")
Back to Journals » International Journal of Nephrology and Renovascular Disease » Volume 9
Independent associations of urine neutrophil gelatinase–associated lipocalin and serum uric acid with interstitial fibrosis and tubular atrophy in primary glomerulonephritis
Authors Lertrit A, Worawichawong S, Vanavanan S, Chittamma A, Muntham D, Radinahamed P, Nampoon A, Kitiyakara C
Received 2 January 2016
Accepted for publication 11 February 2016
Published 20 April 2016 Volume 2016:9 Pages 111—118
DOI https://doi.org/10.2147/IJNRD.S103512
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Pravin Singhal
Amornpan Lertrit,1 Suchin Worawichawong,2 Somlak Vanavanan,2 Anchalee Chittamma,2 Dittapol Muntham,3 Piyanuch Radinahamed,1 Aumporn Nampoon,4 Chagriya Kitiyakara1
1Department of Medicine, 2Department of Pathology, Faculty of Medicine, Ramathibodi Hospital, Mahidol University, Bangkok, 3Section for Mathematics, Faculty of Science and Technology, Rajamangala University of Technology Suvarnabhumi, Phranakhon Si Ayutthaya, 4Research Center, Faculty of Medicine, Ramathibodi Hospital, Mahidol University, Bangkok, Thailand
Abstract: The degree of interstitial fibrosis and tubular atrophy (IFTA) is one of the strongest prognostic factors in glomerulonephritis (GN). In experimental models, high serum uric acid (UA) could contribute to IFTA through direct effects on the renal tubules, but the significance of this process has not been evaluated in patients. Urine neutrophil gelatinase–associated lipocalin (NGAL) is produced by renal tubules following acute or chronic damage. We investigated the relationship between UA and NGAL excretion in primary GN and tested whether these biomarkers are independently associated with IFTA. Urine and blood were collected from patients on the day of kidney biopsy. IFTA was assessed semi-quantitatively. Fifty-one patients with primary GN were enrolled. NGAL/creatinine correlated significantly with proteinuria but not with glomerular filtration rate (GFR). By contrast, UA correlated with GFR but not with proteinuria. NGAL/creatinine did not correlate with UA. Both NGAL/creatinine and UA increased with the severity of IFTA. By multivariate analysis, GFR, NGAL/creatinine, and UA were independently associated with moderate-to-severe IFTA. Combining UA and NGAL/creatinine with classical predictors (proteinuria and GFR) tended to improve discrimination for moderate-to-severe IFTA. Findings that UA was unrelated to urinary NGAL excretion suggest that the two biomarkers reflect different pathways related to the development of IFTA in primary GN. Both NGAL/creatinine and UA were independently associated with moderate-to-severe IFTA.
Keywords: biomarker, fibrosis, glomerulonephritis, kidney, neutrophil gelatinase–associated lipocalin, uric acid
Background
Primary glomerulonephritis (GN) is a heterogeneous group of disorder with diverse etiology that together constitutes one of the leading causes of end-stage renal disease (ESRD) worldwide.1 The pathogenic processes leading to progressive decline in renal function in primary GN are complex involving both systemic and local mediators.2 Once established, proteinuric glomerular injury causes activation of profibrotic pathways leading to subsequent nephron loss.3 Independent of the primary disease, the mechanisms causing progressive nephron loss converge on a common pathway characterized by tubular atrophy and interstitial fibrosis culminating in irreversible ESRD.4
The rates of decline in renal function of individuals with primary GN differ widely with the deterioration often being more accurately associated with the severity of the tubulointerstitial disease than the glomerular lesions.2–4 Severe proteinuria, hypertension, and decreased glomerular filtration rate (GFR) have been shown to be useful clinical predictors of ESRD, but these parameters cannot fully detect early kidney damage because tubulointerstitial damage can occur even when GFR remains in the normal range. At present, the extent of tubular injury and fibrosis can only be assessed by a renal biopsy. Because a biopsy is invasive, it may be delayed and cannot be performed serially to monitor disease progress. Noninvasive biomarkers with close correlations to the pathologic lesions would be useful to guide appropriate management strategies. Currently, there are no universally accepted biomarkers to predict tubulointerstitial lesions,2 but both neutrophil gelatinase–associated lipocalin (NGAL) and serum uric acid (UA) have shown promise as predictors of progression in patients with chronic kidney diseases (CKD)without kidney biopsies, in separate studies.5,6
NGAL is a protein originally purified from human neutrophils and subsequently shown to be highly expressed in renal tubules upon injury.7 Currently, there are considerable data supporting NGAL as a biomarker to detect acute kidney injury (AKI). Recent studies have shown that urine NGAL may also be a useful biomarker for detecting chronic tubulointerstitial damage,8,9 but few studies have examined the relationship between NGAL and the severity of histopathological damage in primary GN.
UA has been shown to be an independent predictor of decline of renal function in CKD.10,11 Lower levels of UA may slow the worsening in renal function implying a direct pathogenic role of UA in renal injury in CKD.12 This is supported by findings that induction of hyperuricemia caused progressive glomerular injury and tubulointerstitial fibrosis in experimental animals.6 While most studies have focused on the adverse role of UA on renal vascular resistance, Ryu et al have recently shown that UA could initiate epithelial–mesenchymal transition in tubular epithelial cells suggesting that UA could act directly on renal tubules to stimulate the fibrogenic process.13 An association between urine NGAL and hyperuricemia was found in adolescents with hypertension raising the possibility that hyperuricemia may be linked to tubular injury.14 The contribution of hyperuricemia to chronic tubular injury in patients with primary GN remains unknown. This study aimed to test the hypothesis that hyperuricemia leads to tubular injury as reflected by parallel increases in NGAL and to determine if urine NGAL and/or UA are independently associated with tubulointerstitial disease in primary GN.
Methods
Patient population
We enrolled patients with primary GN who underwent a renal biopsy at Ramathibodi Hospital. Patients with kidney transplantation, AKI, secondary causes of GN such as diabetic nephropathy, and lupus nephritis were excluded. Written informed consent was obtained and the study was conducted according to the Declaration of Helsinki and approved by the Ethics Committee of the Faculty of Medicine, Ramathibodi Hospital.
Blood and urine sample collections
Blood and second urine samples were collected on the day of the biopsy. Common biochemical parameters were measured in a laboratory in compliance with ISO 15189. Creatinine was measured by enzymatic method. Urine samples were centrifuged at 3,000 rpm for 10 minutes at 4°C and the supernatant was analyzed for NGAL using a chemiluminescent microparticle immunoassay kit (The ARCHITECT Urine NGAL assay, Abbott Laboratories, Abbott Park, IL, USA). Coefficients of variation for the low (20.2 ng/mL), medium (196.7 ng/mL), and high (1,174.4 ng/mL) urine NGAL levels were 4.4%, 3.0%, and 2.2% for intra-assay variation, respectively, while for the inter-assay variation, they were 2.1%, 1.7%, and 1.4%, respectively. Using the same aliquot, urine protein was measured by modified pyrogallol red-molybdate method and urine creatinine by enzymatic method on the Dimension ExL Analyzer (Siemens Healthcare Diagnostics, Newark, DE, USA).
Urine samples were collected from healthy individuals with normal renal function and urinalysis and from patients with AKI (defined by KDIGO guideline 201215) as healthy and AKI controls, respectively. GFR (in mL/min/1.73 m2) was calculated by using the CKD-EPI equation.16 Urine protein was reported as urine protein-to-creatinine ratio (UPCR) (mg/mg Cr). NGAL was reported as absolute concentration (ng/mL) or as a ratio of NGAL to creatinine (ng/mg Cr).
Pathologic studies
Kidney biopsies were fixed in histological fixative (Glyo-Fixx; Thermo Fisher Scientific, Waltham, MA, USA), paraffin embedded, and 2 μm sections were processed for light microscopy (hematoxylin and eosin, periodic acid–Schiff base, Masson’s trichrome, and silver staining) and evaluated by a nephropathologist blinded to the laboratory and NGAL data. The severity of interstitial fibrosis and tubular atrophy (IFTA) was assessed semi-quantitatively according to a proportion relative to the total section area as follows: none (0), <5%; mild (1+), 5%–25%; moderate (2+), 26%–50%; and severe (3+), >50%.
Statistical analysis
The STATA version 13 software was used for analyses. Data are shown as mean ± SD for normally distributed values and median (interquartile range) for non-normally distributed values or percentage as appropriate. Pearson or Spearman coefficients were used to evaluate correlations between two variables. Continuous variables between two groups were compared by unpaired t test or Mann–Whitney U test, and the chi-square test was used for categorical variables. Differences among three groups were analyzed by Kruskal–Wallis test. Receiver-operating characteristic analysis was used to calculate the area under the curve (AUC) for NGAL, NGAL/Cr, or UA in the identification of moderate-to-severe IFTA. The adjusted risk estimates of these variables to identity moderate-to-severe IFTA using optimal cutoff values determined by Youden index were calculated by univariate followed by multivariate Cox regression after adjustments for traditional risk factors. Discrimination, the capacity of models to correctly classify those with and those without moderate-to-severe IFTA, was evaluated by a comparison of concordance statistics (C-statistics) using the Delong test. P-values <0.05 were considered significant.
Results
Patient characteristics
Fifty-one patients with GN (immunoglobulin A [IgA] nephropathy [n=20], focal and segmental glomerulosclerosis [n=12], minimal change disease [n=11], and membranous glomerulopathy [n=8]) were enrolled. Baseline characteristics are shown in Table 1. Age was 39.3±16.3 years and 64.7% were females. Laboratory values were as follows: GFR (mL/min/1.73 m2), 72±31 (mL/min/1.73 m2); UPCR (mg/mg Cr), 3.38±4.1; UA (mg/dL), 6.8±2.0; NGAL (ng/mL), 16.7 (6.7–43.2); NGAL/Cr (ng/mgCr), 19.1 (11.1–47.7). For comparisons, values from normal subjects (n=5) and subjects with AKI (n=19) were obtained: NGAL – healthy controls, 4.4 (3.2–9.3); AKI, 302 (148–1432); NGAL/Cr – healthy controls, 4.3 (3.7–4.6); AKI 608 (228–2616).
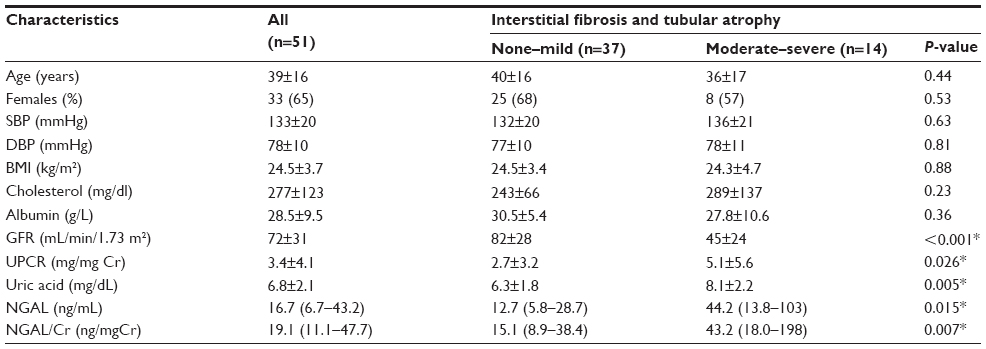 | Table 1 Patient characteristics classified by severity of interstitial fibrosis and tubular atrophy |
Relationship between NGAL, UA and clinical parameters
Overall (Figure 1), both NGAL and NGAL/Cr levels showed a significant positive correlation with UPCR but not with GFR. By contrast, UA showed strong inverse correlation with GFR but no correlations with UPCR. Neither NGAL nor NGAL/Cr correlated with UA. Calculations using log-transformed NGAL and NGAL/Cr did not alter the results (data not shown).
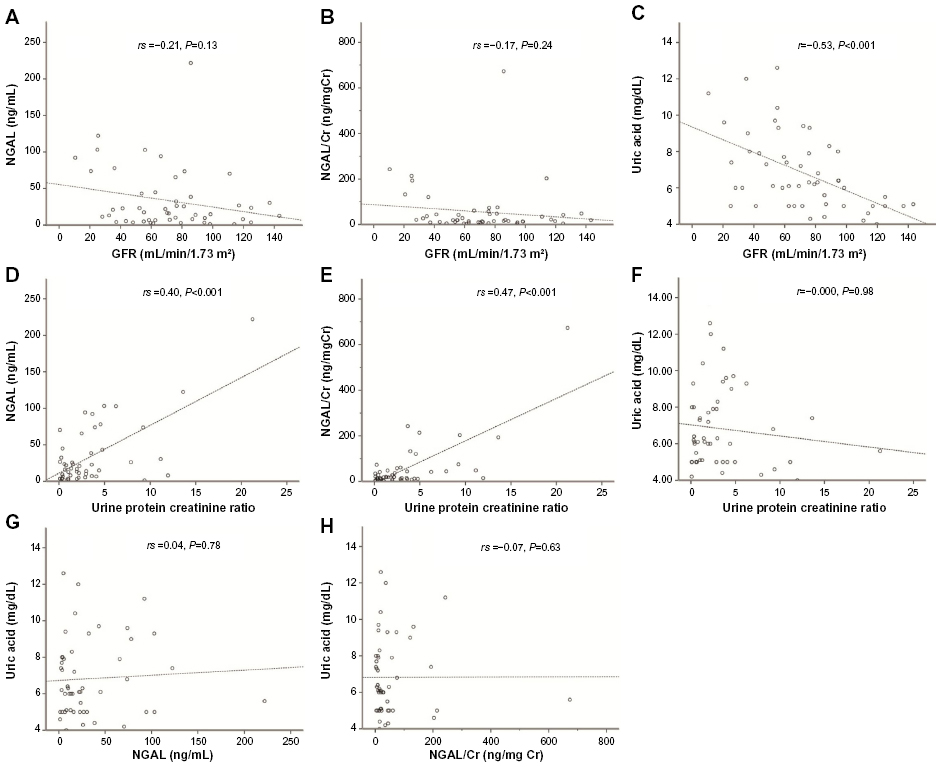 | Figure 1 Correlations between clinical parameters and biomarkers. |
Patients were categorized according to standard proteinuria categories: mild (UPCR <1, n=16), moderate (UPCR 1–3, n=17), and severe (UPCR>3, n=18). NGAL and NGAL/Cr showed significant increase with higher proteinuria, but there was no significant difference in UA levels. (NGAL: mild 9.5 [3.7–26.0] vs moderate 13.2 [6.8–22.1] vs severe 40.9 [13.6–95.0], P=0.011; NGAL/Cr: mild 14.1 [8.6–26.7] vs moderate 19.1 [10.2–32.0] vs severe 46.5 [14.3–195.7], P=0.005; UA: mild 6.1 [5.0–7.1] vs moderate 7.2 [6.0–8.1] vs severe 5.8 [4.9–9.3], P=0.251).
Relationship between IFTA with clinical and biochemical features
IFTA was graded according to severity: none (IFTA=0), n=10; mild (IFTA=1), n=27; moderate (IFTA=2), n=8; and severe (IFTA=3), n=6 (Figure 2). Patients were categorized into three groups after combining moderate and severe IFTA groups (moderate–severe, n=14) because of low numbers. There were no differences in systolic blood pressure (SBP) (mmHg) across IFTA groups (data not shown). UPCR tended to increase with increasing IFTA, whereas GFR decreased with increasing IFTA groups (proteinuria [mg/mg Cr]: none 0.96 [0.11–9.7] vs mild IFTA 1.53[0.41–3.6] vs moderate-to-severe IFTA 3.29 [2.17–5.27], P=0.085; GFR [mL/min/1.73 m2]: none 100 [79–121] vs mild IFTA 72 [58–85] vs moderate-to-severe IFTA 39 [25–61], P<0.001).
 | Figure 2 Degree of interstitial fibrosis and tubular atrophy (IFTA). |
Figure 3 shows the levels of NGAL, NGAL/Cr, and UA according to the severity of IFTA. NGAL/Cr increased with increasing IFTA: none 17.6 (11.4–51.5) vs mild 15.1 (8.6–34.6) vs moderate-to-severe 43.2 (18.0–198), P=0.025. NGAL showed similar trends: none 12.1 (3.6–41.3) vs mild 12.7 (8.6–34.7) vs moderate-to-severe 44.2 (13.8–103), P=0.052. UA also increased with IFTA: none 5.0 (5.0–5.5) vs mild 6.2 (5.5–7.4) vs moderate-to-severe 7.95 (5.9–9.8), P=0.004.
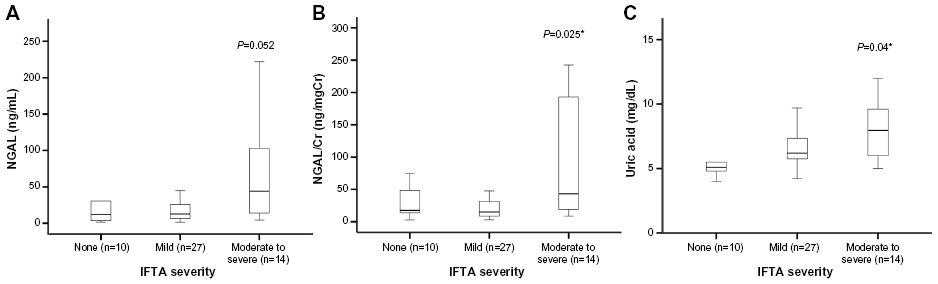 | Figure 3 Levels of biomarkers and severity of interstitial fibrosis and tubular atrophy (IFTA). |
Cutoff values to predict moderate-to-severe IFTA
To evaluate the discriminatory values of NGAL, NGAL/Cr, and UA, patients were classified into two groups according to the severity of IFTA. Grades 0 and 1 were combined into the none-to-mild group and grades 2 and 3 were combined into the moderate-to-severe group. Characteristics for each group are shown in Table 1. GFR, UPCR, NGAL, NGAL/Cr, and UA were significantly different between the two groups.
Receiver-operating characteristic curves to discriminate moderate-to-severe from none-to-mild IFTA were constructed for NGAL, NGAL/Cr, and UA (Figure 4). AUCs (95% confidence interval [CI]) were as follows: NGAL, 0.72 (0.56–0.89), P=0.015; NGAL/Cr, 0.75 (0.59–0.90), P=0.007; UA, 0.74 (0.57–0.90), P=0.005. Cutoff values of NGAL ≥22 ng/mL yielded optimal sensitivity (57%) and specificity (62%), NGAL/Cr ≥20 ng/mgCr yielded optimal sensitivity (71%) and specificity (62%), and UA ≥7 yielded optimal sensitivity (71%) and specificity (73%).
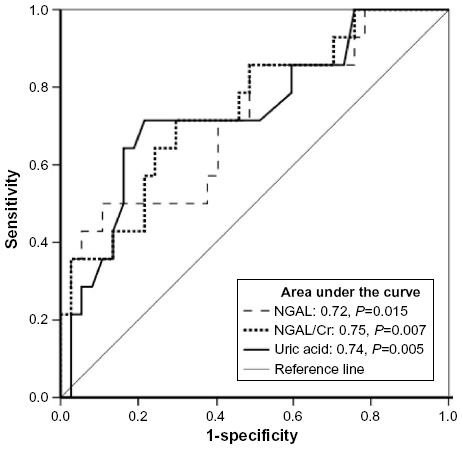 | Figure 4 Receiver-operating curve for discriminating moderate-to-severe interstitial fibrosis and tubular atrophy (IFTA) for NGAL or uric acid. |
NGAL/Cr and UA as independent predictors of moderate-to-severe IFTA
Multivariate analysis was performed using standard clinical parameters known to influence kidney disease pathology (Table 2). Proteinuria was classified into three groups: UPCR <1, UPCR 1–3 and UPCR >3. SBP was classified into two groups: SBP ≤140 and SBP >140. GFR was classified into two groups: GFR >60 and GFR ≤60. NGAL, NGAL/Cr, and UA were classified into two groups using optimum cutoff as defined earlier. Univariate analysis indicated that proteinuria, GFR and UA, and NGAL/Cr were associated with the presence of moderate-to-severe IFTA. By multivariate analysis, GFR, urine NGAL/Cr, and UA were independently associated with moderate-to-severe IFTA.
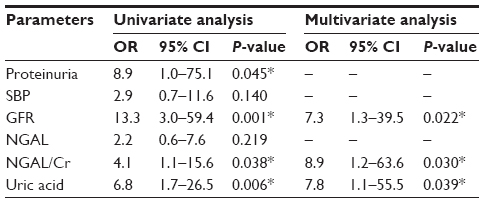 | Table 2 Classical factors and biomarkers as risk factors for moderate-to-severe interstitial fibrosis and tubular atrophy |
Combined NGAL/Cr and UA with classical predictors of moderate-to-severe IFTA
AUC (95%CI) for combined UA+NGAL/Cr was 0.85 (0.73–0.97). Figure 5 shows that combined UA+NGAL/Cr with classical predictors tended to improve discrimination compared to classical predictors alone: AUC GFR+proteinuria 0.84 (0.73–0.94) vs UA+NGAL/Cr+GFR+proteinuria 0.9025 (0.81–0.99), P=0.08.
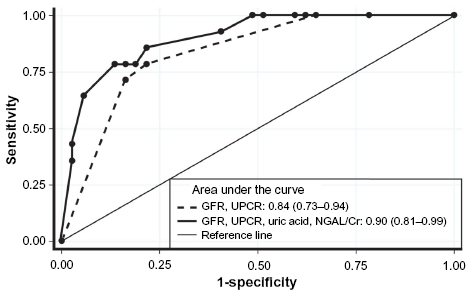 | Figure 5 Receiver-operating curve for discriminating moderate-to-severe interstitial fibrosis and tubular atrophy (IFTA) for combined factors. |
Discussion
The major findings of this study are that both serum UA and urine NGAL/Cr ratio increased with severity of tubulointerstitial lesion in primary GN. UA correlated inversely with GFR and NGAL correlated with proteinuria. Serum UA did not correlate with NGAL. Both high urine NGAL/Cr and high UA along with decreased GFR were independently associated with moderate-to-severe tubulointerstitial injury. Combining NGAL/Cr and UA with high proteinuria and low GFR tended to improve discrimination for moderate to severe tubulointersitial lesions compared to classical predictors alone.
NGAL, a 25 kDa protein produced by injured tubular epithelia, is one of the most promising new markers of AKI. In contrast to serum creatinine, which measures renal excretory function, NGAL is specifically induced in the damaged nephron and then released into the urine.17 Only low levels of NGAL are detectable in the urine of healthy subjects.7 AKI leads to rapid NGAL mRNA upregulation in kidney tubules followed by marked increase in urine NGAL excretion.18 More recently, urine NGAL has been shown to be elevated in patients with chronic tubulointerstitial disease,9,19 and urine NGAL may be predictive of long-term decline in renal function in patients with CKD.5,20,21 Limited data are available for proteinuric GN. Ding et al found increases in urinary, but not serum, NGAL in patients with advanced IgA nephropathy levels consistent with local renal generation as the major source of urinary NGAL.8 Bolignano et al showed that patients with membranous nephropathy had increased urine NGAL compared to controls and that higher urine NGAL predicted worsening of GFR.20 Nishida et al found increased urine NGAL in pediatric patients with nephrotic syndrome.19 Only a few studies have examined the relationship of tubulointerstitial changes and NGAL in primary GN. Ding et al found higher urine NGAL in IgA nephropathy with more severe disease8 and Nickolas et al found close correlations of tubular chronicity scores with NGAL in a variety of kidney diseases including GN.22 Our results extend previous findings by showing that NGAL/Cr levels could identify moderate-to-severe tubulointerstitial disease in primary GN in addition to traditional clinical predictors. The average levels in GN were about five times higher than in normal subjects and 30 fold lower than in AKI. The better association of NGAL/Cr compared to NGAL likely reflects the importance of using urine creatinine to normalize for differences in urine volume/water intake.
Previous studies have shown that urinary NGAL increased in proteinuric patients, which is consistent with our data.8,19,20 Increased tubular production of NGAL by damaged tubules is likely to be an important source of NGAL excretion in GN.21 Previously, tubular NGAL protein expression in patients with active IgA nephropathy was shown to be increased in parallel with elevated urinary NGAL levels.8 Proteinuria has been shown to be directly toxic to tubular cells and may act as a mediator of tubulointerstitial injury and nephron loss in GN.3,4 Augmented production of NGAL by tubular cells may be a defensive compensatory response to prevent tubular cell apoptosis induced by proteinuria.23 The observation that subjects with higher degree of IFTA also have higher degree of proteinuria and NGAL emphasizes the close relationship between these entities. Correlations of urine NGAL and GFR have been observed in previous studies in CKD.5,20,21 A trend for inverse correlation of NGAL with GFR was also observed in our study, but this did not reach statistical significance. Lower numbers of patients with advanced disease and higher mean GFR could account for the differences in the relationship between NGAL with GFR in our study compared to others. A similar lack of correlation between NGAL and GFR was also found in a study of diabetic subjects with relatively preserved GFR emphasizing the potential benefit of NGAL to detect tubular injury before GFR decline is evident.24
UA has been shown to be an independent predictor of kidney disease progression in primary GN.25 The mechanisms by which UA may cause progression in GN are still a matter of debate. In experimental animals, hyperuricemia could induce endothelial dysfunction and vascular smooth muscle proliferation leading to secondary tubulointerstitial injury through reductions in GFR and ischemia.4,10 Recent studies have also suggested that UA may act directly on renal tubules by stimulating cytokines production26 and epithelial-to-mesenchymal transition,13 which are key events in renal fibrosis. A primary role of UA on the renal tubules is further supported by the observation that phenotypic transition of the renal tubules in hyperuricemia occurred before the development of GFR reduction and that significant tubulointerstitial fibrosis could be ameliorated by lowering UA.13 We showed that high UA level was independently associated with tubulointerstitial damage in patients with diverse causes of primary GN consistent with previous studies in IgA nephropathy.27,28 Similar to other studies, we found that UA levels correlated inversely with GFR.10 Unlike NGAL, serum UA did not correlate with proteinuria. Although both UA and NGAL correlated with IFTA, the two parameters did not correlate with one another indicating that they likely reflect different pathogenic pathways involved in tubulointersitial injury. In GN, high UA levels may contribute to tubulointerstitial injury indirectly, largely through UA effects on renal vasculature or through promotion of inflammation rather than direct tubular damage.10,11 By contrast, elevated NGAL levels may reflect proteinuria-induced tubular damage. To our knowledge, only one other study has explored the relationship between UA and urine NGAL. Tomczak et al found the levels of both parameters to be increased in hypertensive adolescents without overt renal disease compared to controls and that serum UA correlated positively with urine NGAL in the hypertensive subjects.14 These subjects did not have macroscopic proteinuria and the levels for NGAL/Cr in the hypertensive children were only marginally elevated. The characteristics of these subjects are dissimilar to our patients and tubular dysfunction may be the primary pathology in these children.
In addition to classical clinical factors such as proteinuria and decreased GFR, the presence of tubulointerstitial lesions may predict progression to ESRD in GN.2–4 This study shows that combining urine NGAL/Cr and UA with classical clinical factors tended to improve discrimination for moderate-to-severe tubulointerstitial disease. However, this study is cross-sectional in design with a limited number of patients. Larger studies with long-term follow-up will be essential to determine the roles of urine NGAL/Cr and UA in predicting tubulointerstitial disease and renal outcomes before their routine use governing therapy in primary GN.
Conclusion
Both UA and NGAL/Cr are independently associated with the severity of tubulointerstitial lesions in primary GN patients, but no relationship exists between the two parameters implying that they reflect different pathogenic processes. Future studies are necessary to determine long-term prognostic values of both UA and NGAL/Cr in primary GN.
Acknowledgments
The authors would like to thank all staff and fellows of the Nephrology Division at Ramathibodi Hospital for inclusion of their patients. This study was supported by grants from the Faculty of Medicine, Ramathibodi Hospital, and the National Science and Technology Development Agency (NSTDA), Thailand.
Disclosure
The authors report no conflicts of interest in this work.
References
Jha V, Garcia-Garcia G, Iseki K, et al. Chronic kidney disease: global dimension and perspectives. Lancet. 2013;382(9888):260–272. | |
Satirapoj B, Nast CC, Adler SG. Novel insights into the relationship between glomerular pathology and progressive kidney disease. Adv Chronic Kidney Disease. 2012;19(2):93–100. | |
Cravedi P, Remuzzi G. Pathophysiology of proteinuria and its value as an outcome measure in chronic kidney disease. Br J Clin Pharmacol. 2013;76(4):516–523. | |
Hodgkins KS, Schnaper HW. Tubulointerstitial injury and the progression of chronic kidney disease. Pediatr Nephrol. 2012;27(6):901–909. | |
Smith ER, Lee D, Cai MM, et al. Urinary neutrophil gelatinase-associated lipocalin may aid prediction of renal decline in patients with non-proteinuric stages 3 and 4 chronic kidney disease (CKD). Nephrol Dial Transplant. 2013;28(6):1569–1579. | |
Kang DH, Nakagawa T, Feng L, et al. A role for uric acid in the progression of renal disease. J Am Soc Nephrol. 2002;13(12):2888–2897. | |
Devarajan P. Neutrophil gelatinase-associated lipocalin: a promising biomarker for human acute kidney injury. Biomarkers Med. 2010; 4(2):265–280. | |
Ding H, He Y, Li K, et al. Urinary neutrophil gelatinase-associated lipocalin (NGAL) is an early biomarker for renal tubulointerstitial injury in IgA nephropathy. Clin Immunol. 2007;123(2):227–234. | |
Wu Y, Yang L, Su T, Wang C, Liu G, Li XM. Pathological significance of a panel of urinary biomarkers in patients with drug-induced tubulointerstitial nephritis. Clin J Am Soc Nephrol. 2010;5(11):1954–1959. | |
Johnson RJ, Nakagawa T, Jalal D, Sanchez-Lozada LG, Kang DH, Ritz E. Uric acid and chronic kidney disease: which is chasing which? Nephrol Dial Transplant. 2013;28(9):2221–2228. | |
Feig DI. Serum uric acid and the risk of hypertension and chronic kidney disease. Curr Opin Rheumatol. 2014;26(2):176–185. | |
Goicoechea M, Garcia de Vinuesa S, Verdalles U, et al. Allopurinol and progression of CKD and cardiovascular events: long-term follow-up of a randomized clinical trial. Am J Kidney Dis. 2015;65(4):543–549. | |
Ryu ES, Kim MJ, Shin HS, et al. Uric acid-induced phenotypic transition of renal tubular cells as a novel mechanism of chronic kidney disease. Am J Physiol Renal Physiol. 2013;304(5):F471–F480. | |
Tomczak J, Wasilewska A, Milewski R. Urine NGAL and KIM-1 in children and adolescents with hyperuricemia. Pediatr Nephrol. 2013;28(9):1863–1869. | |
Khwaja A. KDIGO clinical practice guidelines for acute kidney injury. Nephron Clin Pract. 2012;120(4):179–184. | |
Levey AS, Stevens LA, Schmid CH, et al. A new equation to estimate glomerular filtration rate. Ann Int Med. 2009;150(9):604–612. | |
Singer E, Marko L, Paragas N, et al. Neutrophil gelatinase-associated lipocalin: pathophysiology and clinical applications. Acta Physiol. 2013;207(4):663–672. | |
Mishra J, Ma Q, Prada A, et al. Identification of neutrophil gelatinase-associated lipocalin as a novel early urinary biomarker for ischemic renal injury. J Am Soc Nephrol. 2003;14(10):2534–2543. | |
Nishida M, Kawakatsu H, Okumura Y, Hamaoka K. Serum and urinary neutrophil gelatinase-associated lipocalin levels in children with chronic renal diseases. Pediatr Int. 2010;52(4):563–568. | |
Bolignano D, Coppolino G, Lacquaniti A, Nicocia G, Buemi M. Pathological and prognostic value of urinary neutrophil gelatinase-associated lipocalin in macroproteinuric patients with worsening renal function. Kidney Blood Press Res. 2008;31(4):274–279. | |
Bolignano D, Lacquaniti A, Coppolino G, et al. Neutrophil gelatinase-associated lipocalin (NGAL) and progression of chronic kidney disease. Clin J Am Soc Nephrol. 2009;4(2):337–344. | |
Nickolas TL, Forster CS, Sise ME, et al. NGAL (Lcn2) monomer is associated with tubulointerstitial damage in chronic kidney disease. Kidney Int. 2012;82(6):718–722. | |
Gwira JA, Wei F, Ishibe S, Ueland JM, Barasch J, Cantley LG. Expression of neutrophil gelatinase-associated lipocalin regulates epithelial morphogenesis in vitro. J Biol Chem. 2005;280(9):7875–7882. | |
Nielsen SE, Hansen HP, Jensen BR, Parving HH, Rossing P. Urinary neutrophil gelatinase-associated lipocalin and progression of diabetic nephropathy in type 1 diabetic patients in a four-year follow-up study. Nephron Clin Pract. 2011;118(2):c130–c135. | |
Ohno I, Hosoya T, Gomi H, Ichida K, Okabe H, Hikita M. Serum uric acid and renal prognosis in patients with IgA nephropathy. Nephron. 2001;87(4):333–339. | |
Zhou Y, Fang L, Jiang L, et al. Uric acid induces renal inflammation via activating tubular NF-kappaB signaling pathway. PloS One. 2012;7(6):e39738. | |
Cheng GY, Liu DW, Zhang N, Tang L, Zhao ZZ, Liu ZS. Clinical and prognostic implications of serum uric acid levels on IgA nephropathy: a cohort study of 348 cases with a mean 5-year follow-up. Clin Nephrol. 2013;80(1):40–46. | |
Myllymaki J, Honkanen T, Syrjanen J, et al. Uric acid correlates with the severity of histopathological parameters in IgA nephropathy. Nephrol Dial Transplant. 2005;20(1):89–95. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.