Back to Journals » OncoTargets and Therapy » Volume 7
Induction of multiple drug resistance in HMEC-1 endothelial cells after long-term exposure to sunitinib
Authors Huang L, Hu C, Di Benedetto M, Varin R, Liu J, Wang L, Vannier J, Jin J, Janin A ![]() , Lu H, Li H
, Lu H, Li H
Received 5 May 2014
Accepted for publication 8 July 2014
Published 4 December 2014 Volume 2014:7 Pages 2249—2255
DOI https://doi.org/10.2147/OTT.S67251
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Limin Huang,1,* Chaoquan Hu,2,* Mélanie Di Benedetto,3 Rémi Varin,4 Jielin Liu,2,3 Li Wang,3 Jean-Pierre Vannier,4 Jian Jin,3,5 Anne Janin,3,6,7 He Lu,3,6 Hong Li4
1Department of Oncology, People’s Hospital of Guizhou Province, Guiyang, People’s Republic of China; 2Department of Surgery, Affiliated Hospital, Guiyang Medical University, Guiyang, People’s Republic of China; 3INSERM UMR-S 1165, Paris, France; 4MERCI (EA 3829), Faculté de Médecine et de Pharmacie, Université de Rouen, Rouen, France; 5School of Medicine and Pharmaceutics, Jiangnan University, Wuxi, People’s Republic of China; 6Université Paris Diderot, Laboratoire de Pathologie, Paris, France; 7AP-HP-Hôpital Saint-Louis, Laboratoire de Pathologie, Paris, France
*These authors contributed equally to this study
Abstract: Multiple drug resistance is still an unsolved problem in cancer therapy. Our previous study demonstrated that the chemotherapeutic drug doxorubicin (Dox) induced upregulation of P-glycoprotein (P-gp) in endothelial cells, resulting in a 20-fold increase in drug resistance and reduced efficiency of Dox treatment in a mice tumor model. In this study, we exposed human microvascular endothelial cells (HMEC-1) to sunitinib, a tyrosine kinase receptor inhibitor, to induce drug resistance. The results show that sunitinib treatment induced multiple drug resistance in these cells. They became resistant not only to sunitinib but also to Dox, paclitaxel, and vinblastine. Significant increases in P-gp (9.3-fold), ABCG2 (breast cancer resistance protein, 1.9-fold), and multidrug resistance-associated protein 1 (2.7-fold) gene transcription were found by quantitative polymerase chain reaction quantification, and their protein expression was confirmed by Western blot. These increases gave rise to an approximately five-fold increase in half maximal inhibitory concentration in these cells in response to sunitinib treatment in vitro. The inhibitors of adenosine triphosphate-binding cassette transporters did not reverse the drug resistance in sunitinib-resistant HMEC-1 cells, assumedly because of a blockage of the pump function caused by sunitinib. Our study indicates that the antiangiogenic drug sunitinib induces multiple drug resistance in endothelial cells. The induction of adenosine triphosphate-binding cassette transporters seems not to be responsible for observed multiple drug resistance, and the underlying mechanisms remain unknown.
Keywords: drug resistance, endothelial cells, cancer therapy, ABC family, sunitinib
Introduction
Tumor angiogenesis supports tumor growth and metastasis. Over the last decade, with the fast advances in our understanding of the tumor angiogenesis process and discoveries of molecules inhibiting angiogenesis, the concept of antiangiogenic therapy has been successfully introduced into clinical use to prevent, slow, and arrest tumor growth through vascular regression and tumor starvation. The first antiangiogenic drug, bevacizumab, was a humanized antivascular endothelial growth factor (VEGF) antibody, which received approval in 2004 for combined use with chemotherapy for metastatic colon cancer.1 It was followed by the fast pharmaceutical development of tyrosine kinase inhibitors (TKIs), which target the receptors for the angiogenic factor VEGF (VEGFR). Today, many TKIs have been successfully introduced into clinical anticancer therapy.2,3 However, regardless of the initial success of antiangiogenesis therapies, these inhibitors failed to produce enduring clinical responses. Therapeutic resistance to TKIs has become a practical limitation for drug development.4 It is generally acknowledged that multiple mechanisms are involved in patient resistance to antiangiogenic drugs, including the existence of alternative and redundant angiogenic factors, an amplification of the stem cells that are resistant to hypoxia, the selection by hypoxia of cells with metastatic and invasive potential, and tumor cell dormancy.5
Among clinically approved TKI drugs, sunitinib (Su) is an orally available small molecule multikinase inhibitor.6 This agent potently inhibits VEGFR, the platelet-derived growth factor receptor (PDGFR), and c-Kit in addition to other kinases in in vitro assays. In several relevant preclinical cancer models, Su exerts significant antiangiogenesis and antitumor effects. Transient clinical resistance to Su has been reported, but the mechanisms of resistance to Su and other TKIs that target VEGFRs are largely unknown.7,8 A series of studies showed that Su could be either a substrate or a blocker/chemosentitizer because of its capacity to bind to ABCB1 and ABCG2.9–11
Since antiangiogenic drugs target genetically stable endothelial cells, they are not expected to give rise to drug resistance.12 Antiangiogenic therapy should allow for prolonged treatment. However, high P-glycoprotein (P-gp) expression was recently found in tumor endothelial cells, probably in response to VEGF stimulation.13 Previously, we have shown that the chemotherapeutic agent doxorubicin induces high levels of P-gp in endothelial cells.14 We also showed that acquired drug resistance in endothelial cells attenuated the efficacy of doxorubicin treatment in a mice tumor model. These studies indicated that the acquired drug resistance of tumor vessels plays a critical role in cancer therapy. As ABCB1 and ABCG2 were supposed to functionally affect Su therapy, this study explored ABCB1 and ABCG2 expression in cultured endothelial cells after long-term exposure to Su.
Materials and methods
Materials
The anti-ABCG2, antimultidrug resistance-associated protein 1 (MRP1), and anti-P-gp antibodies were purchased from Abcam Inc., Cambridge, UK. Su was provided by Pfizer (New York, NY, USA). Verapamil was obtained from Merck KGaA, Darmstadt, Germany. Diethylstilbesterol, paclitaxel, cyclosporine A, vinblastine, fumitremorgin C, and MK571 were purchased from Sigma Chemical Co (Saint Louis, MO, USA). Doxorubicin chlorhydrate was from Amersham Pharmacia Biotech (Uppsala, Sweden).
Cell culture and drug resistance induction
Human microvascular endothelial cell (HMEC-1) lines (Dr TL Lawley, Department of Dermatology, Atlanta, GA, USA) were cultured in MCDB-131 medium supplemented with 1 μg/mL hydrocortisone, 10 ng/mL EGF, 2 mM L-glutamine, 100 μg/mL streptomycin, 100 units/mL penicillin, and 10% fetal calf serum, as described previously.15 Su-resistant HMEC-1 cells (HMECsu or Hsu) were obtained by continuously exposing HMEC-1 cells to escalating concentrations from 0.01 μM to 25 μM of Su over a 12-week period. All types of cells were digested with trypsin–ethylenediaminetetraacetic acid (EDTA) once or twice a week and cultured in a 37°C incubator with a 100% humidified atmosphere of 5% CO2. No mutagenic agents were used in the establishment of these sunitinib-resistant HMEC-1 cells (HMECsu). HMECd2 cells were an HMEC-1 subcell line obtained by long-term exposure to doxorubicin.14 Delta–delta Ct was used to calculate the differences in quantitative polymerase chain reaction (qPCR).
MTS cell viability assay
3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) cell proliferation assay (Promega Corporation, Fitchburg, WI, USA) was used to determine cell viability. Briefly, cells were cultured to a confluence of 90% in 75 cm2 cell culture flasks. Then the cells were passed into 96-well plates (7,500 cells/well). Each well contained 100 μL of culture medium in the presence or absence of various concentrations of drugs or dimethyl sulfoxide as a control. After incubation for 24, 48, or 72 hours, 20 μL of the MTS reagent was added to each well. The plates were then put back to the incubator for an additional 2 hours. The optical density was then determined at 492 nm using a microplate reader (LabSystems Multiskan MS). Experiments were conducted in triplicate and repeated at least three times. The half maximal inhibitory concentration (IC50) values were defined as the concentration of drug producing 50% inhibition of cell growth. The resistance index corresponded to the ratio of IC50 values between the resistant and parental cell lines.14
Blocking effect of adenosine triphosphate-binding cassette protein inhibitors
After incubation for 48 or 72 hours, cell viability was measured by MTS kit. Fumitremorgin C at 5 μM or diethylstilbestrol at 0.5 μM was used to block ABCG2, cyclosporine A at 2.5 μM or verapamil at 1 μM was used to block P-gp, and MK571 at 5 μM was used to block MRP1. The reversal fold, obtained by IC50 of cytotoxic drug alone over IC50 of cytotoxic drug in the presence of a modulator, was used to indicate the potency of reversal.16
Quantification of messenger ribonucleic acid by qPCR
Adenosine triphosphate-binding cassette (ABC) protein inhibitors 1 μM verapamil, 2.5 μM cyclosporine A, 0.5 μM diethylstilbestrol, 5 μM fumitremorgin C, or 5 μM MK571 were used to treat HMEC-1 and HMECsu cells for 24 hours. After incubation, the cells were harvested and total ribonucleic acid (RNA) was isolated using the SV Total RNA Isolation System Kit (Promega Corporation), and the purity of total RNA was measured by the ratio of A260/A280 (>1.9) as described previously.14 Total RNA (50 ng) was taken to prepare the first-strand complementary deoxyribonucleic acid (cDNA) in a 20 μL reaction solution (the GoScript Reverse Transcription System Kit; Promega Corporation). cDNA (2 μL) was used for qPCR in triplicate using a TaqMan® gene expression assay. The primers for ABCG2 (Hs01053790_m1), P-gp (Hs01067802_m1), and MRP1 (Hs00219905_m1), and the primers for TATA box binding protein (TBP) (Hs99999910_m1) as controls were used (Applied Biosystems). The qPCR was conducted by 10 minutes of denaturation, 44 cycles of 15 seconds at 95°C and 60 seconds at 60°C in a BioRad CFX96® Real-Time System. The Delta–delta Ct method was used to analyze the qPCR results. For messenger (m)RNA-level normalization, TBP was set as control.
Western blot analysis
Whole cell lysates were used for Western blot. Briefly, the cells were treated by a lysis buffer (1 mM EDTA, 10 mM Tris pH 6.8, 10% NP40, 0.1% sodium dodecyl sulfate, 1 mM phenylmethylsulfonyl fluoride) for 30 minutes on ice and cell debris was removed. Protein concentration was measured using the BCA™ protein assay (Thermo Scientific, Waltham, MA, USA). Each sample (50 μg protein) was loaded on 8% sodium dodecyl sulfate polyacrylamide gel electrophoresis, and the gels were transferred to a PVDF membrane (the iBlot™ dry blotting system; Invitrogen, Carisbad, NM, USA). Five percent nonfat dry milk was used to block the membranes for 1 hour and incubated with anti-MRP1 (ab-32574), anti-ABCG2 (ab-3380), or anti-P-gp (ab-3364) antibodies (Abcam) at 4°C overnight. After washing with Tris-buffered saline–Tween buffer for 1 hour, the membranes were incubated with appropriate secondary horseradish peroxidase-conjugated antibodies (Invitrogen) in blocking buffer for 1 hour at room temperature. Western blotting luminol reagent (Santa Cruz Biotechnology, Dallas, TX, USA) was then applied to the washed membranes and the chemiluminescence was recorded with a Fuji LAS-3000 system. At the end, the membranes were washed with antibody stripping buffer (Gene Bio-Application Ltd, Tel Aviv, Israel) for the staining of antiactin antibody (1:4,000 dilution, Sigma Chemical Co) as a control. The ratio of blot density signal of specific protein bands (ABC transporters) over control actin band density signal is presented in “Results”.
Statistical analyses
One-way analysis of variance or Mann–Whitney U methods were used to analyze the data as appropriate. The qPCR data are presented as mean ± standard error of the mean. Otherwise, other results are presented as mean ± standard deviation. P-values ≤0.05 were considered as statistically significant.
Results
Endothelial cells resistant to antiangiogenesis drugs
HMEC-1 cells are initially sensitive to Su treatment in our experiments. In an attempt to induce drug resistance in endothelial cells, we introduced progressively escalating doses of Su into the cell culture medium for a duration of approximately 12 weeks. When the cells had gradually adapted to the conditions of higher concentrations of Su, the population was maintained in a culture with 15 μM Su. We noticed that the proliferation rate of the cells was slightly slowed (replication time 50 hours for HMECsu cells versus 46 hours for HMEC-1 cells) without obvious changes in morphology. As is shown in Table 1, a 5.49-fold increase in drug resistance in the stabilized subcell lines HMECsu as compared with their parental cells was observed with the MTS assay. We assessed the stability of the Su-resistant phenotype. By culturing HMECsu in the absence of Su for 2 weeks, we found that there was no significant change in the resistance index (5.38 with IC50 =22.6 μM).
 | Table 1 Exposure to sunitinib induces multiple drug resistance |
Multidrug resistance of endothelial cells
We then tested the resistance of these cells to other drugs. The tests with three cytotoxic drugs, vinblastine, doxorubicin, and paclitaxel, showed that compared with parental cells, the Su-resistant endothelial cell lines were also resistant to higher concentrations of these drugs (Table 1).
P-gp, ABCG2, and MRP1 were upregulated in the endothelial cells after long-term exposure to Su
We used qPCR to measure changes in drug efflux transporter protein expression in the HMECsu cells. P-gp, ABCG2, and MRP1 mRNA expression increased significantly in HMECsu cells compared with parental cells (9.3-fold, 1.9-fold, and 2.7-fold increase, respectively) (Figure 1A–C). We confirmed the upregulation of P-gp and ABCG2 gene expression in HMECd2 endothelial cells that had been treated with doxorubicin.14 Furthermore, we also determined the changes in P-gp, ABCG2, and MRP1 mRNA levels with the inhibitors of the three transporters, respectively. We found that the presence of these inhibitors in the culture did not modify the expression of P-gp, ABCG2, and MRP1 genes in HMECsu (Figure 1A–C). There was no statistical difference in ABCG2 expression between HMECsu and the HMECsu plus fumitremorgin C (Figure 1B).
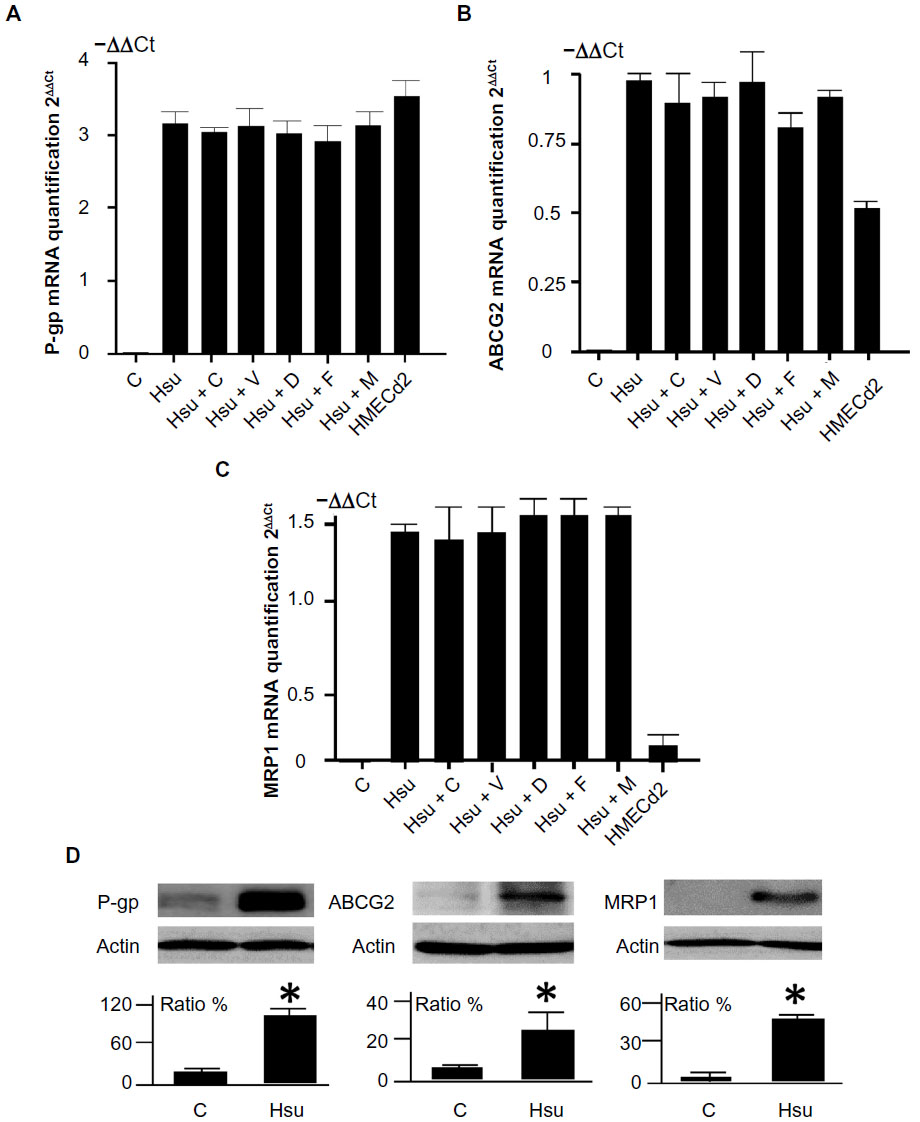 | Figure 1 Expression of ABC transporters in HMEC-1 cells and variant cell lines. |
We confirmed the increase in P-gp, ABCG2, and MRP1 protein expression using Western blotting. The results showed an increase in P-gp, ABCG2, and MRP1 protein in the HMECsu cells (Figure 1D). Thus, our results indicate that Su resistance is associated with upregulated P-gp, ABCG2, and MRP1 expression in HMECsu cells.
Effects of P-gp, ABCG2, and MRP1 inhibitors on the drug resistance of endothelial cells
We evaluated cell survival after Su treatment in the presence of cyclosporine A, verapamil, fumitremorgin C, diethylstilbestrol, and MK571 in the HMECsu cells. The cells were treated with Su in the presence of 1) 2.5 μM cyclosporine A or 1 μM verapamil (which blocks the P-gp function), 2) 0.5 μM diethylstilbestrol or 5 μM fumitremorgin C (which blocks the ABCG2 function), and 3) 5 μM MK571 (which blocks the MRP1 function). The results show that blocking the ABCG2, P-gp, or MRP1 functions did not restore significantly the sensitivity of the HMECsu cells to Su (Table 2). In addition, the simultaneous blockage of P-gp and ABCG2 did not restore the sensitivity of the HMECsu cells to Su.
 | Table 2 Effects of ABC transporter inhibitors on IC50 |
We then checked cell survival after doxorubicin, paclitaxel, and vinblastine treatment, respectively, in the presence of the inhibitors of P-gp, ABCG2, and MRP1 in the HMECsu cells. The results showed that ABC transporter inhibitors failed to significantly restore drug efflux in these cells (Table 2).
Discussion
We previously described the work of obtaining doxorubicin-resistant endothelial cells in vitro.14 As recent studies have reported tumor resistance to Su, we extended our investigation into long-term in vitro exposure of endothelial cells to Su. The results show an acquisition of resistance to Su with upregulated expression levels of P-gp, ABCG2, and MRP1 in HMECsu endothelial cells. We show for the first time that P-gp, ABCG2, and MRP1 expression is upregulated in these stabilized Su-resistant endothelial cells. The protein levels revealed by Western blots were found to have significantly increased in these cells. Similarly, the qPCR experiment demonstrated a 9.3-fold increase in P-gp gene expression, a 1.9-fold increase in ABCG2 gene expression, and a 2.7-fold increase in MRP1 gene expression. It is interesting to note a higher ABCG2 level and high MRP1 expression in HMECsu cells compared with HMECd2 cells. It indicates different mechanisms involved in induced resistance of endothelial cells in response to Su and/or doxorubicin treatment. We further show that the drug resistance induced by Su in endothelial cells has a broad drug spectrum because these cells are also resistant to doxorubicin, paclitaxel, and vinblastine. The overexpression of ABC transporters has been shown to be an adaptation of cells to toxic stress. The mechanisms involved include gene regulation by hormone and xenobiotic-sensing nuclear receptors and epigenetic alternation, which could interfere with multiple drug resistance (MDR)–ABC transcriptional regulation.9,17,18
We found that the use of the inhibitors of ABC transporters, such as the P-gp inhibitors cyclosporine A and verapamil, the ABCG2 inhibitors fumitremorgin C and diethylstilbestrol, and the MRP1 inhibitor MK571 had not affected the multiple drug resistance property in HMECsu. These results are not surprising, because Su has been reported to block ABC transportors’ efflux function.9 Like other cell-penetrating small molecule TKIs, Su has been recognized to be both an ABC transporters substrate and a blocker/chemosensitizer.19 Reasonably, oversaturation of ABC transporters by Su can explain why other blockers showed no effect in our study. The unusual drug resistance property in HMECsu is interesting. It may indicate the involvement of supplementary mechanisms in the drug-resistant property of HMECsu. The functional redundancy of the ABC transporters could be a plausible explanation. Probably, as is suggested by a study using an in ex vivo tumor model, prosurvival pathways might also contribute to the acquisition of Su resistance.20 The elucidation of unknown supplementary mechanisms needs further investigation.
The research on cancer resistance to TKIs has recently been intensified.21,22 Clearly, genetic mutation (in particular, EGFR), alternative/compensatory signaling within the tumor microenvironment, and intratumor heterogeneity may concurrently contribute to cancer resistance to TKIs. The acquisition of multiple drug resistance in endothelial cells in vitro after long-term exposure to Su, as described by this study, indicates the existence of the intrinsic resistance in tumor vessels independent of genetic alternation or tumor heterogeneity. It has been found that exposure to Su induces Su resistance of cancer cells with epigenetic silencing of the PTEN gene and a constitutive activation of RAS/RAF/ERK pathway.23,24 It has also been reported that HGF/c-Met overexpression and fibroblast growth factor 2 stimulation contribute to the acquired resistance to Su.25,26
In a previous study, we showed that acquired multiple drug resistance in endothelial cells could be induced by long-term exposure to chemotherapeutic doxorubicin in vitro and in vivo.14 High P-gp expression was recently found in tumor endothelial cells, probably in response to VEGF stimulation.13,27 Similar mechanisms may occur when antiangiogenic drugs are administered in clinics. Our data give rise to questions about the involvement of acquired multiple resistances of tumor vessels in antiangiogenic cancer therapies such as Su therapy. Great efforts have been made to better understand drug resistance in antiangiogenic therapy.5 Our findings complement these recent advances well. Further investigation is needed to better understand the mechanisms underlying drug resistance to targeted therapy.
Acknowledgments
We thank the Institute of Cancer (INCA, PL06_130) and the Association Ti’toine for their support. We thank Professors J Soria and C Soria for their support. We thank Professor E Cazin and Dr L Legres for their help during the study.
Author contributions
All authors contributed toward data analysis, drafting and revising the paper and agree to be accountable for all aspects of the work.
Disclosure
The authors declare that they have no competing interests in this work.
References
Ferrara N, Hillan KJ, Gerber HP, Novotny W. Discovery and development of bevacizumab, an anti-VEGF antibody for treating cancer. Nat Rev Drug Discov. 2004;3:391–400. | |
Young RJ, Reed MW. Anti-angiogenic therapy: concept to clinic. Microcirculation. 2012;19(2):115–125. | |
Eckstein N, Röper L, Haas B, et al. Clinical pharmacology of tyrosine kinase inhibitors becoming generic drugs: the regulatory perspective. J Exp Clin Cancer Res. 2014;33:15. | |
Engelman JA, Settleman J. Acquired resistance to tyrosine kinase inhibitors during cancer therapy. Curr Opin Genet Dev. 2008;18: 73–79. | |
Giuliano S, Pagès G. Mechanisms of resistance to anti-angiogenesis therapies. Biochimie. 2013;95(6):1110–1119. | |
Rini BI. Sunitinib. Expert Opin Pharmacother. 2007;8(14):2359–2369. | |
Bergers G, Hanahan D. Modes of resistance to anti-angiogenic therapy. Nat Rev Cancer. 2008;8(8):592–603. | |
Bracci R, Maccaroni E, Cascinu S. Transient sunitinib resistance in gastrointestinal stromal tumors. N Engl J Med. 2013;368(21):2042–2043. | |
Shukla S, Robey RW, Bates SE, Ambudkar SV. Sunitinib (Sutent, SU11248), a small-molecule receptor tyrosine kinase inhibitor, blocks function of the ATP-binding cassette (ABC) transporters P-glycoprotein (ABCB1) and ABCG2. Drug Metab Dispos. 2009;37(2):359–365. | |
Dai CL, Liang YJ, Wang YS, et al. Sensitization of ABCG2-overexpressing cells to conventional chemotherapeutic agent by sunitinib was associated with inhibiting the function of ABCG2. Cancer Lett. 2009;279(1):74–83. | |
Pénzváltó Z, Tegze B, Szász AM, et al. Identifying resistance mechanisms against five tyrosine kinase inhibitors targeting the ERBB/RAS pathway in 45 cancer cell lines. PLoS One. 2013;8(3):e59503. | |
Kerbel RS. Inhibition of tumor angiogenesis as a strategy to circumvent acquired resistance to anti-cancer therapeutic agents. Bioessays. 1991;13:31–36. | |
Akiyama K, Ohga N, Hida Y, et al. Tumor endothelial cells acquire drug resistance by MDR1 up-regulation via VEGF signaling in tumor microenvironment. Am J Pathol. 2012;180:1283–1293. | |
Huang L, Perrault C, Coelho-Martins J, et al. Induction of acquired drug resistance in endothelial cells and its involvement in anticancer therapy. J Hematol Oncol. 2013;6(1):49. | |
Trochon-Joseph V, Martel-Renoir D, Mir LM, et al. Evidence of antiangiogenic and antimetastatic activities of the recombinant disintegrin domain of metargidin. Cancer Res. 2004;64(6):2062–2069. | |
Ji BS, He L, Liu GQ. Reversal of p-glycoprotein-mediated multidrug resistance by CJX1, an amlodipine derivative, in doxorubicin-resistant human myelogenous leukemia (K562/DOX) cells. Life Sci. 2005;77:2221–2232. | |
Baker EK, Johnstone RW, Zalcberg JR, El-Osta A. Epigenetic changes to the MDR1 locus in response to chemotherapeutic drugs. Oncogene. 2005;24(54):8061–8075. | |
To KK, Polgar O, Huff LM, Morisaki K, Bates SE. Histone modifications at the ABCG2 promoter following treatment with histone deacetylase inhibitor mirror those in multidrug-resistant cells. Mol Cancer Res. 2008;6(1):151–164. | |
Brózik A, Hegedüs C, Erdei Z, et al. Tyrosine kinase inhibitors as modulators of ATP binding cassette multidrug transporters: substrates, chemosensitizers or inducers of acquired multidrug resistance? Expert Opin Drug Metab Toxicol. 2011;7(5):623–642. | |
Zhou Q, Lv H, Mazloom AR, Xu H, Ma’ayan A, Gallo JM. Activation of alternate prosurvival pathways accounts for acquired sunitinib resistance in U87MG glioma xenografts. J Pharmacol Exp Ther. 2012;343(2):509–519. | |
Rosenzweig SA. Acquired resistance to drugs targeting receptor tyrosine kinases. Biochem Pharmacol. 2012;83(8):1041–1048. | |
Huang M, Shen A, Ding J, Geng M. Molecularly targeted cancer therapy: some lessons from the past decade. Trends Pharmacol Sci. 2014;35(1):41–50. | |
Yang J, Ikezoe T, Nishioka C, et al. Long-term exposure of gastrointestinal stromal tumor cells to sunitinib induces epigenetic silencing of the PTEN gene. Int J Cancer. 2012;130(4):959–966. | |
Piscazzi A, Costantino E, Maddalena F, et al. Activation of the RAS/RAF/ERK signaling pathway contributes to resistance to sunitinib in thyroid carcinoma cell lines. J Clin Endocrinol Metab. 2012;97(6):E898–E906. | |
Shojaei F, Lee JH, Simmons BH, et al. HGF/c-Met acts as an alternative angiogenic pathway in sunitinib-resistant tumors. Cancer Res. 2010;70(24):10090–10100. | |
Welti JC, Gourlaouen M, Powles T, et al. Fibroblast growth factor 2 regulates endothelial cell sensitivity to sunitinib. Oncogene. 2011; 30(10):1183–1193. | |
Hida K, Akiyama K, Ohga N, Maishi N, Hida Y. Tumour endothelial cells acquire drug resistance in a tumour microenvironment. J Biochem. 2013;153(3):243–249. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.