Back to Journals » Drug Design, Development and Therapy » Volume 14
Indole: The After Next Scaffold of Antiplasmodial Agents?
Authors Surur AS ![]() , Huluka SA
, Huluka SA ![]() , Mitku ML, Asres K
, Mitku ML, Asres K
Received 24 August 2020
Accepted for publication 12 October 2020
Published 11 November 2020 Volume 2020:14 Pages 4855—4867
DOI https://doi.org/10.2147/DDDT.S278588
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Tin Wui Wong
Abdrrahman Shemsu Surur,1 Solomon Assefa Huluka,2 Melese Legesse Mitku,3 Kaleab Asres4
1CDT-Africa, Addis Ababa University, Addis Ababa, Ethiopia; 2Department of Pharmacology and Clinical Pharmacy, Addis Ababa University, Addis Ababa, Ethiopia; 3Department of Pharmaceutical Chemistry, University of Gondar, Gondar, Ethiopia; 4Department of Pharmaceutical Chemistry and Pharmacognosy, Addis Ababa University, Addis Ababa, Ethiopia
Correspondence: Abdrrahman Shemsu Surur
CDT-Africa, Addis Ababa University, P. O Box 182932, Addis Ababa, Ethiopia
Tel +251-911171194
Email [email protected]
Abstract: Malaria remains a global public health problem due to the uphill fight against the causative Plasmodium parasites that are relentless in developing resistance. Indole-based antiplasmodial compounds are endowed with multiple modes of action, of which inhibition of hemozoin formation is the major mechanism of action reported for compounds such as cryptolepine, flinderoles, and isosungucine. Indole-based compounds exert their potent activity against chloroquine-resistant Plasmodium strains by inhibiting hemozoin formation in a mode of action different from that of chloroquine or through a novel mechanism of action. For example, dysregulating the sodium and osmotic homeostasis of Plasmodium through inhibition of PfATP4 is the novel mechanism of cipargamin. The potential of developing multi-targeted compounds through molecular hybridization ensures the existence of indole-based compounds in the antimalarial pipeline.
Keywords: indole, antimalarial agents, hemozoin inhibition, PfATP4, multi-target approach
Introduction
Malaria is one of the most prevalent and deadliest diseases in the world. World history tells us malaria has claimed the lives of millions of people in the past, and current reports reveal that malaria remains among the top three persistent foes to people living in Africa. According to world malaria report 2019, 213 million or 93% of malaria cases were in Africa, followed by 3.4% cases from South-East Asia region. Two hundred twenty-eight million malaria cases were reported in 2018 compared to 251 million cases in 2010, clear proof that the reduction in prevalence of malaria has stalled after several years of decline. The plateauing in the number of malaria cases for the past two years and the estimated 405,000 deaths, globally, in 2018 is affirmation of the treatment gap in malaria.1–3
The constitutive element for the rejuvenation of malaria has been the development and spread of drug-resistant Plasmodium strains. Development of effective treatments against drug-resistant Plasmodium strains is the greatest challenge in the chemotherapy of malaria. Poor understanding of mechanisms of emergence of resistance, few chemotypes in the antimalarial pipeline and the associated cross resistance between similar chemotype hampered new antimalarial drug development.4 Recently, resources and spotlights aimed at malaria including initiatives such as Medicines for Malaria Venture and Multilateral Initiative on Malaria have increased. However, the history of inconsistent support of researches and prevention projects in the endemic areas are one of the major determinants for the current resurgence of malaria.5–7
Scaffolds in Currently Available Antimalarial Agents
Quinine, from the bark of Cinchona calisaya tree, was the first widely used antimalarial drug. The quinoline nucleus in quinine (1, Figure 1) is the classical yet the loftiest scaffold that have relieved the human race from the aftermath of Plasmodium infections for more than 400 years. The benefactions from quinoline-based antimalarials was prolonged by synthetic derivatives such as chloroquine (2), which was widely used as the first-line antimalarial agent. The clinical indications of 2, however, is falling at an accelerated rate owning to a rapid surge in resistance. Furthermore, the immediate quinoline-based prospects including amodiaquine are suffering from cross-resistance with their predecessors and development of full resistance is on the horizon.8,9
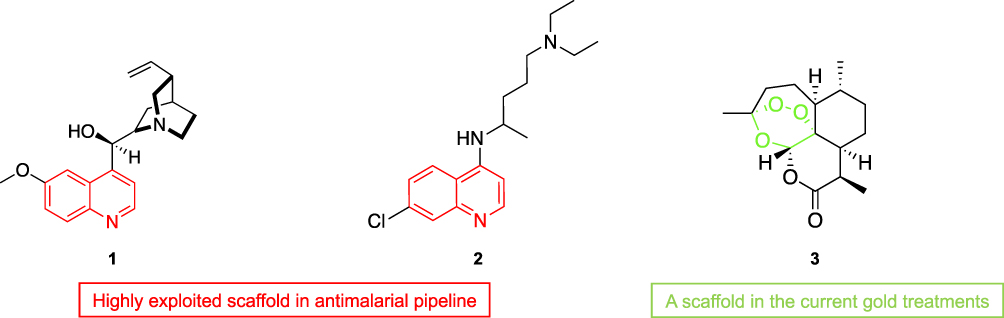 |
Figure 1 The chemical structures of quinoline in quinine (1) and chloroquine (2), and the endoperoxide scaffold in (+)-artemisinin (3). |
The endoperoxide scaffold in (+)-artemisinin (3) and its analogues represents the elite among mid-term prospects. Artemisinin-based combination therapies are currently the mainstay treatments for uncomplicated malaria, albeit they suffer from incomplete oral absorption associated with poor dissolution in the intestinal fluids, short half-lives and emergence of resistant strains of Plasmodium.10 Undoubtedly, new scaffolds capable of filling the highly unmet medical needs in the treatment of malaria are urgently needed. Efficacy, low toxicity and affordability were the most principal virtue for the early bird success of 2. In addition, new drugs should be safe, effective against resistant strains, provide cure within a reasonable period of time, and have appropriate formulations for oral use.9,11 The criteria for new antimalarial agents are likely to be met if attentions are diverted from the established and exceedingly evaluated scaffolds to chart less exploited galaxies of scaffolds in the antimalarial chemical space.12,13 In this review, the indole scaffold as a lead for the development of novel antiplasmodial agents meeting the above-mentioned ideal characteristics are discussed. Though numerous indole-based compounds endowed with antimalarial activity are reported and reviewed,14–16 this mini-review focuses only on recently reported compounds with drug-like properties and promising activity against resistant strains of Plasmodium.
Propitious Indole-Based Antiplasmodial Agents
Diverse indole-based antiplasmodial agents of natural sources or synthetic origin are reported. Piperidine indole, spiroindolone, bisindole, or aminoindole are examples of scaffolds in the novel chemical classes of antiplasmodial agents exhibiting multiple modes of action, of which, targeting the formation of hemozoin in P. falciparum, PfATP4 and melatonin receptor are the most commonly recognized modes of actions. Indole derivatives in respective chemical classes having promising antiplasmodial activity, acceptable drug likeness and preclinical profiles are discussed below.
Piperidine Indoles
Screening of the recently publicized Tres Cantos Antimalarial Set (TCAMS) of GlaxoSmithKline (GSK) led to the discovery of a piperidine indole derivative TCMDC-134281 (4a, Figure 2), which has an EC50 of 0.034 μM against chloroquine (CQ) sensitive P. falciparum 3D7 strain. However, poor drug-like properties and cross-resistance with CQ were demises of 4a, which were addressed by removing one of the piperidin-4-yl and the 4-aminoquinolinyl fragments. The modifications reduced the overall lipophilicity and molecular weight, thereby improving the druglikeness of 4a. Specifically, removal of the 4-aminoquinolonyl fragment, the essential pharmacophore of CQ, is considered responsible for retaining activity against CQ resistant P. falciparum Dd2 strain. Among the simplified analogues, compound 4b has lead-like properties, with cLog P of 2.42 and molecular weight of 305, and overcome the cross-resistance against Dd2 strain. The relative spatial distance between the carbonyl group and the pyridinyl nitrogen atom is required for activity. Compound 4b represents a new chemotype for further optimizations towards novel and affordable antiplasmodial drugs.17
 |
Figure 2 Non-essential structural features in TCMDC-134,281 (4a) which were moved in the design of TCMDC-134,281 (4b). |
Bisindoles
Various compounds, having bisindole moiety embedded somewhere in their structure, have shown potent antiplasmodial properties. Dihydrousamabarensine (5a, Figure 3) isolated from Strychnos usambarensis exhibited more potency against CQ-resistant strains (IC50 = 0.032 μM) than CQ-sensitive strains (IC50 = 0.857 μM). Additional bisindole alkaloids with a moderate activity such as usamabarensine, strychnopentamine and isostrychnopentamine were isolated from other Strychnos species. Pygmies from Cameroon have a tradition of treating malaria fever with the use of S. icaja. An oxygenated bisindole alkaloid with IC50 value of 0.168 μM against CQ-resistant strains, isosungucine (5b), was considered as the underlying compound in the bioactivity of S. icaja.16 Modest basicity is essential for the antiplasmodial activity of Strychnos alkaloids. Monomers of Strychnos alkaloids do not have antiplasmodial properties. However, polymerization increases the basic nature of the monomers and confers antiplasmodial activity.18
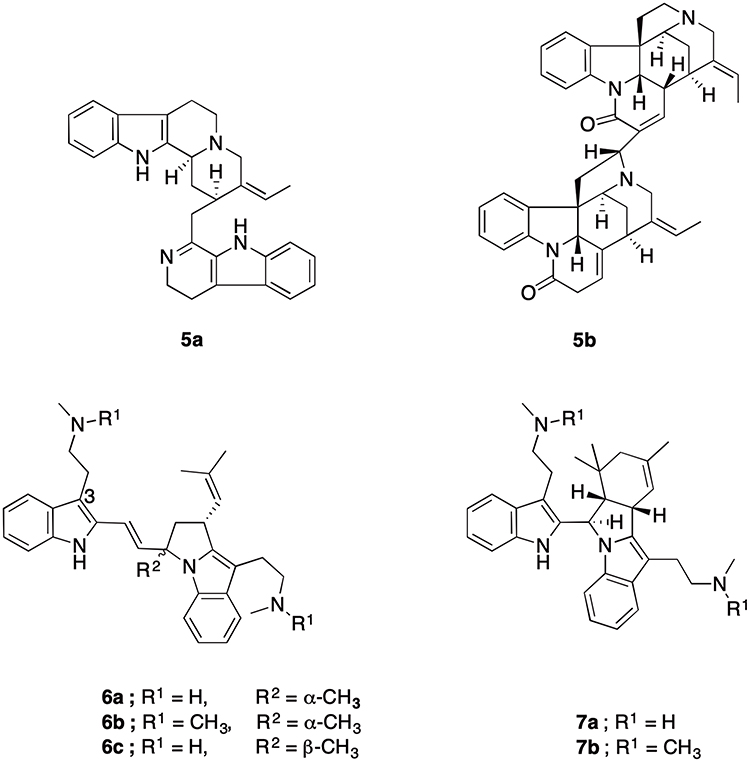 |
Figure 3 Chemical structures of dihydrousamabarensine (5a), isosungucine (5b), flinderoles A–C (6a–6c), isoborreverine (7a) and dimethylisoborreverine (7b). |
A novel bisindole alkaloid, flinderole A (6a), was discovered from the Australian plant Flindersia acuminata. Chemically related and almost equipotent analogues, flinderole B–C (6b–6c), were isolated from the extract of F. amboinensis. Flinderoles 6a–c (Figure 3) have IC50 values of 0.08–1.42 μM against Dd2 P. falciparum strain.19 Methylation of the C-3 ethylamine side chain enhanced the antiplasmodial property of flinderoles. Although in vitro β-hematin (hemozoin) formation assay indicated their direct binding to heme, flinderoles and related bisindole analogues such as isoborreverine (7a) and dimethylisoborreverine (7b) inhibit hemozoin formation in a different way to CQ. P. falciparum trophozoites treated with 7b showed abnormal morphology of the digestive food vacuole and a reduced formation of hemozoin. Interrupting hemozoin degradation through a novel mechanism might explain their activity against CQ resistant strains18,20,21 and highlights the versatile antiplasmodial properties of indole-based compounds.
Spiroindolones
Spiroindolones or spirotetrahydro-β-carbolines are a new class of antimalarial agents identified using a whole cell high-throughput screening. Spiroindolones have a mode of action distinct from clinically available antimalarial agents via inhibition of PfATP4, a plasmodial plasma membrane Na+-ATPase that regulates sodium and osmotic homeostasis by exporting Na+ ion, while simultaneously importing H+ ion. Yeung et al22 identified a racemic spiroazepineindole (8a, Figure 4) with a moderate potency against CQ resistant K1 strain and administration of a single dose of 100 mg kg−1 to P. berghei infected mice reduced parasitemia by 96%. The spirocenter in 8a was found essential for activity and its removal led to almost complete loss of activity. In vitro testing against the wild-type NF54 P. falciparum strain revealed the eutomer, where (1R,3S)-8a was 250-fold more potent than (1S,3R)-8a. Replacing the 5ʹ-bromo with a 5ʹ-chloro moiety promoted optimal balance between potency, favourable pharmacokinetics and synthesis accessibility. Simplification by removing the chiral center at 3-position led to an inactive analogue. However, bulkier substituent in lieu of the 3-methyl decreased activity and only a methyl or trifluoromethyl group was tolerable. Structural manipulations of 8a following the above and more structure-activity relationships led to the synthesis of NITD609 (renamed KAE 609 and now rebranded as cipargamin). The C6 and C7 positions stood as susceptible metabolic positions more prone to oxidation and hence, faster clearance. Accordingly, 6-fluoro and 7-chloro substituents were added in cipargamin (8b). Cipargamin is currently under Phase II clinical trial and meets the general criteria set for new antimalarial drugs; kill parasite blood stage, active against drug resistant parasites, safe and has pharmacokinetic properties that allow once-daily oral dosing.22–24 Cipargamin has a sub-nanomolar potency against blood stage malaria parasite, which is more effective than the current gold standard treatment. Cipargamin is effective against both sexual and asexual stages of P. falciparum. Moreover, as it was revealed in 35S-radiolabeled methionine and cysteine ([35S]-Met/Cys) incorporation assay, 8b blocks protein synthesis in P. falciparum.23
 |
Figure 4 Chemical structures of (1R,3S)-8a and (1S,3R)-8a enantiomers of a spiroazepineindole derivative and cipargamin (8b). |
Conjugated Indole Analogues
Molecular hybridization gave a new impetus to the science of medicinal chemistry and drug design. The methodology involves the rational design of drugs in which two or more different pharmacophoric units are covalently linked into a single entity to form potentially dual-acting compounds.25
Isatin, 1H-indole-2,3-dione, is the most prevalent indole derivatives in natural products and pharmaceuticals. Series of 1H-1,2,3-triazole-tethered isatin-7-chloroquinoline and piperazine-tethered isatin-7-chloroquinoline using a molecular hybridization approach was reported by Raj et al26–28 and the length of the alkyl spacer and a substitution at the C-5 position of the isatin were determinants of antiplasmodial activity. In 1H-1,2,3-triazole-tethered 7-chloroquinoline-isatin hybrids, the most potent derivative 9a (Figure 5) with IC50 of 1.2 μM against the CQ-resistant W2 P. falciparum strain has a propyl spacer and a chloro substituent at C-5 of the isatin moiety. In a series of 1H-1,2,3-triazole-tethered 3-hydroxyl-indole-7-chloroquine, on the other hand, the most active compound 9b with IC50 value of 0.069 μM against W2 strains has a reduced imine and a butyl linker. The most potent compound 9c in piperazine-tethered series of compounds has a pentyl linker and a fluoro substituted isatin moiety.26–28
 |
Figure 5 Chemical structures of conjugated indole analogues; (A) The optimal length of the linker connecting the triazolyl or piperazinyl tether to isatin or 3-hydroxyindole various from ethyl to pentyl alkyl in isatin-chloroquine conjugates (9a–9c); (B) Conjugates of indol-3-yl with quinoline (10a) or quinolinium (10b); (C) The chemical structures of cryptolepine (11a), ellipticine (11b) and 2,7-dibromocryptolepine (11c); (D) The chemical structure of tryptanthrin (12a), and its more soluble 3-methyl ester (12b) and 3-chloro (12c) derivatives. |
Indole-quinoline conjugation with an indol-3-yl linked to the 2-position of a 4-aminoquinoline moiety represents another example of molecular hybridization for novel antimalarial agents with promising activity against P. falciparum. The indole-quinoline conjugate with a quaternary nitrogen on the quinolone, compound 10a (Figure 5), showed an improved activity against K1 strain (IC50 = 0.12 μM) but the corresponding nonquaternerized analogue 10b was found to be relatively less active against the K1 strain. How compound 10a, with a quaternary quinolinium, crosses the cell membrane is interesting. However, the uptake of quinolinium conjugates was thought to be slow since potencies were lowered after a brief exposure to parasites. Like delocalized cationic compounds endowed with a cell-based activity and oral bioavailability, delocalization of net positively charged quinolinium conjugates may retain the membrane permeability.29 Furthermore, alkylation of the 4-amino moiety improved activity against 3D7 strain. A unique mechanism of antiplasmodial activity by compromising mitochondrial function is suggested for indole-quinoline conjugates.30
Structure complexity is a major limitation of indole conjugates mentioned above. Derivatives of cryptolepine and tryptanthrin represent a class of monomeric heterocycle conjugates which are attractive for chemical optimization. Cryptolepine (11a) is an indoloquinoline isolated from the root of Cryptolepis sanguinolenta, a plant frequently used as an antimalarial and febrifuge in Central and Western Africa. Mechanistic studies demonstrated that 11a is capable of inhibiting heme crystal growth and the structurally related analogue, ellipticine (11b), directly interacts with heme to inhibit hemozoin formation. Ellipticine was isolated from the bark of the Amazonian tree Aspidosperma vargasii, which is used as a traditional antimalarial treatment. Ellipticine and cryptolepine are effective against K1 strain. The circumstances that 11a–11b do not exhibit cross-resistance with CQ suggest additional mechanisms, and intercalation with DNA and inhibition of topoisomerase II have surfaced. More active semi-synthetic analogues of 11a (IC50 = 0.114 μM) were reported. Of this, the most interesting 2,7-dibromocryptolepine (11c) is nine times more active against K1 strain than 11a and suppressed 90% of parasitemia in mice infected with P. berghei.16,31 Furthermore, more potent analogues of 11a bearing diamino-alkane side chain at C-11 were synthesized depending on the observation that a basic side chain of CQ was important for accumulation in the acidic vacuole of the Plasmodium species.32
The crystal structure of cryptolepine–DNA complex (Figure 6) identified intercalation into the CG-rich sequence of DNA with non-alternating CC sites. 2,7-Dibromocryptolepine does not appear to intercalate into DNA, albeit cytotoxic activity similar to the parent 11a was reported. An additional mode of action is suggested for 11c since the increased activity compared to 11a was not due to more potent inhibition of hemozoin formation nor its increased accumulation inside the acidic food vacuole.33,34
 |
Figure 6 Intercalation into the CG-rich sequence of DNA by cryptolepine (PyMOL was used to regenerate the picture from the crystal structure with PDB accession number 1K9G). Cyan color: cryptolepine (19). |
Tryptanthrin (12a) is an indoloquinazoline conjugate isolated from different plant sources and has broad biological properties, including anti-tubercular and anti-trypanosomal activities. Unlike 2 that showed 13 fold difference in activity between CQ-sensitive NF54 and CQ-resistant Dd2 strains of P. falciparum, 12a (Figure 5) showed similar activity against NF54 and Dd2 strains with IC50 values of 0.288 μM and 0.114 μM, respectively, though poor solubility has impeded its development. Addition of carboxylic group at position 3 improved solubility but with a costly price of reduced antiplasmodial activity. Inability to cross the biological membrane due to ionization at physiological pH was the reason for the reduced activity. Accordingly, analogues with a carboxylic acid methyl ester such as 12b have regained antiplasmodial activity. Furthermore, substitution by a halogen or nitro moiety at position 8 was critical for improving antiplasmodial activity.16 Onambele et al35 reasoned a further development of a better soluble derivative 12c with IC50 values of 0.033 μM and 0.031 μM against NF54 and Dd2 strains, respectively, which was also considered as a transmission-blocking candidate as it prevented the maturation of early- to late-stage gametocytes and inhibited exflagellation of microgametocytes.
Aminoindoles
Aminoindole is a novel scaffold with a potent activity against P. falciparum.36 Hit screening of almost 70,000 compounds, against Dd2 and 3D7 P. falciparum strains from the Broad Institute and ICCB-L compound collection of Harvard Medical School led to the identification of Genz-644442 (13a, Figure 7) as a promising hit compound, with IC50 values of 0.2 and 0.285 μM against Dd2 and 3D7, respectively. Compound 13a cured mice infected with P. berghei with an efficacy of greater than 99%. In addition to its potent antiplasmodial properties, In vitro ADME assays revealed that 13a has high membrane permeability, high solubility, better stability in liver microsomes and hepatocytes, and low affinities for all CYP450 isozymes. Accordingly, a major hit-to-lead filtering campaign generated 321 analogues of 13a. The enantiomers of 13a which were resolved using supercritical fluid chromatography displayed similar in vitro potency against P. berghei. One of the enantiomer (absolute configuration not disclosed) was far less active against P. berghei in vivo and lower plasma clearance in mice was reported as an underlying reason. Structural manipulations of 13a depending on the structure-activity relationships developed from the 321 analogues yielded a more potent Genz-668,764 (13b) with IC50 of 0.028 μM (against 3D7) and 0.065 μM (against Dd2). Similar to enantiomers of 13a, enantiomers of 13b differed in their in vivo efficacy against P. berghei. The mechanism of action of aminoimidazole is not established at present, though the trophozoite stage appeared to be the target based on the microscopic examinations of synchronized in vitro P. falciparum cultures.15,37
 |
Figure 7 Chemical structures of aminoindole derivatives (13a–13b), indole-3-glyoxyl tyrosine derivatives (14a–14b), prenylated indole alkaloids (15a–15c), and melatonin antagonist (16a–16b). |
Indole-3-Glyoxyl Tyrosine Derivatives
Promoted by the potent antiplasmodial activity of indole-based compounds from GSK, Vasconcelos et al38 synthesized the conjugate of indole with a series of tyrosine residues. Compounds 14a (IC50 = 1.3 μM) and 14b (IC50 = 3.7 μM) displayed potential antiplasmodial property against 3D7 strains. Conformational analysis indicated that potent indole-3-glyoxyl tyrosine derivatives including 14a and 14b (Figure 7) have linear conformation, unlike the U-shaped conformation by less active analogues. Vasconcelos et al38 reported that intramolecular hydrogen bonding between indole and tyrosine residues or hydrophobic interaction of п-п type are considered responsible for the U-conformation.
Prenylated Indole Alkaloids
Plants from the genus Flindersia, including the aforementioned F. acuminata and F. amboinensis, have been a source of antimalarial alkaloids of the indole and quinoline types. Pimentelamine A–C (15a–15c, Figure 7) represent a new class of indole alkaloids isolated from F. pimenteliana with a biosynthetic origin via cyclization of 2-prenyl-N,N-dimethyltryptamine with semidehydroascorbic acid radical. Pimentelamine C (15c) exhibited a moderate activity against 3D7 and Dd2 strains with IC50 values of 3.6 and 2.7 μM, respectively. The polar N-oxide moiety at C3” in 15c may enhance bioactivity and solubility, unlike the inactive analogues 15a–15b, and is responsible for the antiplasmodial activity.39
Indole as a Lead for Multi-Targeted Drugs
The “one target, one drug” approach, where a drug with a high degree of selectivity to a single biological target is designed in order to avoid unwanted side effects, has dominated drug discovery in the past. Genomics has challenged this drug discovery paradigm by unveiling the complexity and interdependence of biological pathways through target-target networks. Consequently, blocking a single process may have a diminishing effect since the system compensates by enhancing alternative pathways.31 In this post-genomic era, the multi-target drug concept is gaining momentum. The complexity of the current incurable pathologies such as cancer and psychosis are witnesses that single-target drugs are inadequate to effect cures. The anti-neoplastic turmeric and the atypical anti-psychotic clozapine are commonly employed examples for multi-target approaches in drug design. Twenty-one percent of new molecular entities approved by FDA from 2015–17 are multi-target drugs, clear proof of the success of this approach.40,41
Multiple modalities of action of indole-based compounds, as alluded to above, is considered responsible for their efficacy against multiple CQ-resistant strains. The resistant Plasmodium spp. are still susceptible to a drug having a different or additional mechanism of action. Antiplasmodial compounds with multiple modes of action are crucial to remain ahead of the relentless Plasmodium spp. in developing resistance to antimalarial treatments. It is important to note that the indole nucleus alone does not guarantee antiplasmodial activity. Indeed, alkaloids from species of Pandaca, Bonafousia or Rauvolfia, for example, contain indole structures, and none are active against Plasmodium parasites.42 Accordingly, designing the indole scaffold to simultaneously consolidate the essential structure-activity relationships for inhibiting hemozoin formation, PfATP4 or melatonin receptors may lead to analogues having multiple modes of action. For example, molecular hybridization in which the pharmacophore units of hemozoin inhibitors and PfATP4 inhibitors are covalently linked to an indole scaffold may lead to dual-acting antiplasmodial agents.
Inhibition of Hemozoin Formation
Inhibiting plasmodial heme detoxification pathway to hemozoin is considered the most promising and ideal target. Hemoglobin decomposition within the parasite digestive food vacuole releases a large quantity of free heme (ferriprotoporphyrin IX). The large quantity of the free heme is thought to be toxic to the Plasmodium through membranes disruptions, lipid peroxidation, and protein and DNA oxidation. Accordingly, Plasmodium spp. use a heme detoxification system called biocrystallization that polymerizes heme into an insoluble hemozoin. Hemozoin is chemically and structurally identical to β-hematin (FeIII-protoporphyrin-IX)2. Hemozoin is believed to consist of strands of FeIII-protoporphyrin units, linked into a polymer through coordination complexes between the carbonyl group of a propionate side chain of one monomer and the Fe3+ atom in the porphyrin ring of another monomer. In addition to the toxic effects of free heme, the ability of the parasite to maintain cationic gradients is impaired by the accumulation of a significant concentration of the free hematin and hematin–antimalarial drug complex, leading to the death of the parasite.34,43
The underlying mechanism for the action of quinoline antimalarial agents against hemozoin formation is not clearly known, though a surface-drug interaction is proposed as the origin of action. The same uncertainty surrounds the mechanism of inhibition of hemozoin formation by indole based antiplasmodial agents, albeit hemozoin inhibition, as mentioned above, was elucidated as a major mechanism for the antiplasmodial properties of indole alkaloids including cryptolepine. Some indole derivatives are capable of directly binding with ferriprotoporphyrin IX, thus disrupting the polymerization of heme to hemozoin. Moreover, indole alkaloids with weak basic properties were active antiplasmodial agents, whereas structurally related alkaloids with different acid–base profiles were inactive, suggesting a similar mode of action to that of quinolines.44,45 Therefore, the above and other pertinent structure-activity relationships of quinolone based antimalarial agents may improve the inhibition of hemozoin formation by indole scaffold, i.e. scaffold hopping.
PfATP4 Inhibition
Ensuing entry, Plasmodium spp. establish a new permeability pathway in the plasma membrane of the host erythrocytes for the uptake of important nutrients, while simultaneously allowing the influx of Na+ ion down the concentration gradient. Despite the increased Na+ ion concentration in the erythrocyte cytosol, the intraerythrocytic Plasmodium maintains a low cytosolic Na+ ion concentration by extruding Na+ ion against the inward gradient via PfATP4, a P-type ATPase of P. falciparum. PfATP4 has a sequence similarity to the Na+ pump of the lower plants’ ENA (exitus natrus) P-type Na+ ATPase. The ENA ATPases are closely related to Sarcoplasmic/Endoplasmic Reticulum Ca2+ ATPases (SERCA) and Plasma Membrane Ca2+ ATPase (PMCA). PfATP4 belongs to the same class (PII class) with SERCA and share a common structural feature of large cytoplasmic domains connected to several transmembrane helices. This superfamily of proteins is restricted to lower eukaryotes, making it an attractive drug target.46–48
PfATP4 is a target for the spiroindolone class of antiplasmodial agents. Spiroindolones inhibit parasite growth via disruption of Na+ homeostasis, that was attenuated in Plasmodium spp. bearing resistance conferring mutations in PfATP4. In addition to spiroindolones, PfATP4 is identified as a major mechanism for the antimalarial activity of novel chemotypes that have progressed to preclinical evaluations, aminopyrazoles and dihydroisoquinolones.49 A homology model of PfATP4 (Figure 8), using SERCA (PDB 3TLM) as a template50,51 and the amino acids sequence of PfATP4 imported from Uniprot,52 is reported, and can be used for the design, evaluation of structure-activity relationships and optimizations of PfATP4 inhibitors. Another homology model using a rabbit Ca2+ ATPase (PDB 2DQS) as a template is reported for the binding interactions of indole-based chiral bicyclic lactam derivatives.53 Structural features essential in the spiroindolones or isoquinoline series of PfATP4 inhibitors and in the quinoline based inhibitors of hemozoin formation can be hybridized for optimization of the indole scaffold as a dual-acting antimalarial agent.
 |
Figure 8 Homology model of PfATP4 regenerated using the crystal structure of SERCA (PDB 3TLM) as a template. |
Melatonin Receptors Modulation
As an additional novel mode of action, targeting melatonin receptor further revealed the potential of developing indole-based multi-targeting drug candidates. Melatonin is established as essential for synchronization of the malaria parasitic cell cycle. Its role in regulating genes involved in ubiquitin/proteasome system is vital to regulate cell cycle and transcriptional activity in Plasmodium and increase its mature schizont percentage.54 Malaria promotes hepatocyte apoptosis by inducing mitochondrial pathology and mitochondrial oxidative stress, an effect prevented by high dose of melatonin.55 Hotta et al40 showed that the indole-based melatonin antagonist, luzindole (16a), was able to inhibit trophozoites by disrupting the rhythmicity of parasite cell cycle. Smear results from mice treated with 16a (Figure 7) exhibited altered percentage of Plasmodium forms with lower number of trophozoites. Those limited number of trophozoites left following luzindole treatment were unable to produce ring forms in their subsequent developmental stage. The antimalarial effect of luzindole was also found to be statistically significant (p < 0.05) in ring stage of the parasite. Further, luzindole significantly abolished the increase in cAMP concentration in both ring form and late trophozoite stage by antagonizing melatonin.56
Besides luzindole, Luthra et al57 reported the promising antimalarial property of C2-arylimino tryptamine derivatives as novel class of melatonin receptor antagonists, of which 16b showed the most potent blockade of trophozoite stage growth via disrupting synchronization of the parasite with a considerable affinity to melatonin receptor. Moreover, compound 16b and luzindole exhibited a 47% and 48% inhibition of parasitemia, respectively, at 5 μM dose. As a novel approach to tackle malaria, harmonized administration of high dose of melatonin and its antagonist is considered.58,59
Further ensuring their future in antimalarial pipeline, indole alkaloids showed synergistic/additive interactions with conventional antimalarial agents. Mice treated with 16a (15mg kg−1) and suboptimal dose of 2 (1.5mg kg−1), for instance, had a drastic reduction in the number of intraerythrocytic parasites.60 In the in vitro interaction assay, 11a exhibited additive effect with 2 and lumefantrine while synergism was noted with amodiaquine.61 This encouraging interaction is augmented by acceptable safety profile of the combinations. Cryptolepine in combination with 4 mg kg−1 dose of artemisinin showed no significant biochemical and histopathology indices variations compared to the control group.62
Preclinical Profiles
Parallel to pharmacological screening, toxicity profiles of indole alkaloids have been heavily investigated. Extracts rich in indole alkaloids revealed the absence of genotoxicity, cardiotoxicity as well as adverse effect in the respiratory system.63 Substantiating this finding, indole analogues such as indole-3-glyoxyl tyrosine derivatives, 11b, 15c and piperidine indoles appeared to have an acceptable safety profile.31,38,63 However, some indole candidates such as 16a and 11a were found to impair cardioprotection and cause cytotoxicity, respectively.64
Cryptolepine, an extensively studied indole alkaloid, was reported to cause in vivo toxicity in mice.65 A recent study,66 moreover, showed that 11a induces malformations in zebrafish embryo. Despite these reported toxicities, Forkuo et al62 reported that liver, spleen, stomach, and kidney histopathology of cryptolepine treated P. berghei infected mice were not different from control mice. Acute toxicity study of indole alkaloids isolated form Alstonia scholaris, with antiplasmodial activity attributed to bisindole alkaloids, was also done on rats, mice and dogs. In dogs, a treatment related short-lasting emesis and reddening of perioral mucosa was noted following a single dose. Due to a possible toxicity to neuromuscular and nervous system, acute dose administration of these indoles caused shortness of breath, wheezing and convulsion in mice but not in rats.67 A one-week treatment of rats with the aminoindole 13a, on the other hand, resulted in a 70% reduction in reticulocytes, which recovered a few days following cessation.37
Cryptolepine was further demonstrated to exert a cytotoxic and genotoxic effect, in vitro. It caused a dose-dependent reduction in viability of the V79 cell line, a Chinese hamster lung fibroblast,68 and non-melanoma skin cell lines, SCC-13 and A431.69 Some bisindole alkaloids were also found to be cytotoxic to MRC-5 cells.70,71 Cytotoxic effect against 3T6 cell line was exerted by piperazine-tethered 7-chloroquinoline-isatin conjugates. Likewise, a derivative of tryptanthrin, 12c, showed a strong inhibition of MCF-7 cells. But, due to its higher selectivity index, similar to the parent molecule, it was considered for further animal studies. Moreover, tryptanthrin and its derivatives did not show any disruption of red blood cell membrane integrity, which is a severe side effect of the conventional antimalarial agents.35,72
It has been demonstrated that cytotoxicity of indole alkaloids is mainly due to their interference with topoisomerase II-DNA complex stability as well as a tendency to intercalate into DNA (Figure 8). It was also noted that indoles can directly inhibit topoisomerase in the absence of DNA. In addition, 11b forms cytochromes P450 and peroxidases mediated DNA adducts.64,73–75
The observed toxicity in this class of compounds partly hindered further preclinical development of indoles.64 Most of the reported side effects are associated with long term exposure to indoles. As a result, chronic toxic effect of indole derivatives could be precluded as far as malarial treatment regimen extends only for a couple of days.61 Moreover, as the antiplasmodial mechanism of most indole alkaloids, exemplified by 11a, is independent of DNA, it is plausible to prepare analogues with less DNA intercalation propensity. Accordingly, several analogues of 11a showed less cytotoxicity and in vivo adverse effects while retaining the antimalarial potency. Of these, 11c displayed an increased potency with apparently no cytotoxicity in mice, evidenced by less affinity to DNA.75,76 Hence, this is proof that such reported toxicity of promising indole alkaloids is amenable to wane through chemical modifications. Modifications such as having an open ring in place of a six-membered oxygen heterocycle in the tertiary bisindole alkaloid longicaudatine, make the derivative 40 times more selective to the parasite than the parent molecule.19
Besides possessing attractive drug-like attributes and comparable efficacy to artemisinin derivatives,23 cipargamin revealed a promising safety profile of indoles, clinically. Safety and tolerability study in healthy male adult patients showed that 8b is generally well tolerated with transient gastrointestinal and genitourinary adverse events of mild to moderate intensity. Cipargamin has a higher selectivity index with a very minimal risk of cardiotoxicity and genotoxicity. Unlike older antimalarials, it did not significantly bind to human proteins such as hERG, suggesting a low potential for arrhythmia.22 Equipped with a remarkable antimalarial potency and low toxicity profile, 8b is on the verge of joining the existing antimalarial armamentarium.
Conclusion and Future Perspectives
Multiple modalities of antimalarial action, including inhibition of hemozoin formation, PfATP4 and melatonin are associated with natural and synthetic compounds having an indole scaffold. This property makes indole a compelling scaffold for the currently trending paradigm in drug discovery, the multi-targeted approach. Synthesis of novel indole analogues bearing pharmacophore units of both hemozoin inhibitors and PfATP4 inhibitors, through molecular hybridization, may be insurmountable to the Plasmodium spp., which are otherwise relentless in developing resistance to drugs with a single mode of action.
Abbreviations
CQ, chloroquine; IC50, half maximal inhibitory concentration; PfATP4, Plasmodium falciparum P-type ATPase; PMCA, plasma membrane Ca2+ ATPase; SERCA, sarcoplasmic/endoplasmic reticulum Ca2+ ATPases; TCAMS, Tres Cantos Antimalarial Set.
Disclosure
The authors declare no conflict of interest.
References
1. World malaria report 2019. 2019. Available from: https://www.who.int/news-room/feature-stories/detail/world-malaria-report-2019.
2. World malaria report 2018. 2019. Available from: https://www.mmv.org/newsroom/publications/world-malaria-report-2018.
3. Kumar S, Singh RK, Patial B, et al. Recent advances in novel heterocyclic scaffolds for the treatment of drug-resistant malaria. J Enzyme Inhib Med Chem. 2016;31(2):173–186. doi:10.3109/14756366.2015.1016513
4. Kumar S, Bhardwaj TR, Prasad DN, et al. Drug targets for resistant malaria: historic to future perspectives. Biomed Pharmacother. 2018;104:8–27. doi:10.1016/j.biopha.2018.05.009
5. Espinoza JL. Malaria resurgence in the Americas: an underestimated threat. Pathogens. 2019;8(1):11. doi:10.3390/pathogens8010011
6. Conrad MD, Rosenthal PJ. Antimalarial drug resistance in Africa: the calm before the storm? Lancet Infect Dis. 2019;19(10):e338–e351. doi:10.1016/S1473-3099(19)30261-0
7. Surur AS, Fekadu A, Makonnen E, et al. Challenges and opportunities for drug discovery in developing countries: the example of cutaneous leishmaniasis. ACS Med Chem Lett. 2020. doi:10.1021/acsmedchemlett.0c00446
8. Achan J, Talisuna AO, Erhart A, et al. Quinine, an old anti-malarial drug in a modern world: role in the treatment of malaria. Malar J. 2011;10(1). doi:10.1186/1475-2875-10-144
9. Fidock DA, Rosenthal PJ, Croft SL, et al. Antimalarial drug discovery: efficacy models for compound screening. Nat Rev Drug Discov. 2004;3(6):509–520. doi:10.1038/nrd1416
10. Suresh N, Haldar K. Mechanisms of artemisinin resistance in Plasmodium falciparum malaria. Curr Opin Pharmacol. 2018;42:46–54. doi:10.1016/j.coph.2018.06.003
11. Tse EG, Korsik M, Todd MH. The past, present and future of anti-malarial medicines. Malar J. 2019;18(1). doi:10.1186/s12936-019-2724-z
12. Surur AS, Schulig L, Link A. Interconnection of sulfides and sulfoxides in medicinal chemistry. Arch Pharm Chem Life Sci. 2019. doi:10.1002/ardp.201800248
13. Kalaria PN, Karad SC, Raval DK. A review on diverse heterocyclic compounds as the privileged scaffolds in antimalarial drug discovery. Eur J Med Chem. 2018;158:917–936. doi:10.1016/j.ejmech.2018.08.040
14. Kumari A, Singh RK. Medicinal chemistry of indole derivatives: current to future therapeutic prospectives. Bioorg Chem. 2019;89:103021. doi:10.1016/j.bioorg.2019.103021
15. Biamonte MA, Wanner J, Le Roch KG. Recent advances in malaria drug discovery. Bioorg Med Chem Lett. 2013;23(10):2829–2843. doi:10.1016/j.bmcl.2013.03.067
16. Frederich M, Tits M, Angenot L. Potential antimalarial activity of indole alkaloids. Trans R Soc Trop Med Hyg. 2008;102(1):11–19. doi:10.1016/j.trstmh.2007.10.002
17. Santos SA, Lukens AK, Coelho L, et al. Exploring the 3-piperidin-4-yl-1H-indole scaffold as a novel antimalarial chemotype. Eur J Med Chem. 2015;102:320–333. doi:10.1016/j.ejmech.2015.07.047
18. Tajuddeen N, Van Heerden FR. Antiplasmodial natural products: an update. Malar J. 2019;18. doi:10.1186/s12936-019-3026-1
19. Fernandez LS, Buchanan MS, Carroll AR, et al. Flinderoles A−C: antimalarial bis-indole alkaloids from Flindersia species. Org Lett. 2009;11(2):329–332. doi:10.1021/ol802506n
20. Fernandez LS, Sykes ML, Andrews KT, et al. Antiparasitic activity of alkaloids from plant species of Papua New Guinea and Australia. Int J Antimicrob Agents. 2010;36(3):275–279. doi:10.1016/j.ijantimicag.2010.05.008
21. Vallakati R, May JA. Biomimetic synthesis of the antimalarial flindersial alkaloids. J Am Chem Soc. 2012;134(16):6936–6939. doi:10.1021/ja301387k
22. Yeung BKS, Zou B, Rottmann M, et al. Spirotetrahydro β-carbolines (spiroindolones): a new class of potent and orally efficacious compounds for the treatment of malaria. J Med Chem. 2010;53(14):5155–5164. doi:10.1021/jm100410f
23. Rottmann M, McNamara C, Yeung BK, et al. Spiroindolones, a potent compound class for the treatment of malaria. Science. 2010;329(5996):1175–1180. doi:10.1126/science.1193225
24. White NJ, Pukrittayakamee S, Phyo AP, et al. Spiroindolone KAE609 for falciparum and vivax malaria. N Engl J Med. 2014;371(5):403–410. doi:10.1056/NEJMoa1315860
25. Muregi FW, Ishih A. Next-generation antimalarial drugs: hybrid molecules as a new strategy in drug design. Drug Dev Res. 2010;71:20–32.
26. Raj R, Biot C, Carrere-Kremer S, et al. 7-chloroquinoline-isatin conjugates: antimalarial, antitubercular, and cytotoxic evaluation. Chem Biol Drug Des. 2014;83(5):622–629. doi:10.1111/cbdd.12273
27. Raj R, Gut J, Rosenthal PJ, et al. 1H-1,2,3-Triazole-tethered isatin-7-chloroquinoline and 3-hydroxy-indole-7-chloroquinoline conjugates: synthesis and antimalarial evaluation. Bioorg Med Chem Lett. 2014;24(3):756–759. doi:10.1016/j.bmcl.2013.12.109
28. Raj R, Singh P, Singh P, et al. Azide-alkyne cycloaddition en route to 1 H -1,2,3-triazole-tetHered 7-cHloroquinoline-isatin cHimeras: syntHesis and antimalarial evaluation. Eur J Med Chem. 2013;62:590–596. doi:10.1016/j.ejmech.2013.01.032
29. Teguh SC, Klonis N, Duffy S, et al. Novel conjugated quinoline–indoles compromise Plasmodium falciparum mitochondrial function and show promising antimalarial activity. J Med Chem. 2013;56(15):6200–6215. doi:10.1021/jm400656s
30. Singh TP, Singh OM. Recent progress in the biological activities of indole and indole alkaloids. Mini Rev Med Chem. 2018;18:9–25. doi:10.2174/1389557517666170807123201
31. Rocha e Silva LF, Montoia A, Amorim RC, et al. Comparative in vitro and in vivo antimalarial activity of the indole alkaloids ellipticine, olivacine, cryptolepine and a synthetic cryptolepine analog. Phytomedicine. 2012;20(1):71–76. doi:10.1016/j.phymed.2012.09.008
32. Lavrado J, Paulo A, Gut JG, Rosenthal J, Moreira R. Cryptolepine analogues containing basic aminoalkyl side-chains at C-11: synthesis, antiplasmodial activity, and cytotoxicity. Bioorg Med Chem Lett. 2008;18(4):1378–1381. doi:10.1016/j.bmcl.2008.01.015
33. Lisgarten JN, Coll M, Portugal J, et al. The antimalarial and cytotoxic drug cryptolepine intercalates into DNA at cytosine-cytosine sites. Nat Struct Biol. 2019;18(1):57–60. doi:10.1038/nsb729
34. Potter BS, Lisgarten JN, Pitts JE, et al. X-ray crystallographic structure of the potent antiplasmodial compound 2,7-dibromocryptolepine acetic acid solvate. J Chem Crystallogr. 2008;38(11):821–825. doi:10.1007/s10870-008-9398-7
35. Onambele LA, Riepl H, Fischer R, et al. Synthesis and evaluation of the antiplasmodial activity of tryptanthrin derivatives. Int J Parasitol Drugs Drug Resist. 2015;5(2):48–57. doi:10.1016/j.ijpddr.2015.03.002
36. Urgaonkar S, Cortese JF, Barker RH, et al. A concise silylamine approach to 2-amino-3-hydroxy-indoles with potent in vivo antimalaria activity. Org Lett. 2010;36(18):3998–4001. doi:10.1016/j.ijantimicag.2010.05.008
37. Barker RH, Urgaonkar S, Mazitschek R, et al. Aminoindoles, a novel scaffold with potent activity against Plasmodium falciparum. Antimicrob Agents Chemother. 2011;55(6):2612–2622. doi:10.1128/AAC.01714-10
38. Vasconcelos SN, Meissner KA, Ferraz WR, et al. Indole-3-glyoxyl tyrosine: synthesis and antimalarial activity against Plasmodium falciparum. Future Med Chem. 2019;11(6):525–538. doi:10.4155/fmc-2018-0246
39. Robertson LP, Duffy S, Wang Y, et al. Pimentelamines A-C, indole alkaloids isolated from the leaves of the Australian tree Flindersia pimenteliana. J Nat Prod. 2017;80:3211–3217.
40. Ramsay RR, Popovic‐Nikolic MR, Nikolic K, et al. A perspective on multi-target drug discovery and design for complex diseases. Clin Transl Med. 2018;7(1). doi:10.1186/s40169-017-0181-2
41. Morphy R, Rankovic Z. Designed multiple ligands. An emerging drug discovery paradigm. J Med Chem. 2005;48(21):6523–6543. doi:10.1021/jm058225d
42. Passemar C, Salery M, Soh PN, et al. Indole and aminoimidazole moieties appear as key structural units in antiplasmodial molecules. Phytomedicine. 2011;18(13):1118–1125. doi:10.1016/j.phymed.2011.03.010
43. Kgokong JL, Smith PP, Matsabisa GM. 1,2,4-Triazino-[5,6b]indole derivatives: effects of the trifluoromethyl group on in vitro antimalarial activity. Bioorg Med Chem. 2005;13(8):2935–2942. doi:10.1016/j.bmc.2005.02.017
44. Herraiz T, Guillén H, Gonzalez-Pena D, et al. Antimalarial quinoline drugs inhibit β-hematin and increase free hemin catalyzing peroxidative reactions and inhibition of cysteine proteases. Sci Rep. 2019;9(1). doi:10.1038/s41598-019-51604-z
45. Kaur K, Jain M, Kaur T, et al. Antimalarials from nature. Bioorg Med Chem. 2009;17(9):3229–3256. doi:10.1016/j.bmc.2009.02.050
46. Spillman NJ, Allen RW, McNamara CW, et al. Na+ regulation in the malaria parasite Plasmodium falciparum involves the cation ATPase PfATP4 and is a target of the spiroindolone antimalarials. Cell Host Microbe. 2013;13(2):227–237. doi:10.1016/j.chom.2012.12.006
47. Flannery EL, McNamara CW, Kim SW, et al. Mutations in the P-type cation-transporter ATPase 4, PfATP4, mediate resistance to both aminopyrazole and spiroindolone antimalarials. ACS Chem Biol. 2015;10(2):413–420. doi:10.1021/cb500616x
48. Ramanathan AA, Morrisey JM, Daly TM, et al. Oligomerization of the antimalarial drug target PfATP4 is essential for parasite survival. bioRxiv. 2019. doi:10.1101/2019.12.12.874826
49. Spillman NJ, Kirk K. The malaria parasite cation ATPase PfATP4 and its role in the mechanism of action of a new arsenal of antimalarial drugs. Int J Parasitol Drugs Drug Resist. 2015;5(3):149–162. doi:10.1016/j.ijpddr.2015.07.001
50. Jimenez-Diaz MB, Ebert D, Salinas Y, et al. (+)-SJ733, a clinical candidate for malaria that acts through ATP4 to induce rapid host-mediated clearance of Plasmodium. PNAS. 2014;111(50):E5455–E5462. doi:10.1073/pnas.1414221111
51. Sacchetto R, Bertipaglia I, Giannetti S, et al. Crystal structure of sarcoplasmic reticulum Ca2+-ATPase (SERCA) from bovine muscle. J Struct Biol. 2012;178(1):38–44. doi:10.1016/j.jsb.2012.02.008
52. PfATP4. 2020. Available from: https://www.uniprot.org/uniprot/?query=PfATP4&sort=score.
53. Dangi P, Jain R, Mamidala R, et al. Natural product inspired novel indole based chiral scaffold kills human malaria parasites via ionic imbalance mediated cell death. Sci Rep. 2019:9. doi:10.1038/s41598-019-54339-z
54. Singh MK, Dias BKDM, Garcia CRS. Role of melatonin in the synchronization of asexual forms in the parasite Plasmodium falciparum. Biomolecules. 2020;10(9):1243. doi:10.3390/biom10091243
55. Dey S, Guha M, Alam A, et al. Malarial infection develops mitochondrial pathology and mitochondrial oxidative stress to promote hepatocyte apoptosis. Free Radic Biol Med. 2009;46(2):271–281. doi:10.1016/j.freeradbiomed.2008.10.032
56. Hotta CT, Gazarini ML, Beraldo FH, et al. Calcium-dependent modulation by melatonin of the circadian rhythm in malarial parasites. Nat Cell Biol. 2000;2(7):466–468. doi:10.1038/35017112
57. Furuyama W, Enomoto M, Mossaad E, et al. An interplay between 2 signaling pathways: melatonin-cAMP and IP3–Ca2+ signaling pathways control intraerythrocytic development of the malaria parasite Plasmodium falciparum.Biochem Biophys Res Commun. 2014;446(1):125–131. doi:10.1016/j.bbrc.2014.02.070
58. Luthra T, Nayak AK, Bose S, et al. Indole based antimalarial compounds targeting the melatonin pathway: their design, synthesis and biological evaluation. Eur J Med Chem. 2019;168:11–27. doi:10.1016/j.ejmech.2019.02.019
59. Srinivasan V, Zakaria R, Mohamed M. Malaria, therapeutic options and melatonin. Austin J Infect Dis. 2014;1:5.
60. Bagnaresi P, Markus RP, Hotta CT, et al. Desynchronizing Plasmodium cell cycle increases chloroquine protection at suboptimal doses. Open Parasitol J. 2008;2(1):55–58. doi:10.2174/1874421400802010055
61. Forkuo AD, Ansah C, Mensah KB, et al. In vitro anti-malarial interaction and gametocytocidal activity of cryptolepine. Malar J. 2017;16. doi:10.2174/1874421400802010055
62. Forkuo AD, Ansah C, Boadu K, et al. Synergistic anti-malarial action of cryptolepine and artemisinins. Malar J. 2016;15. doi:10.2174/1874421400802010055
63. Zhao YL, Su M, Shang JH, et al. Genotoxicity and safety pharmacology studies of indole alkaloids extract from leaves of Alstonia Scholaris. Nat Prod Bioprospect. 2020;10:119–129. doi:10.1007/s13659-020-00242-4
64. Gopalan RC, Emerce E, Wright CW, et al. Effects of the anti-malarial compound cryptolepine and its analogues in human lymphocytes and sperm in the comet assay. Toxicol Lett. 2011;207(3):322–325. doi:10.1016/j.toxlet.2011.09.010
65. Wright CW. Antiprotozoal natural products. In: Evans WC, editor. Trease and Evan’s Pharmacognosy.
66. Mensah KB, Benneh C, Forkuo AD, et al. Cryptolepine, the main alkaloid of the antimalarial Cryptolepis sanguinolenta (Lindl.) schlechter, induces malformations in Zebrafish Embryos. Biochem Res Int. 2019;2019:1–7. doi:10.1155/2019/7076986
67. Zhao YL, Su M, Shang JH, et al. Acute and sub-chronic toxicity of indole alkaloids extract from leaves of Alstonia scholaris (L.) R. Br. in beagle dogs. Nat Prod Bioprospect. 2020;10:209–220. doi:10.1007/s13659-020-00246-0
68. Ansah C, Gooderham NJ. The popular herbal antimalarial, extract of Cryptolepis sanguinolenta, is potently cytotoxic. Toxicol Sci. 2002;70:245–251. doi:10.1093/toxsci/70.2.245
69. Pal HC, Katiyar SK. Cryptolepine, a plant alkaloid, inhibits the growth of non-melanoma skin cancer cells through inhibition of topoisomerase and induction of DNA damage. Molecules. 2016;21(12):1758. doi:10.3390/molecules21121758
70. Muganza DM, Fruth B, Nzunzu JL, et al. In vitro antiprotozoal activity and cytotoxicity of extracts and isolated constituents from Greenwayodendron suaveolens. J Ethnopharmacol. 2016;193:510–516. doi:10.1016/j.jep.2016.09.051
71. Girardot M, Deregnaucourt C, Deville A, et al. Indole alkaloids from Muntafara sessilifolia with antiplasmodial and cytotoxic activities. Phytochemistry. 2012;73:65–73. doi:10.1016/j.phytochem.2011.09.012
72. Dokunmu T, Ahanonu C, Abegunde O, et al. Artemisinin-induced delayed hemolysis after administration of artesunate and artesunate-amodiaquine in malaria-free Wistar rats. Biomed Res Ther. 2017;4(4):1246–1260. doi:10.15419/bmrat.v4i4.160
73. Stiborova M, Cerna V, Moserova M, et al. The anticancer drug ellipticine activated with cytochrome P450 mediates DNA damage determining its pharmacological efficiencies: studies with rats, hepatic cytochrome P450 reductase null (HRN™) mice and pure enzymes. Int J Mol Sci. 2014;16(1):284–306. doi:10.3390/ijms16010284
74. Kundu N, Roy A, Banik D, et al. Unveiling the mode of interaction of berberine alkaloid in different supramolecular confined environments: interplay of surface charge between nano-confined charged layer and DNA. J Phys Chem B. 2016;120(6):1106–1120. doi:10.1021/acs.jpcb.5b10121
75. Li AL, Hao Y, Wang WY, et al. Design, synthesis, and anticancer evaluation of novel indole derivatives of ursolic acid as potential topoisomerase II inhibitors. Int J Mol Sci. 2020;21(8):2876. doi:10.3390/ijms21082876
76. Van Miert S, Jonckers T, Cimanga K, et al. In vitro inhibition of β-haematin formation, DNA interactions, antiplasmodial activity, and cytotoxicity of synthetic neocryptolepine derivatives. Exp Parasitol. 2004;108(3–4):163–168. doi:10.1016/j.exppara.2004.08.006
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.