecules on the endothelial cells, causes their overactivation and shedding of the adhesion molecules in the circulation and organ damage.
NLRP3 inflammasome activation in the endothelial cells during Lactobacillus casei infection causes dysfunction in endothelial cells and increases the upregulation of VCAM-1128 (Figure 3A). ICAM-1 levels are also elevated on endothelial cells upon inflammasome activation129 (Figure 3A). Thus, this increased expression of ICAM-1 and VCAM-1 upon inflammasome stimulation plays an important role in neutrophil adhesion, transcellular migration, and neutrophil-dependent and -independent organ (ie, lungs, liver, heart, etc) damage during sepsis130–134 (Figure 3A). Furthermore, an increased expression of ICAM-1 on the neutrophils is also observed during sepsis, which enhances their phagocytic potential and proinflammatory action.135 However, it would be interesting to explore if the increased expression of ICAM-1 on the neutrophils is also a result of an inflammasome-based event or is independent of inflammasome activation. A potent NLRP3 inflammasome inhibitor called MCC950 was found to decrease the expression of ICAM-1 and VCAM-1 on the endothelial cells136 (Figure 3A). Thus, it would be novel to study the impact of MCC950 on these adhesion molecules’ expression on endothelial cells, circulating adhesion molecules, and MOF during sepsis. It would also be interesting to observe its action on the expression of ICAM-1 on neutrophils isolated from sepsis patients. Human neutrophils also express NLRP3, AIM2 inflammasomes, and CASP1.137 Thus, activation of these inflammasomes on neutrophils during sepsis can provide defense against the invading cytosolic pathogens, along with an increased proinflammatory action via releasing IL-1β and IL-18.137,138 However, if this inflammasome-based protective action gets dysegulated, it may cause collateral organ damage as reactive oxygen species (ROS) generated by neutrophils exert a tissue destructive effect, and also, inflammasomes are further activated by ROS, which may further aggravate the tissue damage by releasing proinflammatory molecules139–141
HMGB1, sepsis, and inflammasomes
HMGB1 is a late mediator of sepsis that is released either actively by activated monocytes/macrophages or passively by cells that died due to necrosis or necroptosis142–145 (Figure 3A). This release of HMGB1 in the circulation during sepsis increases the expression of ICAM-1 and VCAM-1 via binding to the receptor for advanced glycation end products (RAGE).146 This HMGB1–RAGE interaction also enhances the release of TNF-α, IL-8, MCP-1 or CCL2, plasminogen activator inhibitor 1, and tissue plasminogen activator.146–148 The binding of HMGB1 to RAGE on endothelial cells causes transient phosphorylation of MAP kinases, ERK, JNK, and p38 and activation of NF-κB and Sp1 for its proinflammatory action.146 It was found that as sepsis worsens, ROS generation and oxidation of HMGB1 further increase its proinflammatory potential, which aggravates the sepsis-associated inflammatory organ damage including acute kidney injury.149
Therapeutic targeting of HMGB1 during sepsis modulated the proinflammatory cytokine profile in such a way that it improved several clinical features of sepsis and survival among experimental animals.150 Furthermore, the mice that survived the episode of sepsis showed resistance toward developing secondary infections, but most of the patients surviving sepsis become prone to develop secondary infection later.150 However, studies have shown the inflammasome-mediated regulation of HMGB1 release from activated immune cells including macrophages during sepsis.151–154 The deletion of inflammasomes including NLRP3 in mice during endotoxemia and bacterial sepsis decreases HMGB1 production and the associated inflammatory organ damage.151,152 In addition to canonical inflammasomes, the component of non-canonical inflammasome called CASP11 also has a great potential to modulate pyroptosis and the release of HMGB1 in the absence of CASP1.155 This is because a genetic deletion of CASP11, rather than CASP1, in mice protected them from developing severe endotoxemia upon lethal lipopolysaccharide (LPS) challenge.31 One of the contributing factors for the release of HMGB1 is the dead cells. However, it is not released by cells dying due to apoptosis; instead, it is rapidly released by cells dying due to pyroptosis that is induced by inflammasomes.152,156 The HMGB1 released due to pyroptotic cell death is more hyperacetylated at the nuclear localization sequences, as compared to the HMGB1 released due to necrotic cell death.157 The hyperacetylated HMGB1 gets translocated easily and more frequently from the nucleus to the cytoplasm and from the cytoplasm to the secretory lysosomes by a default process; from there, it gets translocated into the extracellular environment upon appropriate stimulus, as compared to the hypoacetylated form.158,159 Further studies have indicated that HMGB1 also activates NLRP3 and CASP8 inflammasome via activating NF-κB.160 Thus, both HMGB1 and inflammasome influence each other and targeting either of these during sepsis will provide a multi-target therapeutic approach that is essentially required. For example, glycyrrhizin-mediated inhibition of HMGB1 caused inhibition of the expression of adhesion molecules and oxidative stress on endothelial cells.161 Thus, it will be interesting to observe the effect of glycyrrhizin on inflammasome activation during experimental sepsis and the associated immune response due to a strong interrelationship between HMGB1 release and inflammasome activation and vice versa.
ROS, oxidative stress, sepsis, and inflammasomes
Phagocytosis of pathogens by immune cells leads to the generation of ROS to kill the pathogen. For example, neutrophils and macrophages responsible for the phagocytic killing of the pathogen also undergo apoptotic cell death due to the higher production of ROS162–164 (Figure 3A). However, this increased generation of ROS also exerts bystander effects on cells responsible for its production along with the adjacent surrounding environment due to very high reactivity of these free radicals causing development of oxidative stress during sepsis.165–167 Circulating ROS responsible for the sepsis-associated oxidative stress induce a profound modification in the erythrocytes causing activation of the circulating leukocytes and maintains the pro-oxidative state even after the clearance of circulating bacteria.168 Additionally, endothelial cells encountering proinflammatory signals (ie, cytokines and MPs released by neutrophils and monocytes/macrophages) and circulating pathogens during sepsis also produce various ROS that cause a profound change in their endothelial vascular function including vascular tone and their morphology, leading to severe coagulating defects and organ dysfunction (ie, cardiac dysfunction, liver and kidney failure, acute lung injury, etc),126,169–171 as shown in Figure 3A. Thus, ROS plays an important role in the pathogenesis of sepsis and associated organ damage. However, a recent study has shown a lower production of ROS during early sepsis does not cause myocardial dysfunction.172 This indicates that ROS generation starts during early sepsis and remains unchecked till late-stage sepsis called severe sepsis and septic shock, causing profound cardiac dysfunction and damage to the other vital organs leading to mortality. The late stage of sepsis is recognized as a stage of immunosuppression/immunoparalysis characterized by the susceptibility of the person to develop secondary infections. ROS generation also plays an important role in the induction of this stage by causing enhanced expression of NRF2-dependent ATF3.173 Although this ROS-mediated induction of ATF3 protected the animals from developing endotoxic shock, it made them prone to develop severe bacterial and fungal infections by suppressing the release of IL-6.173 Thus, the ROS levels impact both organ damage and susceptibility to secondary infections during sepsis.
The intracellular generation of ROS is also known to regulate the inflammasome activity; also, ROS are produced due to the activity of NLRP3 inflammasomes.141,174–176 Thus, ROS generation in the macrophages provides a priming effect to activate NLRP3 inflammasomes required for the efficient clearance of the cytosolic pathogen that is blocked by treatment with ROS inhibitors.141 Thus, it can be inferred that the higher levels of ROS generated during sepsis keep the NLRP3 inflammasomes activated (even after clearance of the infection) and the inflammatory process unchecked, causing the death of these immune cells. This exaggerated death of proinflammatory immune cells including M1 macrophages, CD4+, and CD8+ T-cells causes the development of a stage of immunosuppression.22,177 Additionally, ROS generation in endothelial cells serves as a local endogenous trigger for NLRP3 activation178,179 (Figure 3A). This NLRP3 inflammasome activation causes the release of cytokines IL-1β and IL-18 that act as chemoattractants to further recruit inflammatory immune cells including neutrophils, causing further inflammatory damage to the organs including kidneys.178,179 Furthermore, ROS-induced NLRP3 activity was found to induce acute liver injury.180 Thus, regulating the generation of ROS during sepsis or inflammasome has a potential to target sepsis and associated organ damage effectively.
ATP, adenosine, sepsis, and inflammasomes
A higher plasma level of adenosine 5’-triphosphate (ATP) is reported in sepsis patients and in animal models of sepsis181–183 (Figure 3B). Furthermore, higher intracellular ATP is also observed in neutrophils isolated from sepsis patients.184 Further study in this direction has indicated that higher levels of systemic ATP molecules cause an impairment in neutrophil chemotaxis and host defense.185 The study further emphasizes that inhibition of endogenous ATP decreases the overactivation of neutrophils and the associated organ injury observed during sepsis. However, it exaggerates bacteremia and the spread of infection to distant organs and, thus, increases the mortality associated with sepsis.185 But chelation or removal of systemic ATP enhanced the bacterial clearance from the circulation and increased survival by causing an improvement in chemotaxis of neutrophils at the site of infection.185 The extracellular ATP is recognized by P2 purine receptors including ionotropic P2X receptors (P2X1–P2X7) and G-protein–coupled P2Y receptors (P2Y1, P2Y2, P2Y4, P2Y6, and P2Y11–P2Y14) expressed by various innate immune cells and nonimmune cells.186–188 The recognition of extracellular ATP by P2X7 receptor is involved in several inflammatory diseases by causing the maturation and release of IL-1β189–191 (Figure 1B).
P2X7 receptor-mediated stimulation of NLRP3 inflammasome on the neutrophils was found to secrete IL-1β that may prove detrimental to the host.191,192 However, P2X7 receptor activation on the macrophages and DCs also causes activation of NLRP3 inflammasome and CASP1 activation, resulting in the release of IL-1β and IL-18193–196 (Figure 3B). This P2X7-mediated activation of NLRP3 due to extracellular ATP in these innate immune cells also increases their antimicrobial action against cytosolic pathogens.191,197 The systemic inhibition of P2X7 receptor with its antagonist called A740003 protected animals from sepsis-associated disruption in the intestinal barrier198 (Figure 3B). Thus, the inhibition of ATP-mediated purinergic signaling mediated via P2X7 receptors has a potential inhibitory effect on overactivated inflammasomes during sepsis. Further studies in this direction are required to fill the unknown gaps in the field. The in vivo stimulation of NLRC4/NAIP inflammasome through flagellated pathogens in monocytes, macrophages, and neutrophils was reported to cause severe systemic inflammation as observed in sepsis.199
Adenosine is an endogenous immunomodulatory metabolite produced under diverse inflammatory conditions including sepsis.200–202 Adenosine is constitutively present in the mammalian system in the extracellular environment in very low amount (≤1 µM).200 However, its plasma level (4–10 µM in patients with septic shock) increases during different diseases causing metabolic stress such as acute tissue injury, sepsis, or septic shock203,204 (Figure 3B). Thus, high plasma adenosine levels can be used as a prognostic marker for sepsis and septic shock. However, adenosine acting via adenosine A2A receptor (A2AR) acts as a major regulator of inflammasome activity.205 The higher plasma adenosine level during sepsis aggravates the maximal amplitude and duration of the inflammasome response.205 This inflammasome regulation via adenosine is mediated by downstream signaling pathway initiated by A2AR activation which causes activation of cyclic adenosine monophosphate (cAMP)/PKA/CREB/hypoxia-inducible factor-1α signaling pathway.205 This further causes the upregulation of pro-IL-1β, NLRP3 and increased activity of CASP1. This NLRP3 inflammasome pathway activation causes an increased production of IL-1β, IL-18, and pyroptosis of immune cells including macrophages to further aggravate the inflammatory process observed during sepsis, even in the absence of pathogen or associated PAMPs. This shows that these immune cells (ie, macrophages) do not exhibit tolerance, but remain in longer post-activation stage controlled by adenosine and cAMP.205 Adenosine A2AR and A2BR targeting has been used as an immunomodulatory therapeutic approach to targeting sepsis.206–209 Thus, it will be interesting to observe the impact of this adenosine-based immunomodulatory therapeutic approach on inflammasome during sepsis.
Complement system, sepsis, and inflammasomes
Complement system (CS) is an evolutionarily conserved component of the innate immune system that plays a very important role in the host immune response and its regulation.210–213 Due to its important role in the recognition of potential pathogens, opsonization, directly killing the pathogens via forming membrane attack complex (MAC), and modulation of potential immune response, complement is recognized to play an important role in the pathogenesis of sepsis, septic shock, and associated organ damage and mortality.214–218 However, the CS components also influence NLRP3 inflammasomes.219 For example, phagocytosis of the complement-opsonized pathogens by macrophages stimulates the activation of NLRP3 inflammasomes and CASP1, which cause the release of proinflammatory cytokines (IL-1β and IL-18) and promote leukocyte infiltration at the site of infection (source of sepsis), as shown in Figure 4.220 This inflammasome activation does not occur upon phagocytosis of unopsonized pathogens or other particles opsonized with serum deficient in the terminal components of the CS. Another study has indicated NLRP3 inflammasome and CASP1 activation due to the formation of sublethal MAC, but not by the complement components C3a and C5a during endotoxemia.221 The deposition of sublethal MAC on the cell surface increases the intracellular Ca2+ which accumulates in the mitochondrial matrix and alters the mitochondrial transmembrane potential triggering NLRP3 inflammasome activation and CASP1 activity (Figure 5).222 This intracellular Ca2+ causes increased generation of mitochondrial ROS (mtROS) and the death of mitochondria causes the release of mitochondrial DNA (mtDNA) into the cytosol.223,224 This oxidized mtDNA activates NLRP3 inflammasome during the apoptosis of macrophages and other leukocytes which is a well-recognized phenomenon observed during sepsis or other acute infection.225–227
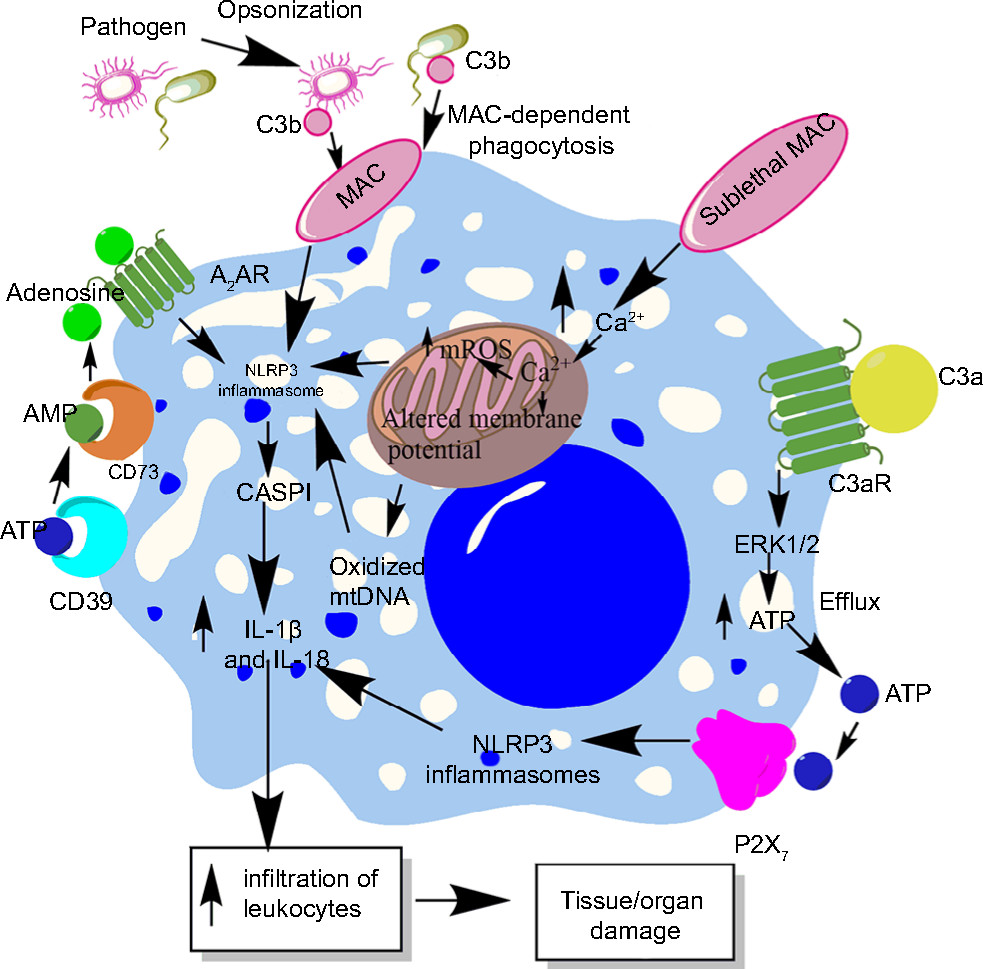 | Figure 4 Role of complement in the activation of inflammasomes in innate immune cells during sepsis. Notes: The complement (C3b)-opsonized bacteria phagocytosed via MAC cause the binding of MAC to the macrophage membrane, which leads to the initiation of bystander damage via the upregulation and activation of NLRP3 inflammasome. The activation of NLRP3 causes activation of CASP1, which leads to the maturation and release of proinflammatory cytokines, IL-1β and IL-18. On the other hand, the sublethal form of the MAC called sublethal MAC also induces the activation of NLRP3 inflammasome and the release of IL-1β and IL-18 via increasing the intracellular Ca2+ which enters into the mitochondrial matrix. The increase of Ca2+ in the mitochondrial matrix leads to increased mtROS generation and alters the mitochondrial membrane potential, causing mitochondrial damage and the release of mitochondrial DNA into the cytosol. All these factors activate NLRP3 inflammasome. Additionally, binding of C3a to its cognate receptor C3aR also activates NLRP3 inflammasome via activating the ERK1/ERK2 pathway which causes the efflux of ATP that activates NLRP3 inflammasome via binding to P2X7. The ATP that gets converted into adenosine via the enzymatic action of CD39 and CD73 also activates NLRP3 inflammasome by binding to A2AR. Abbreviations: A2AR, A2A receptor; ATP, adenosine triphosphate; MAC, membrane attack complex; mtDNA, mitochondrial DNA; mtROS, mitochondrial reactive oxygen species. |
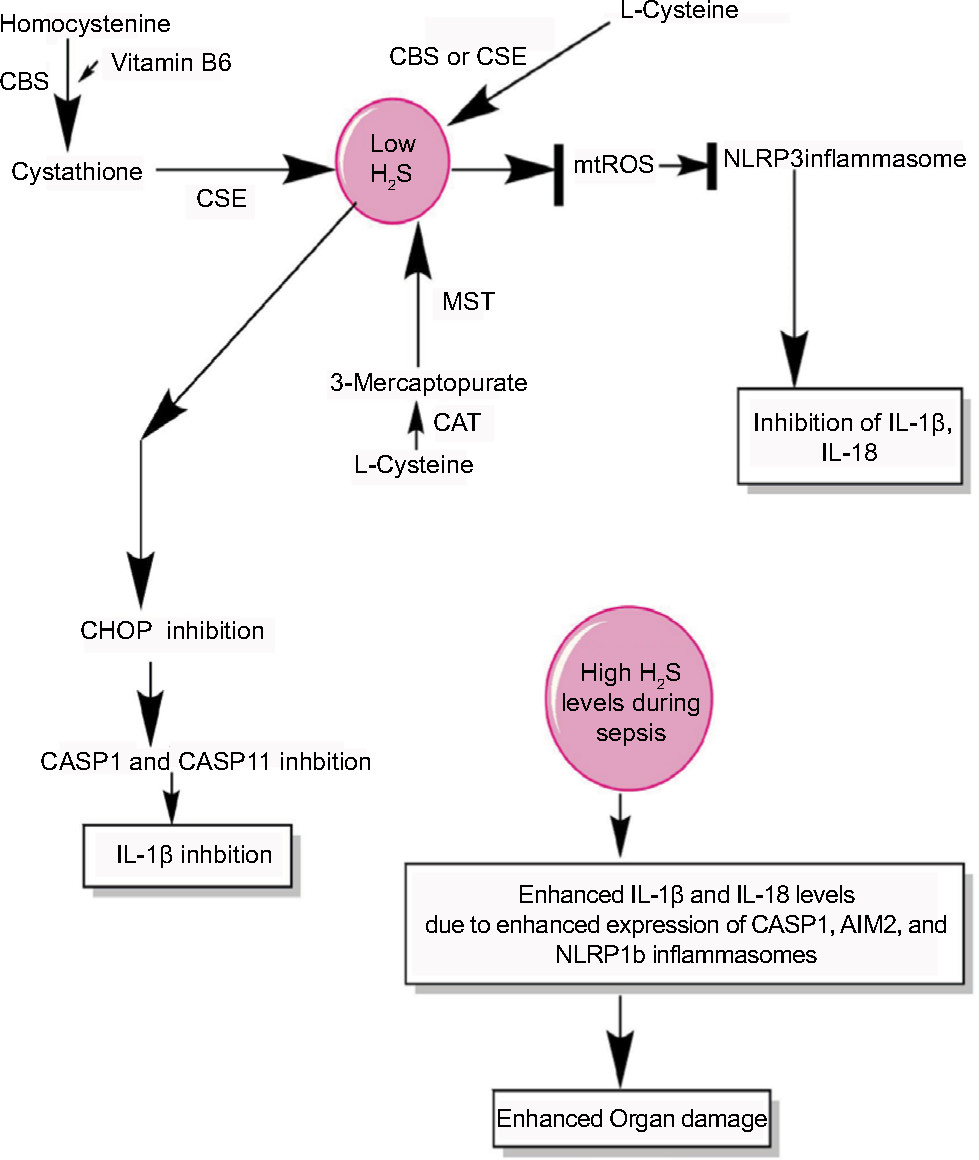 | Figure 5 H2S production during sepsis and its impact on inflammasomes. Notes: H2S is produced by the enzymatic action of CSE, CBS, CAT, and MST on the amino acids l-cysteine and homocysteine (for details, see Wang271). At a lower level, H2S inhibits NLRP3 inflammasome activation and the release of IL-1β and IL-18 via inhibiting the generation of mtROS and CHOP activity required for the activation of CASP1 and CASP11. However, at its higher levels observed during severe sepsis or septic shock, H2S exerts proinflammatory action via enhancing the levels of IL-1β and IL-18 through activating the CASP1, AIM2, and NLRP1b inflammasomes. Abbreviation: mtROS, mitochondrial reactive oxygen species. |
Intracellular CS components, that is, C3aR and C5aR1, are shown to regulate the Th1 (CD4+ T-cells) immune response via generating intracellular ROS and effectively upregulating NLRP3 inflammasome, causing the release of IL-1β and IFN-γ in response to the pathogen.228,229 Studies have indicated early death and an alteration in the function of CD4+ and CD8+ T-cells during sepsis.16,22,230 Thus, it will be interesting to study the intracellular C5aR1 and C3aR and NLRP3 inflammasome in the T-cell population during early sepsis. The complement component C5a also plays an important role in the activation of NLRP3 inflammasomes and IL-1β release.231 The cecal ligation and puncture (CLP) model of sepsis in C5aR1 or C5aR2 (C5L2)-/- mice showed decreased activation of NLRP3 inflammasome and decreased production of IL-1β.231 Further study indicates an increased production of ROS (both cytosolic and mitochondrial) in cardiomyocytes upon challenging with recombinant C5a, an inducer of NLRP3 inflammasome.231 The NLRP3-/- mice showed decreased defect in echo/Doppler parameters in the heart after CLP; thus, complement-mediated activation of NLRP3 inflammasome plays an important role in cardiac dysfunction observed during sepsis.231
Furthermore, C3a component of CS also enhances the production of IL-1β from the macrophages and DCs treated with LPS via regulating the efflux of ATP and subsequent activation of NLRP3 (Figure 4).232 The binding of C3a to C3aR on macrophages induces ERK-1/2 phosphorylation which further promotes the efflux of ATP from the macrophages (Figure 4).232,233 Thereafter, the binding of extracellular ATP to a P2X7 receptor or the conversion of ATP into adenosine via CD39 and CD73 and its binding to adenosine A2 receptors both cause activation of NLRP3 inflammasome and CASP1 during sepsis.191,200,205 The C1q component of CS acts as a negative regulator of inflammasome activation during the uptake of apoptotic cells and directs macrophage polarization via decreasing the cleavage of proCASP1 to CASP1 required for the release of IL-1β.234 Thus, the CS components influence inflammasome during sepsis in a wide variety of immune cells, and therapies targeting CS in a cell-specific manner have the potential to target sepsis via inflammasome modulation.
Autophagy, sepsis, and inflammasomes
Autophagy is considered one of the crucial innate immune mechanisms against infectious agents invading the host and plays an important role in inflammation.235,236 The major intracellular signaling mechanisms controlling the induction of autophagy comprise AMPK, JNK/p38 MAPK pathways generating ROS and regulating NF-κB transcription factor regulated by TLRs (ie, TLR4, TLR9, etc).237–239 The induction of autophagy during sepsis provides protection to the host against multiorgan dysfunction syndrome by preventing apoptotic cell death of immune cells, maintaining the homeostatic cytokine balance between the production of pro- and anti-inflammatory cytokines, and preserving mitochondrial functions,240–243 whereas any decline in the process of autophagy during sepsis is associated with an increased tissue and organ injury (ie, liver, brain, kidneys, etc).244,245 The polymorphisms of the autophagy-related loci in the IRGM gene are associated with high mortality in sepsis.246 However, the induction of autophagy starts at the beginning of sepsis, but its flux is lost after a short duration of hyperactivity.241,247 Various strategies are used to restore the immune homeostasis via modulating the autophagy during sepsis.248,249 Thus, a regulated process of autophagy is essential to prevent exaggerated organ damage during sepsis.
It was found that loss of Atg16L1 protein required for the initiation of autophagy causes an increased production of IL-1β and IL-18 by the macrophages upon LPS challenge or during endotoxemia, due to overactivation of NLRP3 inflammasome and CASP1 in the presence of higher ROS and oxidized mtDNA.225,250–252 Thus, increased autophagy prevents an upregulated inflammasome activity responsible for proinflammatory damage observed during sepsis. For example, the inflammasome components, including NLRP3, NLRP1, and pro-caspase 1, can also be degraded via the process of autophagy through TRIM20 in the macrophages stimulated with IFN-γ.253 Autophagy also controls the release of IL-1β via targeting the degradation of pro-IL-1β as it sequesters into autophagosomes, and further increasing the process of autophagy via rapamycin (an mTOR inhibitor) caused its degradation and inhibited the release of mature IL-1β during endotoxemia.254,255 The inhibition of autophagy increased the release of IL-1β in an NLRP3 inflammasome-dependent manner due to increased generation of ROS.254,255 Also, activation of NLRP3 and NLRP4 inflammasomes inhibits autophagy in a beclin1- and PINK1-dependent manner during group A streptococcal infection responsible for sepsis in humans and hyperoxia that causes an increased ROS generation.256–258
An enhanced activation of NLRP3 inflammasome during Pseudomonas aeruginosa infection in human macrophages prevents their intracellular killing without affecting the generation of antimicrobial peptides, ROS, and NO.259 This pathogen escape via activation of NLRP3 involves an increase in LC-3-II protein and the formation of autophagosomes leading to enhanced autophagy.259 The deficiency of Atg7 during P. aeruginosa-induced abdominal infection causes defective bacterial clearance, increased mortality, bacteremia, and development of sepsis.260 The deficiency of Atg7 causes an aggravated release of IL-1β and pyroptosis of macrophages due to the upregulation of NLRC4 inflammasome and its activity.260 NLRP6 inflammasome deficiency was also found to cause defective autophagy among intestinal epithelial cells and goblet cells, making them more susceptible to persistent Citrobacter rodentium infection and their dysbiosis to peripheral circulation causing sepsis.261 SESN2 or Sestrin 2, a stress-inducible protein, suppresses sepsis by inhibiting the NLRP3 inflammasome activation via inducing mitophagy through mitochondrial priming to clear damaged mitochondria in activated macrophages.262 SESN2 promotes the perinuclear clustering of mitochondria via mediating the aggregation of SQSTM1 and its binding to Lys63-linked ubiquitins on the mitochondrial surface.262 Thereafter, SESN2 activates the specific autophagic machinery to degrade the primed mitochondria through an increased expression of ULK1 protein, a mitophagy inducer. The increased SESN2 expression during sepsis/endotoxemia is further enhanced by NO production through iNOS2 in the macrophages. Thus, NO production during sepsis exerts an inhibitory action of NLRP3 inflammasomes in both human and mice myeloid immune cells via induction of SESN2.262,263 SESN2−/- mice subjected to sepsis exhibit defective mitophagy which causes increased activation of inflammasomes and enhanced mortality.262 Additionally, RIPK2 was also found to exert a negative impact on NLRP3 inflammasome activation during acute infections via inducing autophagy of mitochondria (mitophagy) in a kinase-dependent manner by phosphorylating ULK1 protein and reducing CASP1 activation.264 Thus, autophagy and inflammasome activation are interrelated processes playing an important role in the pathogenesis of sepsis and its outcome.265,266 For example, autophagy inhibits inflammasome activation and inflammasome activation inhibits autophagy.267–269 Therefore, therapeutics enhancing autophagy may indirectly decrease overactivation of the inflammatory function of inflammasomes or inhibiting inflammasomes during sepsis may enhance autophagy to maintain the immune homeostasis during sepsis. This area needs an urgent research priority to target sepsis.
H2S, sepsis, and inflammasomes
H2S is a gaseous transmitter/gasotransmitter that is produced endogenously in many cell types, including the macrophages and endothelial cells, from cysteine due to the activity of enzymes called CBS, CSE, CAT, and MST.270,271 H2S plays an important role in the modulation of cardiovascular function including hypertension and ischemia–reperfusion injury.272,273 For example, H2S protects from ischemia–reperfusion injury via preserving the structure and function of myocardial mitochondria and decreasing the neutrophil infiltration, IL-1β secretion, and cellular necrosis.272 However, the cardiac dysfunction is a major problem in sepsis patients who had undergone septic shock.274–278 H2S is shown to protect the mitochondrial function via inhibiting the generation of mtROS that activates the NLPR3 inflammasome279 (Figure 5). The decrease in mtROS by H2S involves S-sulfhydration of c-Jun that enhances its transcriptional activity of SIRT3 and p62. The H2S-mediated anti-inflammatory action in both murine and human macrophages involves the inhibition of NLPR3 inflammasome via the inhibition mtROS production and ASC oligomerization in vitro and in a mouse model of peritonitis280 (Figure 5). H2S also inhibits the TLR4/NF-κB activation-mediated inflammatory pathway, along with reducing the expression of NLRP3, ASC, pro-caspase-1, CASP-1, IL-1β, IL-18, and CASP-3 that is regulated by TLR4 signaling.281 Thus, H2S exerts its protective action via inhibiting NLRP3 inflammasome-mediated proinflammatory action.
In animal model of sepsis, the inhibition of H2S or its activation is proven beneficial to the hosts in terms of their survival and reduced tissue/organ injury, including myocardial injury, via improving neutrophil infiltration, K+ATP channel activation, reducing TNF-α, and increasing IL-10 levels.282–284 The CLP-induced sepsis in mice causes a significant increase in the plasma level of H2S along with its synthesis in the liver 8 hours post-CLP.282 Thus, it can be inferred that the beneficial and harmful effects of H2S depend on its concentration and duration or stage of sepsis. The beneficial effect of H2S during sepsis depends on the inhibition of CHOP, which is a transcription factor and mediates the unfolded protein response that plays a major role in the induction of apoptosis.285–287 CHOP is also involved in the generation of IL-1β during endotoxemia or sepsis via regulating the induction of CASP11 that is involved in the maturation of pro-IL-1β via CASP1.288,289 Thus, CHOP inhibition via H2S is also another mechanism of inflammasome inhibition during sepsis to prevent exaggerated inflammation and organ damage. Hence, H2S inhibits NLRP3 activation via inhibiting mtROS generation and CHOP. However, at later stages of sepsis including septic shock, its circulating levels are increased due to the hepatic dysfunction causing an impaired oxygen (O2) availability to the tissues.290–292 The acute exposure of higher amount of H2S causes an increased expression of IL-18, MyD88, and TLR9 in addition to a decreased expression of CCL12 and CXCR4.293 Furthermore, this acute expression of H2S increases the expression of genes associated with inflammasome, including IL-1β, CASP-1, and AIM2 (Figure 5). However, NLRP3 expression was decreased, but NLRP1b was upregulated upon H2S treatment.293 H2S has a great potential to act as a prognostic marker of septic shock and its severity level becomes higher in septic patients within 12 hours of onset of sepsis.294,295 Even H2S can be detected in the exhaled pulmonary gases of patients with sepsis.296 Thus, H2S negatively regulates NLRP3 at all concentrations, but increases the proinflammatory action of NLRP1b inflammasome at its higher level, aggravating the sepsis-associated organ damage. Thus, H2S acts as a novel gasotransmitter regulating the function of inflammasomes during sepsis at all levels. Future studies will prove beneficial in this direction as H2S donors have therapeutic potential for both acute and chronic inflammatory diseases.297
Prostanoids, eicosanoids, inflammasomes, and sepsis
Prostanoids are lipid mediators synthesized from a 20-carbon unsaturated fatty acid called arachidonic acid by the action of enzymes called cyclooxygenases (COX-1 and COX-2) or PG G/H synthase.298,299 COX-1 plays an important role in the generation of prostanoids during homeostasis, while COX-2 is induced by proinflammatory cytokines, shear stress, and tumor promoters and serves as a major source for prostanoid biosynthesis during inflammation.299 Sepsis is shown to alter vasoconstriction via modulating prostanoids’ levels (Figure 6).300 The generation of prostanoids is also regulated by IL-1β generated due to the activation of inflammasomes, and mice lacking COX-2 are defective in producing IL-18 cytokine.69,301 Bacterial flagellin or anthrax lethal toxin-mediated systemic activation of NAIP5/NLRC4 inflammasomes causes dysregulated production of eicosanoids called eicosanoid storm (a profound or pathological release of signaling lipids, including PGs and leukotrienes, causing a rapid initiation of inflammation and vascular fluid loss), which proves highly lethal to the host due to the rapid loss of vascular fluid into the intestine and peritoneal cavity (Figure 6).302,303 The eicosanoid generation due to the inflammasome activation occurs due to the activation of cytosolic PLA2 in the peritoneal macrophages.303 The NLRP3 inflammasome activation is also shown to activate leukotriene B4 that further activates PGE2 via IL-1β/IL-1R signaling.304 However, an exogenous administration of leukotriene B4 inhibits NLRP3 inflammasome and IL-1β release.304 Furthermore, 15-deoxy-Δ12,14-PGJ2 and related cyclopentenone PGs inhibit CASP1 activation via inhibiting NLRP1 and NLRP3 inflammasome activation through preventing the autoproteolytic cleavage of CASP-1 and, thus, the maturation of IL-1β through the induction of a cellular state inhibitory to CASP-1 proteolytic function.305 Thus, cyclopentenone PGs acts as anti-inflammatory molecules via inhibiting CASP1, NLRP1, and NLRP3 inflammasomes. The PGE2 via PGE2 receptor E-prostanoid 4 enhances the activity of PKA that directly inhibits NLRP3 inflammasome activation via direct phosphorylation of cytoplasmic NLRP3 at Ser295 and inhibits its ATPase activity (Figure 6).306 Additionally, PGE2 via E-prostanoid 4 also increases the intracellular levels of cAMP which further decreases NLPR3 inflammasome activation.307 Thus, eicosanoids both inhibit and enhance inflammasome activation and this mechanism needs to be identified clearly.
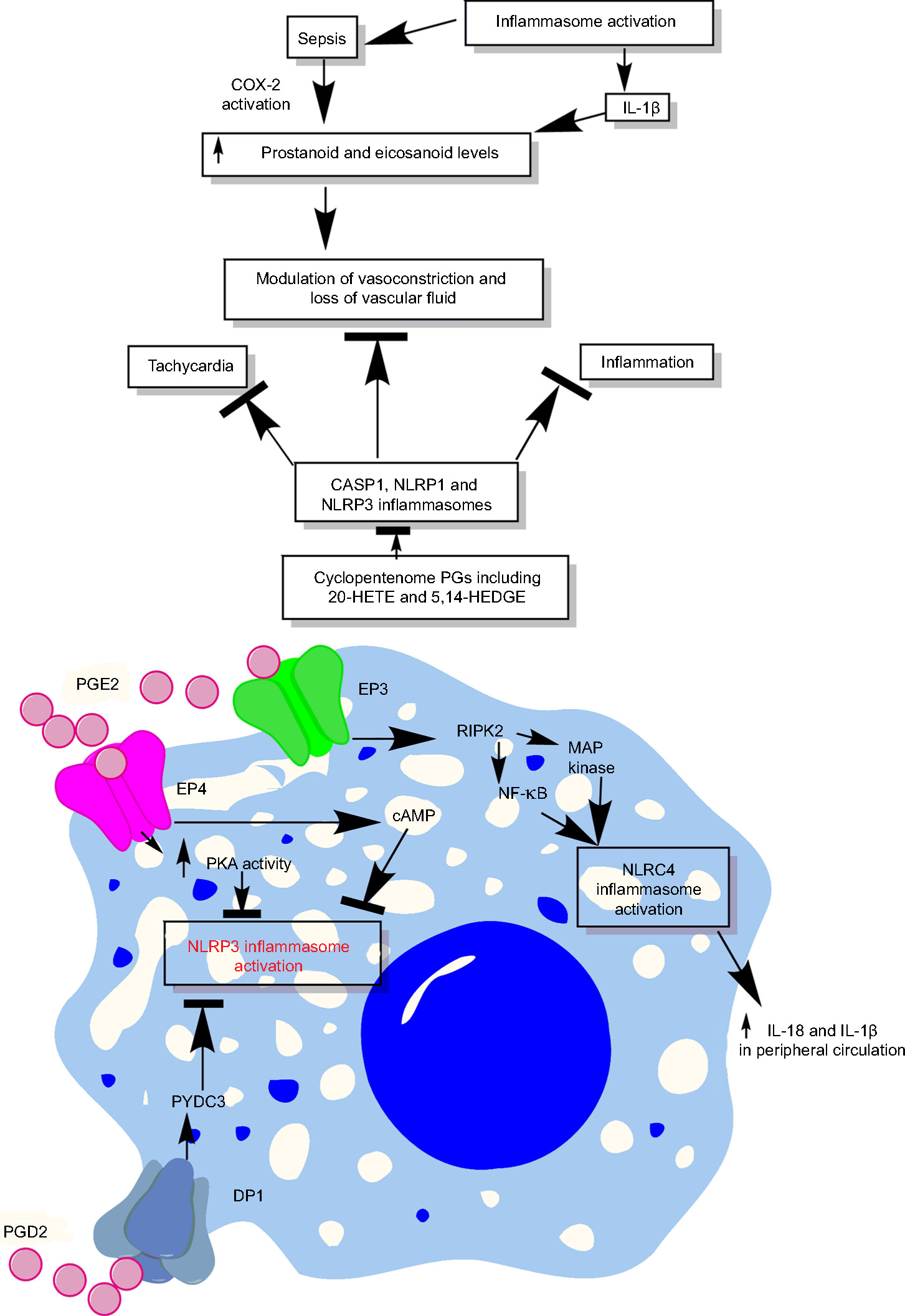 | Figure 6 Schematic representation of the relationship between prostanoids, eicosanoids, sepsis, and inflammasomes. Notes: Overactivation of inflammasomes is seen in sepsis, which also exhibits overproduction of prostanoids and eicosanoids. These prostanoids and eicosanoids exert their impact on vascular tone and vascular contractility and induce vascular leakage. Additionally, at the immune cell level, a prostanoid called PGE2 causes induction of PKA via acting through EP4 receptors expressed on the macrophages, which directly inhibits NLRP3 inflammasome by binding to Ser295. Also, PGE2 activates RIPK2 via EP3, which activates MAP kinases and NF-κB causing activation of NLRC4 inflammasomes. The activation of NLRC4 inflammasome increases the systemic levels of IL-18 and IL-1β cytokines, while PGD2, via acting through DP1, induces the production of PYDC3 protein that directly inhibits NLRP3 inflammasome overactivation. See text for detail. Abbreviations: 5,14-HEDGE, N-[20-hydroxyeicosa-5(Z),14(Z)-dienoyl]glycine; 20-HETE, 20-hydroxyeicosatetraenoic acid; DP1, D-prostanoid receptor 1; PG, prostaglandin. |
The PG E2-E type PG receptor 3 (PGE2-EP3) signaling is shown to activate NLRC4 inflammasome and the secretion of IL-1β and IL-18 via RIPK2 that activates NF-κB signaling and MAP kinases upon infection.301,308 In addition, the COX-2-dependent generation of prostanoids in endothelial cells (ECs) plays an important role in the vascular functions of ECs, which gets dysregulated during sepsis and results in septic shock and organ damage.309 Thus, it will be interesting to delineate the relationship between inflammasome activation and prostanoid generation during sepsis to develop future therapeutic to prevent vascular dysfunction or vascular leakage seen during sepsis. Inhibition of prostanoids by 20-hydroxyeicosatetraenoic acid (an ω-hydroxylation product of arachidonic acid that is produced by CYP/CYP450 [CYP] enzymes, mainly by the CYP4A and CYP4F isoforms, in the kidney, heart, liver, brain, lung, and vasculature) and N-[20-hydroxyeicosa-5(Z),14(Z)-dienoyl]glycine during sepsis is shown to inhibit vasodilation, hypotension, tachycardia, and inflammation in a rat model of septic shock.310,311 This can be attributed to the inhibition of NLRP3 and NLRC4 inflammasome also and needs to be investigated. Another study has indicated the negative effect of PG D2 on inflammasome activation during certain infections via D-prostanoid receptor 1 that stimulates the induction of a putative inflammasome inhibitor called PYDC3 in CD11b+ myeloid cells including monocytes/macrophages and microglial cells (Figure 6).312 PYDC3 acts as PYD only protein lacking other ligand-binding domains and inhibits the process of inflammasome nucleation via direct binding.312 In addition to PYDC3, POP1 and POP3 (pyrin domain-only proteins) also act as negative regulators of NLRP3 and ALR inflammasomes and prevent their overactivation.313,314 POP1 inhibits ASC-dependent inflammasome assembly through prevention of inflammasome nucleation, which interferes with CASP-1 activation, IL-1β and IL-18 release, pyroptosis, and the release of ASC particles.313 POP1 expression is regulated by other inflammatory signals mediated by TLR and IL-1R signaling that also affect prostanoid or PG synthesis. Thus, it will be interesting to observe the impact of prostanoids on POP1 and POP3 expression during sepsis. It can be suggested that inflammasomes and prostanoids impact each other in both directions and the regulation of one affects the other. The relationship between inflammasomes and prostanoids needs to be understood for forming better therapeutics to manage sepsis.
Immunosuppression during sepsis and inflammasomes
Sepsis-associated immunosuppression/immunoparalysis proves detrimental to the host and makes it prone to develop secondary infections even after full recovery.8,23,315 This is because it lasts for longer duration due to severe damage acquired by the host immune system, including paralyzed DCs and macrophages.316,317 Activation of inflammasomes is a major regulator of the release of proinflammatory cytokines IL-1β and IL-18 and the induction of death of macrophages and DCs via pyroptosis or necroptosis. However, the death of these immune cells contributes to the development of immunosuppression/immunoparalysis observed during the late stage of sepsis.8 Additionally, conventional T-cells (CD4+, CD8+, and CD4+CD8+ T-cells) are also lost during early sepsis and further aggravate the immunoparalysis.318 Also, an upregulation and profound activation of Tregs at the later stages of sepsis is the major contributor to the development of immunosuppression.319,320 A recent study has identified the existence of NLRP3 inflammasome and its activation for the normal Th1 immune response mediated by CD4+ T-cells.229 Another study has indicated the role of NLRP3 inflammasome activation in the migration of CD4+ T-cells to the distant organs due to the upregulation of chemotactic proteins including osteopontin, CCR2, and CXCR6.124 Thus, it would be interesting to investigate the role of the inflammasome in T-cell function, migration, and their death during sepsis.
Another mechanism that can contribute to the sepsis-associated immunosuppression involves the Foxp3− type one regulatory (Tr1) cells which suppress NLRP3 activation via an IL-10–dependent mechanism.321 The activation of Tr1 cells causes the release of IL-10 that, in turn, inhibits the release of maturation of pro-IL-1β, release of mature IL-1β, and inflammasome-mediated activation of CASP1 in macrophages stimulated with LPS and ATP.321 It should be noted that IL-10 is a major immunosuppressive cytokine that gets elevated during sepsis.322–325 Also, a higher level of IL-10 is shown to regulate the transition from early reversible sepsis to the late phase of irreversible septic shock in laboratory animals.326 Thus, inflammasomes play an essential role during both the stages of immune response (ie, exaggerated activation of immune response and immunosuppression) observed during sepsis.
Therapeutic targeting of inflammasomes to manage sepsis
Inflammasomes play an important role in inflammation via regulating the initiation of the inflammatory cascade through regulating the release of IL-1β and IL-18 cytokines, immune cell (ie, neutrophils, monocytes etc) chemotaxis and infiltration, and direct recognition of cytosolic pathogens and their PAMPs. Thus, molecules targeting inflammasomes will prove beneficial to control the exaggerated systemic inflammation and inflammatory organ damage during sepsis. In addition to increasing the release of IL-1β and IL-18 cytokines via activating CASP1, the NLRP3/ASC inflammasome complex inhibits the maturation and release of IL-33 cytokine.327,328 Thus, IL-33 levels are decreased in the circulation during sepsis. IL-33 works by binding to IL1RL1 or ST2 that belongs to IL-1 superfamily.329,330 The binding of IL-33 to membrane-bound ST2 initiates MyD88/NF-κB signaling which increases the mast cell function and Th2- and Treg-mediated immune response.330,331 However, this protective action of IL-33 is lost due to the inhibition of its maturation and release by NLRP3 inflammasome-mediated activation of CASP1 that inactivates IL-33. This is because activation of mast cells causes prompt release of TNF-α due to their property of storing presynthesized cytokine during acute bacterial peritonitis.332 In addition to a decrease in circulating IL-33 during sepsis, the serum levels of soluble ST2 also increases, which acts as decoy receptor for IL-33 that further lowers IL-33 levels by sequestering it.330,333–335 Exogenous administration of soluble ST2 in septic mice leads to decreased IL-6 and TNF-α levels,336 which further indicates that the IL-33–mediated activation of mast cells is inhibited giving an initial proinflammatory signal via releasing presynthesized TNF-α and IL-6. IL-33 cytokine therapy in a mouse model of sepsis exhibits protective action.337 IL-33–treated animals exhibited increased neutrophil infiltration that is required to kill the pathogens efficiently when their number can be easily controlled. This can be explained as the IL-33–mediated activation of peritoneal mast cells that initiate the very first signal for neutrophil infiltration via releasing the presynthesized TNF-α and IL-6.332,338 Furthermore, IL-33 treatment decreased systemic inflammation, leaving the local proinflammatory response intact, which is required to kill the pathogen at its niche acting as a source for bacteremia and, thus, the sepsis. The increased neutrophil infiltration into the peritoneum can also be expected due to the upregulation of CXCR2 on neutrophils as a result of inhibition of GRK2 expression.337 GRK2 is a serine–threonine protein kinase that induces internalization of chemokine receptors. However, it does not activate an immunologic shift from a Th1 immune response to a Th2 immune response.337 The activation of peritoneal mast cells during bacterial peritonitis inhibits the phagocytic capacity of peritoneal macrophages via releasing prestored IL-4.339 Thus, it will be interesting to observe the effect of IL-33 treatment on the levels of IL-4.
Various phytochemicals and chemicals (MCC950) have been identified and developed that target inflammasomes during diverse sterile inflammatory conditions mentioned.340–342 For example, MCC950 is shown to control the endothelial overexpression of adhesion molecules during the inflammatory process observed during sepsis.136 Furthermore, MCC950 regulates inflammation via targeting both NLRP3-dependent canonical CASP1 and non-canonical activation of CASP11 required for IL-1β release and pyroptosis during endotoxemia in mice.343 Thus, this molecule has a great potential to be used during sepsis for controlling severe systemic inflammation and inflammatory organ damage. In addition, a ketone body called β-hydroxybutyrate is also shown to inhibit NLRP3 inflammasome via inhibiting K+ efflux and ASC oligomerization and prevent the synthesis and release of proinflammatory cytokines IL-1β and IL-18.344 Ketone bodies are small lipid-derived molecules that serve as a circulating energy source for tissues during shortage of energy, that is, due to fasting or prolonged exercise.345,346 An altered ketone body metabolism and changes in their levels in the systemic circulation due to its impaired synthesis by the liver during sepsis are observed.347–349 These ketone bodies exert anti-inflammatory action under diverse inflammatory conditions via different mechanisms including targeting and modulating HDACs and activating GPCRs and PPAR-α.346
PPARs are nuclear receptors that have three isoforms (ie, PPAR-α, PPAR-β, and PPAR-γ) and play an active role in various cellular processes (ie, lipid metabolism, glucose homeostasis, implantation, embryonic development, bone formation, adipogenesis, insulin sensitivity, inflammation, etc).350 The activation of PPAR-α and PPAR-γ via drugs such as fenofibrate and pioglitazone (PIO) has been found to inhibit NLRP3 inflammasome and CASP1 activation under diverse inflammatory conditions.351,352 Previously, it was found that the activation of PPAR-γ with rosiglitazone and PIO during sepsis prevented cardiac dysfunction and mortality, along with increasing bacterial clearance.353–356 A reduced expression of PPAR-α is associated with decreased survival and increased bacterial load in a polymicrobial sepsis model.357 This can be attributed to the decreased activation of NLRP3 inflammasome and CASP1 required for IL-1β and clearance of intracellular bacteria.357 Thus, β-hydroxybutyrate analogs, MCC950, and PPAR agonists including fenofibrate and PIO have a novel potential to be used during sepsis to prevent inflammatory organ damage and mortality.
Thalidomide has been found to exhibit protective action in a mouse model of sepsis and pneumonia due to its immunomodulatory action alone or in combination with antibiotics.358–361 A recent study has also shown that thalidomide exerts its immunomodulatory action by inhibiting NLRP3 inflammasome and CASP1 required to release IL-1β and IL-18.362 Thus, thalidomide is a broad-spectrum immunomodulatory agent that also inhibits inflammation via targeting NLRP3 inflammasome and CASP1 activation. Another molecule called cytokine release inhibitory drug 3 (CP-456773) blocks ASC oligomerization, a crucial step required for the activation of canonical inflammasome signaling pathway, and is also found to exert immunomodulatory action.363,364 Recently, a new molecule called OLT1177 (β-sulfonyl nitrile compound) was found to be orally active in inhibiting NLRP3 inflammasome oligomerization and activation through inhibiting the interaction between NLRP3 and ASC and the interaction between NLRP3 and CASP1.365 Treatment of a mouse model of endotoxemia/sepsis with the compound caused a significant decrease in IL-1β and IL-18 levels in tissue homogenates, oxidative stress, and an enhanced oxidative metabolism in the muscle tissues.365 Interestingly, this compound is considered safe in humans.
Shikonin is a highly lipophilic naphthoquinone derivative derived from the roots of the plant called Lithospermum erythrorhizon belonging to the Boraginaceae family.366 Shikonin and its derivatives are the major active components in the Chinese traditional medicine called Zicao which is used due to its antibacterial, antiviral, anti-inflammatory, and antipyretic action. A study by the authors has shown that shikonin inhibits the NLRP3 inflammasome-dependent maturation of proIL-1β as well as AIM2 inflammasome.366 Thus, it inhibits the process of ASC oligomerization along with directly inhibiting the activation of CASP1. Another plant extract derived from the plant Artemisia princeps of the Asteraceae family has also shown NLRP3 and AIM2 inhibitory action via reducing ASC phosphorylation, its oligomerization, and speck formation required for the activation of canonical inflammasome signaling.367,368 Treatment of animals with A. princeps extract also reduced the levels of the proinflammatory cytokines including IL-1β in a mouse model of peritonitis.368 Additionally, another naturally occurring compound known as sulforaphane isolated from the vegetables of Brassicaceae or Cruciferae was also found to inhibit NLRC4 and NLRP3 inflammasomes without affecting AIM2 in a mouse model of peritonitis.369 Sulforaphane directly inhibits the expression of NLRP3 gene and proIL-1β during the priming step and further inhibits the secretion of IL-1β and mtROS generation.369 Thus, the continuous development in molecular biology and immunology tools along with the advances in pharmacology and natural chemistry have led to the development and identification of novel inflammasome inhibitors with negligible toxic effects. Hence, molecules directly targeting inflammasomes can be ascribed as future therapeutic agents to target sepsis-associated inflammation and inflammatory organ damage, alone or in combination with other potential drugs and antibiotics. Inflammasome targeting comprises an area of sepsis research with great therapeutic potential.
Future perspective
Sepsis is a disease of dysregulation of immune response against an infection that can be easily contained in humans with normal immune response, but not in newborns or older group of people or persons having some chronic diseases (ie, type 2 diabetes mellitus [T2DM], obesity, etc) where it impacts the effectiveness of the innate immune response.370–374 Both diabetes and obesity are also associated with impaired functions of inflammasomes. For example, an overactivated NLRP3 inflammasome in the immune cells is associated with both obesity and insulin resistance and T2DM.375–380 Thus, one can speculate that pre-existing dysregulated inflammasome function may predispose these persons to develop sepsis from an infection that can be easily taken care of in otherwise healthy persons. One reason can be decreased autophagy during obesity and T2DM that has been observed in both conditions.381–385 Even the activation of FXR or BAR is shown to negatively regulate NLRP3 inflammasome and CASP1 activation via physical interaction during cholestasis-associated sepsis.386 The targeting of FXR (Fexaramine) also has a great potential to target obesity that have potential to impact NLRP3.387 Thus, these FXR agonists can be used to target NLRP3 inflammasome due to the negative effect of FXR activation on NLRP3 inflammasome and CASP1. For example, GW4064 is a molecular agonist for FXR and inhibits NLRP3 inflammasome in FXR-independent manner.388 GW4064 inhibits the oligomerization and ubiquitination of ASC, a critical step for NLRP3 inflammasome activation, and partially protects from the symptoms of NLRP3-dependent inflammatory diseases including sepsis in animal models. Hence, targeting specific inflammasomes, FXR, and autophagy during sepsis can be a future patient-based specific therapeutic approach. In addition to autophagy, other specific molecules or mediators released during early or late stages (ie, C5a, C3a, adenosine, ROS, HMGB1, H2S, prostanoids, eicosanoids, etc) of sepsis have great potential to impact inflammasome function and vice versa during the pathogenesis of sepsis.
Inflammasomes were first discovered by Dr Jürg Tschopp in the year 2002 to identify the LPS (responsible for endotoxemia and endotoxic shock during gram-negative bacterial sepsis)-mediated activation of CASP1 as an inflammatory complex playing a role in the release of IL-1β. Since their discovery, inflammasomes have been studied in detail in various chronic sterile inflammatory conditions including different tumors, neurodegenerative diseases, atherosclerosis, and autoimmune diseases such as systemic lupus erythematosus, rheumatoid arthritis, juvenile idiopathic arthritis, experimental autoimmune encephalomyelitis, and multiple sclerosis in humans. But continuous research in the field targeting host–pathogen interaction has also established them as potent intracellular PRRs for a wide variety of pathogens that can lead to the development of sepsis if remained unchecked without an effective clearance. Thus, a perfect time has come where we are equipped with advanced molecular biology, omics, and immunology tools for closing the Pandora’s box of inflammasomes to understand more precisely their role in the immunopathogenesis of sepsis and its targeting. For example, NLRP3 inflammasome-deficient mice show resistance toward polymicrobial sepsis due to increased production of proresolving lipid mediator lipoxin B4 that exerts anti-inflammatory and anti-pyroptotic effects.389 Future studies in the direction are immediately required as targeting inflammasomes has a novel potential to serve as a panacea for sepsis.
Disclosure
The author reports no conflicts of interest in this work.
References
Geroulanos S, Douka ET. Historical perspective of the word “sepsis”. Intensive Care Med. 2006;32:2077. | ||
Funk DJ, Parrillo JE, Kumar A. Sepsis and septic shock: a history. Crit Care Clin. 2009;25(83-101). | ||
Rittirsch D, Flierl MA, Ward PA. Harmful molecular mechanisms in sepsis. Nat Rev Immunol. 2008;8(10):776–787. | ||
Budelmann G. Hugo Schottmuller, 1867–1936. The problem of sepsis. Internist. 1969;10:92–101. | ||
Bone RC. Definitions for sepsis and organ failure. Crit Care Med. 1992;20:724–726. | ||
Bone RC, Balk RA, Cerra FB, et al. Definitions for sepsis and organ failure and guidelines for the use of innovative therapies in sepsis. The ACCP/SCCM Consensus Conference Committee. American College of Chest Physicians/Society of Critical Care Medicine. Chest. 1992;101:1644–1655. | ||
Singer M, Deutschman CS, Seymour CW, et al. The third international consensus definitions for sepsis and septic shock (sepsis-3). JAMA. 2016;315(8):801–810. | ||
Hotchkiss RS, Monneret G, Payen D. Immunosuppression in sepsis: a novel understanding of the disorder and a new therapeutic approach. Lancet Infect Dis. 2013a;13:260–268. | ||
Hotchkiss RS, Monneret G, Payen D. Sepsis-induced immunosuppression: from cellular dysfunctions to immunotherapy. Nat Rev Immunol. 2013;13(12):862–874. | ||
Hotchkiss RS, Moldawer LL, Opal SM, Reinhart K, Turnbull IR, Vincent JL. Sepsis and septic shock. Nat Rev Dis Primers. 2016;2:16045. | ||
Gül F, Arslantaş MK, Cinel İ, Kumar A. Changing definitions of sepsis. Turk J Anaesthesiol Reanim. 2017;45:129–138. | ||
Martin GS. Sepsis, severe sepsis and septic shock: changes in incidence, pathogens and outcomes. Expert Rev Anti Infect Ther. 2012;10(6):701–706. | ||
Angus DC, van der Poll T. Severe sepsis and septic shock. N Engl J Med. 2013;369:840–851. | ||
Colling KP, Banton KL, Beilman GJ. Vasopressors in sepsis. Surg Infect (Larchmt). 2018;19:202–207. | ||
Stearns-Kurosawa DJ, Osuchowski MF, Valentine C, Kurosawa S, Remick DG. The pathogenesis of sepsis. Annu Rev Pathol. 2011;6:19–48. | ||
Cabrera-Perez J, Condotta SA, Badovinac VP, Griffith TS. Impact of sepsis on CD4 T cell immunity. J Leukoc Biol. 2014;96:767–777. | ||
Wiersinga WJ, Leopold SJ, Cranendonk DR, van der Poll T. Host innate immune responses to sepsis. Virulence. 2014;5(1):36–44. | ||
Cao C, Ma T, Chai YF, Shou ST. The role of regulatory T cells in immune dysfunction during sepsis. World J Emerg Med. 2015;6(1):5–9. | ||
Delano MJ, Ward PA. The immune system’s role in sepsis progression, resolution, and long-term outcome. Immunol Rev. 2016;274(1):330–353. | ||
Delano MJ, Ward PA. Sepsis-induced immune dysfunction: can immune therapies reduce mortality? J Clin Invest. 2016;126(1):23–31. | ||
van der Poll T, van de Veerdonk FL, Scicluna BP, Netea MG. The immunopathology of sepsis and potential therapeutic targets. Nat Rev Immunol. 2017;17(7):407–420. | ||
Kumar V. T cells and their immunometabolism: a novel way to understanding sepsis immunopathogenesis and future therapeutics. Eur J Cell Biol. 2018a. | ||
Sundar KM, Sires M. Sepsis induced immunosuppression: implications for secondary infections and complications. Indian J Crit Care Med. 2013:17:162–169. | ||
Danahy DB, Strother RK, Badovinac VP, Griffith TS. Clinical and experimental sepsis impairs CD8 T-cell-mediated immunity. Crit Rev Immunol. 2016;36:57–74. | ||
Lamkanfi M, Dixit VM. Inflammasomes: guardians of cytosolic sanctity. Immunol Rev. 2009;227:95–105. | ||
Franchi L, Munoz-Planillo R, Reimer T, Eigenbrod T, Nunez G. Inflammasomes as microbial sensors. Eur J Immunol. 2010;40:611–615. | ||
Lamkanfi M, vande Walle L, Kanneganti TD. Deregulated inflammasome signaling in disease. Immunol Rev. 2011;243:163–173. | ||
Franchi L, Munoz-Planillo R, Nunez G. Sensing and reacting to microbes through the inflammasomes. Nat Immunol. 2012;13:325–332. | ||
Zmora N, Levy M, Pevsner-Fishcer M, Elinav E. Inflammasomes and intestinal inflammation. Mucosal Immunol. 2017;10:865–883. | ||
Alhallaf R, Agha Z, Miller CM, et al. The NLRP3 inflammasome suppresses protective immunity to gastrointestinal helminth infection. Cell Rep. 2018;23:1085–1098. | ||
Kayagaki N, Warming S, Lamkanfi M, et al. Non-canonical inflammasome activation targets caspase-11. Nature. 2011;479:117–121. | ||
Broz P, Monack DM. Noncanonical inflammasomes: caspase-11 activation and effector mechanisms. PLoS Pathog. 2013;9:e1003144. | ||
Latz E, Xiao TS, Stutz A. Activation and regulation of the inflammasomes. Nat Rev Immunol. 2013;13(6):397–411. | ||
von Moltke J, Ayres JS, Kofoed EM, Chavarría-Smith J, Vance RE. Recognition of bacteria by inflammasomes. Annu Rev Immunol. 2013;31:73–106. | ||
Monie TP. The canonical inflammasome: a macromolecular complex driving inflammation. Subcell Biochem. 2017;83:43–73. | ||
Pellegrini C, Antonioli L, Lopez-Castejon G, Blandizzi C, Fornai M. Canonical and non-canonical activation of NLRP3 inflammasome at the crossroad between immune tolerance and intestinal inflammation. Front Immunol. 2017;8(8):36. | ||
Próchnicki T, Latz E. Inflammasomes on the crossroads of innate immune recognition and metabolic control. Cell Metab. 2017;26(1):71–93. | ||
Kerur N, Fukuda S, Banerjee D, et al. cGAS drives noncanonical-inflammasome activation in age-related macular degeneration. Nat Med. 2018;24:50–61. | ||
Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell. 2002;10:417–426. | ||
Singhal G, Jaehne EJ, Corrigan F, Toben C, Baune BT. Inflammasomes in neuroinflammation and changes in brain function: a focused review. Front Neurosci. 2014;8(3). | ||
Freeman LC, Ting JP. The pathogenic role of the inflammasome in neurodegenerative diseases. J Neurochem. 2016;136(Suppl 1):29–38. | ||
Guo B, Fu S, Zhang J, Liu B, Li Z. Targeting inflammasome/IL-1 pathways for cancer immunotherapy. Sci Rep. 2016;6:36107. | ||
Nikolett L, David B, Ádám D. Inflammasomes link vascular disease with neuroinflammation and brain disorders. J Cereb Blood Flow Metab. 2016;36:1668–1685. | ||
Saresella M, La Rosa F, Piancone F, et al. The NLRP3 and NLRP1 inflammasomes are activated in Alzheimer’s disease. Mol Neurodegener. 2016;11(1):23. | ||
Kantono M, Guo B. Inflammasomes and cancer: the dynamic role of the inflammasome in tumor development. Front Immunol. 2017;8:1132. | ||
Lin C, Zhang J. Inflammasomes in inflammation-induced cancer. Front Immunol. 2017;8(Pt 1):271. | ||
Mamik MK, Power C. Inflammasomes in neurological diseases: emerging pathogenic and therapeutic concepts. Brain. 2017;140(9):2273–2285. | ||
Elinav E, Thaiss CA, Flavell RA. Analysis of microbiota alterations in inflammasome-deficient mice. Methods Mol Biol. 2013;1040:185–194. | ||
Sester DP, Sagulenko V, Thygesen SJ, et al. Deficient NLRP3 and AIM2 inflammasome function in autoimmune NZB mice. J Immunol. 2015;195(3):1233–1241. | ||
Thygesen SJ, Sester DP, Cridland SO, Wilton SD, Stacey KJ. Correcting the NLRP3 inflammasome deficiency in macrophages from autoimmune NZB mice with exon skipping antisense oligonucleotides. Immunol Cell Biol. 2016;94(5):520. | ||
Kim YM, Brinkmann MM, Paquet ME, Ploegh HL. UNC93B1 delivers nucleotide-sensing toll-like receptors to endolysosomes. Nature. 2008;452:234–238. | ||
Mogensen TH. Pathogen recognition and inflammatory signaling in innate immune defenses. Clin Microbiol Rev. 2009;22(2):240–273. | ||
Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell. 2010;140(6):805–820. | ||
Wang Y, Yang C, Mao K, Chen S, Meng G, Sun B. Cellular localization of NLRP3 inflammasome. Protein Cell. 2013;4(6):425–431. | ||
Lamkanfi M, Dixit VM. Mechanisms and functions of inflammasomes. Cell. 2014;157:1013–1022. | ||
Man SM, Kanneganti T-D. Regulation of inflammasome activation. Immunol Rev. 2015;265(1):6–21. | ||
de Zoete MR, Palm NW, Zhu S, Flavell RA. Inflammasomes. Cold Spring Harb Perspect Biol. 2014;6:a016287. | ||
Rathinam VAK, Fitzgerald KA. inflammasome complexes: emerging mechanisms and effector functions. Cell. 2016;165(4):792–800. | ||
Lopez-Castejon G, Brough D. Understanding the mechanism of IL-1β secretion. Cytokine Growth Factor Rev. 2011;22(4):189–195. | ||
Kang S, Fernandes-Alnemri T, Rogers C, et al. Caspase-8 scaffolding function and MLKL regulate NLRP3 inflammasome activation downstream of TLR3. Nat Commun. 2015;6:7515. | ||
Zhang X, Fan C, Zhang H, et al. MLKL and FADD are critical for suppressing progressive lymphoproliferative disease and activating the NLRP3 inflammasome. Cell Rep. 2016;16(12):3247–3259. | ||
Conos SA, Chen KW, de Nardo D, et al. Active MLKL triggers the NLRP3 inflammasome in a cell-intrinsic manner. Proc Natl Acad Sci U S A. 2017;114:E961–e969. | ||
Gutierrez KD, Davis MA, Daniels BP, et al. MLKL activation triggers NLRP3-mediated processing and release of IL-1beta independently of gasdermin-D. J Immunol. 2017;198:2156–2164. | ||
Weinlich R, Oberst A, Beere HM, Green DR. Necroptosis in development, inflammation and disease. Nat Rev Mol Cell Biol. 2017;18(2):127–136. | ||
Sharma AA, Jen R, Kan B, et al. Impaired NLRP3 inflammasome activity during fetal development regulates IL-1β production in human monocytes. Eur J Immunol. 2015;45(1):238–249. | ||
Stouch AN, Mccoy AM, Greer RM, et al. IL-1β and inflammasome activity link inflammation to abnormal fetal airway development. J Immunol. 2016;196(8):3411–3420. | ||
Arango Duque G, Descoteaux A. Macrophage cytokines: involvement in immunity and infectious diseases. Front Immunol. 2014;5:491. | ||
Dinarello CA, Cannon JG, Mier JW, et al. Multiple biological activities of human recombinant interleukin 1. J Clin Invest. 1986;77:1734–1739. | ||
Radeke HH, Martin M, Topley N, Kaever V, Resch K. Differential biological activities of human interleukin-1 alpha and interleukin-1 beta. Eur Cytokine Netw. 1991;2:51–59. | ||
Reznikov LL, Kim S-H, Westcott JY, et al. IL-18 binding protein increases spontaneous and IL-1-induced prostaglandin production via inhibition of IFN-gamma. Proc Natl Acad Sci U S A. 2000;97(5):2174–2179. | ||
Lee JK, Kim SH, Lewis EC, Azam T, Reznikov LL, Dinarello CA. Differences in signaling pathways by IL-1beta and IL-18. Proc Natl Acad Sci U S A. 2004;101:8815–8820. | ||
Keyel PA. How is inflammation initiated? Individual influences of IL-1, IL-18 and HMGB1. Cytokine. 2014;69:136–145. | ||
Gatti S, Beck J, Fantuzzi G, Bartfai T, Dinarello CA. Effect of interleukin-18 on mouse core body temperature. Am J Physiol Regul Integr Comp Physiol. 2002;282:R702–709. | ||
Komai-Koma M, Gracie JA, Wei X-Q, et al. Chemoattraction of human T cells by IL-18. J Immunol. 2003;170:1084–1090. | ||
Gutcher I, Becher B. APC-derived cytokines and T cell polarization in autoimmune inflammation. J Clin Invest. 2007;117:1119–1127. | ||
Netea MG, Joosten LAB, Lewis E, et al. Deficiency of interleukin-18 in mice leads to hyperphagia, obesity and insulin resistance. Nat Med. 2006;12(6):650–656. | ||
Zorrilla EP, Sanchez-Alavez M, Sugama S, et al. Interleukin-18 controls energy homeostasis by suppressing appetite and feed efficiency. Proc Natl Acad Sci U S A. 2007;104:11097–11102. | ||
Gagliani N, Palm NW, de Zoete MR, Flavell RA. Inflammasomes and intestinal homeostasis: regulating and connecting infection, inflammation and the microbiota. Int Immunol. 2014;26:495–499. | ||
Rathinam VAK, Chan FK-M. Inflammasome, inflammation, and tissue homeostasis. Trends Mol Med. 2018;24(3):304–318. | ||
Elinav E, Strowig T, Kau AL, et al. NLRP6 inflammasome regulates colonic microbial ecology and risk for colitis. Cell. 2011;145:745–757. | ||
Levy M, Thaiss CA, Zeevi D, et al. Microbiota-modulated metabolites shape the intestinal microenvironment by regulating NLRP6 inflammasome signaling. Cell. 2015;163:1428–1443. | ||
Lobo LA, Benjamim CF, Oliveira AC. The interplay between microbiota and inflammation: lessons from peritonitis and sepsis. Clin Transl Immunology. 2016;5(7):e90. | ||
Cabrera-Perez J, Badovinac VP, Griffith TS. Enteric immunity, the gut microbiome, and sepsis: rethinking the germ theory of disease. Exp Biol Med (Maywood). 2017;242:127–139. | ||
Lei-Leston AC, Murphy AG, Maloy KJ. Epithelial cell inflammasomes in intestinal immunity and inflammation. Front Immunol. 2017;8. | ||
Medzhitov R. Pattern recognition theory and the launch of modern innate immunity. J Immunol. 2013;191(9):4473–4474. | ||
Janeway CA., Jr. Approaching the asymptote? Evolution and revolution in immunology. Cold Spring Harb Symp Quant Biol. 1989;54(Pt 1):1–13. | ||
Hart SP, Smith JR, Dransfield I. Phagocytosis of opsonized apoptotic cells: roles for ‘old-fashioned’ receptors for antibody and complement. Clin Exp Immunol. 2004;135:181–185. | ||
Taylor PR, Martinez-Pomares L, Stacey M, Lin HH, Brown GD, Gordon S. Macrophage receptors and immune recognition. Annu Rev Immunol. 2005;23:901–944. | ||
Chen G, Shaw MH, Kim YG, Nunez G. NOD-like receptors: role in innate immunity and inflammatory disease. Annu Rev Pathol. 2009;4:365–398. | ||
Martinon F, Mayor A, Tschopp J. The inflammasomes: guardians of the body. Annu Rev Immunol. 2009;27(1):229–265. | ||
Nakhaei P, Genin P, Civas A, Hiscott J. RIG-I-like receptors: sensing and responding to RNA virus infection. Semin Immunol. 2009;21(4):215–222. | ||
Jacobs SR, Damania B. NLRs, inflammasomes, and viral infection. J Leukoc Biol. 2012;92:469–477. | ||
Schmidt A, Rothenfusser S, Hopfner K-P. Sensing of viral nucleic acids by RIG-I: from translocation to translation. Eur J Cell Biol. 2012;91(1):78–85. | ||
Bauernfeind F, Hornung V. Of inflammasomes and pathogens – sensing of microbes by the inflammasome. EMBO Mol Med. 2013;5:814–826. | ||
Takeda K, Akira S. Toll-like receptors. Curr Protoc Immunol. 2015;10912.(14):11–10. | ||
Satoh T, Akira S. Toll-like receptor signaling and its inducible proteins. Microbiol Spectr. 2016;4. | ||
Tartey S, Takeuchi O. Pathogen recognition and Toll-like receptor targeted therapeutics in innate immune cells. Int Rev Immunol. 2017;36(2):57–73. | ||
Zhang H, Luo J, Alcorn JF, et al. AIM2 inflammasome is critical for influenza-induced lung injury and mortality. J Immunol. 2017;198(11):4383–4393. | ||
Kumar V, Sharma A. Neutrophils: Cinderella of innate immune system. Int Immunopharmacol. 2010;10(11):1325–1334. | ||
Kumar V, Sharma A. Mast cells: emerging sentinel innate immune cells with diverse role in immunity. Mol Immunol. 2010a;48:14–25. | ||
Kumar V, Ahmad A. Role of MAIT cells in the immunopathogenesis of inflammatory diseases: new players in old game. Int Rev Immunol. 2018;37:90–110. | ||
Boyd JH, Divangahi M, Yahiaoui L, Gvozdic D, Qureshi S, Petrof BJ. Toll-like receptors differentially regulate CC and CXC chemokines in skeletal muscle via NF-κB and calcineurin. Infect Immun. 2006;74:6829–6838. | ||
Salomão R, Martins PS, Brunialti MKC, et al. TLR signaling pathway in patients with sepsis. Shock. 2008;30(Suppl 1):73–77. | ||
Tsujimoto H, Ono S, Efron PA, Scumpia PO, Moldawer LL, Mochizuki H. Role of Toll-like receptors in the development of sepsis. Shock. 2008;29(3):315–321. | ||
Savva A, Roger T. Targeting Toll-like receptors: promising therapeutic strategies for the management of sepsis-associated pathology and infectious diseases. Front Immunol. 2013;18(4):387. | ||
Turner MD, Nedjai B, Hurst T, Pennington DJ. Cytokines and chemokines: at the crossroads of cell signalling and inflammatory disease. Biochim Biophys Acta. 2014;1843(11):2563–2582. | ||
Franchi L, Eigenbrod T, Muñoz-Planillo R, Nuñez G. The inflammasome: a caspase-1 activation platform regulating immune responses and disease pathogenesis. Nat Immunol. 2009;10:241. | ||
Taxman DJ, Huang MT, Ting JP. Inflammasome inhibition as a pathogenic stealth mechanism. Cell Host Microbe. 2010;8(1):7–11. | ||
Menu P, Vince JE. The NLRP3 inflammasome in health and disease: the good, the bad and the ugly. Clin Exp Immunol. 2011;166(1):1–15. | ||
Chavarria-Smith J, Vance RE. The NLRP1 inflammasomes. Immunol Rev. 2015;265:22–34. | ||
Zhao Y, Shao F. The NAIP–NLRC4 inflammasome in innate immune detection of bacterial flagellin and type III secretion apparatus. Immunol Rev. 2015;265:85–102. | ||
Krueger M, Heinzmann A, Nauck M. Adhesion molecules in pediatric intensive care patients with organ dysfunction syndrome. Intensive Care Med. 2007;33:359–363. | ||
Zonneveld R, Martinelli R, Shapiro NI, Kuijpers TW, Plötz FB, Carman CV. Soluble adhesion molecules as markers for sepsis and the potential pathophysiological discrepancy in neonates, children and adults. Crit Care. 2014;18:204–204. | ||
Amalakuhan B, Habib SA, Mangat M, et al. Endothelial adhesion molecules and multiple organ failure in patients with severe sepsis. Cytokine. 2016;88:267–273. | ||
Whalen MJ, Doughty LA, Carlos TM, Wisniewski SR, Kochanek PM, Carcillo JA. Intercellular adhesion molecule-1 and vascular cell adhesion molecule-1 are increased in the plasma of children with sepsis-induced multiple organ failure. Crit Care Med. 2000;28(7):2600–2607. | ||
Sosa-Bustamante GP, Amador-Licona N, Barbosa-Sabanero G, et al. Intercellular adhesion molecules and mortality for sepsis in infants younger than 1 year of life. Rev Invest Clin. 2011;63:601–606. | ||
Su CM, Cheng HH, Tsai TC, et al. Elevated serum vascular cell adhesion molecule-1 is associated with septic encephalopathy in adult community-onset severe sepsis patients. Biomed Res Int. 2014;2014:10. | ||
Sager HB, Dutta P, Dahlman JE, et al. RNAi targeting multiple cell adhesion molecules reduces immune cell recruitment and vascular inflammation after myocardial infarction. Sci Transl Med. 2016;8342(342):ra380:. | ||
Hildebrand F, Pape H-C, Harwood P, et al. Role of adhesion molecule ICAM in the pathogenesis of polymicrobial sepsis. Exp Toxicol Pathol. 2005;56:281–290. | ||
Chi Z, Melendez AJ. Role of cell adhesion molecules and immune-cell migration in the initiation, onset and development of atherosclerosis. Cell Adh Migr. 2007;1:171–175. | ||
Chae YK, Choi WM, Bae WH, et al. Overexpression of adhesion molecules and barrier molecules is associated with differential infiltration of immune cells in non-small cell lung cancer. Sci Rep. 2018;8:1023. | ||
Bauer PR. Microvascular responses to sepsis: clinical significance. Pathophysiology. 2002;8:141–148. | ||
Reinhart K, Bayer O, Brunkhorst F, Meisner M. Markers of endothelial damage in organ dysfunction and sepsis. Crit Care Med. 2002;30(Supplement):S302–S312. | ||
Inoue M, Williams KL, Gunn MD, Shinohara ML. NLRP3 inflammasome induces chemotactic immune cell migration to the CNS in experimental autoimmune encephalomyelitis. Proc Natl Acad Sci U S A. 2012;109:10480–10485. | ||
Wang JG, Williams JC, Davis BK, et al. Monocytic microparticles activate endothelial cells in an IL-1β-dependent manner. Blood. 2011;118(8):2366–2374. | ||
Boisrame-Helms J, Kremer H, Schini-Kerth V, Meziani F. Endothelial dysfunction in sepsis. Curr Vasc Pharmacol. 2013;11:150–160. | ||
Ince C, Mayeux PR, Nguyen T, et al. The endothelium in sepsis. Shock. 2016;45:259–270. | ||
Chen Y, Li X, Boini KM, et al. Endothelial Nlrp3 inflammasome activation associated with lysosomal destabilization during coronary arteritis. Biochim Biophys Acta. 2015;1853:396–408. | ||
Luo S-F, Chin C-Y, Ho L-J, Tseng W-Y, Kuo C-F, Lai J-H. Monosodium urate crystals induced ICAM-1 expression and cell–cell adhesion in renal mesangial cells: implications for the pathogenesis of gouty nephropathy. J Microbiol Immunol Infect. 2018;S1684–1182(18)30003 | ||
Hewett JA, Schultze AE, Vancise S, Roth RA. Neutrophil depletion protects against liver injury from bacterial endotoxin. Lab Invest. 1992;66:347–361. | ||
Tsuji C, Minhaz MU, Shioya S, Fukahori M, Tanigaki T, Nakazawa H. The importance of polymorphonuclear leukocytes in lipopolysaccharide-induced superoxide anion production and lung injury: ex vivo observation in rat lungs. Lung. 1998;176(1):1–13. | ||
Raeburn CD, Calkins CM, Zimmerman MA, et al. ICAM-1 and VCAM-1 mediate endotoxemic myocardial dysfunction independent of neutrophil accumulation. Am J Physiol Regul Integr Comp Physiol. 2002;283(2):R477–R486. | ||
Yang L, Froio RM, Sciuto TE, Dvorak AM, Alon R, Luscinskas FW. ICAM-1 regulates neutrophil adhesion and transcellular migration of TNF-alpha-activated vascular endothelium under flow. Blood. 2005;106(2):584–592. | ||
Xu R, Huang H, Zhang Z, Wang FS. The role of neutrophils in the development of liver diseases. Cell Mol Immunol. 2014;11(3):224–231. | ||
Woodfin A, Beyrau M, Voisin MB, et al. ICAM-1-expressing neutrophils exhibit enhanced effector functions in murine models of endotoxemia. Blood. 2016;127(7):898–907. | ||
van der Heijden T, Kritikou E, Venema W. NLRP3 inflammasome inhibition by MCC950 reduces atherosclerotic lesion development in apolipoprotein E–deficient mice. Arterioscler Thromb Vasc Biol. 2017;37(8):1457–1461. | ||
Bakele M, Joos M, Burdi S, et al. Localization and functionality of the inflammasome in neutrophils. J Biol Chem. 2014;289:5320–5329. | ||
Pérez-Figueroa E, Torres J, Sánchez-Zauco N, Contreras-Ramos A, Alvarez-Arellano L, Maldonado-Bernal C. Activation of NLRP3 inflammasome in human neutrophils by Helicobacter pylori infection. Innate Immun. 2016;22(2):103–112. | ||
Alfonso-Loeches S, Ureña-Peralta JR, Morillo-Bargues MJ, Oliver-de La Cruz J, Guerri C. Role of mitochondria ROS generation in ethanol-induced NLRP3 inflammasome activation and cell death in astroglial cells. Front Cell Neurosci. 2014;1(8):216. | ||
Harijith A, Ebenezer DL, Natarajan V. Reactive oxygen species at the crossroads of inflammasome and inflammation. Front Physiol. 2014;5:352. | ||
Abais JM, Xia M, Zhang Y, Boini KM, Li P-L. Redox regulation of NLRP3 inflammasomes: ROS as trigger or effector? Antioxid Redox Signal. 2015;22:1111–1129. | ||
Andersson U, Tracey KJ. HMGB1 in sepsis. Scand J Infect Dis. 2003;35:577–584. | ||
Huang W, Tang Y, Li L. HMGB1, a potent proinflammatory cytokine in sepsis. Cytokine. 2010;51:119–126. | ||
Kaczmarek A, Vandenabeele P, Krysko Dv. Necroptosis: the release of damage-associated molecular patterns and its physiological relevance. Immunity. 2013;38:209–223. | ||
Zhao H, Jaffer T, Eguchi S, Wang Z, Linkermann A, Ma D. Role of necroptosis in the pathogenesis of solid organ injury. Cell Death Dis. 2015;6:e1975. | ||
Fiuza C, Bustin M, Talwar S, et al. Inflammation-promoting activity of HMGB1 on human microvascular endothelial cells. Blood. 2003;101:2652–2660. | ||
Asavarut P, Zhao H, Gu J, Ma D. The role of HMGB1 in inflammation-mediated organ injury. Acta Anaesthesiol Taiwan. 2013;51:28–33. | ||
Lui G, Wong CK, Ip M, et al. HMGB1/RAGE signaling and pro-inflammatory cytokine responses in non-HIV adults with active pulmonary tuberculosis. PLoS One. 2016;11(7):e0159132. | ||
Abdulmahdi W, Patel D, Rabadi MM, et al. HMGB1 redox during sepsis. Redox Biol. 2017;13:600–607. | ||
Stevens NE, Chapman MJ, Fraser CK, Kuchel TR, Hayball JD, Diener KR. Therapeutic targeting of HMGB1 during experimental sepsis modulates the inflammatory cytokine profile to one associated with improved clinical outcomes. Sci Rep. 2017;7(1):5850. | ||
Willingham SB, Allen IC, Bergstralh DT, et al. NLRP3 (NALP3, Cryopyrin) facilitates in vivo caspase-1 activation, necrosis, and HMGB1 release via inflammasome-dependent and -independent pathways. J Immunol. 2009;183(3):2008–2015. | ||
Lamkanfi M, Sarkar A, vande Walle L, et al. Inflammasome-dependent release of the alarmin HMGB1 in endotoxemia. J Immunol. 2010;185:4385–4392. | ||
vande Walle L, Kanneganti TD, Lamkanfi M. HMGB1 release by inflammasomes. Virulence. 2011;2(2):162–165. | ||
Leblanc PM, Doggett TA, Choi J, et al. An immunogenic peptide in the A-box of HMGB1 protein reverses apoptosis-induced tolerance through RAGE receptor. J Biol Chem. 2014;289:7777–7786. | ||
Lu B, Wang H, Andersson U, Tracey KJ. Regulation of HMGB1 release by inflammasomes. Protein Cell. 2013;4(3):163–167. | ||
Scaffidi P, Misteli T, Bianchi ME. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature. 2002;418(6894):191–195. | ||
Lu B, Nakamura T, Inouye K, et al. Novel role of PKR in inflammasome activation and HMGB1 release. Nature. 2012;488(7413):670–674. | ||
Bonaldi T, Talamo F, Scaffidi P, et al. Monocytic cells hyperacetylate chromatin protein HMGB1 to redirect it towards secretion. EMBO J. 2003;22:5551–5560. | ||
Evankovich J, Cho SW, Zhang R, et al. High mobility group box 1 release from hepatocytes during ischemia and reperfusion injury is mediated by decreased histone deacetylase activity. J Biol Chem. 2010;285:39888–39897. | ||
Chi W, Chen H, Li F, Zhu Y, Yin W, Zhuo Y. HMGB1 promotes the activation of NLRP3 and caspase-8 inflammasomes via NF-kappaB pathway in acute glaucoma. J Neuroinflammation. 2015;12:137. | ||
Abu El-Asrar AM, Alam K, Garcia-Ramirez M, et al. Association of HMGB1 with oxidative stress markers and regulators in PDR. Mol Vis. 2017;23:853–871. | ||
Watson RW, Redmond HP, Wang JH, Condron C, Bouchier-Hayes D. Neutrophils undergo apoptosis following ingestion of Escherichia coli. J Immunol. 1996;156(10):3986–3992. | ||
Deleo FR. Modulation of phagocyte apoptosis by bacterial pathogens. Apoptosis. 2004;9:399–413. | ||
Wilkie RP, Vissers MC, Dragunow M, Hampton MB. A functional NADPH oxidase prevents caspase involvement in the clearance of phagocytic neutrophils. Infect Immun. 2007;75(7):3256–3263. | ||
Victor VM, Rocha M, de La Fuente M. Immune cells: free radicals and antioxidants in sepsis. Int Immunopharmacol. 2004;4(3):327–347. | ||
Víctor VM, Espulgues JV, Hernández-Mijares A, Rocha M. Oxidative stress and mitochondrial dysfunction in sepsis: a potential therapy with mitochondria-targeted antioxidants. Infect Disord Drug Targets. 2009;9(4):376–389. | ||
Bime C, Zhou T, Wang T, Slepian MJ, Garcia JGN, Hecker L. Reactive oxygen species–associated molecular signature predicts survival in patients with sepsis. Pulm Circ. 2016;6:196–201. | ||
Oliveira Ypade, Pontes-de-Carvalho LC, Couto RD, Noronha-Dutra AA. Oxidative stress in sepsis. Possible production of free radicals through an erythrocyte-mediated positive feedback mechanism. Braz J Infect Dis. 2017;21(1):19–26. | ||
Cepinskas G, Wilson JX. Inflammatory response in microvascular endothelium in sepsis: role of oxidants. J Clin Biochem Nutr. 2008;42:175–184. | ||
Huet O, Dupic L, Harrois A, Duranteau J. Oxidative stress and endothelial dysfunction during sepsis. Front Biosci (Landmark Ed). Vol. 16; 2011:1986–1995. | ||
Potz BA, Sellke FW, Abid MR. Endothelial ROS and impaired myocardial oxygen consumption in sepsis-induced cardiac dysfunction. J Intensive Crit Care. 2016;02(01):20. | ||
Léger T, Charrier A, Moreau C, et al. Early sepsis does not stimulate reactive oxygen species production and does not reduce cardiac function despite an increased inflammation status. Physiol Rep. 2017;5:e13231. | ||
Hoetzenecker W, Echtenacher B, Guenova E, et al. ROS-induced ATF3 causes susceptibility to secondary infections during sepsis-associated immunosuppression. Nat Med. 2011;18:128. | ||
Cruz CM, Rinna A, Forman HJ, Ventura AL, Persechini PM, Ojcius DM. ATP activates a reactive oxygen species-dependent oxidative stress response and secretion of proinflammatory cytokines in macrophages. J Biol Chem. 2007;282:2871–2879. | ||
Martinon F. Signaling by ROS drives inflammasome activation. Eur J Immunol. 2010;40(3):616–619. | ||
He Y, Hara H, Núñez G. Mechanism and regulation of NLRP3 inflammasome activation. Trends Biochem Sci. 2016;41:1012–1021. | ||
Kumar V. Targeting macrophage immunometabolism: dawn in the darkness of sepsis. Int Immunopharmacol. 2018;58:173–185. | ||
Abais JM, Zhang C, Xia M, et al. NADPH oxidase-mediated triggering of inflammasome activation in mouse podocytes and glomeruli during hyperhomocysteinemia. Antioxid Redox Signal. 2013;18:1537–1548. | ||
Boini KM, Xia M, Abais JM, et al. Activation of inflammasomes in podocyte injury of mice on the high fat diet: effects of ASC gene deletion and silencing. Biochim Biophys Acta. 2014;1843:836–845. | ||
Han S, Cai W, Yang X, et al. ROS-mediated NLRP3 inflammasome activity is essential for burn-induced acute lung injury. Mediators Inflamm. 2015;2015:16. | ||
Sumi Y, Woehrle T, Chen Y, et al. Plasma ATP is required for neutrophil activation in a mouse sepsis model. Shock. 2014;42(2):142–147. | ||
Ledderose C, Li X, Bao Y, Sumi Y, Shapiro N, Junger W. Systemic ATP levels suppress the function of CD4+ T cells in sepsis by impairing autocrine purinergic signaling. FASEB J. 2015;29:972–976. | ||
Ledderose C, Bao Y, Kondo Y, et al. Purinergic signaling and immune responses in sepsis. Clin Ther. 2016;38:1054–1065. | ||
Sueyoshi K, Sumi Y, Inoue Y, et al. Fluorescence imaging of ATP in neutrophils from patients with sepsis using organelle-localizable fluorescent chemosensors. Ann Intensive Care. 2016;6(1):64. | ||
Li X, Kondo Y, Bao Y, et al. Systemic adenosine triphosphate impairs neutrophil chemotaxis and host defense in sepsis. Crit Care Med. 2017;45(1):e97–e104. | ||
Dubyak GR. Signal transduction by P2-purinergic receptors for extracellular ATP. Am J Respir Cell Mol Biol. 1991;4:295–300. | ||
Buzzi N, Bilbao PS, Boland R, de Boland AR. Extracellular ATP activates MAP kinase cascades through a P2Y purinergic receptor in the human intestinal Caco-2 cell line. Biochim Biophys Acta. 2009;1790:1651–1659. | ||
Säve S, Persson K. Extracellular ATP and P2Y receptor activation induce a proinflammatory host response in the human urinary tract. Infect Immun. 2010;78(8):3609–3615. | ||
Riteau N, Gasse P, Fauconnier L, et al. Extracellular ATP is a danger signal activating P2X 7 receptor in lung inflammation and fibrosis. Am J Respir Crit Care Med. 2010;182(6):774–783. | ||
Garg AD, Krysko DV, Vandenabeele P, Agostinis P. Extracellular ATP and P2X7 receptor exert context-specific immunogenic effects after immunogenic cancer cell death. Cell Death Dis. 2016;7:e2097. | ||
di Virgilio F, dal Ben D, Sarti AC, Giuliani AL, Falzoni S. The P2X7 receptor in infection and inflammation. Immunity. 2017;47:15–31. | ||
Karmakar M, Katsnelson MA, Dubyak GR, Pearlman E. Neutrophil P2X7 receptors mediate NLRP3 inflammasome-dependent IL-1β secretion in response to ATP. Nat Commun. 2016;7:10555. | ||
di Virgilio F. Liaisons dangereuses: P2X(7) and the inflammasome. Trends Pharmacol Sci. 2007;28:465–472. | ||
Coutinho-Silva R, Ojcius DM. Role of extracellular nucleotides in the immune response against intracellular bacteria and protozoan parasites. Microbes Infect. 2012;14:1271–1277. | ||
Gombault A, Baron L, Couillin I. ATP release and purinergic signaling in NLRP3 inflammasome activation. Front Immunol. 2012;3:414. | ||
Idzko M, Ferrari D, Eltzschig HK. Nucleotide signalling during inflammation. Nature. 2014;509:310–317. | ||
Csoka B, Nemeth ZH, Toro G, et al. Extracellular ATP protects against sepsis through macrophage P2X7 purinergic receptors by enhancing intracellular bacterial killing. FASEB J. 2015;29:3626–3637. | ||
Wu X, Ren J, Chen G, et al. Systemic blockade of P2X7 receptor protects against sepsis-induced intestinal barrier disruption. Sci Rep. 2017;7(1):4364. | ||
Nichols RD, von Moltke J, Vance RE. NAIP/NLRC4 inflammasome activation in MRP8+ cells is sufficient to cause systemic inflammatory disease. Nat Commun. 2017;8(1):2209. | ||
Kumar V, Sharma A. Adenosine: an endogenous modulator of innate immune system with therapeutic potential. Eur J Pharmacol. 2009;616:7–15. | ||
Kumar V. Adenosine as an endogenous immunoregulator in cancer pathogenesis: where to go? Purinergic Signal. 2013;9:145–165. | ||
Bao R, Hou J, Li Y, et al. Adenosine promotes Foxp3 expression in Treg cells in sepsis model by activating JNK/AP-1 pathway. Am J Transl Res. 2016;8:2284–2292. | ||
Martin C, Leone M, Viviand X, Ayem M-L, Guieu R. High adenosine plasma concentration as a prognostic index for outcome in patients with septic shock. Crit Care Med. 2000;28(9):3198–3202. | ||
Sperlagh B, Doda M, Baranyi M, Hasko G. Ischemic-like condition releases norepinephrine and purines from different sources in superfused rat spleen strips. J Neuroimmunol. 2000;111:45–54. | ||
Ouyang X, Ghani A, Malik A, et al. Adenosine is required for sustained inflammasome activation via the A2A receptor and the HIF-1α pathway. Nat Commun. 2013;4(1):2909. | ||
Kumar V, Chhibber S. Intravenous 2-chloroadenosine protects BALB/c mice from Klebsiella pneumoniae B5055-induced sepsis by modulating the pro-inflammatory immune response. J Chemother. 2009;21:639–645. | ||
Csoka B, Nemeth ZH, Rosenberger P, et al. A2B adenosine receptors protect against sepsis-induced mortality by dampening excessive inflammation. J Immunol. 2010;185:542–550. | ||
Kumar V, Harjai K, Chhibber S. 2-Chloroadenosine (2-CADO) treatment modulates the pro-inflammatory immune response to prevent acute lung inflammation in BALB/c mice suffering from Klebsiella pneumoniae B5055-induced pneumonia. Int J Antimicrob Agents. 2010;35(6):599–602. | ||
Sivak KV, Vasin AV, Egorov VV, et al. Adenosine A2A receptor as a drug target for treatment of sepsis. Mol Biol. 2016;50(2):200–212. | ||
Nonaka M, Kimura A. Genomic view of the evolution of the complement system. Immunogenetics. 2006;58(9):701–713. | ||
Nonaka M. Evolution of the complement system. Subcell Biochem. 2014;80:31–43. | ||
Merle NS, Church SE, Fremeaux-Bacchi V, Roumenina LT. Complement system part I – molecular mechanisms of activation and regulation. Front Immunol. 2015;6(4):262. | ||
Merle NS, Noe R, Halbwachs-Mecarelli L, Fremeaux-Bacchi V, Roumenina LT. Complement system part II: role in immunity. Front Immunol. 2015;6(180):257. | ||
Ward PA. The dark side of C5a in sepsis. Nat Rev Immunol. 2004;4(2):133. | ||
Gressner OA, Koch A, Sanson E, Trautwein C, Tacke F. High C5a levels are associated with increased mortality in sepsis patients-no enhancing effect by actin-free Gc-globulin. Clin Biochem. 2008;41:974–980. | ||
Markiewski MM, Deangelis RA, Lambris JD. Complexity of complement activation in sepsis. J Cell Mol Med. 2008;12(6a):2245–2254. | ||
Charchaflieh J, Wei J, Labaze G, et al. The role of complement system in septic shock. Clin Dev Immunol. 2012;2012:8. | ||
Yan C, Gao H. New insights for C5a and C5a receptors in sepsis. Front Immunol. 2012;3:368. | ||
Ricklin D, Reis ES, Lambris JD. Complement in disease: a defence system turning offensive. Nat Rev Nephrol. 2016;12(7):383–401. | ||
Suresh R, Chandrasekaran P, Sutterwala FS, Mosser DM. Complement-mediated ‘bystander’ damage initiates host NLRP3 inflammasome activation. J Cell Sci. 2016;129(9):1928–1939. | ||
Laudisi F, Spreafico R, Evrard M, et al. Cutting edge: the NLRP3 inflammasome links complement-mediated inflammation and IL-1β release. J Immunol. 2013;191:1006–1010. | ||
Triantafilou M, Hughes TR, Morgan BP, Triantafilou K. Complementing the inflammasome. Immunology. 2016;147(2):152–164. | ||
Murakami T, Ockinger J, Yu J, et al. Critical role for calcium mobilization in activation of the NLRP3 inflammasome. Proc Natl Acad Sci U S A. 2012;109(28):11282–11287. | ||
Horng T. Calcium signaling and mitochondrial destabilization in the triggering of the NLRP3 inflammasome. Trends Immunol. 2014;35:253–261. | ||
Shimada K, Crother TR, Karlin J, et al. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity. 2012;36(3):401–414. | ||
Luan Y-Y, Yao Y-M, Xiao X-Z, Sheng Z-Y. Insights into the apoptotic death of immune cells in sepsis. J Interferon Cytokine Res. 2015;35(1):17–22. | ||
Fang C, Wei X, Wei Y. Mitochondrial DNA in the regulation of innate immune responses. Protein Cell. 2016;7:11–16. | ||
Liszewski MK, Kolev M, LeFriec G, et al. Intracellular complement activation sustains T cell homeostasis and mediates effector differentiation. Immunity. 2013;39(6):1143–1157. | ||
Arbore G, West EE, Spolski R, et al. T helper 1 immunity requires complement-driven NLRP3 inflammasome activity in CD4+ T cells. Science. 2016;352(6292):aad1210-aad1210. | ||
Iwasaka H, Noguchi T. Th1/Th2 balance in systemic inflammatory response syndrome (SIRS). Nihon Rinsho. 2004;62:2237–2243. | ||
Kalbitz M, Fattahi F, Grailer JJ, et al. Complement-induced activation of the cardiac NLRP3 inflammasome in sepsis. FASEB J. 2016;30:3997–4006. | ||
Asgari E, Le Friec G, Yamamoto H, et al. C3a modulates IL-1beta secretion in human monocytes by regulating ATP efflux and subsequent NLRP3 inflammasome activation. Blood. 2013;122:3473–3481. | ||
Dinarello CA. The C3a receptor, caspase-1, and release of IL-1beta. Blood. 2013;122:3394–3395. | ||
Benoit ME, Clarke EV, Morgado P, Fraser DA, Tenner AJ. Complement protein C1q directs macrophage polarization and limits inflammasome activity during the uptake of apoptotic cells. J Immunol. 2012;188:5682–5693. | ||
Nakagawa I, Amano A, Mizushima N. Autophagy defends cells against invading group A Streptococcus. Science. 2004;306(5698):1037–1040. | ||
Matsuzawa-Ishimoto Y, Hwang S, Cadwell K. Autophagy and inflammation. Annu Rev Immunol. 2018;36(1):73–101. | ||
Djavaheri-Mergny M, Amelotti M, Mathieu J, Besancon F, Bauvy C, Codogno P. Regulation of autophagy by NFkappaB transcription factor and reactive oxygen species. Autophagy. 2007;3:390–392. | ||
Xu Y, Jagannath C, Liu XD, Sharafkhaneh A, Kolodziejska KE, Eissa NT. Toll-like receptor 4 is a sensor for autophagy associated with innate immunity. Immunity. 2007;27(1):135–144. | ||
Wu HM, Wang J, Zhang B, Fang L, Xu K, Liu RY. CpG-ODN promotes phagocytosis and autophagy through JNK/P38 signal pathway in Staphylococcus aureus-stimulated macrophage. Life Sci. 2016;161:51–59. | ||
Piquereau J, Godin R, Deschênes S, et al. Protective role of PARK2/Parkin in sepsis-induced cardiac contractile and mitochondrial dysfunction. Autophagy. 2013;9(11):1837–1851. | ||
Takahashi W, Watanabe E, Fujimura L, et al. Kinetics and protective role of autophagy in a mouse cecal ligation and puncture-induced sepsis. Crit Care. 2013;17(4):R160. | ||
Yen YT, Yang HR, Lo HC, et al. Enhancing autophagy with activated protein C and rapamycin protects against sepsis-induced acute lung injury. Surgery. 2013;153(5):689–698. | ||
Ho J, Yu J, Wong SH, et al. Autophagy in sepsis: degradation into exhaustion? Autophagy. 2016;12:1073–1082. | ||
Hsiao HW, Tsai KL, Wang LF, et al. The decline of autophagy contributes to proximal tubular dysfunction during sepsis. Shock. 2012;37:289–296. | ||
Su Y, Qu Y, Zhao F, Li H, Mu D, Li X. Regulation of autophagy by the nuclear factor κB signaling pathway in the hippocampus of rats with sepsis. J Neuroinflammation. 2015;12:116. | ||
Kimura T, Watanabe E, Sakamoto T, et al. Autophagy-related IRGM polymorphism is associated with mortality of patients with severe sepsis. PLoS One. 2014;9:e91522. | ||
Lin C-W, Lo S, Perng D-S, et al. Complete activation of autophagic process attenuates liver injury and improves survival in septic mice. Shock. 2014;41(3):241–249. | ||
Zhang L, Ai Y, Tsung A. Clinical application: restoration of immune homeostasis by autophagy as a potential therapeutic target in sepsis. Exp Ther Med. 2016;11(4):1159–1167. | ||
Ren C, Zhang H, Wu T-T, Yao Y-M. Autophagy: a potential therapeutic target for reversing sepsis-induced immunosuppression. Front Immunol. 2017;8. | ||
Saitoh T, Fujita N, Jang MH, et al. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1β production. Nature. 2008;456(7219):264–268. | ||
Zhou R, Tardivel A, Thorens B, Choi I, Tschopp J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat Immunol. 2010;11:136–140. | ||
Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature. 2011;469:221–225. | ||
Kimura T, Jain A, Choi SW, et al. TRIM-mediated precision autophagy targets cytoplasmic regulators of innate immunity. J Cell Biol. 2015;210:973–989. | ||
Harris J, Hartman M, Roche C, et al. Autophagy controls IL-1beta secretion by targeting pro-IL-1beta for degradation. J Biol Chem. 2011;286:9587–9597. | ||
Harris J, Lang T, Thomas JPW, Sukkar MB, Nabar NR, Kehrl JH. Autophagy and inflammasomes. Mol Immunol. 2017;86:10–15. | ||
Suzuki T, Franchi L, Toma C, et al. Differential regulation of caspase-1 activation, pyroptosis, and autophagy via Ipaf and ASC in Shigella-infected macrophages. PLoS Pathog. 2007;3(8):e111. | ||
Jounai N, Kobiyama K, Shiina M, Ogata K, Ishii KJ, Takeshita F. NLRP4 negatively regulates autophagic processes through an association with beclin1. J Immunol. 2011;186:1646–1655. | ||
Zhang Y, Sauler M, Shinn AS, et al. Endothelial PINK1 mediates the protective effects of NLRP3 deficiency during lethal oxidant injury. J Immunol. 2014;192(11):5296–5304. | ||
Deng Q, Wang Y, Zhang Y, et al. Pseudomonas aeruginosa triggers macrophage autophagy to escape intracellular killing by activation of the NLRP3 inflammasome. Infect Immun. 2016;84:56–66. | ||
Pu Q, Gan C, Li R, et al. Atg7 deficiency intensifies inflammasome activation and pyroptosis in Pseudomonas sepsis. J Immunol. 2017;198(8):3205–3213. | ||
Wlodarska M, Thaiss CA, Nowarski R, et al. NLRP6 inflammasome orchestrates the colonic host–microbial interface by regulating goblet cell mucus secretion. Cell. 2014;156(5):1045–1059. | ||
Kim MJ, Bae SH, Ryu JC, et al. SESN2/sestrin2 suppresses sepsis by inducing mitophagy and inhibiting NLRP3 activation in macrophages. Autophagy. 2016;12(8):1272–1291. | ||
Mao K, Chen S, Chen M, et al. Nitric oxide suppresses NLRP3 inflammasome activation and protects against LPS-induced septic shock. Cell Res. 2013;23(2):201–212. | ||
Lupfer C, Thomas PG, Anand PK, et al. Receptor interacting protein kinase 2–mediated mitophagy regulates inflammasome activation during virus infection. Nat Immunol. 2013;14(5):480–488. | ||
Saitoh T, Akira S. Regulation of inflammasomes by autophagy. J Allergy Clin Immunol. 2016;138(1):28–36. | ||
Qian S, Fan J, Billiar TR, Scott MJ. Inflammasome and autophagy regulation: a two-way street. Mol Med. 2017;23:188–195. | ||
Shi C-S, Shenderov K, Huang N-N, et al. Activation of autophagy by inflammatory signals limits IL-1β production by targeting ubiquitinated inflammasomes for destruction. Nat Immunol. 2012;13(3):255–263. | ||
Yuk JM, Jo EK. Crosstalk between autophagy and inflammasomes. Mol Cells. 2013;36(5):393–399. | ||
Takahama M, Akira S, Saitoh T. Autophagy limits activation of the inflammasomes. Immunol Rev. 2018;281(1):62–73. | ||
Shibuya N, Mikami Y, Kimura Y, Nagahara N, Kimura H. Vascular endothelium expresses 3-mercaptopyruvate sulfurtransferase and produces hydrogen sulfide. J Biochem. 2009;146(5):623–626. | ||
Wang R. Physiological implications of hydrogen sulfide: a whiff exploration that blossomed. Physiol Rev. 2012;92(2):791–896. | ||
Elrod JW, Calvert JW, Morrison J, et al. Hydrogen sulfide attenuates myocardial ischemia–reperfusion injury by preservation of mitochondrial function. Proc Natl Acad Sci U S A. 2007;104:15560–15565. | ||
Yang G, Wu L, Jiang B, et al. H2S as a physiologic vasorelaxant: hypertension in mice with deletion of cystathionine gamma-lyase. Science. 2008;322(5901):587–590. | ||
Fernandes CJ., Jr, de Assuncao MS. Myocardial dysfunction in sepsis: a large, unsolved puzzle. Crit Care Res Pract. 2012;2012:896430. | ||
Antonucci E, Fiaccadori E, Donadello K, Taccone FS, Franchi F, Scolletta S. Myocardial depression in sepsis: from pathogenesis to clinical manifestations and treatment. J Crit Care. 2014;29:500–511. | ||
Kakihana Y, Ito T, Nakahara M, Yamaguchi K, Yasuda T. Sepsis-induced myocardial dysfunction: pathophysiology and management. J Intensive Care. 2016;4:22. | ||
Lv X, Wang H. Pathophysiology of sepsis-induced myocardial dysfunction. Mil Med Res. 2016;3(1):30. | ||
Vallabhajosyula S, Pruthi S, Shah S, Wiley BM, Mankad SV, Jentzer JC. Basic and advanced echocardiographic evaluation of myocardial dysfunction in sepsis and septic shock. Anaesth Intensive Care. 2018;46(1):13–24. | ||
Lin Z, Altaf N, Li C, et al. Hydrogen sulfide attenuates oxidative stress-induced NLRP3 inflammasome activation via S-sulfhydrating c-Jun at Cys269 in macrophages. Biochim Biophys Acta. 2018:1864(9 Pt B):2890–2900. | ||
Castelblanco M, Lugrin J, Ehirchiou D, et al. Hydrogen sulfide inhibits the NLRP3 inflammasome and reduces cytokine production both in vitro and in a mouse model of inflammation. J Biol Chem. 2018;293(7):2546–2557. | ||
Huang Z, Zhuang X, Xie C, et al. Exogenous hydrogen sulfide attenuates high glucose-induced cardiotoxicity by inhibiting NLRP3 inflammasome activation by suppressing TLR4/NF-kappaB pathway in H9c2 Cells. Cell Physiol Biochem. 2016;40:1578–1590. | ||
Zhang H, Zhi L, Moore PK, Bhatia M. Role of hydrogen sulfide in cecal ligation and puncture-induced sepsis in the mouse. Am J Physiol Lung Cell Mol Physiol. 2006;290(6):L1193–1201. | ||
Spiller F, Orrico MI, Nascimento DC, et al. Hydrogen sulfide improves neutrophil migration and survival in sepsis via K+ATP channel activation. Am J Respir Crit Care Med. 2010;182(3):360–368. | ||
Li X, Cheng Q, Li J, He Y, Tian P, Xu C. Significance of hydrogen sulfide in sepsis-induced myocardial injury in rats. Exp Ther Med. 2017;14(3):2153–2161. | ||
Oyadomari S, Mori M. Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ. 2004;11(4):381–389. | ||
Tabas I, Ron D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat Cell Biol. 2011;13(3):184–190. | ||
Ferlito M, Wang Q, Fulton WB, et al. Hydrogen sulfide increases survival during sepsis: protective effect of CHOP inhibition. J Immunol. 2014;192:1806–1814. | ||
Endo M, Oyadomari S, Suga M, Mori M, Gotoh T. The ER stress pathway involving CHOP is activated in the lungs of LPS-treated mice. J Biochem. 2005;138:501–507. | ||
Endo M, Mori M, Akira S, Gotoh T. C/EBP homologous protein (CHOP) is crucial for the induction of caspase-11 and the pathogenesis of lipopolysaccharide-induced inflammation. J Immunol. 2006;176:6245–6253. | ||
Hui Y, du J, Tang C, Bin G, Jiang H. Changes in arterial hydrogen sulfide (H(2)S) content during septic shock and endotoxin shock in rats. J Infect. 2003;47:155–160. | ||
Norris E, Narasimhan S, Culberson C, Clemens MG. The liver is a critical site for hydrogen sulfide clearance in sepsis. FASEB J. 2011;25:1117–1118. | ||
Coletta C, Szabo C. Potential role of hydrogen sulfide in the pathogenesis of vascular dysfunction in septic shock. Curr Vasc Pharmacol. 2013;11:208–221. | ||
Kim D, Anantharam PV, Whitley EM, et al. Acute exposure to high level of hydrogen sulfide induces neurodegeneration via activation of inflammasomes in brain. FASEB J. 2016a;30:1191–1195. | ||
Gaddam RR, Chambers S, Murdoch D, Shaw G, Bhatia M. Circulating levels of hydrogen sulfide and substance P in patients with sepsis. J Infect. 2017;75:293–300. | ||
Košir M, Podbregar M. Advances in the diagnosis of sepsis: hydrogen sulfide as a prognostic marker of septic shock severity. EJIFCC. 2017;28:134–141. | ||
Bee N, White R, Petros AJ. Hydrogen sulfide in exhaled gases from ventilated septic neonates and children: a preliminary report. Pediatr Crit Care Med. 2017;18:e327–e332. | ||
Beltowski J. Hydrogen sulfide in pharmacology and medicine – an update. Pharmacol Rep. 2015;67:647–658. | ||
Tilley SL, Coffman TM, Koller BH. Mixed messages: modulation of inflammation and immune responses by prostaglandins and thromboxanes. J Clin Invest. 2001;108(1):15–23. | ||
Smyth EM, Grosser T, Wang M, Yu Y, Fitzgerald GA. Prostanoids in health and disease. J Lipid Res. 2009;50(Supplement):S423–S428. | ||
Kawabe T, Harris PD, Zakaria EL, Garrison RN. Sepsis alters vessel contraction by adrenoceptor-induced nitric oxide and prostanoid. J Surg Res. 2003;110:352–359. | ||
Wang X, Shaw DK, Hammond HL, et al. The prostaglandin E2-EP3 receptor axis regulates Anaplasma phagocytophilum-mediated NLRC4 inflammasome activation. PLoS Pathog. 2016;12(8):e1005803. | ||
Ricciotti E, Fitzgerald GA. Prostaglandins and inflammation. Arterioscler Thromb Vasc Biol. 2011;31(5):986–1000. | ||
von Moltke J, Trinidad NJ, Moayeri M, et al. Rapid induction of inflammatory lipid mediators by the inflammasome in vivo. Nature. 2012;490(7418):107–111. | ||
Zoccal KF, Sorgi CA, Hori JI, et al. Opposing roles of LTB4 and PGE2 in regulating the inflammasome-dependent scorpion venom-induced mortality. Nat Commun. 2016;7:10760. | ||
Maier NK, Leppla SH, Moayeri M. The cyclopentenone prostaglandin 15d-PGJ2 inhibits the NLRP1 and NLRP3 inflammasomes. J Immunol. 2015;194(6):2776–2785. | ||
Mortimer L, Moreau F, Macdonald JA, Chadee K. NLRP3 inflammasome inhibition is disrupted in a group of auto-inflammatory disease CAPS mutations. Nat Immunol. 2016;17(10):1176–1186. | ||
Sokolowska M, Chen L-Y, Liu Y, et al. Prostaglandin E2 inhibits NLRP3 inflammasome activation through EP4 receptor and intracellular cyclic AMP in human macrophages. J Immunol. 2015;194(11):5472–5487. | ||
Ting JP, Duncan JA, Lei Y. How the noninflammasome NLRs function in the innate immune system. Science. 2010;327(5963):286–290. | ||
Li Z, Zhang Y, Liu B, Luo W, Li H, Zhou Y. Role of E-type prostaglandin receptor EP3 in the vasoconstrictor activity evoked by prostacyclin in thromboxane-prostanoid receptor deficient mice. Sci Rep. 2017;7(1):42167. | ||
Tunctan B, Korkmaz B, Sari AN, et al. A novel treatment strategy for sepsis and septic shock based on the interactions between prostanoids, nitric oxide, and 20-hydroxyeicosatetraenoic acid. Antiinflamm Antiallergy Agents Med Chem. 2012;11(2):121–150. | ||
Tunctan B, Korkmaz B, Sari AN, et al. Contribution of iNOS/sGC/PKG pathway, COX-2, CYP4A1, and gp91(phox) to the protective effect of 5,14-HEDGE, a 20-HETE mimetic, against vasodilation, hypotension, tachycardia, and inflammation in a rat model of septic shock. Nitric Oxide. 2013;33:18–41. | ||
Vijay R, Fehr AR, Janowski AM, et al. Virus-induced inflammasome activation is suppressed by prostaglandin D2/DP1 signaling. Proc Natl Acad Sci U S A. 2017;114(27):E5444–e5453. | ||
Khare S, Ratsimandresy RA, de Almeida L, et al. The PYRIN domain-only protein POP3 inhibits ALR inflammasomes and regulates responses to infection with DNA viruses. Nat Immunol. 2014;15:343–353. | ||
de Almeida L, Khare S, Misharin AV, et al. The PYRIN domain-only protein POP1 inhibits inflammasome assembly and ameliorates inflammatory disease. Immunity. 2015;43:264–276. | ||
Angus DC, Opal S. Immunosuppression and secondary infection in sepsis: part, not all, of the story. JAMA. 2016;315:1457–1459. | ||
Fattahi F, Ward PA. Understanding immunosuppression after sepsis. Immunity. 2017;47:3–5. | ||
Roquilly A, Mcwilliam HEG, Jacqueline C, et al. Local modulation of antigen-presenting cell development after resolution of pneumonia induces long-term susceptibility to secondary infections. Immunity. 2017;47(1):e135–147. | ||
Jensen IJ, Sjaastad FV, Griffith TS, Badovinac VP. Sepsis-induced T cell immunoparalysis: the ins and outs of impaired T cell immunity. J Immunol. 2018;200:1543–1553. | ||
Cao C, Ma T, Chai Y-F, Shou S-T. The role of regulatory T cells in immune dysfunction during sepsis. World J Emerg Med. 2015a;6:5–9. | ||
Tatura R, Zeschnigk M, Hansen W, et al. Relevance of Foxp3+ regulatory T cells for early and late phases of murine sepsis. Immunology. 2015;146(1):144–156. | ||
Yao Y, Vent-Schmidt J, Mcgeough MD, et al. Tr1 cells, but not Foxp3+ regulatory T cells, suppress NLRP3 inflammasome activation via an IL-10-dependent mechanism. J Immunol. 2015;195(2):488–497. | ||
Friedman G, Jankowski S, Marchant A, Goldman M, Kahn RJ, Vincent JL. Blood interleukin 10 levels parallel the severity of septic shock. J Crit Care. 1997;12:183–187. | ||
Gogos CA, Drosou E, Bassaris HP, Skoutelis A. Pro- versus anti-inflammatory cytokine profile in patients with severe sepsis: a marker for prognosis and future therapeutic options. J Infect Dis. 2000;181:176–180. | ||
Pileri D, Accardo Palombo A, D’Amelio L, et al. Concentrations of cytokines Il-6 and Il-10 in plasma of burn patients: their relationship to sepsis and outcome. Ann Burns Fire Disasters. 2008;21:182–185. | ||
Surbatovic M, Popovic N, Vojvodic D, et al. Cytokine profile in severe Gram-positive and Gram-negative abdominal sepsis. Sci Rep. 2015;5:11355. | ||
Latifi SQ, O’Riordan MA, Levine AD. Interleukin-10 controls the onset of irreversible septic shock. Infect Immun. 2002;70:4441–4446. | ||
Cayrol C, Girard J-P. The IL-1-like cytokine IL-33 is inactivated after maturation by caspase-1. Proc Natl Acad Sci U S A. 2009;106:9021–9026. | ||
Madouri F, Guillou N, Fauconnier L, et al. Caspase-1 activation by NLRP3 inflammasome dampens IL-33-dependent house dust mite-induced allergic lung inflammation. J Mol Cell Biol. 2015;7(4):351–365. | ||
Tominaga SI, Ohta S, Tago K. Soluble form of the ST2 gene product exhibits growth promoting activity in NIH-3T3 cells. Biochem Biophys Rep. 2016;5:8–15. | ||
Griesenauer B, Paczesny S. The ST2/IL-33 axis in immune cells during inflammatory diseases. Front Immunol. 2017;8. | ||
Kakkar R, Lee RT. The IL-33/ST2 pathway: therapeutic target and novel biomarker. Nat Rev Drug Discov. 2008;7:827–840. | ||
Echtenacher B, Mannel DN, Hultner L. Critical protective role of mast cells in a model of acute septic peritonitis. Nature. 1996;381:75–77. | ||
Hoogerwerf JJ, Tanck MWT, van Zoelen Mad, Wittebole X, Laterre P-F, van der Poll T. Soluble ST2 plasma concentrations predict mortality in severe sepsis. Intensive Care Med. 2010;36:630–637. | ||
Parenica J, Malaska J, Jarkovsky J, et al. Soluble ST2 levels in patients with cardiogenic and septic shock are not predictors of mortality. Exp Clin Cardiol. 2012;17:205–209. | ||
Hur M, Kim H, Kim HJ, et al. Soluble ST2 has a prognostic role in patients with suspected sepsis. Ann Lab Med. 2015;35:570–577. | ||
Sweet MJ, Leung BP, Kang D, et al. A novel pathway regulating lipopolysaccharide-induced shock by ST2/T1 via inhibition of Toll-like receptor 4 expression. J Immunol. 2001;166(11):6633–6639. | ||
Alves-Filho JC, Sônego F, Souto F, et al. Interleukin-33 attenuates sepsis by enhancing neutrophil influx to the site of infection. Nat Med. 2010. | ||
Sutherland RE, Olsen JS, Mckinstry A, Villalta SA, Wolters PJ. Mast cell IL-6 improves survival from Klebsiella pneumonia and sepsis by enhancing neutrophil killing. J Immunol. 2008;181(8):5598–5605. | ||
Dahdah A, Gautier G, Attout T, et al. Mast cells aggravate sepsis by inhibiting peritoneal macrophage phagocytosis. J Clin Invest. 2014;124:4577–4589. | ||
Netea MG, Joosten LAB. Inflammasome inhibition: putting out the fire. Cell Metab. 2015;21(4):513–514. | ||
Shao B-Z, Xu Z-Q, Han B-Z, Su D-F, Liu C. NLRP3 inflammasome and its inhibitors: a review. Front Pharmacol. 2015;6(378). | ||
Jahan S, Kumar D, Chaturvedi S, et al. Therapeutic targeting of NLRP3 inflammasomes by natural products and pharmaceuticals: a novel mechanistic approach for inflammatory diseases. Curr Med Chem. 2017;24:1645–1670. | ||
Coll RC, Robertson Aab, Chae JJ, et al. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat Med. 2015;21:248. | ||
Youm YH, Nguyen KY, Grant RW, et al. The ketone metabolite β-hydroxybutyrate blocks NLRP3 inflammasome-mediated inflammatory disease. Nat Med. 2015;21(3):263. | ||
Newman JC, Verdin E. Ketone bodies as signaling metabolites. Trends Endocrinol Metab. 2014;25(1):42–52. | ||
Grabacka M, Pierzchalska M, Dean M, Reiss K. Regulation of ketone body metabolism and the role of PPARα. Int J Mol Sci. 2016;17:2093. | ||
Ohtoshi M, Jikko A, Asano M, Uchida K, Ozawa K, Tobe T. Ketogenesis during sepsis in relation to hepatic energy metabolism. Res Exp Med. 1984;184(4):209–219. | ||
Lanza-Jacoby S, Rosato E, Braccia G, Tabares A. Altered ketone body metabolism during gram-negative sepsis in the rat. Metabolism. 1990;39:1151–1157. | ||
Levy B, Sadoune LO, Gelot AM, Bollaert PE, Nabet P, Larcan A. Evolution of lactate/pyruvate and arterial ketone body ratios in the early course of catecholamine-treated septic shock. Crit Care Med. 2000;28:114–119. | ||
Guan Y, Breyer MD. Peroxisome proliferator-activated receptors (PPARs): novel therapeutic targets in renal disease. Kidney Int. 2001;60:14–30. | ||
Deng Y, Han X, Yao Z, et al. PPARalpha agonist stimulated angiogenesis by improving endothelial precursor cell function via a NLRP3 inflammasome pathway. Cell Physiol Biochem. 2017;42:2255–2266. | ||
Wang Y, Yu B, Wang L, et al. Pioglitazone ameliorates glomerular NLRP3 inflammasome activation in apolipoprotein E knockout mice with diabetes mellitus. PLoS One. 2017;12(7):e0181248. | ||
Aronoff DM, Serezani CH, Carstens JK, et al. Stimulatory effects of peroxisome proliferator-activated receptor-γ on Fcγ receptor-mediated phagocytosis by alveolar macrophages. PPAR Res. 2007;2007:8. | ||
Drosatos K, Khan RS, Trent CM, et al. PPARγ activation prevents sepsis-related cardiac dysfunction and mortality in mice. Circ Heart Fail. 2013;6:550–562. | ||
Kaplan J, Nowell M, Chima R, Zingarelli B. Pioglitazone reduces inflammation through inhibition of NF-κB in polymicrobial sepsis. Innate Immun. 2014;20:519–528. | ||
Reddy AT, Lakshmi SP, Reddy RC. PPAR γ in bacterial infections: a friend or foe? PPAR Res. 2016;(3):7963540–7. | ||
Standage SW, Caldwell CC, Zingarelli B, Wong HR. Reduced peroxisome proliferator-activated receptor α expression is associated with decreased survival and increased tissue bacterial load in sepsis. Shock. 2012;37(2):164–169. | ||
Kumar V, Chhibber S. Anti-inflammatory effect of thalidomide alone or in combination with augmentin in Klebsiella pneumoniae B5055 induced acute lung infection in BALB/c mice. Eur J Pharmacol. 2008;592:146–150. | ||
Kumar V, Harjai K, Chhibber S. A combination of thalidomide and augmentin protects BALB/c mice suffering from Klebsiella pneumoniae B5055-induced sepsis. J Chemother. 2009;21:159–164. | ||
Kumar V, Harjai K, Chhibber S. Thalidomide treatment modulates macrophage pro-inflammatory function and cytokine levels in Klebsiella pneumoniae B5055 induced pneumonia in BALB/c mice. Int Immunopharmacol. 2010;10(7):777–783. | ||
Kumar V, Chhibber S. Thalidomide: an old drug with new action. J Chemother. 2011;23:326–334. | ||
Keller M, Sollberger G, Beer HD. Thalidomide inhibits activation of caspase-1. J Immunol. 2009;183:5593–5599. | ||
López-Castejón G, Pelegrín P. Current status of inflammasome blockers as anti-inflammatory drugs. Expert Opin Investig Drugs. 2012;21(7):995–1007. | ||
Ludwig-Portugall I, Bartok E, Dhana E, et al. An NLRP3-specific inflammasome inhibitor attenuates crystal-induced kidney fibrosis in mice. Kidney Int. 2016;90(3):525–539. | ||
Marchetti C, Swartzwelter B, Gamboni F, et al. OLT1177, a β-sulfonyl nitrile compound, safe in humans, inhibits the NLRP3 inflammasome and reverses the metabolic cost of inflammation. Proc Natl Acad Sci U S A. 2018;115(7):E1530–E1539. | ||
Zorman J, Sušjan P, Hafner-Bratkovič I. Shikonin suppresses NLRP3 and AIM2 inflammasomes by direct inhibition of caspase-1. PLoS One. 2016;11:e0159826. | ||
Hara H, Tsuchiya K, Kawamura I, et al. Phosphorylation of the adaptor ASC acts as a molecular switch that controls the formation of speck-like aggregates and inflammasome activity. Nat Immunol. 2013;14:1247–1255. | ||
Kwak S-B, Koppula S, In E-J, et al. Artemisia extract suppresses NLRP3 and AIM2 inflammasome activation by inhibition of ASC phosphorylation. Mediators Inflamm. 2018;2018:6054069. | ||
Lee J, Ahn H, Hong EJ, An BS, Jeung EB, Lee GS. Sulforaphane attenuates activation of NLRP3 and NLRC4 inflammasomes but not AIM2 inflammasome. Cell Immunol. 2016:30653–30760. | ||
Yende S, van der Poll T. Diabetes and sepsis outcomes –it is not all bad news. Crit Care. 2009;13(1):117. | ||
Koh GCKW, Peacock SJ, van der Poll T, Wiersinga WJ. The impact of diabetes on the pathogenesis of sepsis. Eur J Clin Microbiol Infect Dis. 2012;31:379–388. | ||
Kolyva AS, Zolota V, Mpatsoulis D, et al. The role of obesity in the immune response during sepsis. Nutr Diabetes. 2014;4:e137. | ||
Papadimitriou-Olivgeris M, Aretha D, Zotou A, et al. The role of obesity in sepsis outcome among critically ill patients: a retrospective cohort analysis. Biomed Res Int. 2016;2016(5):9–9. | ||
Ng PY, Eikermann M. The obesity conundrum in sepsis. BMC Anesthesiol. 2017;17(1):147. | ||
Dixit VD. Nlrp3 inflammasome activation in type 2 diabetes: is it clinically relevant? Diabetes. 2013;62:22–24. | ||
Lee H-M, Kim J-J, Kim HJ, Shong M, Ku BJ, Jo E-K. Upregulated NLRP3 inflammasome activation in patients with type 2 diabetes. Diabetes. 2013;62:194–204. | ||
Volpe CM, Anjos PM, Nogueira-Machado JA. Inflammasome as a new therapeutic target for diabetic complications. Recent Pat Endocr Metab Immune Drug Discov. 2016;10(1):56–62. | ||
Rheinheimer J, de Souza BM, Cardoso NS, Bauer AC, Crispim D. Current role of the NLRP3 inflammasome on obesity and insulin resistance: a systematic review. Metabolism. 2017;74:1–9. | ||
Sepehri Z, Kiani Z, Afshari M, Kohan F, Dalvand A, Ghavami S. Inflammasomes and type 2 diabetes: an updated systematic review. Immunol Lett. 2017;192:97–103. | ||
Zhang X, Dai J, Li L, Chen H, Chai Y. NLRP3 inflammasome expression and signaling in human diabetic wounds and in high glucose induced macrophages. J Diabetes Res 2017. 2017b;7. | ||
Fujitani Y, Kawamori R, Watada H. The role of autophagy in pancreatic beta-cell and diabetes. Autophagy. 2009;5:280–282. | ||
Nuñez CE, Rodrigues VS, Gomes FS, et al. Defective regulation of adipose tissue autophagy in obesity. Int J Obes. 2013;37(11):1473–1480. | ||
Barlow AD, Thomas DC. Autophagy in diabetes: beta-cell dysfunction, insulin resistance, and complications. DNA Cell Biol. 2015;34:252–260. | ||
Sarparanta J, García-Macia M, Singh R. Autophagy and mitochondria in obesity and type 2 diabetes. Curr Diabetes Rev. 2017;13(4):352–369. | ||
Yang JS, Lu CC, Kuo SC, et al. Autophagy and its link to type II diabetes mellitus. Biomedicine. 2017;7(2):8. | ||
Hao H, Cao L, Jiang C, et al. Farnesoid X receptor regulation of the NLRP3 inflammasome underlies cholestasis-associated sepsis. Cell Metab. 2017;25(856-867):e855. | ||
de Magalhaes Filho CD, Downes M, Evans RM. Farnesoid X receptor an emerging target to combat obesity. Dig Dis. 2017;35:185–190. | ||
Xie S, Guo C, Chi Z, et al. A rapid administration of GW4064 inhibits the NLRP3 inflammasome activation independent of farnesoid X receptor agonism. FEBS Lett. 2017;591(18):2836–2847. | ||
Lee S, Nakahira K, Dalli J, et al. NLRP3 inflammasome deficiency protects against microbial sepsis via increased lipoxin B4 synthesis. Am J Respir Crit Care Med. 2017;196:713–726. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.