Back to Journals » Clinical Interventions in Aging » Volume 15
Increased Osteoblastic Cxcl9 Contributes to the Uncoupled Bone Formation and Resorption in Postmenopausal Osteoporosis
Authors Liu Z ![]() , Liang W
, Liang W ![]() , Kang D, Chen Q, Ouyang Z, Yan H
, Kang D, Chen Q, Ouyang Z, Yan H ![]() , Huang B
, Huang B ![]() , Jin D
, Jin D ![]() , Chen Y, Li Q
, Chen Y, Li Q
Received 27 March 2020
Accepted for publication 29 May 2020
Published 20 July 2020 Volume 2020:15 Pages 1201—1212
DOI https://doi.org/10.2147/CIA.S254885
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Zhi-Ying Wu
Zezheng Liu,1,* Wenquan Liang,1,* Dawei Kang,1 Qingjing Chen,1 Zhicong Ouyang,1 Huibo Yan,1 Bin Huang,1 Dadi Jin,1 Yinkui Chen,2 Qingchu Li1
1Academy of Orthopedics, Guangdong Province, Department of Spine Surgery, The Third Affiliated Hospital of Southern Medical University, Guangzhou, People’s Republic of China; 2Department of Oncology, The Third Affiliated Hospital of Southern Medical University, Guangzhou, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Qingchu Li
Department of Spine Surgery, The Third Affiliated Hospital of Southern Medical University, Guangzhou, People’s Republic of China
Email [email protected]
Yinkui Chen
Department of Oncology, The Third Affiliated Hospital of Southern Medical University, Guangzhou, People’s Republic of China
Email [email protected]
Introduction: Estrogen deficiency leads to bone loss in postmenopausal osteoporosis, because bone formation, albeit enhanced, fails to keep pace with the stimulated osteoclastic bone resorption. The mechanism driving this uncoupling is central to the pathogenesis of postmenopausal osteoporosis, which, however, remains poorly understood. We previously found that Cxcl9 secreted by osteoblasts inhibited osteogenesis in bone, while the roles of Cxcl9 on osteoclastic bone resorption and osteoporosis are unclear.
Materials and Methods: Postmenopausal osteoporosis mouse model was established by bilateral surgical ovariectomy (OVX). In situ hybridization was performed to detect Cxcl9 mRNA expression in bone. ELISA assay was conducted to assess Cxcl9 concentrations in bone and serum. Cxcl9 activity was blocked by its neutralizing antibody. Micro-CT was performed to determine the effects of Cxcl9 neutralization on bone structure. Cell Migration and adhesion assay were conducted to evaluate the effects of Cxcl9 on osteoclast activity. TRAP staining and Western blot were performed to assess osteoclast differentiation. CXCR3 antagonist NBI-74,330 or ERK antagonist SCH772984 was administered to osteoclast to study the effects of Cxcl9 on CXCR3/ERK signaling.
Results: Cxcl9 was expressed and secreted increasingly in OVX mice bone. Neutralizing Cxcl9 in bone marrow prevented bone loss in the mice by facilitating bone formation as well as inhibiting bone resorption. In vitro, Cxcl9 secreted from osteoblasts facilitated osteoclast precursors adhesion, migration and their differentiation into mature osteoclasts. The positive role of osteoblastic Cxcl9 on osteoclasts was eliminated by blocking CXCR3/ERK signaling in osteoclasts. Estrogen negatively regulated Cxcl9 expression and secretion in osteoblasts, explaining the increased Cxcl9 concentration in OVX mice bone.
Conclusion: Our study illustrates the roles of Cxcl9 in inhibiting bone formation and stimulating bone resorption in osteoporotic bone, therefore providing a possible therapeutic target to the treatment of postmenopausal osteoporosis.
Keywords: postmenopausal osteoporosis, bone resorption, osteoclast, Cxcl9
Introduction
Maintenance of bone mass depends on balanced activities between new bone formation by osteoblasts and old bone resorption by osteoclasts.1,2 In postmenopausal women, however, estrogen deficiency causes higher bone resorption levels than those of bone formation. These women exhibit osteoporosis with increased bone fragility and are susceptible to bone fractures.3 In the worldwide, about 100 million people are suffering from postmenopausal osteoporosis.4 To treat osteoporosis, several drugs have been developed to prevent bone resorption or promote bone formation,5 whereas modulating of only one of the two processes (bone resorption and bone formation) limits the efficacy of these drugs. The mechanism driving uncoupling is central to the pathogenesis of postmenopausal osteoporosis and vital for development of new drugs to restore the remodeling balance, which, however, remains poorly understood.
CXCL9, which is also called MIG (monokine induced by interferon-γ (IFN-γ)), is a member of the CXC chemokine family. CXCL9 is mainly produced by activated macrophages.6 We previously found that Cxcl9 is constitutively expressed and secreted by osteoblasts in the bone marrow microenvironment. Cxcl9 abrogates osteogenesis by inhibiting differentiation of osteoblast as well as bone marrow stem cells (BMSCs).7
Recently, researchers identified a unique vascular subtype, called type H vessels, which is characterized by high expression of endothelial markers CD31 and endomucin (CD31hiEmcnhi).8,9 This particular vascular subtype decreases with age, which is consistent with a decrease in the number of osteoprogenitor cells and a decrease in bone mass. Our previous study showed that Cxcl9 secreted by osteoblasts also attenuates type H vessels formation in bone.7 However, the effects of Cxcl9 on osteoclast bone resorption and bone loss associated with estrogen deficiency have not been illustrated.
In this study, we found that chemokine Cxcl9 is up-regulated by estrogen deficiency in osteoblasts of ovariectomized (OVX) mice. Neutralization of Cxcl9 alleviated bone loss in the mice. Further studies revealed that Cxcl9 inhibited osteoblastic bone formation while stimulated osteoclast adhesion, migration and differentiation. Mechanistically, Cxcl9 facilitated activity of osteoclast by binding and activating CXCR3/ERK signaling. We propose a novel model, whereby upregulation of Cxcl9 leads to suppression of bone formation, while simultaneously repressing osteoclast differentiation and activity. Therefore, reducing Cxcl9 concentration in bone marrow might be beneficial for developing novel drugs to treat osteoporosis.
Materials and Methods
Animal
12-week female C57BL/6 mice were purchased from the Laboratory Animal Research Center of the Southern Medical University. The mice were randomly divided into sham, OVX, OVX+Veh (Vehicle) and OVX+Ab (Cxcl9 antibody) groups. Under general anesthesia, the mice were subjected to Sham surgery or bilateral surgical ovariectomy (OVX) by dorsal approach.10 Bone loss was observed in them 2 months after OVX. Mice in the OVX+Veh or OVX + Ab group (n=5) were injected subcutaneously with saline or anti-Cxcl9 (R&D System, #AF-492-NA, 1μg/50 μL) every other day for 2 months and then sacrificed for further analysis. The treatment was conducted with the first injection at the same day of OVX or 2 months after OVX. All procedures involving the mice were approved by the Southern Medical University Animal Care and Use Committee and were conducted according to the recommendations of “Guide for the Care and Use of Laboratory Animals, 8th edition”.
Cells
Primary osteoblastic cells were prepared from the calvaria of newborn mice as described previously11,12 and were maintained in α-MEM (Gibco) supplemented with 10% fetal bovine serum (Gibco), 100 U/mL penicillin, and 100 mg/mL streptomycin sulfate, at 37°C with 5% CO2. After reaching confluence in 60 mm culture dishes, the medium was replaced with α-MEM (Gibco) supplemented with 1% bovine serum albumin and the cells were cultured for 16 h before harvesting the conditional medium.
Primary osteoclast precursors were generated from mouse bone marrow cells as previously described.13 We flushed tibia and femora of 6- to 7- week-old mice to obtain bone marrow cells. Then we suspended the cells in α-minimum essential medium (α-MEM) containing 10% fetal bovine serum, 100 U/mL penicillin, 100μg/mL streptomycin, 5 ng/mL M-CSF. After 3 days of culture, we removed the floating cells. Adherent cells were used as osteoclast cell precursors. To induce osteoclastic differentiation, we plated osteoclast precursors in 96-well plates at the concentration of 2×103 cells per well in DMEM with 75 ng/mL recombinant RANKL and 15 ng/mL M-CSF.
Cell Staining
Before cell staining, we fixed the cells in 4% paraformaldehyde for 20 minutes at room temperature before ALP staining of the osteogenic differentiated osteoblasts. We then washed the cells with PBS, incubated them with ALP staining buffer (NBT-BCIP, Sigma-Aldrich) at 37°C for 30 minutes, and washed them with PBS again to remove excess dye. Before TRAP staining of osteoclastic differentiated cells, we fixed the cells 60% citrate buffered acetone to for 30 s. Then we stained the cells for TRAP with 0.1 M acetate solution (PH 5.0) containing 6.76 mM sodium tartrate, 0.12 mg/mL naphthol AS-MX phosphate, and 0.07 mg/mL of fast Garnet GBC solution as described in the manufacturer’s instruction (Waltham, MA, USA). We counted osteoclasts as TRAP-positive multinucleated cells (MNCs) containing more than three nuclei.
Migration Assay
Cell migration assays were performed as previously described.14 Firstly, we cultured osteoclast precursors were in 50ng/mL M-CSF. After 60 hours, we treated the cells with cell dissociation solution (Sigma, St Louis, MO), harvested and resuspended them in serum-free α-MEM. Next, we seeded the cells in the upper well of the Boyden chamber with polycarbonate filters containing 8-μm pore membranes (Corning Costar, Cambridge, MA). We loaded the lower well with conditional medium collected from osteoblast. After 6 to 8 hours of incubation, we fixed the migrated cells in 3.7% formaldehyde for 10 minutes, stained them with DAPI and counted the migrated cells.
Adhesion Assay
First, we coated 96-well microtiter plates with 20 μg/mL fibronectin in PBS for 16 hours at 37°C. Then washed the plates with PBS and incubated them with PBS containing 0.2% BSA for 1 hour at 37°C. Next, we suspended osteoclast precursors in serum-free α-MEM seeded them to each well of fibronectin-coated 96-well plates and incubated for 10 minutes at 37°C in the presence of conditional medium collected from osteoblast.
Micro-CT Scanning and Quantitative Analysis
Femurs were wiped off all surrounding soft tissue, fixed for 48 h in 4% paraformaldehyde and analyzed at 12 μm resolution on a micro-CT Scanner (Viva CT40; Scanco Medical AG, Bassersdorf, Switzerland). The femurs were scanned at the lower growth plate and extended proximally for 300 slices. Morphometric analysis was started with the first slice in which the femoral condyles were fully merged and extended for 100 slices proximally. We segmented the trabecular bone from the cortical shell manually on key slices using a contouring tool, and morphed the contours automatically to segment the trabecular bone on all slices. The three-dimensional structure and morphometry were constructed and analyzed for trabecular bone volume fraction (BV/TV), trabecular thickness (Tb. Th), trabecular number (Tb. N) and trabecular separation (Tb. Sp).
Decalcified Sections, Histochemistry and Immunohistochemistry (IHC)
After micro-CT scanning and analysis, we decalcified the femurs in 15% EDTA (pH 7.4) at 4°C for 14 days. Then we embedded the decalcified tissues in paraffin and prepared 5 μm sagittal-oriented sections for histological analyses. After removal of the paraffin, we performed Tartrate-resistant acid phosphatase (TRAP) staining according to a standard protocol (Sigma-Aldrich). For immunohistochemistry, the sections were incubated primary antibodies recognizing mouse Runx2 (Cell Signaling, #12556S, 1:100) and osteocalcin (Abcam, 1:500, ab93876) overnight at 4°C. Then the sections were washed with PBS and sequentially incubated with fluorescently-labeled secondary antibodies for 1 h at room temperature. All sections were observed and photographed on an Olympus BX51 microscope. Immunohistochemical staining was evaluated by positive cell number per bone perimeter (B.Pm).
ELISA Analysis
Using Mouse CXCL9/MIG (Monokine induced by interferon-gamma) ELISA Kit (Elabscience, # E-EL-M0020), we analyzed Cxcl9 concentrations in serum, bone marrow supernatant and conditional medium respectively. We performed the ELISA analysis according to the manufacturer’s instructions.
Real-Time Reverse Transcription-Polymerase Chain Reaction
We extracted total RNA from cell pellets with TRIzol Reagent (Life Technologies, #15,596-018). Total RNA was then reverse transcribed using PrimeScript Reverse Transcriptase (Takara, #2680B). A volume of 2 μL of cDNA (corresponding to 100 ng of total RNA) was used for real-time PCR using SYBR Premix Ex Taq (Takara, #RR420A). The primer sequences used are as follows: Cxcl9 forward, GGAGTTCGAGGAACCCTAGTG and reverse GGGATTTGTAGTGGATCGTGC. PCR amplification program was 94 °C for 3 minutes, 94 °C for 15 seconds, and 60 °C for 30 seconds, 40 cycles. Relative gene expression was determined using the ΔΔ-ct method versus the housekeeping gene GAPDH.
Western Blot Assay
We first lysed cells in 2% sodium dodecyl sulfate with 2 M urea, 10% glycerol, 10 mM Tris-HCl (pH 6.8), 10 mM dithiothreitol and 1 mM phenylmethylsulfonyl fluoride. We then centrifuged the lysates and separated the supernatants by SDS-PAGE and blotted them onto a nitrocellulose (NC) membrane (Bio-Rad Laboratories). We next incubated the membrane with specific antibodies to Runx2 (Cell Signaling Technology, #12556S, 1:1000), Osteocalcin (Abcam, #ab76690, 1:1000), NFATc1 (Cell Signaling Technology, # 8032S, 1:1000), c-FOS (Cell Signaling Technology, #2250, 1:2000), CTSK (Abcam, # ab19027, 1:2000), phospho-Akt (Ser473) (Cell Signaling Technology, #4060, 1:1000), phospho-Src (Tyr416) (Cell Signaling Technology, #2101, 1:1000), phospho-ERK1/2 (Thr202/Tyr204) (Cell Signaling Technology, #4370, 1:1000), ERK1/2 (Abclonal Technology, #A0229, 1:1000). The membrane was then visualized by enhanced chemiluminescence (ECL Kit, Amersham Biosciences).
siRNA Knockdown
We transiently transfected cells with Cxcl9 siRNA using Lipofectamine RNAi MAX (Invitrogen, Carlsbad, CA, USA) in Opti-MEM medium (Invitrogen), according to the manufacturer’s instructions. The efficiency of transfection was measured by Western blot. The sequence of Cxcl9 siRNA was as follows: 5′-GUUUGUAAGCACGAACUUUA-3′ (GenePharma, Shanghai, China).
Statistics
All results are presented as the mean ± S.D. Curve analysis was determined using Prism (GraphPad). All data demonstrated a normal distribution and similar variation between groups. The data in each group were analyzed using unpaired, two-tailed Student’s t-test. The level of significance was set at P < 0.05.
Results
Osteoblasts Secrete Increased Cxcl9 in OVX Mice
First, 12-week old C57BL/6 mice were subjected to sham surgery (Sham) or OVX, and were sacrificed 8 weeks later, a length of time used by others to investigate the acute phase of bone loss induced by OVX.15 We first measured the uterine wet weight to confirm the estrogen status in the OVX or sham-operated mice. Uterine weight of mice significantly decreased by 80% after ovariectomy (Figure S1). Expectedly, OVX mice exhibited significant bone loss as compared with their Sham control (Figure 1A), as characterized by lower trabecular bone volume (BV/TV), number (Tb.N) and thickness (Tb.Th) and increased trabecular separation (Tb.Sp) 8 weeks after OVX when compared to sham mice (Figure 1B). In consistent with previous reports, OVX mice presented significantly reduced type H vessels in the metaphysis of the femur as compared with the sham group (Figure S2). Moreover, an increase in osteoclast number was also witnessed in OVX mice (Figure 1C and D). Then we sought to determine the role of osteoblastic Cxcl9 in osteoporosis induced by OVX. As we reported previously, Cxcl9 exhibited specific expression in osteoblasts in bone and showed increased expression in osteoblasts in OVX mice (Figure 1E and F). We also detected secretion of Cxcl9 into bone and blood in these mice by ELISA assay, results from which confirmed that osteoblasts secreted more Cxcl9 in OVX mice (Figure 1G).
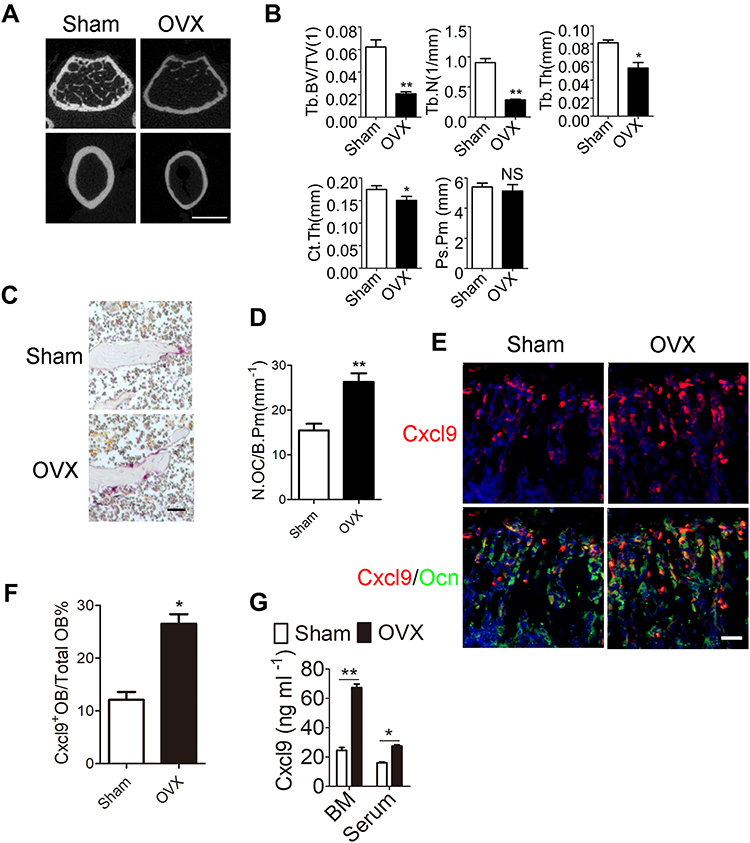 |
Figure 1 Osteoblasts secrete increased Cxcl9 in OVX mice. (A) Representative µCT images of metaphyseal trabecular bone and cortical bone in femur. Scale bar, 1 mm. (B) Quantitative µCT analysis of the trabecular bone volume/total volume (Tb. BV/TV), trabecular number (Tb. N), trabecular thickness (Tb. Th), cortical thickness (Ct. Th) and periosteal perimeter (Ps. Pm) of femora. n=5 per group. (C) TRAP staining of distal femur from Sham and OVX mice. Scale bar, 100 μm. (D) The number of osteoclasts (N.OC) on bone surface (/B.Pm) was measured. (E) Representative images of in situ hybridization of Cxcl9 mRNA in conjunction with immunostaining of Ocn in femur sections. Scale bar, 50 μm. (F) Cxcl9+ osteoblasts out of total osteoblasts were also quantified. (G) Cxcl9 concentrations assessed by ELISA in bone marrow (BM) and serum. n=5 per group. Data are shown as mean ± s.d. *P < 0.05, **P < 0.01 (Student’s t-test). |
Neutralizing Cxcl9 in Bone Marrow Alleviates Bone Loss in OVX Mice
To investigate the role of the increased Cxcl9 in development of OVX-induced osteoporosis, we subcutaneously injected neutralizing antibody against Cxcl9 into OVX mice to block Cxcl9 in vivo with the first injection on the same day as OVX. Treatment of Cxcl9 antibody substantially increased trabecular bone volume and both the cortical bone thickness and perimeter in OVX mice (Figure 2A and B). These results show that inhibition of Cxcl9 can prevent bone loss in OVX mice. Furthermore, we established another group of OVX mice and treated them with Cxcl9 antibody 2 months after OVX, when bone loss has occurred, to determine the therapeutic role of anti-Cxcl9 in OVX-induced bone loss. After 2 months, anti-Cxcl9 caused a significant regain of bone mass in OVX mice (Figure S3), from which we inferred that suppression of Cxcl9 leads to the recovery of bone mass. Together, these results proved that the increased Cxcl9 contributes to bone loss in OVX mice and suggested a preventative as well as a therapeutic role of Cxcl9 suppression in postmenopausal osteoporosis.
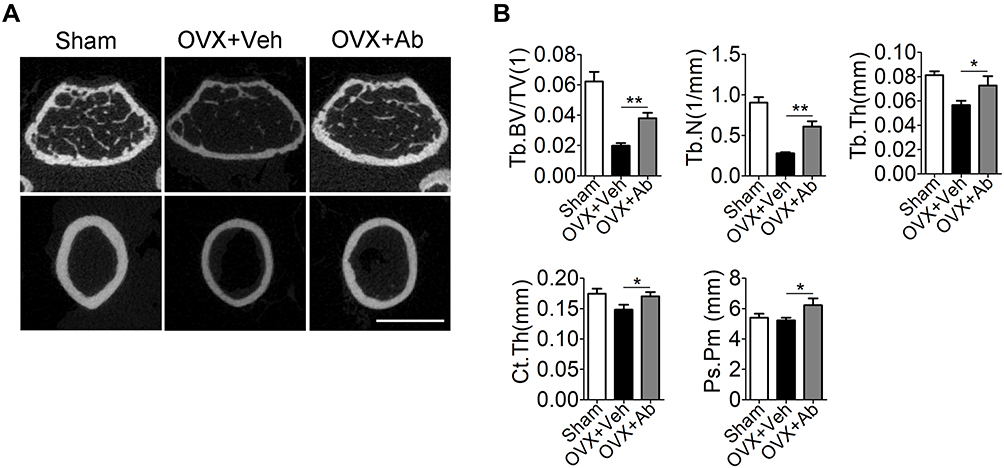 |
Figure 2 Neutralization of endogenous Cxcl9 alleviates bone loss in OVX mice. (A) Representative micro-computed tomography (µCT) images of metaphyseal trabecular bone and cortical bone in the distal femur of OVX mice treated with vehicle (OVX+Veh) or Cxcl9–neutralizing antibody (OVX+Ab) (1μg/50 μL) every other day for 2 months with the first injection at the same day of OVX. Scale bar, 1 mm. (B) Quantitative µCT analysis of the trabecular bone fraction (Tb. BV/TV), trabecular number (Tb.N), trabecular thickness (Tb.Th), cortical bone thickness (Ct. Th) and periosteal perimeter (Ps. Pm) of femora. n=5 per group. Data are shown as mean ± s.d. *P < 0.05, **P < 0.01 (Student’s t-test). |
Cxcl9 Inhibits Bone Formation in vivo and in vitro
We then explored how anti-Cxcl9 alleviated bone loss in OVX mice. We found that type H vessels density was significantly elevated in OVX mice injected with Cxcl9 antibody (Figure S4). We then detected numbers of osteoblast, the chief bone-making cells, in bone. Immunostaining of osteocalcin (Figure 3A), marker of mature osteoblasts, showed that anti-Cxcl9 increased osteoblast number in OVX bone (Figure 3B). These results suggested that Cxcl9 inhibited osteoblast formation in bone. To reveal the underlying mechanism, we firstly studied the effect of Cxcl9 on osteoblast proliferation using BrdU assay. As shown in Figure 3C and D, Cxcl9 markedly inhibited proliferation of mouse primary calvarial osteoblasts. We then induced osteoblastic differentiation of the cells. When Cxcl9 was added to the osteogenic medium, the alkaline phosphatase (ALP) staining showed a decrease in comparison with the untreated cells (Figure 3E). Moreover, Cxcl9 impaired expression of Runx2 and osteocalcin in osteoblasts (Figure 3F). These results proved that Cxcl9 attenuated bone formation in vivo and in vitro.
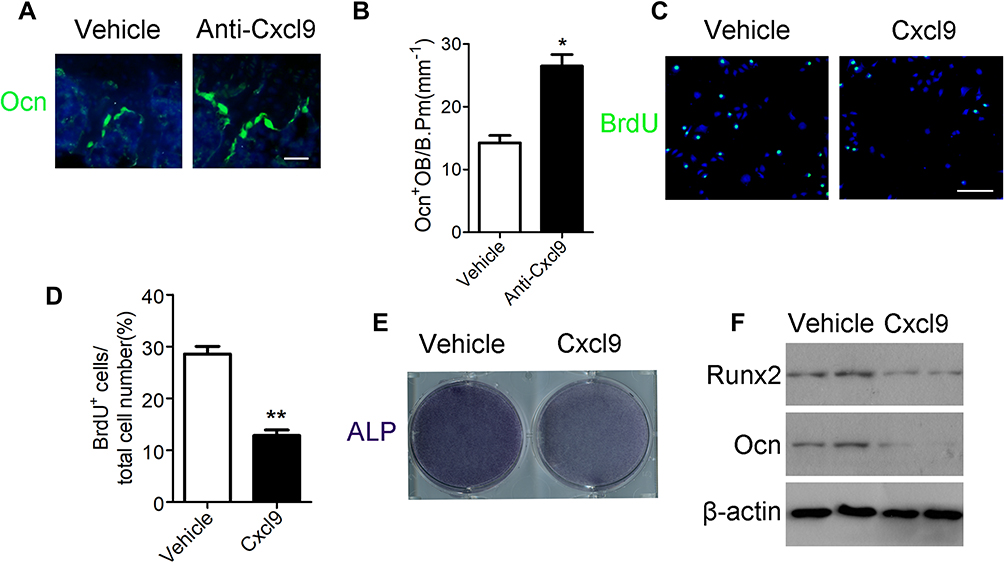 |
Figure 3 Cxcl9 attenuates bone formation in vivo and in vitro. (A) Immunofluorescent staining of Ocn in the distal femur of OVX mice treated with vehicle or Cxcl9–neutralizing antibody (Anti-Cxcl9) with the first injection at the same day of OVX. Scale bar, 50 μm. (B) Number of osteocalcin positive osteoblasts (Ocn+ OB) on the bone surface was measured as cells per millimeter of perimeter in sections (/B.Pm). n=9 per group. (C) Representative confocal images of immunostaining of BrdU (green) in mouse primary calvarial osteoblasts. Scale bar, 100 μm. (D) Quantitative analysis of BrdU+ cells over total cells. n=9 per group. (E) ALP staining differentiated calvarial osteoblasts on the 7th day of osteogenic induction. (F) Western blot analysis of osteoblastic marker Runx2 and Ocn expression in differentiated calvarial osteoblasts on the 7th day. Data are shown as mean ± s.d. *P < 0.05, **P < 0.01 (Student’s t-test). |
Cxcl9 Promotes Bone Resorption in vivo and in vitro
We then investigated whether Cxcl9 affected bone resorption in OVX mice. We performed TRAP staining of tibia slices to detect osteoclasts in OVX mice bone, results from which clearly showed that anti-Cxcl9 reduced osteoclast numbers in OVX mice (Figure 4A and B). As we previously showed that Cxcl9 in the bone marrow mainly derived from osteoblasts, these results suggested that osteoblasts might secrete Cxcl9 to promote osteoclast formation. In order to test this hypothesis, we treated osteoclast precursors with conditional medium (CM) collected from osteoblasts interfered with Cxcl9 expression. It has been reported that Cxcl9 facilitates the adhesion of hematopoietic progenitor cells,16 we firstly examined whether osteoblastic Cxcl9 affects adhesion of osteoclast precursors. Osteoclast precursor cells were allowed to attach to the fibronectin-coated substratum in the presence of osteoblasts CM. The CM of osteoblasts interfered with Cxcl9 decreased the adhesion of osteoclast precursors (Figure 4C), indicating that osteoblastic Cxcl9 promoted adhesion of osteoclasts. Osteoclasts are recruited from bone marrow or peripheral circulation and migrate to a local site to resorb bone. We then performed migration assays to test whether Cxcl9 could induce the migration of osteoclasts. As shown in Figure 4D, osteoclast migration was significantly decreased on treatment with CM of osteoblasts with impaired Cxcl9 expression. This suggested that osteoblastic Cxcl9 facilitated osteoclast migration. We then induced osteoclastic differentiation of the cells in the presence of osteoblasts CM and observed that TRAP+ mature osteoclast formation was significantly impaired when cultured in CM from osteoblasts with decreased Cxcl9 expression (Figure 4E). Moreover, osteoclast markers (NFATc1, c-FOS and CTSK) expression were all reduced by this osteoblast CM (Figure 4F). These results suggested that osteoblastic Cxcl9 enhanced osteoclast differentiation.
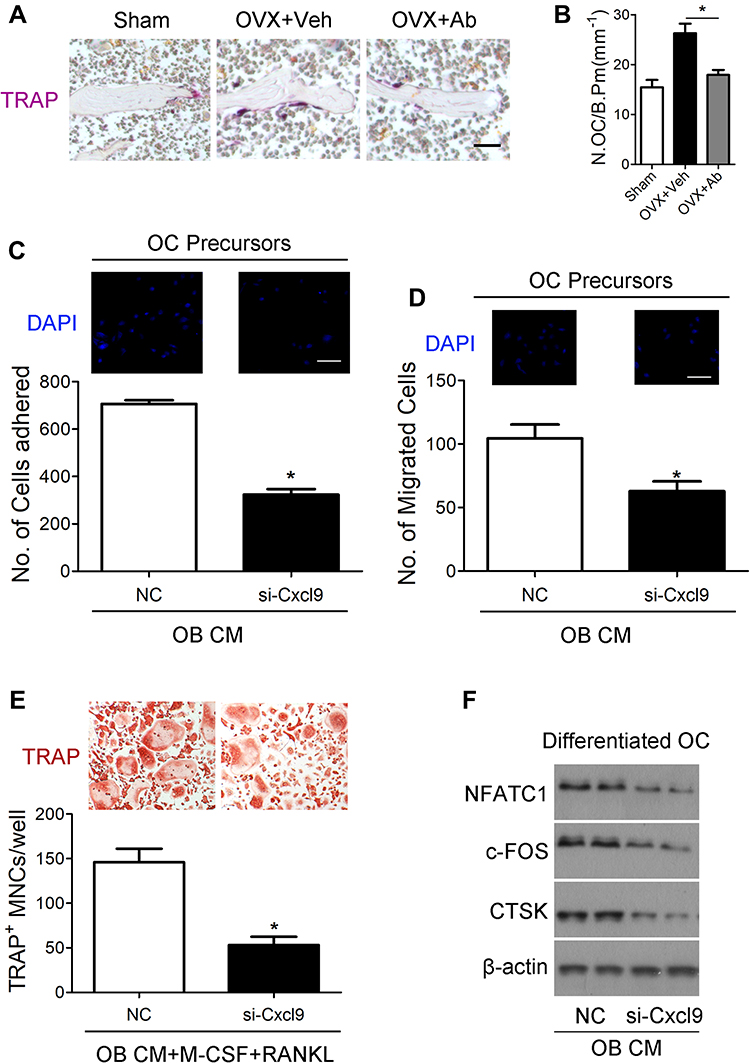 |
Figure 4 Cxcl9 facilitates bone resorption in vivo and in vitro. (A) TRAP staining of tibia sections from OVX mice treated with vehicle or Cxcl9–neutralizing antibody (Anti-Cxcl9) with the first injection at the same day of OVX. Scale bar, 100 μm. (B) The number of osteoclasts (N.OC) on bone surface (/B.Pm) was measured. (C) Osteoclast precursors (Bone Marrow-Derived Macrophages (BMM)) were treated with 50 ng/mL M-CSF for 60 hours. Cells were incubated for 10 minutes on fibronectin-coated culture plates supplemented with the indicated conditional medium from osteoblasts (OB CM) pretreated with Cxcl9 siRNA (si-Cxcl9) or negative control (NC). Nonadherent cells were washed with PBS, and adherent cells were stained with DAPI and counted under a fluorescence microscope. Scale bar, 50 μm. (D) Osteoclast precursors were cultured in the presence 50 ng/mL M-CSF plus 100 ng/mL RANKL for 60 hours. Cells were washed with PBS, suspended in serum-free α-MEM, and loaded to the upper well of transwell chambers. The lower well contained conditional medium from osteoblasts (OB CM) pretreated with Cxcl9 siRNA (si-Cxcl9) or negative control (NC). After 6 hours, cells migrated onto the lower well were stained with DAPI and counted under a fluorescence microscope. Scale bar, 50 μm. (E) BMMs were incubated with conditional medium from osteoblasts (OB CM) pretreated with Cxcl9 siRNA (si-Cxcl9) or negative control (NC) with supplementary M-CSF and RANKL. After 72 h, osteoclast formation was analyzed by TRAP staining and numbers of osteoclasts were counted as TRAP-positive multinucleated cells (TRAP+ MNCs). (F) Immunoblotting was carried out to detect expression of osteoclast markers in the differentiated BMMs. Data are shown as mean ± s.d. *P < 0.05 (Student’s t-test). |
Cxcl9 Facilitates Osteoclast Activity via CXCR3/ERK Signaling Pathway
We then investigated the mechanisms responsible for the impaired osteoclast activity by Cxcl9. CXCR3 is a well-known receptor bound with high affinity by Cxcl9. We asked whether Cxcl9 activates CXCR3 in osteoclast. We first investigated by immunofluorescence whether osteoclast precursors express this chemokine receptor. As shown in Figure 5A and B, osteoclast precursor cells cultured in vitro and CTSK positive osteoclasts in mice bone clearly presented expression of CXCR3. Moreover, Srk/ERK signaling pathways, downstream of CXCR3,17 was activated in osteoclast precursors treated with Cxcl9 (Figure 5C, Figure S5A). However, the cells pretreated with CXCR3 antagonist did not exhibit activation of Srk/ERK signaling pathway upon Cxcl9 treatment (Figure 5D, Figure S5B). Therefore, Cxcl9 activates CXCR3/ERK signaling in osteoclasts. We next investigated whether CXCR3/ERK signaling mediated the effects of Cxcl9 on osteoclast. Indeed, the positive roles of Cxcl9 on osteoclast precursors adhesion (Figure 6A), migration (Figure 6B) and differentiation (Figure 6C–E) were markedly abrogated by supplement of CXCR3 antagonist NBI-74,330 or ERK antagonist SCH772984. Totally, these results proved that Cxcl9 facilitates osteoclast activity via CXCR3/ERK signaling pathway.
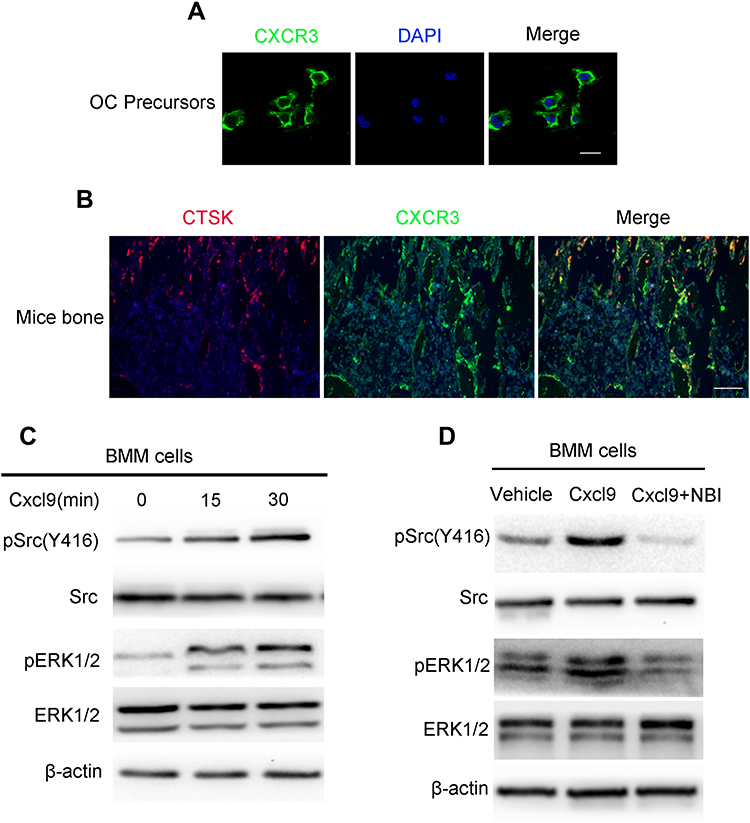 |
Figure 5 Cxcl9 activates CXCR3/ERK signaling in osteoclasts. (A) Representative photomicrographs of immunostaining of CXCR3 in cultured BMMs. Scale bar, 20 μm. (B) Representative photomicrographs of immunostaining of CXCR3 in CTSK+ osteoclasts in mice bone. (C) Serum-starved BMM cells were incubated with 100 ng/mL Cxcl9 for the indicated time points. Total cell lysates were immunoblotted with antibodies specifically recognizing the phosphorylated form of Src and ERK. (D) Serum-starved BMM cells were incubated with Cxcl9 (100 ng/mL) or Cxcl9 plus NBI-74,330 (CXCR3 antagonist) as indicated for 10 min. Total cell lysates were immunoblotted with antibodies specifically recognizing the phosphorylated form of Src and ERK. |
 |
Figure 6 Cxcl9 facilitates osteoclast activity via CXCR3/ERK signaling pathway. (A) BMM cells were treated with 50 ng/mL M-CSF for 60 hours, and were then incubated for 10 minutes on fibronectin-coated culture plates supplemented with Cxcl9, Cxcl9 plus NBI-74,330 or Cxcl9 plus SCH772984 (ERK antagonist) as indicated. Nonadherent cells were washed with PBS, and adherent cells were stained with DAPI and counted under a fluorescence microscope. (B) BMM cells were cultured in the presence 50 ng/mL M-CSF plus 100 ng/mL RANKL for 60 hours. Cells were washed with PBS, suspended in serum-free α-MEM, and loaded to the upper well of transwell chambers. The lower well contained Cxcl9, Cxcl9 plus NBI-74,330 or Cxcl9 plus SCH772984 as indicated. After 6 hours, cells migrated onto the lower well were stained with DAPI and counted under a fluorescence microscope. BMMs were incubated with Cxcl9, Cxcl9 plus NBI-74,330 or Cxcl9 plus SCH772984 as indicated with supplementary M-CSF and RANKL. After 72 h, osteoclast formation was analyzed by TRAP staining (C) and numbers of osteoclasts were counted as TRAP-positive multinucleated cells (TRAP+ MNCs) (D). (E) Immunoblotting was carried out to detect expression of osteoclast markers in the differentiated BMMs. Data are shown as mean ± s.d. *P < 0.05, **P < 0.01 (Student’s t-test). |
Estrogen Negatively Regulates Cxcl9 Secretion in Osteoblasts
Finally, we investigated how Cxcl9 secretion is elevated in osteoblasts in OVX mice bone. Bone phenotypes of OVX mice are mainly caused by estrogen deficiency, we thus determined whether estrogen regulate Cxcl9 expression and secretion in osteoblasts and contributes to the increased Cxcl9 secretion in OVX mice bone. We treated mouse primary osteoblasts with estradiol and fulvestrant (estrogen receptor antagonist) respectively and examined Cxcl9 expression and secretion. Cxcl9 mRNA (Figure 7A), protein (Figure 7B) and secretion (Figure 7C) were all significantly increased upon estradiol treatment while reduced by fulvestrant. These results suggested that estrogen negatively regulates Cxcl9 expression and secretion in osteoblasts.
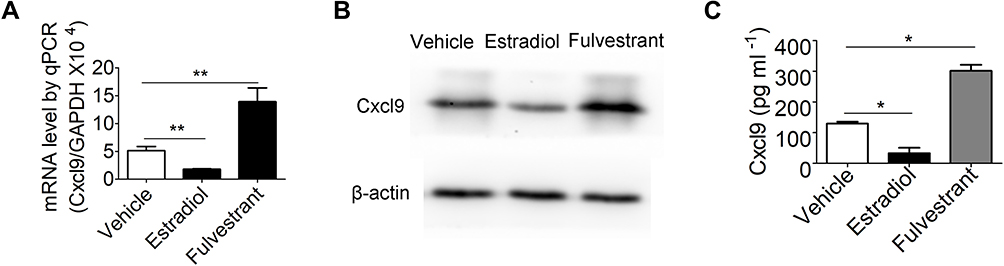 |
Figure 7 Estrogen negatively regulates Cxcl9 secretion in osteoblasts. (A) qPCR analysis of Cxcl9 mRNA in primary osteoblasts treated with vehicle, estradiol or fulvestrant (estrogen receptor antagonist). (B) Western blot of Cxcl9 in primary osteoblasts. (C) Cxcl9 concentrations assessed by ELISA in supernatant of primary osteoblasts treated with vehicle, estradiol or fulvestrant. n=5 per group. Data are shown as mean ± s.d. *P < 0.05, **P < 0.01 (Student’s t-test). |
Discussion
In women with postmenopausal osteoporosis, the withdrawal of estrogen leads to bone loss because bone formation, albeit enhanced, fails to keep pace with the stimulated osteoclastic bone resorption,4 a phenomenon known as uncoupling.18 The mechanisms driving this uncoupling, however, is not yet fully demonstrated. In this study, we examined the role of Cxcl9 in the development of postmenopausal osteoporosis in ovariectomized (OVX) mice and investigated the mechanisms by which Cxcl9 uncouples bone formation from bone resorption. We provide demonstrations that estrogen deficiency upregulates the levels of Cxcl9 in the bone marrow, and that Cxcl9 neutralization in vivo alleviates bone loss after OVX. These observations demonstrate that Cxcl9 plays a pivotal role in the pathogenesis of postmenopausal osteoporosis.
Bone remodeling depends a tight balance of the activities of bone formation and resorption. Various types of cells and factors are involved in the process of bone remodeling.19 Osteoblast and osteoclast are the two main cells participating in those progresses.20 Moreover, it has been shown that osteoblast and osteoclast communicate with each other to keep the balance of the bone formation and resorption through direct cell-cell contact, cytokine or cell-bone matrix.21 Several mechanisms have been reported to be utilized by osteoblasts to control osteoclastogenesis, among which the RANKL/OPG axis is the best studied pathway.22,23 Here, we describe Cxcl9 as novel mediator of interactions between osteoblasts and osteoclasts and illustrate its function on bone formation and resorption. Phosphorylation of MAPKs (ERK, JNK, and p38) in BMMs is the early event of RANKL/RANK signaling activation that controls osteoclastogenesis. Mechanism research in this study showed that stimulation of osteoclast formation by Cxcl9 also depends on ERK phosphorylation in BMMs, indicating synergy of Cxcl9/CXCR3 and RANKL/RANK signaling in osteoclast activation.
We previously found that Cxcl9 inhibited proliferation and osteoblastic differentiation of bone marrow stem cells (BMSCs) and osteoblast precursors.7 Therefore, the finding that treatment with anti–Cxcl9 prevents bone loss by enhancing bone formation was not unexpected. However, we provide additional evidence that Cxcl9 also influences osteoclast adhesion, migration and differentiation by stimulating CXCR3/ERK signaling pathway. In accordance with this, CXCR3 signaling has been reported to activate osteoclastogenesis by other studies.24,25 However, the effects of Cxcl9 on osteoblasts are independent on its established receptor CXCR3, since CXCR3 antagonist did not reversed the inhibitory role of Cxcl9 on osteoblasts. Instead, Cxcl9 exerts its function by binding with VEGF and abrogated its osteogenesis stimulation.7 It is interesting that Cxcl9 acts on osteoblasts and osteoclasts through different mechanisms. We suspected that the different origins of the two cell types (osteoblasts from mesenchyme stem cells26 and osteoclasts from bone marrow hematopoietic stem cells27) might contribute to the different mechanism of Cxcl9 action.
Our data revealed that Cxcl9 contributes to the uncoupled bone formation and resorption in postmenopausal osteoporosis by affecting osteoblasts as well as osteoclasts. However, we could not rule out that effects of Cxcl9 on other cells might contribute to the bone phenotypes observed in OVX mouse model. For instance, Cxcl9 is a chemokine that is known to recruit T-cells which have a key role in OVX-induce bone loss.28–30 Blockade of Cxcl9 might disrupt the trafficking and co-localization of T-cells to the bone remodeling unit and affect the onset of osteoporosis. However, osteoblasts and osteoclasts cultured in vitro were subjected to the same effects as those in bone marrow when exposed directly to Cxcl9, which consolidated our conclusion that Cxcl9 affects bone formation as well as resorption and contributes to the bone loss in postmenopausal osteoporosis.
Several drugs have been developed to treat osteoporosis, many of which inhibit bone resorption and only a few drugs can promote bone formation.3,31 Administration of antiresorptive agents such as bisphosphonates increases bone mass by suppression of bone remodeling in osteoporosis patients.32,33 However, bone quality is reduced in these patients due to accumulation of microfractures and reduced restructuring of bone architecture resulted by suppressed bone remodeling.34,35 Therefore, understanding mechanisms of the uncoupling between osteoclast and osteoblast is essential to design drugs to gain bone mass as well as maintain bone quality. This study establishes Cxcl9 as a novel factor contributing the uncoupling of bone formation and resorption in postmenopausal osteoporosis. Drugs coordinating Cxcl9 expression and secretion in osteoblasts might be beneficial for osteoporosis treatment.
Acknowledgment
This work was supported by grants 81672120, 81700783 from National Natural Science Foundation of China and 2018A030313937, 2019A1515011876 from Guangdong Natural Science Fund Management Committee.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Manolagas SC, Jilka RL, Epstein FH. Bone marrow, cytokines, and bone remodeling-emerging insights into the pathophysiology of osteoporosis. N Engl J Med. 1995;332(5):305–311. doi:10.1056/NEJM199502023320506
2. Chen X, Wang Z, Duan N, Zhu G, Schwarz EM, Xie C. Osteoblast-osteoclast interactions. Connect Tissue Res. 2018;59(2):99–107. doi:10.1080/03008207.2017.1290085
3. Black DM, Rosen CJ, Solomon CG. Clinical practice. Postmenopausal osteoporosis. N Engl J Med. 2016;374(3):254–262. doi:10.1056/NEJMcp1513724
4. Klibanski A, Campbell LA, Bassford T, et al. NIH consensus development panel on osteoporosis prevention D, therapy. Osteoporosis prevention, diagnosis, and therapy. JAMA. 2001;285(6):785–795. doi:10.1001/jama.285.6.785
5. Rodan GA, Martin TJ. Therapeutic approaches to bone diseases. Science. 2000;289(5484):1508–1514. doi:10.1126/science.289.5484.1508
6. Goldberg MF, Roeske EK, Ward LN, et al. Salmonella persist in activated macrophages in T cell-sparse granulomas but are contained by surrounding CXCR3 ligand-positioned Th1 cells. Immunity. 2018;49(6):1090–102.e7. doi:10.1016/j.immuni.2018.10.009
7. Huang B, Wang W, Li Q, et al. Osteoblasts secrete Cxcl9 to regulate angiogenesis in bone. Nat Commun. 2016;7:13885. doi:10.1038/ncomms13885
8. Kusumbe AP, Ramasamy SK, Adams RH. Coupling of angiogenesis and osteogenesis by a specific vessel subtype in bone. Nature. 2014;507(7492):323–328. doi:10.1038/nature13145
9. Ramasamy SK, Kusumbe AP, Wang L, Adams RH. Endothelial notch activity promotes angiogenesis and osteogenesis in bone. Nature. 2014;507(7492):376–380. doi:10.1038/nature13146
10. Li J-Y, Tawfeek H, Bedi B, et al. Ovariectomy disregulates osteoblast and osteoclast formation through the T-cell receptor CD40 ligand. Proc Natl Acad Sci U S A. 2011;108(2):768–773. doi:10.1073/pnas.1013492108
11. Rao LG, Ng B, Brunette DM, Heersche JN. Parathyroid hormone- and prostaglandin E1-response in a selected population of bone cells after repeated subculture and storage at −80C. Endocrinology. 1977;100(5):1233–1241. doi:10.1210/endo-100-5-1233
12. Bhargava U, Bar-Lev M, Bellows CG, Aubin JE. Ultrastructural analysis of bone nodules formed in vitro by isolated fetal rat calvaria cells. Bone. 1988;9(3):155–163. doi:10.1016/8756-3282(88)90005-1
13. Lee SE, Chung WJ, Kwak HB, et al. Tumor necrosis Factor-α supports the survival of osteoclasts through the activation of Akt and ERK. J Biol Chem. 2001;276(52):49343–49349. doi:10.1074/jbc.M103642200
14. Kwak HB, Lee SW, Jin HM, et al. Monokine induced by interferon- is induced by receptor activator of nuclear factor B ligand and is involved in osteoclast adhesion and migration. Blood. 2005;105(7):2963–2969. doi:10.1182/blood-2004-07-2534
15. Nakamura T, Imai Y, Matsumoto T, et al. Estrogen prevents bone loss via estrogen receptor alpha and induction of Fas ligand in osteoclasts. Cell. 2007;130(5):811–823. doi:10.1016/j.cell.2007.07.025
16. Jinquan T, Quan S, Jacobi HH, et al. CXC chemokine receptor 3 expression on CD34(+) hematopoietic progenitors from human cord blood induced by granulocyte-macrophage colony-stimulating factor: chemotaxis and adhesion induced by its ligands, interferon gamma-inducible protein 10 and monokine induced by interferon gamma. Blood. 2000;96(4):1230–1238.
17. Bonacchi A, Romagnani P, Romanelli RG, et al. Signal transduction by the chemokine receptor CXCR3: activation of Ras/ERK, Src, and phosphatidylinositol 3-kinase/Akt controls cell migration and proliferation in human vascular pericytes. J Biol Chem. 2001;276(13):9945–9954. doi:10.1074/jbc.M010303200
18. Weitzmann MN, Roggia C, Toraldo G, Weitzmann L, Pacifici R. Increased production of IL-7 uncouples bone formation from bone resorption during estrogen deficiency. J Clin Invest. 2002;110(11):1643–1650. doi:10.1172/JCI0215687
19. Xiao W, Wang Y, Pacios S, Li S, Graves DT. Cellular and molecular aspects of bone remodeling. Front Oral Biol. 2016;18:9–16.
20. Matsuo K, Irie N. Osteoclast–osteoblast communication. Arch Biochem Biophys. 2008;473(2):201–209. doi:10.1016/j.abb.2008.03.027
21. Tamma R, Zallone A. Osteoblast and osteoclast crosstalks: from OAF to Ephrin. Inflamm Allergy Drug Targets. 2012;11(3):196–200. doi:10.2174/187152812800392670
22. Nakahama K-I. Cellular communications in bone homeostasis and repair. Cell Mol Life Sci. 2010;67(23):4001–4009. doi:10.1007/s00018-010-0479-3
23. Sims NA, Walsh NC. Intercellular cross-talk among bone cells: new factors and pathways. Curr Osteoporos Rep. 2012;10(2):109–117. doi:10.1007/s11914-012-0096-1
24. Lee J-H, Kim H-N, Kim K-O, et al. CXCL10 promotes osteolytic bone metastasis by enhancing cancer outgrowth and osteoclastogenesis. Cancer Res. 2012;72(13):3175–3186. doi:10.1158/0008-5472.CAN-12-0481
25. Lee J-H, Kim B, Jin WJ, Kim -H-H, Ha H, Lee ZH. Pathogenic roles of CXCL10 signaling through CXCR3 and TLR4 in macrophages and T cells: relevance for arthritis. Arthritis Res Ther. 2017;19(1):163. doi:10.1186/s13075-017-1353-6
26. Matic I, Matthews BG, Wang X, et al. Quiescent bone lining cells are a major source of osteoblasts during adulthood. Stem Cells. 2016;34(12):2930–2942. doi:10.1002/stem.2474
27. Hanaoka H. The origin of the osteoclast. Clin Orthop Relat Res. 1979;(145):252–263.
28. Pacifici R. Role of T cells in ovariectomy induced bone loss–revisited. J Bone Miner Res. 2012;27(2):231–239. doi:10.1002/jbmr.1500
29. Cline-Smith A, Axelbaum A, Shashkova E, et al. Ovariectomy activates chronic low-grade inflammation mediated by memory T cells, which promotes osteoporosis in mice. J Bone Miner Res. 2020. doi:10.1002/jbmr.3966
30. Tyagi AM, Srivastava K, Mansoori MN, Trivedi R, Chattopadhyay N, Singh D. Estrogen deficiency induces the differentiation of IL-17 secreting Th17 cells: a new candidate in the pathogenesis of osteoporosis. PLoS One. 2012;7(9):e44552. doi:10.1371/journal.pone.0044552
31. Wang T, Liu Q, Tjhioe W, et al. Therapeutic potential and outlook of alternative medicine for osteoporosis. Curr Drug Targets. 2017;18(9):1051–1068. doi:10.2174/1389450118666170321105425
32. Gennari L, Rotatori S, Bianciardi S, Nuti R, Merlotti D. Treatment needs and current options for postmenopausal osteoporosis. Expert Opin Pharmacother. 2016;17(8):1141–1152. doi:10.1080/14656566.2016.1176147
33. Anastasilakis AD, Polyzos SA, Makras P. THERAPY OF ENDOCRINE DISEASE: denosumab vs bisphosphonates for the treatment of postmenopausal osteoporosis. Eur J Endocrinol. 2018;179(1):R31–R45. doi:10.1530/EJE-18-0056
34. Shibahara T. Antiresorptive agent-related osteonecrosis of the jaw (ARONJ): a twist of fate in the bone. Tohoku J Exp Med. 2019;247(2):75–86. doi:10.1620/tjem.247.75
35. Lems WF, Saag K. Bisphosphonates and glucocorticoid-induced osteoporosis: cons. Endocrine. 2015;49(3):628–634. doi:10.1007/s12020-015-0639-1
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.