")
Back to Journals » Clinical, Cosmetic and Investigational Dermatology » Volume 16
Incontinentia Pigmenti: A Case Report of Early Clinical Symptoms in a Lack of Family Inheritance Positive Result
Authors Yuan F, Zhu FN, Liu XJ, Li J, Xu HT
Received 21 February 2023
Accepted for publication 1 May 2023
Published 9 May 2023 Volume 2023:16 Pages 1209—1214
DOI https://doi.org/10.2147/CCID.S407506
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Jeffrey Weinberg
Fang Yuan,1,* Feng-Na Zhu,2,* Xiao-Juan Liu,1,* Jun Li,3 Hong-Tao Xu1
1Department of Pediatrics, General Hospital of Central Theater Command of the People’s Liberation Army, Wuhan, 430070, People’s Republic of China; 2Department of Neonatology, Maternal and Child Health Hospital of Hubei Province, Wuhan, 430070, People’s Republic of China; 3Department of Blood Purification, General Hospital of Central Theater Command of the People’s Liberation Army, Wuhan, 430070, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Jun Li; Hong-Tao Xu, General Hospital of Central Theater Command of the People’s Liberation Army, 627#, Wuluo Road, Wuchang District, Wuhan, Hubei Province, 430070, People’s Republic of China, Tel +86-13659846692, Fax +86-27-50773333, Email [email protected]; [email protected]
Background: Incontinentia pigmenti (IP), also known as Bloch-Sulzberger syndrome, is an X-linked dominant genetic disease involving multiple systems. Previous literature has not reported the case of parents with negative genetic test results, and typical early clinical symptoms and auxiliary inspection results were also lacking.
Case Report: A female child was found to have broken skin immediately after birth with no family inheritance disease, and the area of the broken skin increased. Immediately afterward, Head magnetic resonance imaging (MRI) showed multiple blood lesions in the brain. Then, the wide-angle digital retinal imaging system suggesting that fundus fluorescein angiography showed fundus vascular loop-like changes. And blood genetic testing showed that exons 4– 10 of the NEMO gene located in Xq28 were deleted. The patient was eventually diagnosed with IP. However, her parents were a non-consanguineous healthy couple, with no specific skin, oral, or perineal diseases. And her parents’ blood genetic testing showed that the parents and sisters of the patient did not have the NEMO gene exon deletion of Xq28.
Conclusion: This case demonstrates the process from suspected neonatal IP cases without familial inheritance to diagnosis, which showed the typical early clinical symptoms and auxiliary inspection results. This case showed that the parents of patients with IP do not necessarily have clinical symptoms and positive symptoms of genetic testing results.
Keywords: incontinentia pigment, genodermatosis, Bloch-Sulzberger syndrome, skin disease, NEMO gene
Introduction
Previous reports have shown that incontinentia pigmenti (IP; OMIM 308300) is a very rare genetic disease with skin lesions as a typical feature, accompanied by multisystem developmental disorders, and can involve teeth, eyes, the central nervous system and other organs.1,2 The incidence of this disease is 1.2 per 100,000 births. Because male patients often die in the uterus, the incidence of pigment incontinence is concentrated in female patients.1–3 The typical clinical manifestations of the skin are divided into four periods, erythema and blisters, verrucous keratotic papules, plaques, pigmentation (Blaschko lines).4,5 However, in some occult cases, the skin symptoms are not obvious, and the genetic test results of the parents are not necessarily positive. Only suggestive tests, such as retinal examination and cranial magnetic resonance, can be used. Pathological examination and genetic testing may confirm the diagnosis. To this end, we report the diagnostic process of this case of neonatal pigment incontinence without familial inheritance.
Case Report
The female child, was her mother’s third fetus and second birth, with a gestational age of 38+2 weeks, and was delivered by cesarean section due to dystocia. Her parents were a non-consanguineous healthy couple, with no specific skin, oral, or perineal diseases. Her birth weight was 2980g, and her Apgar score was 6 (A+P+G+A+R=1+1+1+1+2) after 1 min. After wiping the fetal fat, it was found that her skin was slightly damaged, which did not attract enough attention. In the following two days, the aforementioned skin damage progressed to scattered coin-sized desquamation and yellow blisters. Among them, the two upper limbs were the weight, and several skin damages were observed together (See Figure 1). Scattered yellow blisters was not easily broken, and the blister fluid is turbid and purulent. There was no family history with the same features. The patient did not have fever, screaming, irritability, etc. Vital signs showed a blood pressure of 85/50 mmHg, a pulse rate of 120 beats/minute, a respiratory rate of 32 breaths/minute, and a body temperature of 38.2°C. Her body weight, height, and head circumference were all. Her laboratory results such as complete blood cell count of WBC (white blood cell) 28.0×109/L, mainly neutrophils, accompanied by increased eosinophils. RBC (red blood cell count) 4.08×1012/L, PLT (platelet) 433×109/L, liver panel: ALT (alanine aminotransferase) 19 U/L, AST (aspartate aminotransferase) 34 U/L, kidney panel: creatinine 47 μmol/L) were within normal. Her limits and respiratory etiology test results (including tuberculosis) were negative. No abnormalities were detected in the TORCH (T: Toxoplasma gondii; R: Rubella virus; C: Cytomegalovirus; H: Herpes simplex virus; O: Other pathogens), immunization series and autoimmune series examinations. No bacteria or fungi were detected in blister smears, cultures, or blood cultures. The blister culture was sterile. Convulsions occurred on the third day after birth. Head magnetic resonance imaging (MRI) showed multiple blood lesions in the brain (Figure 2). Combined with the patient’s medical history, the diagnosis was considered IP. To further clarify and seek sufficient evidence, the patient was examined by a wide-angle digital retinal imaging system, suggesting that fundus fluorescein angiography showed fundus vascular loop-like changes, consistent with the fundus vascular manifestations of pigmented incontinence (Figure 3). In the meantime, the patient underwent skin biopsy, which showed that more eosinophils were aggregated in the epidermis, a small number of dyskeratocytes were scattered in the surrounding epidermis, and a small number of eosinophils and lymphocytes were infiltrated in the superficial dermis (Figure 4). To further clarify the genetic status of the patient’s family, the parents and older sister of the patient underwent blood genetic testing. The results suggested that the patient had a heterozygous loss in exons 4–10 of the IKBKG gene, but the parents and older sister were normal (Figure 5).
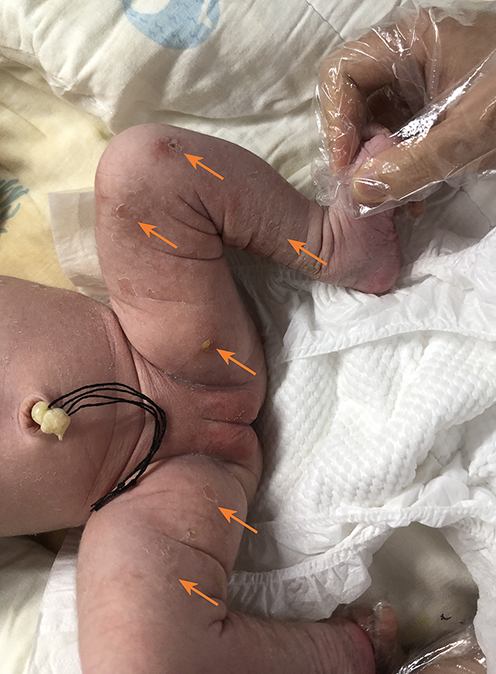 |
Figure 1 Typical yellow blisters on the infant’s skin (see arrow). |
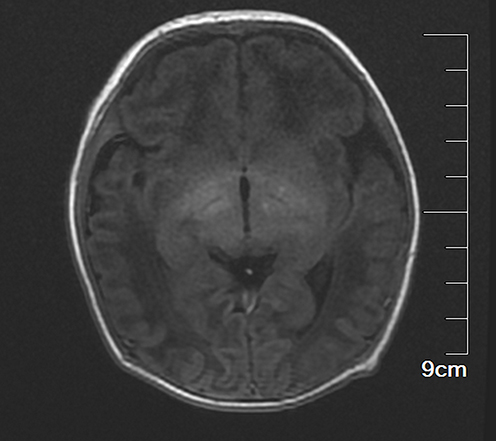 |
Figure 2 Cranial magnetic resonance imaging. |
 |
Figure 3 Fundus photography showed incomplete peripheral vascularization and foveal hypoplasia (A) Left eye; (B) right eye). |
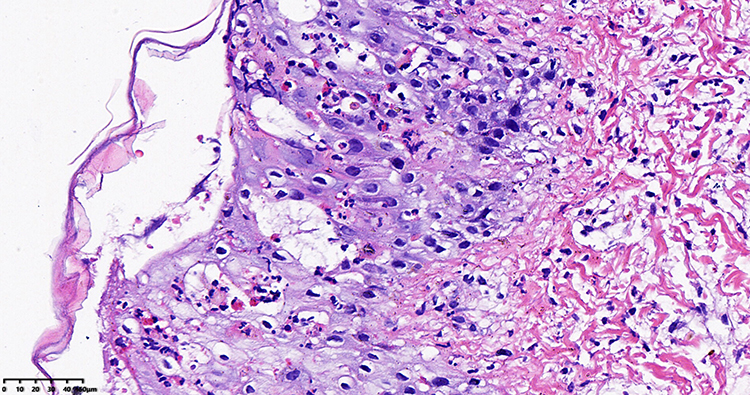 |
Figure 4 Anatomopathological exam of yellow blisters (hematoxylin-eosin stained, original×400). |
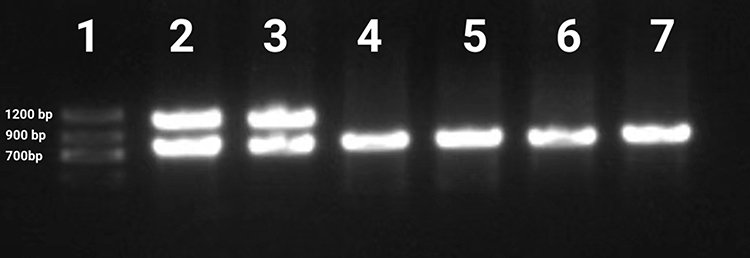 |
Figure 5 Mutational analysis of the IKBKG gene with PCR analysis of blood samples. Lane 1, Marker; Lane 2, IKBKG deletion exon 4‐10 control; Lane 3, patient; The IKBKG deletion was detected. Lanes 4 and 5 are the father and mother. Lanes 6, big sister; Lanes 7, healthy control. |
Discussion
At present, the history of disease, the characteristic manifestations and evolution of skin lesions are the internationally recognized standards for the diagnosis of IP.6 Typical skin symptoms such as typical erythroid blisters in the neonatal period, and the blisters contain eosinophilia; Pigmentation with a linear distribution on the trunk, and Linear skin atrophy. These are the main criteria for diagnosis for traditional IP.4,6,7 Therefore, characteristic skin changes are the main clinical manifestations of this disease. The typical clinical manifestations of the skin are divided into four periods. The first period is usually between birth and 2 weeks, is also known as lupus erythematosus blister.4,6–8 These blisters are usually characterized by pimples, erythema, blisters, rash along the inside of the limbs and the trunk with a linear lateral distribution.4,7 The blister fluid contains no bacteria, but it does contain eosinophils in large numbers. Skin histopathological findings suggest typical eosinophilic sponge edema.6,7 The second stage involves verrucous keratosis papules and plaques, which are distributed on the extremities and appear after 2–6 weeks of birth.4,6,7 The third stage involves pigmentation, which is distributed in stripes or vortices along the Blaschko line on the trunk at 12 to 26 weeks of birth.4,6,7 The fourth stage of pale atrophic plaques or hypopigmentation can occur at any stage from early adolescence to adulthood.4,7,8 These four stages of skin lesions can occur simultaneously or sequentially.
Besides the typical skin symptoms of IP, positive head imaging has been reported in the literature.9 30% of IP patients present with neurological involvement.9,10 Based on conventional MRI in patients with IP, the dominant lesions were widespread with sometimes confluent punctate and patchy changes in the periventricular and subcortical white matter.10 A characteristic feature of DWI abnormalities was punctate lesions spread throughout the white matter, often associated with changes to the corpus callosum.10 Changes in SWI are not as extensive as those in DWI, but there are better hints for IP patients with spotty bleeding.9,10 Unlike other neurological conditions, IP neurological lesions does not develop new neurological lesions or change over time.9,10 Therefore, cranial magnetic resonance examination has a certain significance for neonatal pigment incontinence.
Approximately one-third (range: 20–77%) of IP patients have ophthalmologic abnormalities, especially retinopathy. These ophthalmologic lesions are mainly retinal vascular abnormalities, including peripheral retinal avascular areas, abnormal vascular anastomosis, and peripheral peripherals.11 Vascular loop-like changes have also been reported that in addition to changes in peripheral blood vessels, patients often have decreased blood flow density in the macular area.12 Other eye diseases include strabismus, cataracts, optic nerve atrophy, retinal pigment epithelial abnormalities, retinal detachment, and microphthalmia.13
IP is a rare ectodermal dysplastic disorder involving multiple organ systems.14,15 The disease is a rare type of X-linked dominant genetic disease. The IKBKG gene, located at Xq28, causes this disorder, which encodes a component of the nuclear factor kappa B (NF-κB) signaling pathway.15,16 This disease has characteristic skin lesions and can also involve teeth, eyes, hair, the central nervous system and the musculoskeletal system. It can lead to blindness, convulsions and mental retardation.17,18
However, in this case, the parents and big sister of the patient underwent blood genetic testing, and the results showed that there was no abnormality in exons 4–10 of the IKBKG gene. The current literature has not found a reason why the parents of the affected child have negative genetic tests. Only a few reports have speculated that it might be somatic mosaicism or mild gene mutations.1 So, for those with low incomes, genetic testing is not necessary due to high cost.
In addition, the rash in the early stage of the patient was not typical, with skin lesions and yellow blisters and no typical manifestations of rashes such as erythema, blisters, papules, and verrucous plaques distributed along the Blaschko line. In the auxiliary examination, the white blood cell count was 13.4*10E9/L, the absolute number of neutrophils was 7.72*10E9/L, the percentage of eosinophils was 14.6%, the absolute number of eosinophils was 1.96*10E9/L, and the absolute number of basophils was 0.09*10E9/L. The above results can easily lead to misdiagnosis of herpes. After positive evidence was obtained from clinical manifestations, magnetic resonance imaging (MRI) and retinal examination were performed in a timely manner. Then, invasive tissue biopsy and genetic testing of the pedigree were performed to accurately diagnose the disease.
Conclusions
In summary, for IP patients with unobvious symptoms and clinical suspicion, magnetic resonance imaging (MRI) and retinal examination can be considered. After positive evidence is obtained, invasive tissue biopsy and family genetic testing can be performed to accurately diagnose the disease. At the same time, our case also reflects that patients with pigment incontinence have early retina and head damage. In addition, although the disease has been confirmed to be an X-linked dominant genetic disease, the relatives of our case did not have a positive result of the relevant family member genetic gene. This will provide a reference for future genetic testing. Such as genetic testing is not necessarily for those with low incomes with IP. After all, genetic testing is too expensive.
Ethical Approval
Informed consent was obtained from the patients’ parents. The written informed consent was signed by the patients’ parents to have the case details and any accompanying images published. This report complies with the guidelines for human studies and is in accordance with the Declaration of Helsinki. The ethics Review Committee of General Hospital of Central Theater Command of the Chinese people’s liberation army general hospital approved the use of clinical data of these patients in this study.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work. These authors contributed equally to this work and should be considered as co-first authors: Fang Yuan, Feng-Na Zhu and Xiao-Juan Liu.
Funding
This study was not supported by any external funds.
Disclosure
The authors declare that they have no conflicts of interest in this work.
References
1. Kawai M, Sugimoto A, Ishihara Y, Kato T, Kurahashi H. Incontinentia pigmenti inherited from a father with a low level atypical IKBKG deletion mosaicism: a case report. BMC Pediatr. 2022;22(1):378. doi:10.1186/s12887-022-03444-6
2. How KN, Leong HJY, Pramono ZAD, Leong KF, Lai ZW, Yap WH. Uncovering incontinentia pigmenti: from DNA sequence to pathophysiology. Front Pediatr. 2022;10:900606. doi:10.3389/fped.2022.900606
3. Fusco F, Pescatore A, Steffann J, et al. Clinical utility gene card: for incontinentia pigmenti. Eur J Hum Genet. 2019;27(12):1894–1900. doi:10.1038/s41431-019-0463-9
4. Li WC, Li ML, Ding JW, et al. Incontinentia pigmenti with intracranial arachnoid cyst: a case report. World J Clin Cases. 2022;10(23):8352–8359. doi:10.12998/wjcc.v10.i23.8352
5. Liang L, Yang Y, Bu S, Lu F. Case report: a case of cotton-wool spots after intravitreal injection of conbercept in an infant with incontinentia pigmenti. Front Med. 2021;8:761398. doi:10.3389/fmed.2021.761398
6. Ni Y, Huang X, Ruan L, et al. Intravitreal injection of ranibizumab in severe retinopathy of incontinentia pigmenti. J AAPOS. 2018;22(4):325–327.e3. doi:10.1016/j.jaapos.2018.01.008
7. Swinney CC, Han DP, Karth PA. Incontinentia pigmenti: a comprehensive review and update. Ophthalmic Surg Lasers Imaging Retina. 2015;46(6):650–657. doi:10.3928/23258160-20150610-09
8. Nirmalasari DA, Tabri F, Waspodo N, Rimayani S, Adriani A. Incontinentia pigmenti / Bloch-Sulzberger syndrome: a case report. Acta Dermatovenerol Alp Pannonica Adriat. 2022;31(1):39–41.
9. Hsieh DT, Chang T. Incontinentia pigmenti: skin and magnetic resonance imaging findings. Arch Neurol. 2011;68(8):1080. doi:10.1001/archneurol.2011.164
10. Soltirovska Salamon A, Lichtenbelt K, Cowan FM, et al. Clinical presentation and spectrum of neuroimaging findings in newborn infants with incontinentia pigmenti. Dev Med Child Neurol. 2016;58(10):1076–1084. doi:10.1111/dmcn.13140
11. Huang SY, Chen LJ, Chiu SC. A 7-year-old female child of incontinentia pigmenti presenting with vitreous hemorrhage. Indian J Ophthalmol. 2017;65(6):533–535. doi:10.4103/ijo.IJO_560_16
12. O’Doherty M, Mc Creery K, Green AJ, Tuwir I, Brosnahan D. Incontinentia pigmenti--ophthalmological observation of a series of cases and review of the literature. Br J Ophthalmol. 2011;95(1):11–16. doi:10.1136/bjo.2009.164434
13. Fusco F, Paciolla M, Conte MI, et al. Incontinentia pigmenti: report on data from 2000 to 2013. Orphanet J Rare Dis. 2014;9:93. doi:10.1186/1750-1172-9-93
14. Bodemer C, Diociaiuti A, Hadj-Rabia S, et al. Multidisciplinary consensus recommendations from a European network for the diagnosis and practical management of patients with incontinentia pigmenti. J Eur Acad Dermatol Venereol. 2020;34(7):1415–1424. doi:10.1111/jdv.16403
15. Wright JT, Fete M, Schneider H, et al. Ectodermal dysplasias: classification and organization by phenotype, genotype and molecular pathway. Am J Med Genet A. 2019;179(3):442–447. doi:10.1002/ajmg.a.61045
16. Cai YR, Liang Y, Zhong X. Late contralateral recurrence of retinal detachment in incontinentia pigmenti: a case report. World J Clin Cases. 2022;10(13):4171–4176. doi:10.12998/wjcc.v10.i13.4171
17. Fusco F, Conte MI, Diociaiuti A, et al. Unusual father-to-daughter transmission of incontinentia pigmenti due to mosaicism in IP males. Pediatrics. 2017;140(3):e20162950. doi:10.1542/peds.2016-2950
18. Islam YFK, Khurshid SG. Incontinentia pigmenti and the eye. Curr Opin Ophthalmol. 2022;33(6):525–531. doi:10.1097/ICU.0000000000000863
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.