")
Back to Journals » International Journal of Nanomedicine » Volume 15
Inclusion of IR-820 into Soybean-Phosphatides-Based Nanoparticles for Near-Infrared-Triggered Release and Endolysosomal Escape in HaCaT Keratinocytes at Insignificant Cytotoxic Level
Authors Homsirikamol C , Suvanasuthi S, Viravaidya-Pasuwat K
Received 24 June 2020
Accepted for publication 22 September 2020
Published 6 November 2020 Volume 2020:15 Pages 8717—8737
DOI https://doi.org/10.2147/IJN.S267119
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Thomas Webster
Chaiyarerk Homsirikamol,1 Saroj Suvanasuthi,2 Kwanchanok Viravaidya-Pasuwat1,3
1Mammalian Cell Culture Laboratory, Biological Engineering Program, Faculty of Engineering, King Mongkut’s University of Technology Thonburi, Bangkok, Thailand; 2Hair Diseases and Hair Transplantation Division, Faculty of Medicine, Siriraj Hospital, Mahidol University, Bangkok, Thailand; 3Department of Chemical Engineering, Faculty of Engineering, King Mongkut’s University of Technology Thonburi, Bangkok, Thailand
Correspondence: Kwanchanok Viravaidya-Pasuwat
Department of Chemical Engineering, Faculty of Engineering, King Mongkut’s University of Technology Thonburi, Bangkok 10140, Thailand
Tel +66 2 470 9221-30 Ext 205
Fax +66 2 428 3534
Email [email protected]
Purpose: The degradation of drugs within endolysosomes has been widely addressed as a cause of poor bioavailability. One of the strategies to allow molecules to escape from a destructive fate is to introduce a photosensitizing moiety into a drug carrier enabling the permeabilization of endosomes and endolysosomes upon irradiation. This paper presents an alternative delivery nanosystem composed of cost-effective soybean phosphatides mixed with IR-820, a near-infrared (NIR) sensitizer, to load various active compounds and trigger an endolysosomal escape with a low cytotoxic effect.
Methods: IR-820-incorporated phosphatides-based nanoparticles were formulated using a thin-film hydration method to encapsulate different molecular probes and a drug model. The nanoparticles were characterized in vitro using dynamic light scattering, transmission electron microscopy, as well as ultraviolet–visible and fluorescence spectroscopy techniques. The NIR-corresponding generation of the photochemical products, the content release, and the cytotoxicity toward the HaCaT keratinocyte cell line were evaluated. The cellular internalization and endolysosomal escape were monitored using a cytochemical marker and fluorescent probes with a colocalization analysis.
Results: The IR-820-combined nanoparticles revealed the NIR-triggered changes in the singlet oxygen presence, nanoparticle architecture, and release rate without being cytotoxic. Additionally, the nanoplatform appeared to enhance cellular uptake of the macromolecules. The localization of the cytochemical marker and the colocalization analysis on the fluorescence signals of the encapsulated fluorophore and the lysosome-labeling reporter implied the transient endolysosomal escape of the cargo within the HaCaT cells after NIR irradiation.
Conclusion: The inclusion of IR-820 into a soybean-phosphatides base ingredient provides NIR responsiveness, particularly the endolysosomal escape of the payload, to the formulated nanoparticles, while preserving the beneficial properties as a drug carrier. This alternative delivery nanomedicine system has future potential to provide high bioavailability of cytosolic drugs utilizing time- and spatial-controllable NIR triggerability as well as the synergistic therapeutic effects with NIR-biomodulation.
Keywords: IR-820, soybean phosphatides, NIR-responsive nanoparticles, endolysosomal escape
Plain Language Summary
Several research groups have developed lipid-based nanocarriers to deliver drugs into cells and further enable the escape of the drugs from the endolysosomes where the degradation of the drugs occurs within the cells. One of the effective delivery methods is to engineer the delivery system to initiate an endolysosomal escape when exposed to light. With this purpose, the researchers put more effort into the chemical modification, separation, and purification of the materials. Despite the proven efficacy, the lengthy process and the use of single pure and low-yield materials might result in costly products. In this paper, the authors suggested the combination of crude soybean phosphatides and a small amount of a near-infrared (NIR) sensitive substance; namely, IR-820, as the alternative base ingredients for the preparation of the nanoparticles. In addition, the experiments were conducted to study the attributes of the nanoparticles and confirm the escape of the loaded substances from the endolysosomes within the skin cells. The results demonstrated that the IR-820-incorporated phosphatides-based nanoparticles responded to the NIR illumination causing the chemical action of the NIR substance, shape alteration, and the accelerated release of the payload. Moreover, the nanoparticles could prevent the aggregation of a drug model in water, increase cellular uptake, and exhibit low toxicity. More importantly, the encapsulated compounds, usually trapped inside the endolysosomes, could escape from these organelles only after the NIR illumination. Thus, the simple mixture of soybean phosphatides and IR-820 could be a promising drug delivery system that could maintain high drug efficacy, while giving synergistic therapeutic effects with NIR.
Introduction
Lysosomal degradation of therapeutic molecules has been addressed as a challenge in delivering cytosolic drugs to obtain high bioavailability within cells.1 Abundant hydrolases in acidic lysosomes not only deteriorate invading pathogens and provide biological elements by catabolizing biomolecules but, unfortunately, also degrade remedial substances in the endo-lysosomal pathway during endocytosis. One particular route for the introduction of lysosomal hydrolases to the payloads packaged in late endosomes is complete fusion between the late endosomes and lysosomes2 resulting in a hybrid organelle; namely, endolysosomes where the degradation of the payloads occurs. This degradation would be amplified when the lysosomal marker and the number of active lysosomes increase, especially in some diseases, eg, psoriasis3 and cancer.4 Thus, to enhance the bioavailability of the cytosolic drugs applied to the above-mentioned diseases, letting the molecules escape from the endolysosomal enclosure and translocate into the cytosol is a strategy of interest.
The drug delivery systems, both passively and actively, triggering the endolysosomal escape of the encapsulated substances have been developed in various studies with different mechanisms; for example, pH buffering/proton sponge,5–7 osmotic pressure shock,8 the flip-flop approach,9 and photochemical/photophysical internalization.10 The photochemical/photophysical internalization is defined by the incorporation of a photosensitive moiety into a carrier, and letting cargoes flee from the endolysosomes upon light-activated chemical reactions and/or physical alterations. This light-triggering mechanism is spatially and time controllable. One desirable activating wavelength range is near-infrared (NIR) due to the benign absorption of biological media in an optical window range of 650 nm to 950 nm11 as well as the potential photobiomodulation effects.12,13 IR-820 or new indocyanine green, an NIR photosensitizer, provides the chemical moiety that is able to absorb NIR and transfer the energy to triplet oxygen yielding singlet oxygen.14 Thus, the simple inclusion of IR-820 into the drug delivery system would, supposedly, disrupt the casing, as a result of the singlet oxygen generation upon the NIR irradiation. This hypothesis would be investigated in this paper.
Phosphatides, essential components of a biological membrane, have been widely used for the preparation of drug delivery systems owing to their biocompatibility, non-immunogenic properties, and flexibility.15,16 Soybean is a plant-based natural source of phosphatides whose acetone-insoluble fraction consists of phosphatidylcholine, phosphatidylethanolamine, phosphatidylserine, N-acylphosphatidylethanolamine, and phosphatidylinositol with mainly C16–18 saturated and unsaturated fatty acids.17 Since the soybean phosphatides contain allylic sites that can be reacted with singlet oxygen,18 they, presumably, allow for the triggered release as engineered and can be served as a more economical ingredient compared to a highly purified single lipid. As a result, soybean phosphatides were selected as a biomaterial of the present drug delivery system. In addition to the material, the construction of phosphatides-based drug carriers in nano-sized particles helped to facilitate the incorporation of the delivery vehicles into the cells, as the subcellular components, readily interacting with the biological environments, also possessed nano-sized attributes,19 and the large surface area to the volume ratio of the nanoparticles resulted in high surface reactivity.20
In this proof of concept study, the authors demonstrated that IR-820-embedded phosphatides-based nanoparticles enabled the endolysosomal escape upon the NIR (830 nm) illumination. The physical and photochemical properties of the NIR-responsive nanoparticles incorporated with different molecular probes or a drug model were characterized. The drug model selected for this study was a crude ethyl acetate extract of Phyllanthus emblica L. fruit (PEm) based on its typical complexity, ie, containing diverse hydrophilic and hydrophobic phenolic compounds21 with different polarities. Besides, PEm could potentially give a variety of therapeutic effects as reportedly possessed by emblica fruit.21 The investigation of the cell internalization and translocation of the cargo utilized a spontaneously immortalized aneuploid human keratinocyte (HaCaT) cell line that was carried out to examine the potential of the nanoparticles for prospective topical delivery.
Materials and Methods
Chemicals
The following chemicals were of either an analytical or reagent grade and used without further pretreatment. IR-820 dye (CAS No 172616–80-7) denoted as “IR”, tetramethylrhodamine isothiocyanate-dextran (TRITC-Dex40; MW = 40kDa), phenol (CAS No 108–95-2), 5-bromo-4-chloro-3-indolyl phosphate disodium salt (BCIP; CAS No 38404–93-2), and Triton® X-100 (CAS No 9002–93-1) were purchased from Sigma-Aldrich Co (Missouri, USA). Soybean lecithin (CAS No 8002–43-5), cholesterol (CAS No 57–88-5), sodium deoxycholate (CAS No 302–95-4), 9,10-dimethylanthracene (DMA; CAS No 781–43-1), and gallic acid hydrate (CAS No 5995–86-8) were obtained from Tokyo Chemical Industry Co, Ltd (Tokyo, Japan). Ammonium ferrous sulfate hexahydrate (CAS No 7783–85-9), butylated hydroxytoluene (BHT; CAS No 128–37-0), and xylenol orange (CAS No 3618–43-7) were obtained from Loba Chemie Pvt Ltd (Mumbai, India). Acetone (CAS No 67–64-1), chloroform (CAS No 67–66-3), and methanol (CAS No 67–56-1) were supplied from VWR International Company (Pennsylvania, USA), RCI Labscan Limited. (Bangkok, Thailand), and Fisher Scientific Co LLC (Pennsylvania, USA), respectively. Type 1 (ultrapure) water was produced by using an arium® pro ultrapure water system (Sartorius AG, Germany) to maintain the electrical conductivity at 0.055 μS/cm at room temperature and was used for all the experiments.
Development and in vitro Evaluation of the Infrared-Responsive Nanoparticles
Thin-Film Hydration
Phosphatides-based nanoparticles were prepared according to the thin-film hydration or the Bangham method.22 Acetone-insoluble phosphatides were separated from crude soybean lecithin as reported in Vandana et al,23 aliquoted, and kept at −20ºC in amber containers subsequently purged with nitrogen and sealed until used. The organic-phase components (Table 1) for each formulation were completely dissolved with a mixture of chloroform and methanol (2:1 v/v) in a round-bottom flask and then evaporated under stepwise pressure reduction to 25 mbar by using a RC 900 rotary evaporator (KNF Neuberger Inc, USA) to maintain the rotational speed at 100 rpm for three hours, resulting in the formation of the lipid film at the bottom of the flask. During the rotary evaporation, the temperature was set at higher than the gel-to-liquid crystalline phase transition temperature of the mixture of the soybean phosphatides, sodium deoxycholate, and cholesterol (formulation NP in Table 1), which was determined by differential scanning calorimetry. Afterwards, the lipid film was rehydrated with the hydrating medium, which was sterile phosphate-buffered saline (PBS) (pH 7.4) comprising the aqueous-phase components (Table 1), and rotated at 60 rpm for an hour, while keeping the temperature similar to the previous step. The nanoparticles were refrigerated at 4°C overnight and subsequently sonicated at a 30% duty cycle for 3×20 seconds pulses with one-minute intervals between pulses using a Sonifier® SFX150 probe sonicator (Branson Ultrasonics, USA). Next, the size exclusion chromatography (SEC) was performed to exclude the free molecules using the column packed with Sepharose® 4B (Sigma-Aldrich, USA). The translucent nanoparticle suspension was collected and kept at 4ºC without any exposure to light. The chemical structures of some components incorporated in the nanoparticle formulations are shown in Figures S1A–F.
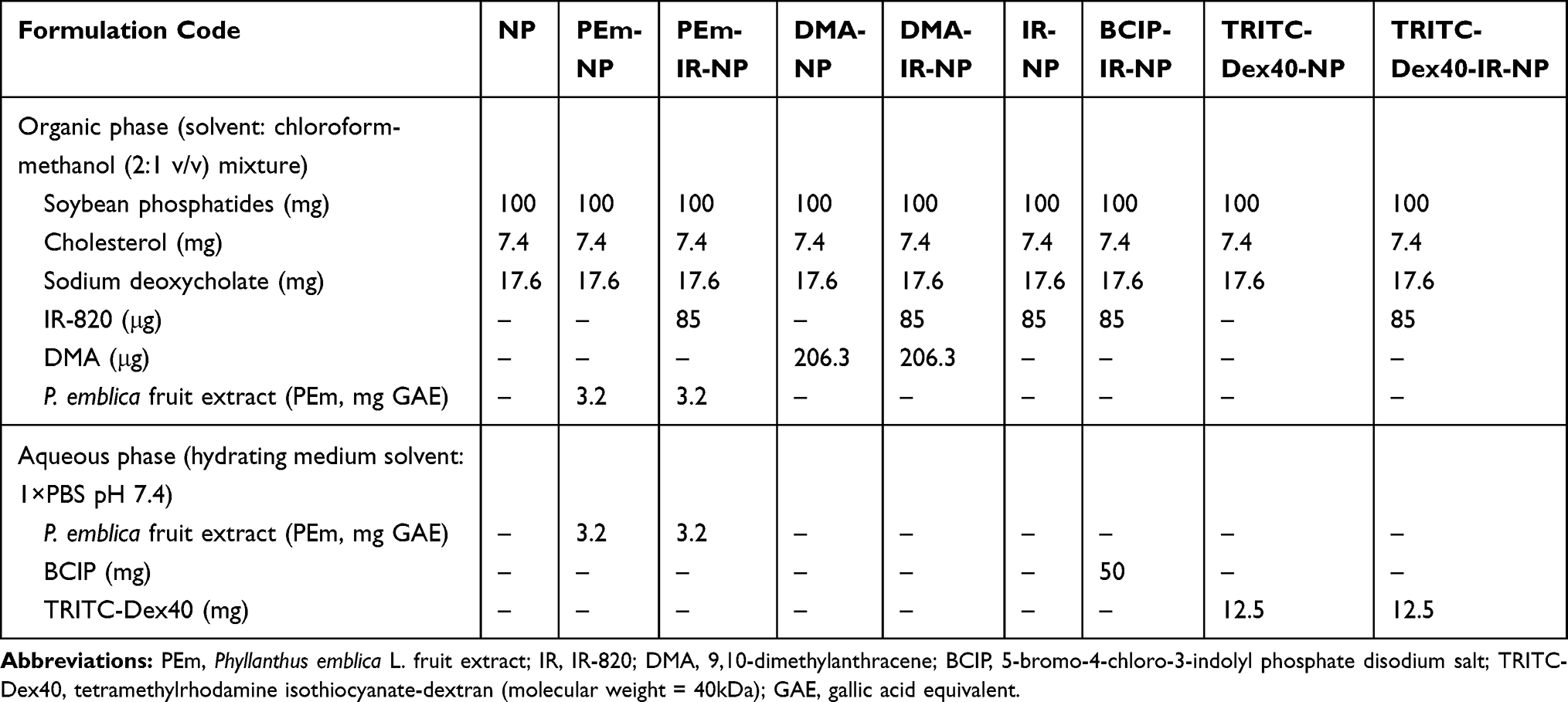 |
Table 1 Nanoparticle Formulations with the Hydrating Medium Volume of 10 mL |
Crude Phyllanthus emblica L. fruit extract (PEm) was prepared by soxhlet extraction of the dried Phyllanthus emblica L. fruit powder obtained as fresh fruit from a local fruit orchard in Nakhon Pathom province, Thailand. The fresh Phyllanthus emblica L. fruit was cut into small pieces, dried in an oven at 60°C for 48 hours, ground into powder, and kept dry in a desiccator cabinet at room temperature. Then, the fruit powder was filled into a soxhlet thimble and extracted with ethyl acetate maintaining the ratio of the fruit powder to ethyl acetate at 1:20 w/v and the temperature at 85°C to obtain approximately one cycle per hour for six hours. Subsequently, the ethyl acetate extract was filtered through quantitative ashless filter paper Grade 15 (pore size 2–3 μm; Filtres Fioroni, France), evaporated under vacuum overnight, aliquoted, and kept in the nitrogen-purged vials at 4°C.
Differential Scanning Calorimetry
Differential scanning calorimetry (DSC) was conducted to determine the transition temperature between the gel and liquid crystalline phase of the NP lipid mixture. Approximately 10 mg of the dried NP formulation was weighed and analyzed by using a DSC 204 F1 Phoenix® differential scanning calorimeter (Netzsch, Germany). The heating rate was set at 5°C/minute over a temperature range of −60 to 67.9°C operating under a nitrogen purge at a flow rate of 50 mL/minute.
Determination of the Phospholipid Concentration
The phospholipid concentrations in the nanoparticle suspensions were measured based on the Stewart assay.24 A certain volume of the nanoparticle suspension was mixed with 2 mL of the standard ammonium ferrothiocyanate solution before adding 2 mL of chloroform. An attained biphasic system was then vortexed for a minute and left undisturbed until complete separation. The lower chloroform layer was carefully aspirated and filled into an optical glass cuvette with a 10 mm light path before its absorbance measurement at 488 nm using a Thermo Scientific GENESYS 20 spectrophotometer (Thermo Fisher Scientific Inc, USA). The phospholipid concentration was calculated from the absorbance using the calibration curve generated from the soybean phosphatide standard and PBS as blank.
Determination of the Total Phenolic Content
The total phenolic content (TPC) of the PEm was evaluated using the Folin-Ciocalteu method25 with some modifications. One mL of samples was mixed with 100 μL of the Folin-Ciocalteu phenol reagent (Merck Millipore Co, USA) and then shaken for five minutes using a benchtop shaker at room temperature. After that, 1 mL of 7% w/v sodium carbonate solution was added to the reaction mixture, diluted with ultrapure water to obtain 2.5 mL of the final volume, and incubated at room temperature for a further 90 minutes. One hundred μL of the solution was pipetted into a transparent culture 96-well plate, and its absorbance was measured at 550 nm using an Infinite® 200 PRO multimode (Tecan Group Ltd, Switzerland). Standard gallic acid solutions were used for the preparation of the calibration curve showing the linear relationship between the absorbance at 550 nm and the TPC in the gallic acid equivalent (GAE) concentration.
Hydrodynamic Size, Size Distribution, and Zeta Potential
To evaluate the hydrodynamic size and polydispersity index (PDI), the nanoparticle suspension was filled into a disposable polystyrene micro cuvette subsequently placed into a Zetasizer Nano ZSP (Malvern Panalytical, The Netherlands) maintaining the temperature at 25°C and the counting rate at a range of 100–500 kilo counts per second (kcps) by diluting the samples with PBS (pH 7.4). The laser intensity and wavelength were 4 mW and 633 nm, respectively, while 173° back-angle scattering was recorded and compiled.
To determine the zeta potential (ζ), the nanoparticle suspension was injected into a disposable folded capillary cell and analyzed the zeta potential with the Zetasizer Nano ZSP. The maintenance of the counting rate at the range of 100–500 kcps as well as the suppression of the ionic conductivity at lower than 5 mS/cm was conducted by diluting the samples with 0.1× PBS (pH 7.4) to compromise between the electrode polarization and joule heating.
The reflective indices of the lipid nanoparticles and PBS were entered into the Zetasizer software at 1.45026,27 and 1.332,28 respectively.
Transmission Electron Microscope
The morphology of the nanoparticles negatively stained with 1% phosphotungstate (PTA) solution was investigated under a JEM-1400 transmission electron microscope (TEM) (JEOL, USA). The samples were dropped onto a formvar-coated copper grid, which was then left undisturbed for five minutes, blotted around the edge of the grid with a filter paper, and air-dried for two minutes. Following that, 1% PTA solution was dropped onto the grid, left intact for two minutes, blotted with filter paper, and kept dry in a desiccator cabinet until investigation under the accelerating voltage of 100 kV.
Encapsulation Efficiency
The encapsulation efficiencies (EEs) of the nanoparticle incorporating substances were calculated according to Equation 1. The encapsulated quantities of IR-820, the TPC, and BCIP were assessed based on the UV-Vis-NIR spectrometry and normalized with the concentration of the phospholipids while TRITC-Dex40 was quantified using a fluorescence spectroscopy technique. The identity wavelengths and the disrupting solvents for each substance are specified in Table S1.

Detection of Singlet Oxygen Formation
DMA, a hydrophobic singlet oxygen probe, was incorporated into the nanoparticle formulations, called DMA-NP and DMA-IR-NP (Table 1). One mL of DMA-NP and DMA-IR-NP was added into each well of six-well culture plates and then irradiated with an 830-nm light-emitting-diode (LED) array providing the irradiance of 0.0118 W/cm2 at a working distance of 4 cm for different irradiation periods up to 60 minutes. The resultant NIR fluences at different irradiation times are specified in Table S2. The samples’ absorption spectra in the range of 320 nm to 450 nm and their emission spectra in the range of 400 nm to 500 nm at the excitation wavelength of 375 nm were measured using an Infinite® 200 PRO multimode (Tecan Group Ltd, Switzerland). The absorbance and the fluorescence intensity were normalized with those of NP maintaining the concentration of the phospholipids at 2.5 mg/mL.
Determination of the Lipid Hydroperoxides
FOX2 assay developed by Woff29 was modified and applied to detect the lipid peroxides in this study. FOX2 reagent was prepared by dissolving 100 μM xylenol orange, 250 μM ammonium ferrous sulfate, 4 mM BHT, and 25 mM sulfuric acid with 90% methanol. Then, 18 μL of the samples and 342 μL of the FOX2 reagent were vortexed and incubated for 30 minutes at room temperature. After that, any aggregates were excluded by centrifugation at 2000 rpm for 15 minutes, and the absorbance of the supernatant was measured at 560 nm using Infinite® 200 PRO multimode microplate readers (Tecan Group Ltd, Switzerland) against the reaction solution of the PBS used as a blank. The standard solutions of hydrogen peroxide were used for the preparation of a calibration curve.
Release Profile of the Total Phenolic Content
A certain volume of the PEm-IR-NP to obtain 125 μg GAE of the TPC was filled into the CelluSep® dialysis tubing T1 3.5 kDa molecular weight cut-off (MWCO) (Interchim Inc, France), which was previously pretreated according to the supplier’s protocol and equilibrated with the release medium, specifically the simulated sweat solution, consisting of NaCl 2.92 mg/mL, CaCl2 0.166 mg/mL, MgSO4 0.12 mg/mL, and KH2PO4 1.02 mg/mL (pH 5.4),30 for at least three hours or until use. The dialysis bag was subsequently immersed into a 500-fold volume of the simulated sweat solution and kept stirring at 250 rpm and 30–35°C. The release medium was collected and replaced with an equal volume of fresh medium at different time points. The trace amount of the TPC in the collected release medium was concentrated and quantified. During the first 30 minutes, a setup of drug release testing was continuously illuminated with 830-nm infrared at the irradiance of 0.0118 W/cm2. The release profiles of the TPC were analyzed to determine the times to reach 25%, 50%, and 75% cumulative TPC release (t25%, t50%, and t75%). Also, the mean dissolution time (MDT) and the dissolution efficiency at time t (DEt) were calculated from Equations 2 and 3, respectively, by using DDSolver add-in program.31
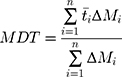
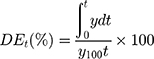
where n is the number of data points; t̄i is the time at the midpoint between ti and ti-1; ΔMi is the increased amount of TPC released between ti and ti-1; y is the cumulative percentage release of TPC at time t; and y100 is the maximum percentage release of TPC within the range of time from 0 to t.
Cytotoxicity Test
The HaCaT cells, originally purchased from American Type Culture Collection (ATCC, Manassas, VA, USA), were provided from the Nanomedicine Research Unit, Chulalongkorn University, Thailand and maintained in a high glucose DMEM medium (Gibco, Life Technologies, USA) supplemented with 10% fetal bovine serum (FBS) (Gibco, Life Technologies, USA) and 1% Antibiotic-Antimycotic 100× (Gibco, Life Technologies, USA) under humidified air composed of 5% CO2 at 37ºC. After reaching the exponential phase of growth, the HaCaT keratinocytes (passage number = 25–30) were seeded into 96-well culture plates at a density of 10,000 cells per well on Day 0 and incubated for a day. The HaCaT cells were then washed and treated with NP, PEm-NP, IR-NP, and PEm-IR-NP maintaining the concentration of the phospholipids at 0.95 mg/mL, PEm at 28.8 μg GAE/mL, or IR-820 at 600 nM. Complete DMEM medium was used as a negative control while the positive controls included 10−4 M clobetasol propionate and 0.1% phenol, respectively. At six hours after the treatment, the HaCaT cells were illuminated with the 830-nm LED panel at the fluence of 20 J/cm2 and further incubated overnight. On Day 2, the HaCaT keratinocytes were labeled with MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) solution (Vybrant® MTT Cell Proliferation Assay Kit, Molecular Probe, Inc, USA), and the formazan product was solubilized with dimethyl sulfoxide (DMSO) according to the supplier’s protocol. The absorbance of the solutions was measured at 540 nm using the Infinite® 200 PRO multimode (Tecan Group Ltd, Switzerland). The negative control was set as 100% cell viability, and the cell viability percentage below 70% was considered as a cytotoxic condition, similar to the threshold specified in ISO 10993–5.32
Cellular Uptake
The HaCaT cells (passage numbers = 25–30) were seeded into 12-well culture plates at a density of 80,000 cells per well on Day 0. On Day 2, the HaCaT keratinocytes were washed, fed with fresh complete medium, and treated with free TRITC-Dex40, TRITC-Dex40-NP, or TRITC-Dex40-IR-NP maintaining the concentration of TRITC-Dex40 at 9.4 μg/mL or IR-820 at 600 nM. The TRITC-Dex40 fluorescence in the living cells was monitored at 0, 2, and 6 hours after the treatment using a CytellTM Cell Imaging System (GE Healthcare Corp, USA). After investigation at six hours, the HaCaT cells were washed with cold PBS three times and lysed with 1% Triton® X-100. The fluorescence of the lysates was measured at λex/em = 551/582 nm using the Infinite® 200 PRO multimode (Tecan Group Ltd, Switzerland) for the relative quantification of the TRITC-Dex40.
Monitoring of the Endolysosomal Escape
On Day 0, the HaCaT keratinocytes (passage numbers = 25–30) were seeded into Lab-TekTM II eight-well chamber slides (Thermo Fisher Scientific, USA) at a density of 10,000 cells per well and incubated overnight. The lysosomes were then labeled using CellLight™ Lysosomes-GFP BacMam 2.0 (Thermo Fisher Scientific, USA) on Day 1. On Day 2, TRITC-Dex40-NP or TRITC-Dex40-IR-NP was added into the well to attain 9.4 μg/mL of TRITC-Dex40 or 600 nM of IR-820 for six hours before 830-nm illumination at 20 J/cm2. The intracellular locations of TRITC-Dex40 and lysosomal-associated membrane protein 1 (LAMP1)-green fluorescent protein (GFP) were investigated under an FV10i confocal laser scanning microscope (CLSM) (Olympus Corporation, USA) at 0, 2, and 4 hours after the infrared illumination.
Colocalization Analysis
The images acquired from the CLSM were deconvoluted using an ImageJ plugin, DeconvolutionLab2.33 Two-dimensional point spread functions (PSFs) were generated by using Diffraction PSF 3D, and the parameters were specified as follows: refractive index of medium = 1.515; numerical aperture = 1.35; emission wavelengths = 510 nm for GFP and 578 nm for TRITC. Richardson–Lucy deconvolution34,35 was performed by setting the number of iterations at 50. Then, the deconvoluted images were analyzed for the colocalization of the TRITC-Dex40 and lysosomes using JACoP v.2.036 and EzColocalization ImageJ plugins.37 The colocalization metrics and parameters are specified in Table S3. For the two fluorophores, the intensity correlation quotient (ICQ) was defined by the total number of pixels (the smallest units of an image) where the intensities for both signal channels were either together above or below the mean intensity of each channel over the total number of pixels, and subtracted by 0.5.38 ICQ had a range from −0.5 (anti-colocalization) to 0.5 (colocalization). The threshold overlap score at the highest threshold (TOSh), in this work, represented the observed overlapping signal above the 10% highest threshold for each channel normalized with the corresponding expected overlap when the signals were distributed independently and uniformly.39 TOSh had a range from −1 (anti-colocalization) to 1 (colocalization). TOSh was reported to exhibit a favorable receiver operating characteristic due to its high true positive as well as low false negative rates.39
Tracking of the Nanoparticle Decomposition
The HaCaT cells (passage numbers = 25–30) were seeded into 75-cm2 cell culture flasks at a density of 93,333 cells/cm2 on Day 0 and allowed to attach overnight. On Day 1, the cells were washed and treated with IR-NP or BCIP-IR-NP to maintain the concentration of the phospholipids at 1 mg/mL for two hours. The cells treated with BCIP-IR-NP were irradiated with the treatment for two hours using the 830-nm LED panel to obtain the fluence of 20 J/cm2 and incubated for a further two hours. The cell monolayers were washed twice with ice-cold PBS, fixed with 2.5% glutaraldehyde in a 0.1M phosphate buffer (pH 7.4) for four hours at 4ºC, washed with PBS, and subsequently collected by using cell scrapers. The collected cells were pelleted, stained with 1% osmium tetroxide in the 0.1 M phosphate buffer for two hours, washed, and then mixed with 2% agar, which was pre-melted and warmed at 45–50ºC. After solidification, the agar blocks were cut into 0.5-mm3 cubes and serially dehydrated at 35%, 50%, 70%, 95%, respectively and absolute ethanol for 15 minutes in each sequence. The dehydration with absolute ethanol was repeated twice before incubating the cells in propylene oxide for 15 minutes for three times. The infiltration with Spurr’s resin40 was accomplished by incubating the cells with Spurr’s resin and propylene oxide with the increasing ratios from 1:3, 1:1, 3:1, and 1:0, respectively overnight at each ratio and repeating the infiltration with pure Spurr’s resin twice overnight. Then, the resin was cured in the oven maintaining the temperature at 70ºC for eight hours and cut into ultra-thin sections (60–90 nm thickness) using an Ultramicrotome Leica EM UC7 (Leica Microsystems GmbH, Germany). The ultra-thin cell sections, placed onto the copper grid, were further stained with uranyl acetate, followed by lead citrate staining before being investigated under a JEM-1400 transmission electron microscope (JEOL, USA).
Statistical Analysis
For single-factor experiments with more than two levels, since the assumption of either the normality or equal variance was violated based on the Kolmogorov–Smirnov test or Levene’s test, respectively, the experimental data were analyzed by performing a Kruskal–Wallis test with a post hoc analysis according to Conover.41 For single-factor experiments with two levels, the differences between the non-parametric dependent data were assessed using the Wilcoxon test. For two-factor experiments with non-parametric repeated measures, the significant difference was tested by the aligned ranks transformation ANOVA,42 followed by post hoc Tukey multiple comparisons. Normally- and non-normally-distributed data were expressed as the mean ± standard deviation and median (Q1 – Q3), respectively.
Results and Discussion
Near-Infrared-Triggered Generation of Singlet Oxygen and Photobleaching
As an important parameter for the preparation of the nanoparticles, the transition temperature between the gel and liquid crystalline phase of the major components, the mixture of soybean phosphatides, cholesterol, and sodium deoxycholate, was determined and set as a floor temperature during the rehydration step to let the hydrocarbon chains have sufficient fluidity and disperse homogenously in the hydration medium.43 Additionally, maintaining the nanoparticles at the transition temperature should be avoided during storage to prevent their collapse. Initially, the soybean phosphatides were derived from crude soybean lecithin by collecting the deoiled acetone-insoluble fraction whose yield was 62.2 ± 1.0% by weight and subsequently formed into a thin film in combination with cholesterol and sodium deoxycholate according to the NP formulation, which was the organic-phase base for all the other formulations (Table 1). The DSC curve of the NP lipid film represented two endothermic peaks at −51°C and −5.6°C while there was no apparent latent-heat-associated event above 4°C (Figure S2). Thus, the NP-based formulations could be stored at 4°C without the adverse effect of the phase alteration and could be rehydrated at any temperature above 4°C. The phosphatides-based nanoparticles combining IR-820 were then detected for the generation and presence of singlet oxygen within the hydrophobic domain upon the NIR irradiation by further incorporating an hydrophobic selective singlet oxygen probe; namely, 9,10-dimethylanthracene (DMA) into the nanoparticle formulations, which were DMA-NP and DMA-IR-NP.
The oxidation of the fluorescent DMA with singlet oxygen would yield non-fluorescent endoperoxide.44 Thus, the generation of singlet oxygen could be indicated by the decreases in the absorbance and fluorescence intensity of the DMA. Continuous 830-nm NIR illumination toward the DMA-NP at 0.0118 W/cm2 had a benign effect on the absorption spectra (Figure 1A) using NP as a blank, while the inclusion of IR-820 into the DMA-IR-NP brought about a decrease in the absorbance at the peak wavelengths of the DMA at 380 nm and 402 nm (Figure 1B), as well as a decrease in the fluorescence intensity at the peak emission wavelength at 429 nm as the irradiation time increased (Figure 1D). When comparing with the absorbance and fluorescence intensity of the DMA-NP at each irradiation time point, both the absorbance and the fluorescence intensity of the DMA-IR-NP at the peak wavelengths decreased by approximately 30% after 60 minutes of illumination (Figure 1C and E). This revealed that the simple inclusion of IR-820 into the nanoparticles led to the photochemical production of singlet oxygen (Figure 1F) in an NIR-fluence-dependent manner. In addition, the existence of singlet oxygen was signified specifically in the hydrophobic domain of the nanoparticles where the DMA was supposed to be located, as its octanol/water partition coefficient was documented at 5.69.45
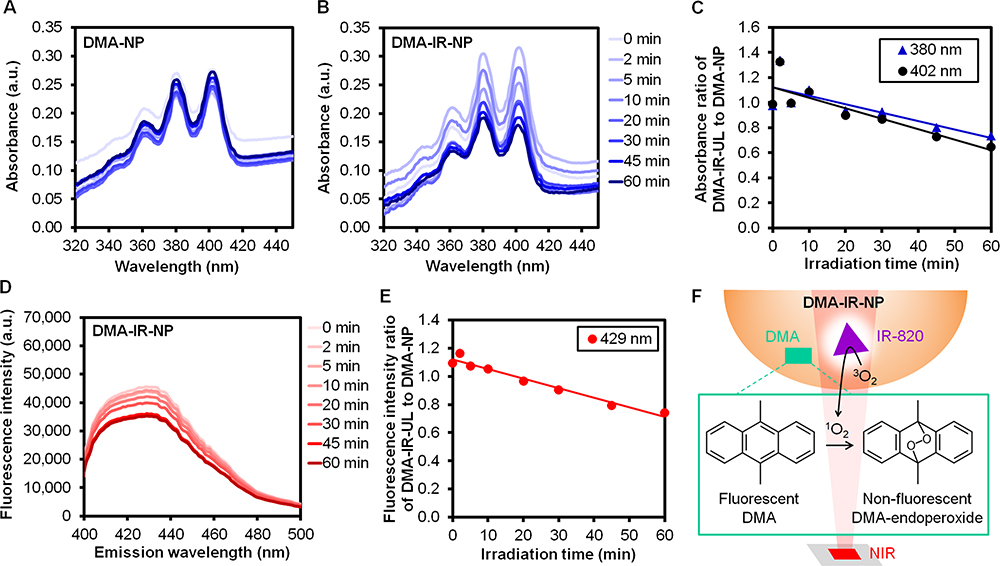 |
Figure 1 Absorption spectra of (A) DMA-NP, (B) DMA-IR-NP, (C) Absorbance ratio of DMA-IR-NP to DMA-NP at the peak wavelengths of 380 and 402 nm, (D) Fluorescence spectra of DMA-IR-NP setting the excitation wavelength at 375 nm, (E) Fluorescence intensity ratio of DMA-IR-NP to DMA-NP at the peak emission wavelength of 429 nm after being exposed to 830-nm NIR at 0.0118 W/cm2 at different time points, and (F) Explanatory illustration of the scenario implied by (C, E). |
The generated singlet oxygen might oxidize nearby IR-820 leading to photobleaching (Figure S3A), which would be the loss of absorbance and emission over a range of a particular wavelength. The photobleaching of IR-820 that would conform with the NIR irradiation time was investigated by monitoring the absorption spectra of IR-NP. The results are shown in Figure S3B. The absorbance of IR-NP decreased from 0.92 to 0.51 at 830 nm after 60 minutes of the NIR irradiation or at the total fluence of 42.48 J/cm2. The absorbance of IR-820 in IR-NP at 830 nm was decreased by almost 60% and 100% after being exposed to the NIR fluences of 20 and 40 J/cm2, respectively. This approximation was conducted by correcting the baseline with the absorbance of NP and setting the absorbance of IR-820 before the irradiation as 100% relative absorbance (Figure S3C). This result suggested the limit of the continuous NIR fluence to provide a photosensitizing effect and the necessity to avoid exposing the nanoparticle samples to NIR prior to use.
Morphological Alteration of Nanoparticles Corresponding to Near-Infrared Illumination
The phosphatides-based nanoparticles loaded with both IR-820 and the PEm extract, a mixture of hydrophobic and hydrophilic compounds as a drug model, were characterized and applied to study the morphological change upon the NIR irradiation. From the TEM images (Figure 2A–C), the shapes of NP, PEm-NP, and PEm-IR-NP were found to be spherical after the SEC. As a result, the determination of the hydrodynamic size and size distribution by using a dynamic light scattering (DLS) technique was considered valid, as this technique recognized an object as a spherical particle. Based on the TEM images, the diameters of NP, PEm-NP, and PEm-IR-NP were 92.2 ± 41.5 nm, 25.0 ± 8.8 nm, and 24.0 ± 8.1 nm, respectively, while the DLS revealed the Z-average hydrodynamic diameters of 151.3 nm (PDI = 0.434), 196.0 nm (PDI = 0.344), and 157.8 nm (PDI = 0.252) in Figure 2D, respectively. Negligible fluctuations at the baseline of the correlograms were referred to having no large aggregates (Figure 2E). The zeta potentials of NP, PEm-NP, and PEm-IR-NP were about the same at ~ −30 mV indicating nearly moderate colloidal stability.46 The absorption spectra of NP, PEm-NP, and PEm-IR-NP after the preparation without the adjustment of the phospholipids concentration verified the ability of PEm-IR-NP to absorb 830-nm NIR due to the inclusion of IR-820 (Figure 2F). The EE of the TPC into PEm-NP was 37.8 ± 3.1%, whereas for the PEm-IR-NP, the EEs of the TPC and IR-820 were 47.4 ± 6.2% and 62.9 ± 4.6%, respectively.
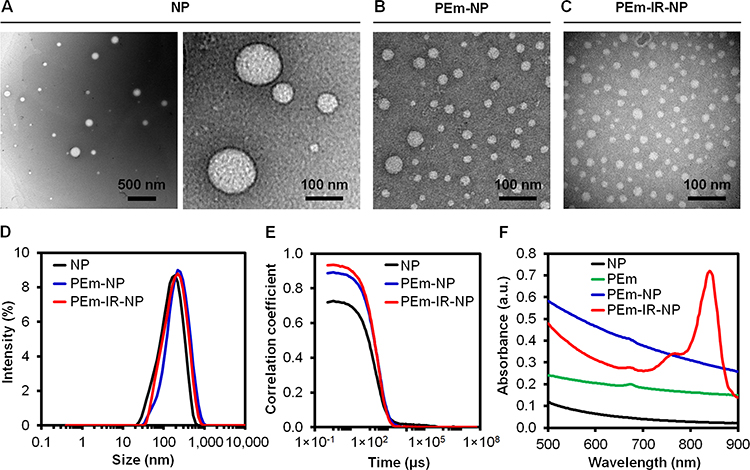 |
Figure 2 Negative-stained TEM images of (A) NP, (B) PEm-NP, (C) PEm-IR-NP, (D) corresponding intensity-weighted hydrodynamic size distribution, (E) correlograms, and (F) absorption spectra. The scale bars are displayed on each image. |
Only PEm-IR-NP demonstrated a noticeable change of morphology under the TEM as the NIR fluence was varied in a range of 0–20 J/cm2 (Figure 3A). From 0 to 2 J/cm2, the PEm-IR-NP nanoparticles were symmetrically enlarged since the circularities determined by using ImageJ remained insignificantly different (P-value = 0.6250) between 0.9540 (0.9273–0.9613) at 0 J/cm2 and 0.9470 (0.9310–0.9500) at 2 J/cm2 based on the Wilcoxon test. From 5 to 20 J/cm2, the nanoparticles appeared to be disrupted, fused, and formed into multivesicular bodies and a long fibrous shape. Almost no spherical nanoparticles remained after the 830-nm illumination at 20 J/cm2. However, it should be noted that the TEM images might represent artifacts caused by the staining and drying processes. Figure 3B illustrates the possible outcomes of the PEm-IR-NP nanoparticles irradiated with increasing NIR fluence. It was assumed that the morphological transformation led to the accelerated release of the encapsulated contents, and the disruption of the nanoparticles was a consequence of singlet oxygen reacting on the allylic positions of the fatty acids. To prove the oxidation of the fatty acids, the development of lipid hydroperoxides upon irradiation was thus subsequently monitored.
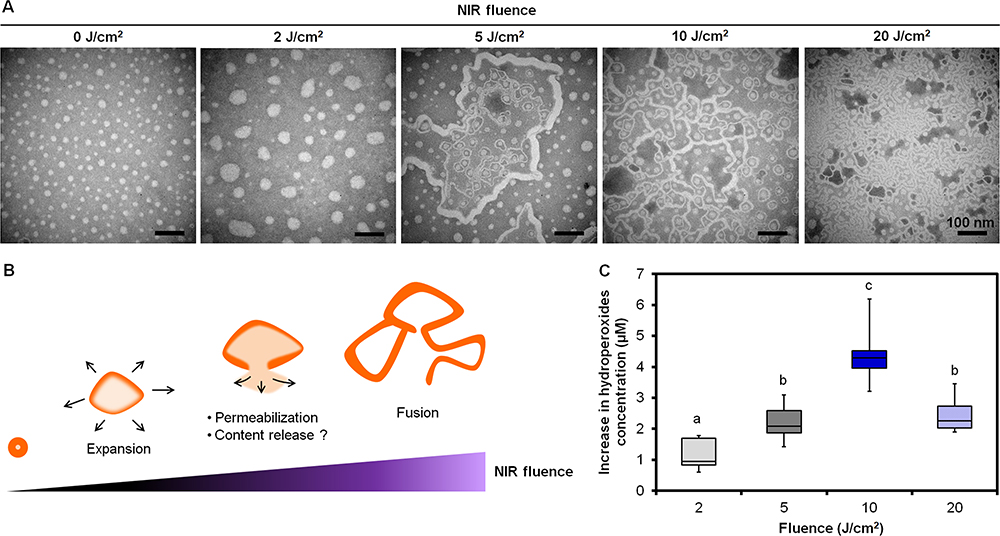 |
Figure 3 (A) Negative-stained TEM images of PEm-IR-NP irradiated at different fluences of 830-nm NIR, (B) A schematic illustration representing the consequences on PEm-IR-NP corresponding to the NIR fluence, and (C) The increases in the lipid hydroperoxides in PEm-IR-NP (phospholipids concentration = 4 mg/mL) irradiated with various fluences of 830-nm NIR when normalized with the concentration of the lipid hydroperoxides in PEm-IR-NP before irradiation. Different letters above the box plots indicate the statistically significant differences (P < 0.001) and n = 8. Scale bar = 100 nm. |
The quantification of lipid hydroperoxides by the FOX2 assay was performed immediately after irradiating PEm-IR-NP at various fluences of 830-nm NIR and corrected with the amount of the lipid hydroperoxides derived from PEm-IR-NP prior to the irradiation as a baseline level. Figure 3C shows that the levels of the hydroperoxides significantly increased from 0.952 (0.833–1.696) µM at 0 J/cm2 to 4.286 (3.958–4.524) µM at 10 J/cm2 and then decreased to 2.262 (2.024–2.738) µM at 20 J/cm2. The decline of the hydroperoxides at a high fluence (> 10 J/cm2) might be because the hydroperoxides were gradually converted into decomposition products, eg, aldehydes, ketones, acids, and epoxides,18 and the formation of the lipid hydroperoxides was retarded as IR-820 that evidently experienced photobleaching (Figure S3B and C). The authors speculated that the introduction of the hydroperoxide (-OOH) moiety into the fatty acid chains made it difficult for the neighboring phospholipids to be packed closely together, hence reducing the nanoparticle’s structural integrity. Furthermore, the conversion of the lipid hydroperoxides into decomposition products might disrupt the structure of the nanoparticles. The decrease in the integrity of the nanoparticles along with the structural breakage would assumingly accelerate the content’s release upon the irradiation, and this assumption would be justified in the following investigation.
Release Profile of the TPCs Upon Near-Infrared Irradiation
The expansion and disruption of the nanoparticles in response to the NIR irradiation likely accelerated the release of the encapsulated contents. Here, PEm was used as a drug model to examine the NIR-stimulated release, and the fruit extract was quantified using its TPC. The release of the TPC from PEm-IR-NP into a simulated sweat solution maintained at a vigorously-stirred sink condition at 32°C was investigated with and without 830-nm irradiation in the first 30 minutes, which was equivalent to 21.24 J/cm2. By plotting the cumulative release of the TPC over 24 hours, the results showed that the NIR illumination accelerated the release of the TPC before its concentration became constant after two hours, while without the NIR illumination, the release of the TPC was prolonged for longer than eight hours (Figure 4). To understand the changes in the TPC release upon the irradiation, the release profiles of the TPC were then analyzed to obtain the times required for 25%, 50%, and 75% cumulative TPC release (t25%, t50%, and t75%), the mean dissolution time (MDT), and the dissolution efficiency at 2 hours (DE2hr) where the cumulative release of TPC appeared steady at ~75% under the irradiation condition. These model-independent parameters have been applied to characterize the release rate and to compare the profiles extensively.47,48 The model dependent approach, ie, the curve fitting with release kinetics models, might be unjustified to evaluate and compare the release parameter, particularly the diffusion coefficient, in this study. The explanation is that the derived diffusion coefficient would be the average value among TPC components in inexact proportions over the time regardless the concentration dependence of the diffusion coefficient for a multicomponent system49 at different time points. The model-independent parameters were, in this case, better suited and used for describing the TPC release profiles.
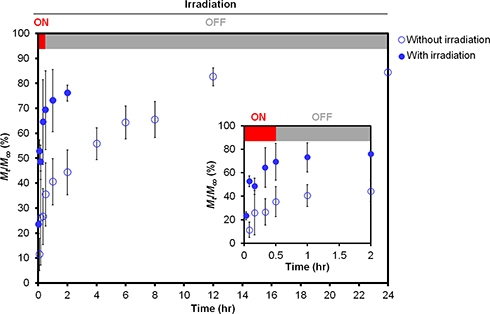 |
Figure 4 The release profile of the TPC from PEm-IR-NP into the stirred simulated sweat solution (pH 5.4) maintained at 32°C with and without 830-nm irradiation at 0.0118 W/cm2 for the first 30 minutes. |
Table 2 displays that NIR irradiation reduced the time required for successive 25%, 50%, and 75% cumulative TPC release from PEm-IR-NP by 4.5, 16.3, and 6.2 times, respectively, when compared to the non-irradiation experimental condition. This demonstrated the clear influence of the initial single NIR illumination throughout the different stages of TPC release. Furthermore, without NIR irradiation, the MDT was approximately 18-fold longer and the DE2hr was about half of that with NIR. The longer MDT and the lower DE2hr could indicate the lower rate of TPC release. The results consistently confirmed the accelerated TPC release upon the irradiation. Despite being able to estimate the fold changes of the MDT and the DE2hr, the authors were aware that the observed cumulative TPC release curves became unchanged at almost but less than 90%. Therefore, some release patterns might not be taken into account but might be sufficient to signify the NIR-responsive accelerated release of TPC from IR-NP loading the complex mixture of the phenolic compositions.
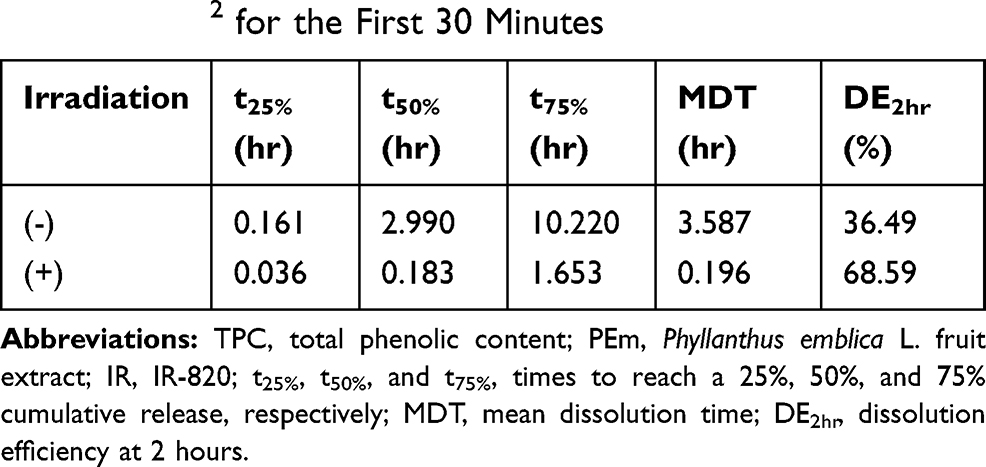 |
Table 2 Characterization of the Release Profile Curves of TPCs from PEm-IR-NP into the Stirred Simulated Sweat Solution (pH 5.4) Maintained at 32°C with and without 830-nm Irradiation at 0.0118 W/cm2 for the First 30 Minutes |
Cytotoxicity of the Nanoparticle Formulations Accompanying Near-Infrared Irradiation
The generation of singlet oxygen and photochemical-induced moderate changes to the total lysosomal rupture might cause cell death, which could be beneficially synergized with chemotherapy to treat cancer,50,51 but could be detrimental to normal cells. Lysosomal membrane permeabilization potentially provoked the release of cathepsins,52 regulators of cell death and inflammation, inducing apoptotic and necrotic outcomes50 depending on the degree of lysosomal membrane permeabilization. Thus, to enhance the bioavailability of the therapeutic molecules while preserving the favorable cell viability of normal cells, the amount of a photosensitizer and light fluence should be modulated to sufficiently trigger the endolysosomal escape, ideally early and late endosomal escape, but not too much lysosomal membrane permeabilization. An in vitro applicable concentration of IR-820 combined with an NIR fluence was suggested and its cytotoxicity was tested accordingly.
The cell viability of the HaCaT keratinocyte cell line after the treatment with various nanoparticle formulations is shown in Figure 5. The MTT tests revealed that NP, PEm-NP, PEm-IR-NP, and IR-NP either with or without 20 J/cm2 of 830-nm irradiation had no cytotoxic effect on the HaCaT cells, according to the similar threshold (cell viability > 70%) reported in ISO 10993,32 while the positive controls, including 10−4 M of clobetasol propionate and 0.1% phenol, resulted in complete cell death. This result suggested the in vitro use of the IR-NP-based nanoparticles by limiting the final concentration of IR-820 to about 600 nM to avoid significant cytotoxicity for the exposure toward the HaCaT cells.
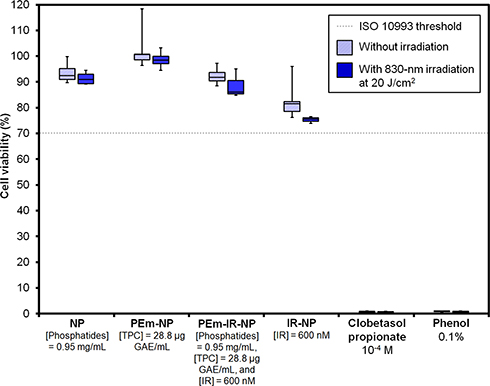 |
Figure 5 Cell viability of the HaCaT cells after being treated with various nanoparticle formulations and positive controls including clobetasol propionate as well as phenol solutions with and without 830-nm irradiation at 20 J/cm2 (n = 4). |
It should be noted that PEm itself was found to have in vitro cytotoxic potential when applied to the HaCaT cells in our experimental setup. This was due to the poor solubility of some components of PEm in the complete DMEM medium resulting in the precipitates covering the cell monolayer, which consequently led to cell death (Figure S4A) regardless the actual cellular responses to PEm. The size distribution and the correlogram of PEm also revealed large aggregates when PEm was suspended in PBS as the fluctuation of the correlogram baseline (Figures S4B and C). The IR-NP-based nanoparticles were able to incorporate PEm and increase the solubility of PEm as no precipitates were observed when incubating PEm-IR-NP in the DMEM medium for one day (Figure S4D). This finding points to the usefulness of soybean phosphatides combined with IR-820 as the base ingredients to encapsulate and prevent the precipitation of the complex mixture, which contains some water-soluble components and the others capably dissolved in a lipidic environment.
Cellular Uptake of a Nanoparticle Incorporated Large Macromolecule
High molecular weight dextran conjugated with tetramethylrhodamine isothiocyanate was used as a cargo to study the cellular uptake since dextran was neither able to be diffused across the biological membrane nor able to be bound to any compartment within the cells.53 Thus, the endocytosis of this macromolecule appeared to involve membrane vesicular trafficking and membrane-bounded endosomes. Formed by encapsulating hydrophilic TRITC-Dex40 into the IR-820-included phosphatides-based components, the TRITC-Dex40-IR-NP particles derived from the first pink translucent fraction eluted from the SEC was shown under the TEM in Figure 6A while the second pink transparent SEC fraction, supposed to be free TRITC-Dex40, was represented in Figure 6B. It was possible that several nanoparticles had fused to enclose and even be embedded into the TRITC-Dex40 network to package a comparably large TRITC-Dex40 instead of organizing into lipid bilayer vesicles as generally assumed. Under the TEM, the diameter of the TRITC-Dex40-IR-NP was 764.9 ± 180.7 nm, while the DLS result revealed two peaks of hydrodynamic size distribution of the TRITC-Dex40-IR-NP after the SEC, which were 295.3 nm and 68.1 nm (Figure 6C), probably because the DLS recognized both the separate and fused nanoparticles. The correlograms shown in Figure 6D implied the negligible aggregates of TRITC-Dex40-IR-NP. The ability of the TRITC-Dex40-IR-NP nanoparticles to absorb 830-nm NIR was proven by the absorption spectra (Figure 6E). The EEs of TRITC-Dex40 were 3.7 ± 0.5%, and 9.0 ± 1.2% for TRITC-Dex40-NP and TRITC-Dex40-IR-NP, respectively, and the EE of IR-820 was 72.0 ± 8.9% for TRITC-Dex40-IR-NP.
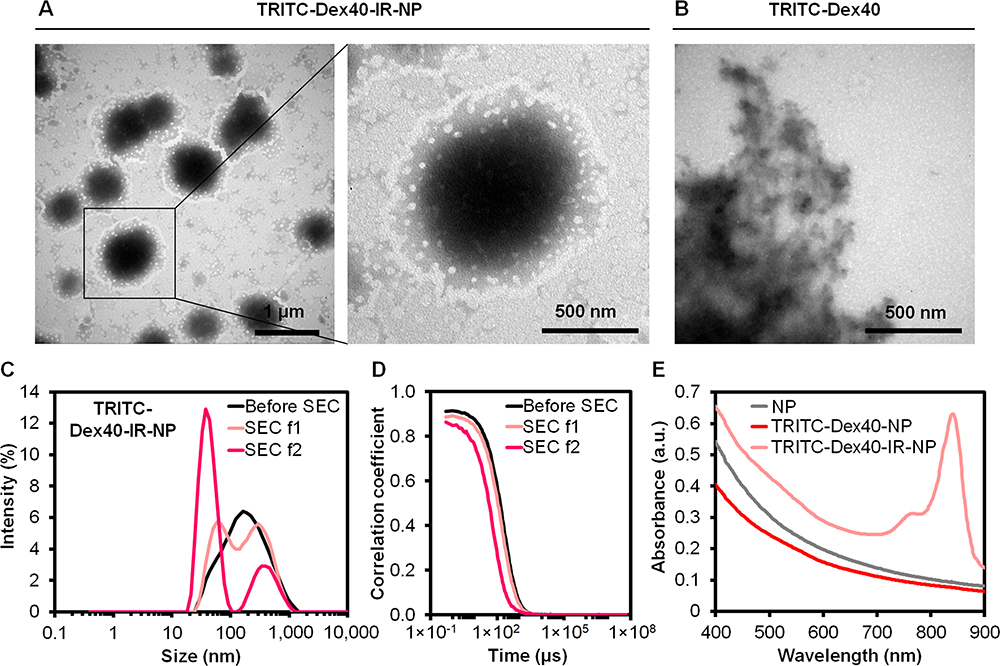 |
Figure 6 TEM images of (A) TRITC-Dex40-IR-NP, (B) TRITC-Dex40, (C) Intensity-weighted hydrodynamic size distribution, (D) Correlograms of TRITC-Dex40-IR-NP before and after SEC, and (E) Absorption spectra of NP, TRITC-Dex40-NP, and TRITC-Dex40-IR-NP. SEC f1 and SEC f2 are the first and second fractions eluted from SEC, respectively. The scale bars are displayed on each image. |
The intracellular fluorescence signals of TRITC after the HaCaT keratinocytes, which were treated with free TRITC-Dex40, TRITC-Dex40-NP, and TRITC-Dex40-IR-NP, maintaining the TRITC-Dex40 concentration at 9.4 μg/mL or IR-820 at 600 nM, for 0, 2, and 6 hours were investigated. The bright-field microscopy together with the fluorescence images revealed noticeable TRITC-Dex40 fluorescence within the HaCaT cells after a six-hour treatment with TRITC-Dex40-NP and TRITC-Dex40-IR-NP, while almost no intracellular signal of TRITC was observed even after a six-hour incubation with free TRITC-Dex40 (Figure 7A). Following the lysis of the plasma membrane with 1% Triton X-100, the fluorescence intensity from the cell lysates were quantified relative to that of the control, which was the HaCaT cells without the treatment. The result in Figure 7B demonstrates that the relative fold changes in the fluorescence intensity of TRITC-Dex40 extracted from the HaCaT cells treated with TRITC-Dex40-NP and TRITC-Dex40-IR-NP were 11.5 (8.5–12.0) and 12.5 (9.4–13.1), respectively, and were significantly higher than that of free TRITC-Dex40, which was 1.8 (1.4–1.9). This result indicated that the NP nanoparticles could effectively enhance the cellular uptake and that the incorporation of IR-820 into the nanoparticles did not affect the internalization degree of TRITC-Dex40.
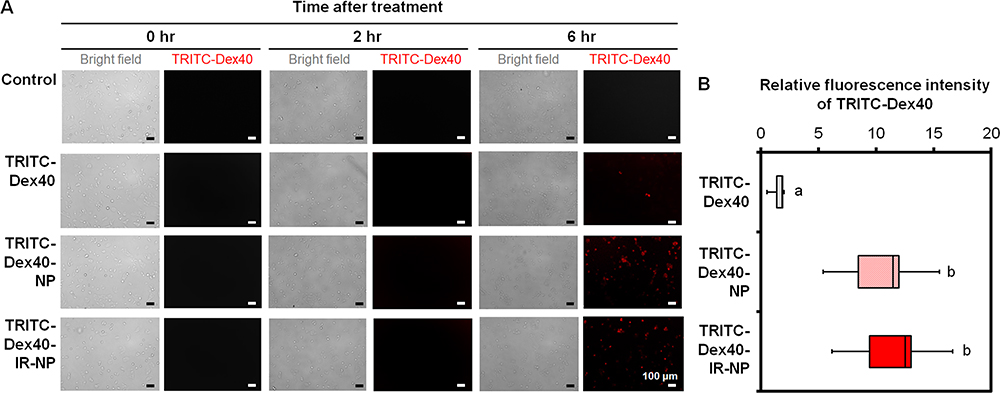 |
Figure 7 (A) Bright-field and fluorescent images of the HaCaT cells at 0, 2, and 6 hours post treatment with free TRITC-Dex40, TRITC-Dex40-NP, or TRITC-Dex40-IR-NP maintaining the concentration of TRITC-Dex40 at 9.4 μg/mL or IR-820 at 600 nM, and (B) Relative fold change in the fluorescence intensity of TRITC-Dex40 in the lysate extracted from the treated HaCaT cells. Different letters above the box plots indicate the statistically significant differences (P < 0.05) and n = 5. Scale bar = 100 µm. |
Near-Infrared Triggered Endolysosomal Escape Based on Fluorescence Colocalization Analysis
The endolysosomal escape was monitored based on both the qualitative and quantitative colocalization analyses of the fluorescence images, which were acquired to meet the Nyquist criteria according to Scriven et al,54 by using different colocalization metrics including Van Steensel’s Cross Correlation Function (CCF),55 ICQ,38 and TOSh39 to verify one another. Non-colocalization (no tendency to exist or not exist in the same pixel) or anti-colocalization (tendency not to exist in the same pixel) of the fluorescence signals between the TRITC-Dex40 cargo and lysosomes-labelling LAMP1-GFP was used as an indicator of the endolysosomal escape in the present study.
After six hours of post-treatment with TRITC-Dex40-NP or TRITC-Dex40-IR-NP, the HaCaT cells were either non-irradiated or irradiated with 830-nm NIR at 20 J/cm2 and the fluorescence images of the living cells were taken at 0, 2, and 4 hours after the completion of the irradiation as illustrated in Figure 8A. Each single-color image was deblurred according to the Richardson–Lucy deconvolution. It was revealed that the signal-to-noise ratios of the deconvoluted images increased and became unchanged after three iterations of the deconvolution although 50 iterations were performed on all images. In the deconvoluted fluorescence images (Figure 8B–E), a yellow pixel represented a colocalized pixel between the green signal of LAMP1-GFP and red signal of TRITC-Dex40 meaning that TRITC-Dex40 was trapped in the endolysosomes. The absence of the yellow fluorescence would indicate that TRITC-Dex40 efficiently escaped from the endolysosomal compartments. Seemingly, the fluorescence images of the TRITC-Dex40-IR-NP-treated HaCaT keratinocytes at 2 hours after irradiation showed fewer yellow pixels over the number of the red and green pixels (Figure 8E) when compared to the non-irradiated condition at the same time point (Figure 8D). However, based on the results in Figure 8B–E, it was not possible to identify the colocalization events with the naked eye. Hence, an image analysis was required for more conclusive proof of the endolysosomal escape.
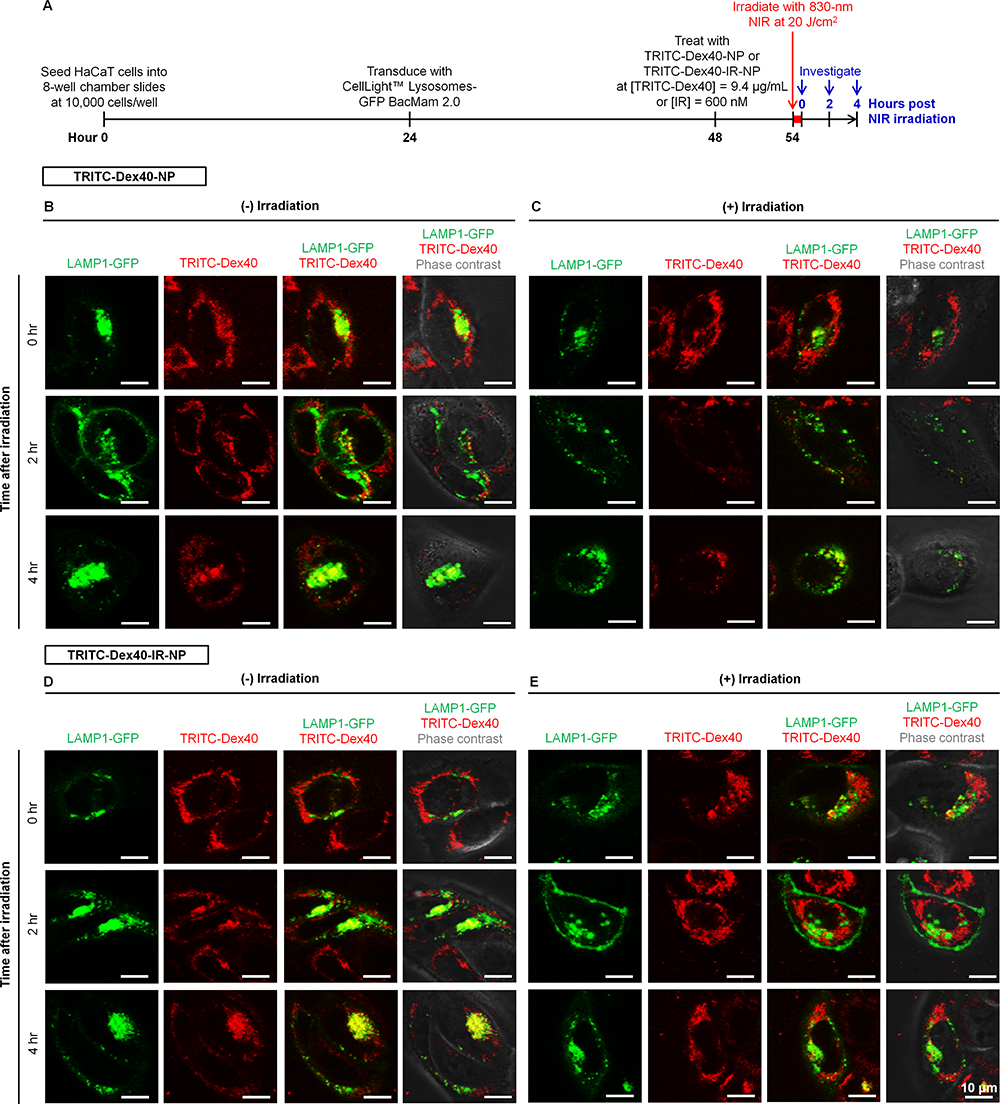 |
Figure 8 (A) Timeline of the experiments for monitoring endolysosomal escape and deconvoluted confocal microscopy images of the HaCaT keratinocytes at 0, 2, and 4 hours post 6-hour treatment with (B, C) TRITC-Dex40-NP and (D, E) TRITC-Dex40-IR-NP (B, D) without and (C, E) with 830-nm irradiation at fluence of 20 J/cm2. Scale bar = 10 µm. |
The intensity scatter plots without thresholding (Figure S5) exhibited non-monotonic and non-linear monotonic relationships between the red and green signals. Furthermore, the red-to-green bleedthrough, ie, the deviation of the fluorescence-intensity distribution toward the green axis, appeared in almost all conditions. As a commonly used colocalization metric to hint the covariance between two color channels over the multiplication of their standard deviations, Pearson’s correlation coefficient (PPC)56,57 was, in this case, inappropriate to quantify the colocalization since the linear correlation between the signals was violated, and bleedthrough occurred. However, PPC could still provide a qualitative idea of whether the two channels were colocalized, non-colocalized, or anti-colocalized via a metric called Van Steensel’s CCFs. This metric functions by shifting one channel onto the other channel in a horizontal or x-direction, eg, moving the green-channel onto the red-channel images, while calculating the PPC, which were called CCFs, of each pixel shift and plotting the CCFs against the pixel shift in the x-direction (Δx). As shown in Figure 9A, all displayed bell-shaped curves that indicated colocalizing events or the entrapment of TRITC-Dex40 within the endolysosomes. However, only the fluorescence image of the TRITC-Dex40-IR-NP-pretreated HaCaT cells at 2 hours post NIR irradiation exhibited the partial anti-colocalization suggesting the escape of TRITC-Dex40 to some degree from the endolysosomes. To further test the significant differences on the colocalization events, ICQ and TOSh were determined as recommended for the non-linear monotonic and non-monotonic relationships between the signal intensities,37 respectively.
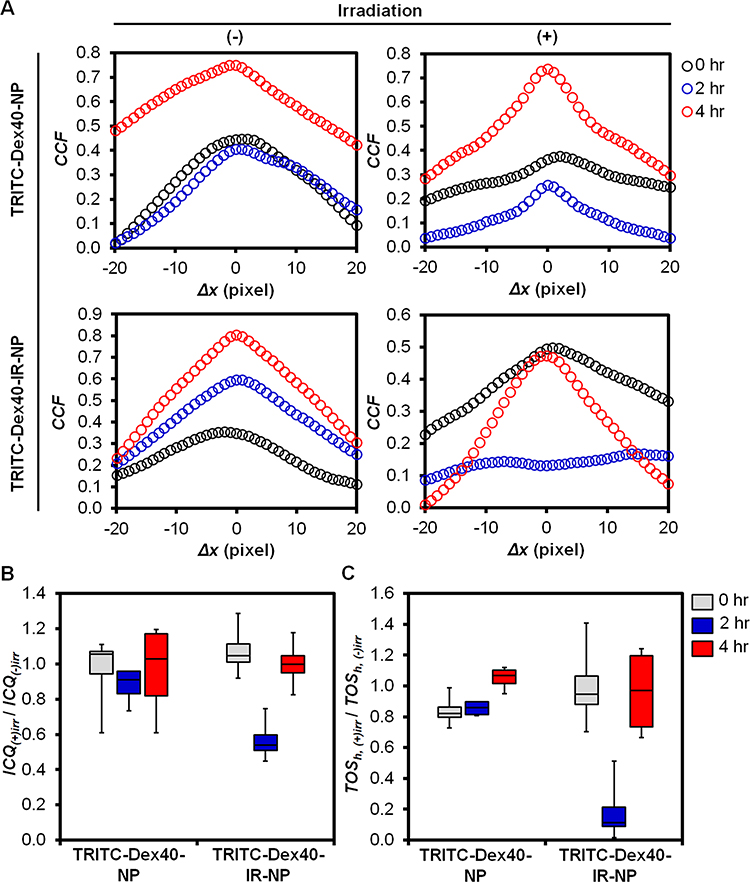 |
Figure 9 (A) Van Steensel CCFs, (B) ICQ, and (C) TOSh ratios (irradiated to non-irradiated conditions) of the fluorescent images acquired from the CellLightTM-lysosome-GFP-transfected HaCaT cells at 0, 2, and 4 hours post-treatment with TRITC-Dex40-IR-NP or TRITC-Dex40-NP for six hours before 830-nm irradiation at a fluence of 20 J/cm2. n = 4 for (B, C). |
The ratios of ICQ and TOSh under the irradiated to non-irradiated conditions were determined at different time points of post irradiation with and without the incorporated IR-820 into the nanoparticles, respectively (Figure 9B and C). The values of the raw ICQ and TOSh with and without NIR irradiation were all positive, hence giving the positive values of their ratios and implying non-colocalizing to colocalizing events. Thus, the endolysosomal escape of TRITC-Dex40 upon irradiation would be indicated by the significant decrease of the ratios of ICQ and TOSh while the ratios at ~1 specified a benign effect of NIR on the colocalization or the escape scenario at a certain time of post irradiation. To inspect the conditions where the ratios significantly decreased, the aligned ranks transformation ANOVA and post hoc test results were presented in Tables S4 and S5.
The time following irradiation (0, 2, and 4 hours) and the interplay between the time after irradiation and the formulation (TRITC-Dex40-NP and TRITC-Dex40-IR-NP) statistically and significantly affected the ratio of TOSh (P < 0.001), but the interplay was less significant toward the ratio of ICQ (P = 0.0569) (Table S4). Additionally, in examining the post hoc results of the interactions on the ratio of TOSh (Table S5), the inclusion of IR-820 led to a significantly lower ratio of TOSh at 2 hours after irradiation (P = 0.0114) compared to that without IR-820 but insignificantly different at 0 hours (P = 0.1723) and 4 hours (P = 0.9759) after the irradiation. This highlighted that the endolysosomal escape of TRITC-Dex40 at 2 hours was caused by the action of IR-820 together with the incubation time after the NIR irradiation. However, when focusing on TRITC-Dex40-IR-NP from 0 to 2 hours after irradiation, the ratios of TOSh were not statistically different (P = 0.0654). Based on this finding, it seemed that the endolysosomal escape of TRITC-Dex40-IR-NP remained unchanged from 0 to 2 hours after irradiation. Thus, this finding raised the question of why the endolysosomal escape was significant when comparing between TRITC-Dex40-IR-NP and TRITC-Dex40-NP at 2 hours post irradiation. The explanation was that the ratio of TOSh for TRITC-Dex40-NP was significantly increased (P = 0.0340) from 0 to 2 hours post irradiation inferring that TRITC-Dex40-NP was further entrapped into the endolysosomes at 2 hours post irradiation in the experimental setup. Thus, an even less significant decrease of the ratio of TOSh for TRITC-Dex40-IR-NP from 0 to 2 hours resulted in a significant endolysosomal escape for TRITC-Dex40-IR-NP when compared to TRITC-Dex40-NP at 2 hours post NIR irradiation.
Moreover, the ratios of TOSh for TRITC-Dex40-IR-NP and TRITC-Dex40-NP at 4 hours after irradiation were not significantly different (P = 0.9759), conceivably revealing the recovery of the colocalizing event at 4 hours post irradiation. This could be possibly explained by following coinciding scenarios. Firstly, the NIR-triggered lysosomal membrane permeabilization might induce lysosome biogenesis.58 Simultaneously, released TRITC-Dex40 as well as remaining intact TRITC-Dex40-IR-NP in the culture medium were further internalized and colocalized with newly generated endolysosomes. The other possibility would be the recycling of the lysosomal membrane proteins as evidenced in Eriksson et al.59 The moderate-to-severe lysosomal membrane damage caused not only the escape of the cargo but also the release of the lysosomal membrane proteins, potentially including the labelled, in the form of lysosomal membrane protein complexes into cytosol. Then, these probe-containing complexes might contribute to the repair of minorly-damaged endolysosomes to maintain functioning lysosomes and cell survival. Consequently, the rest of TRITC-Dex40 became enclosed and the colocalizing event was resumed at 4 hours post irradiation. The observed endolysosomal escape at 2 hours for TRITC-Dex40-IR-NP appeared to be transient, yet sufficient to allow translocation of a portion of the cargo into cytosol.
One question has been raised: How were the endolysosomes and/or endosomes permeabilized resulting in the endolysosomal escape by internalized TRITC-Dex40-IR-NP after the NIR irradiation? The authors hypothesized that the nanoparticles interacted with the cells by (1) fusing with the biological membrane during endocytosis, and/or (2) exchanging the hydrophobic contents with the vesicular membrane engulfing the nanoparticles. Both scenarios introduced IR-820 and the allylic sites to the endolysosomes and/or endosomes. As a result, the integrity of the endolysosomes and/or endosomes was compromised upon the NIR irradiation by the generation of lipid hydroperoxides within the vesicular shell. Although the exchange between the contents of the lipid-based nanoparticles and the surroundings, eg, biological membrane and bulk solutions, was also mentioned elsewhere,60–62 certain mechanisms triggering the endolysosomal escape remain uninvestigated.
Near-Infrared Triggered the Endolysosomal Escape Evidenced by the Localization of a Cytochemical Marker
In addition to the fluorescent labeling along with the colocalization analysis to explore the endolysosomal escape, IR-NP encapsulating a cytochemical marker; namely, BCIP was prepared to probe the fate of the nanoparticles toward the endolysosomal pathway and the escape event. Within the lysosomes and other cellular compartments, BCIP could react with the alkaline phosphatase to form blue 5,5′-dibromo-4,4′-dichloro-indigo precipitates, which could be used to track the location of BCIP.63 Figure 10A and B reveal the diameters of IR-NP and BCIP-IR-NP of 160.8 ± 68.4 nm and 97.1 ± 28.8 nm, respectively. The DLS results show the Z-average hydrodynamic diameters of 101.3 nm (PDI = 0.271, ζ = −33.5 ± 0.5 mV) and 112.9 nm (PDI = 0.392, ζ = −34.2 ± 0.4 mV) for IR-NP and BCIP-IR-NP without large aggregates (Figure 10C). Also, the absent fluctuations at the large delay time in the correlograms (Figure 10D) verified the insignificant sedimentation of the nanoparticles. The absorption spectra of IR-NP and BCIP-IR-NP highlighted the absorbance at the triggering wavelength of 830 nm (Figure 10E). The EE of BCIP into BCIP-IR-NP was 0.3% presenting a limitation of the IR-820-incorporating phosphatides-based nanoparticles to load this hydrophilic substance.
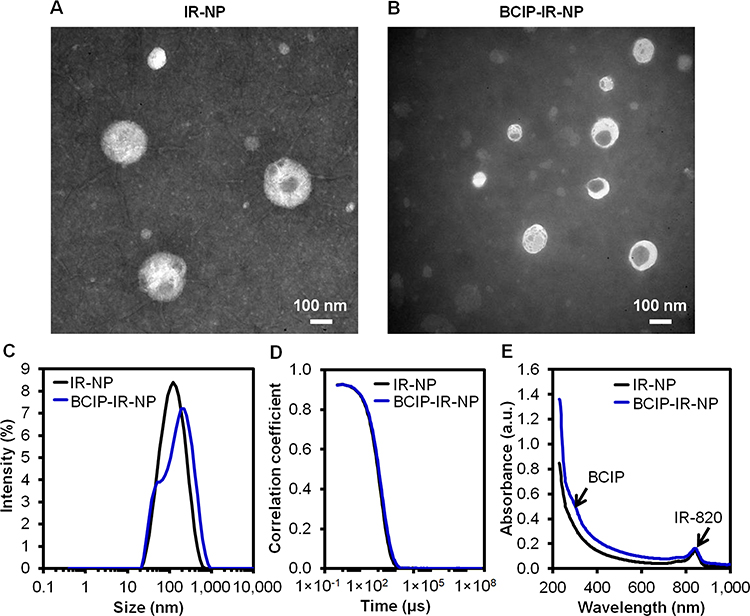 |
Figure 10 Negative-stained TEM images of (A) IR-NP, (B) BCIP-IR-NP, (C) corresponding intensity-weighted hydrodynamic size distribution, (D) correlograms, and (E) absorption spectra. Scale bar = 100 nm. |
In this study, the HaCaT keratinocytes were initially treated with the nanoparticles maintaining the phospholipids concentration at 1 mg/mL for two hours. Afterwards, the treated cells were either irradiated or not irradiated with 830-nm NIR at 20 J/cm2, followed by further incubation for two hours. The TEM images of the ultra-thin sections of the HaCaT keratinocytes without the treatment with the nanoparticles and without irradiation (the control) revealed the baseline images of the lysosomes and endolysosomes (Figure 11). Lysosomes, which contained abundant enzymes, were electron-dense organelles enclosing minute granules; therefore, the lysosomes were shown as dark vesicles under the TEM. The vesicles with packaged partial electron-dense region might represent endolysosomes at the ultrastructure level. Similar to the control, the HaCaT keratinocytes treated with IR-NP had no apparent precipitates within the cells because of the absence of BCIP in the nanoparticle formulation, and their lysosomes or endolysosomes appeared to be the same. For the BCIP-IR-NP treatment without the NIR irradiation, the precipitates were found in the endolysosomes and the lysosomes, as indicated by the blue arrows (Figure 11), while some precipitates were scattered within the cytosol, which might have resulted from small free BCIP molecules simply diffusing into the cells. This indicated the endocytosis of BCIP-IR-NP via the endolysosomal pathway and the decomposition of the nanoparticles inside the endolysosomes while the cargoes were confined within the endolysosomal vesicles. For the treatment with BCIP-IR-NP and the NIR illumination, the precipitates considerably dispersed within the cytosol, and some clusters of precipitates were still attached to the edge of the vesicular organelles possibly implying the endolysosomal escape of the cargoes. However, these findings need to be interpreted with caution since the qualitative identity of those precipitates remains unknown, and the NIR irradiation might have destabilized the nanoparticles within the culture medium resulting in a higher amount of free BCIP diffusing into cells.
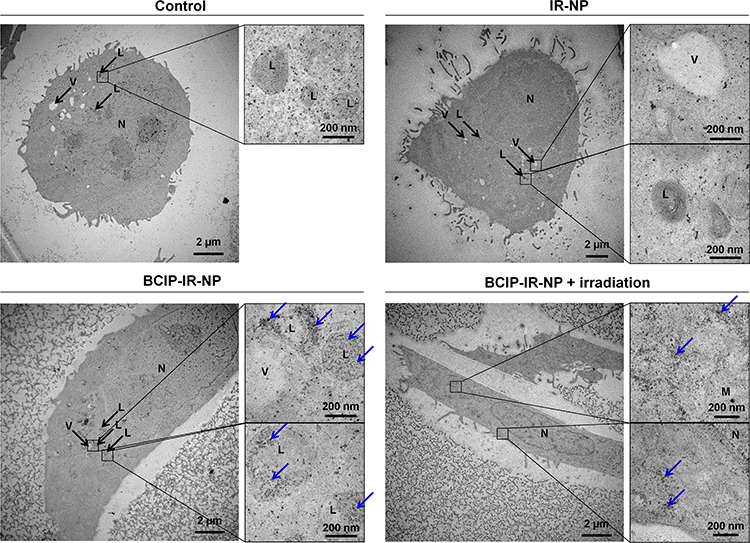 |
Figure 11 Ultra-thin section of the TEM images of the HaCaT keratinocytes treated with IR-NP and BCIP-IR-NP with and without 830-nm irradiation at 20 J/cm2 post-treatment. Possible dehydroindigo precipitates are indicated by blue arrows. The scale bars are displayed on each image. |
Conclusion
This proof of concept study demonstrated the successful NIR-triggered endolysosomal escape using soybean-phosphatides-based nanoparticles incorporating IR-820, an NIR sensitizer, while conserving other benefits, eg, enhanced internalization and biocompatibility, as a delivery nanocarrier towards HaCaT keratinocytes. The results from this study suggest a positive correlation among NIR fluence, the generation of singlet oxygen, lipid hydroperoxides production, morphological change of the nanoparticles, and TPC release whose time required for 50% release reduced by 16 times with a single NIR irradiation at 21.24 J/cm2 in vitro. However, NIR fluence negatively correlated with NIR absorbance of the nanoparticles possibly due to photobleaching. The findings also support that formulated nanoparticles with moderate charge stability increase the water solubility of a natural complex mixture or PEm, enhance the cellular uptake of a macromolecule, specifically TRITC-Dex40, by 6.4 to 6.9 times, and display low cytotoxicity even after NIR irradiation. The visualization of the NIR-triggerable particles reveals two possible nanoplatforms including fused and spherical vesicles when loading relatively large TRITC-Dex40 and small PEm or BCIP, respectively. The fluorescent labeling of the living HaCaT cells and the colocalization analysis highlighted the endolysosomal escape upon the NIR irradiation at 20 J/cm2 depending on the interaction between the inclusion of IR-820 and the post-irradiation time at 2 hours. The authors also confirmed the evidence of the endocytosis and endolysosomal escape of the NIR-triggerable nanoparticle formulation using BCIP as a cargo.
The present work has important implications for the prospective development of an economical transdermal and topical delivery system providing high bioavailability, time-, and spatial-controllable release upon NIR by simply mixing IR-820 into a formulation. IR-820-incorporated soybean-phosphatides based nanoparticles could potentially be useful for delivering cytosolic therapeutics into cells having high lysosomal activity with benign cytotoxicity. Moreover, the authors hope that further investigations would discover the synergistic therapeutic effects between the carrier and NIR.
Acknowledgments
The authors would like to thank the Nanomedicine Research Unit, Chulalongkorn University, Bangkok, Thailand for providing the HaCaT keratinocyte line, as well as acknowledge the support and knowledge transfer from the Molecular Bionics Laboratory, University College London, UK.
Funding
This study was supported by the Petchra Pra Jom Klao Ph.D. Research Scholarship of King Mongkut’s University of Technology Thonburi (KMUTT) and the Capacity Building Program for New Researcher 2017 from the National Research Council of Thailand (NRCT).
Disclosure
Chaiyarerk Homsirikamol reports grants from National Research Council of Thailand (NRCT), during the conduct of the study. The authors report no other potential conflicts of interest in this work.
References
1. Paillard A, Hindré F, Vignes-Colombeix C, Benoit J-P, Garcion E. The importance of endo-lysosomal escape with lipid nanocapsules for drug subcellular bioavailability. Biomaterials. 2010;31(29):7542–7554. doi:10.1016/j.biomaterials.2010.06.024
2. Sharma A, Vaghasiya K, Ray E, Verma RK. Lysosomal targeting strategies for design and delivery of bioactive for therapeutic interventions. J Drug Target. 2018;26(3):208–221. doi:10.1080/1061186X.2017.1374390
3. Bocheńska K, Moskot M, Malinowska M, et al. Lysosome alterations in the human epithelial cell line HaCaT and skin specimens: relevance to psoriasis. Int J Mol Sci. 2019;20(9):2255. doi:10.3390/ijms20092255
4. Marques AR, Saftig P. Lysosomal storage disorders–challenges, concepts and avenues for therapy: beyond rare diseases. J Cell Sci. 2019;132:2. doi:10.1242/jcs.221739
5. Lale SV, Kumar A, Naz F, Bharti AC, Koul V. Multifunctional ATRP based pH responsive polymeric nanoparticles for improved doxorubicin chemotherapy in breast cancer by proton sponge effect/endo-lysosomal escape. Polym Chem. 2015;6(11):2115–2132. doi:10.1039/C4PY01698J
6. Nel AE, Mädler L, Velegol D, et al. Understanding biophysicochemical interactions at the nano–bio interface. Nat Mater. 2009;8(7):543–557. doi:10.1038/nmat2442
7. Jin M, Jin G, Kang L, Chen L, Gao Z, Huang W. Smart polymeric nanoparticles with pH-responsive and PEG-detachable properties for co-delivering paclitaxel and survivin siRNA to enhance antitumor outcomes. Int J Nanomedicine. 2018;13:2405–2426. doi:10.2147/IJN.S161426
8. Lomas H, Massignani M, Abdullah KA, et al. Non-cytotoxic polymer vesicles for rapid and efficient intracellular delivery. Faraday Discuss. 2008;139:143–159. doi:10.1039/b717431d
9. Xu Y, Szoka FC. Mechanism of DNA release from cationic liposome/DNA complexes used in cell transfection. Biochem. 1996;35(18):5616–5623. doi:10.1021/bi9602019
10. Yaghini E, Dondi R, Edler KJ, Loizidou M, MacRobert AJ, Eggleston IM. Codelivery of a cytotoxin and photosensitiser via a liposomal nanocarrier: a novel strategy for light-triggered cytosolic release. Nanoscale. 2018;10(43):20366–20376. doi:10.1039/C8NR04048F
11. Sordillo LA, Pu Y, Pratavieira S, Budansky Y, Alfano RR. Deep optical imaging of tissue using the second and third near-infrared spectral windows. J Biomed Opt. 2014;19(5):056004. doi:10.1117/1.JBO.19.5.056004
12. Hamblin MR. Mechanisms and applications of the anti-inflammatory effects of photobiomodulation. AIMS Biophys. 2017;4(3):337–361. doi:10.3934/biophy.2017.3.337
13. Shiva S, Gladwin MT. Shining a light on tissue NO stores: near infrared release of NO from nitrite and nitrosylated hemes. J Mol Cell Cardiol. 2009;46(1):1–3.
14. Delaey E, van Laar F, De Vos D, Kamuhabwa A, Jacobs P, de Witte P. A comparative study of the photosensitizing characteristics of some cyanine dyes. J Photochem Photobiol B. 2000;55(1):27–36. doi:10.1016/S1011-1344(00)00021-X
15. Ishii F, Nii T. Lipid emulsions and lipid vesicles prepared from various phospholipids as drug carriers. In: Ohshima H, Makino K, editors. Colloid and Interface Science in Pharmaceutical Research and Development. New York: Elsevier; 2014:469–501.
16. van Hoogevest P, Fahr A. Phospholipids in cosmetic carriers. In: Cornier J, Keck CM, Van de Voorde M, editors. Nanocosmetics: From Ideas to Products. New York: Springer International Publishing; 2019:95–140.
17. Scholfield C. Composition of soybean lecithin. J Am Oil Chem Soc. 1981;58(10):889–892. doi:10.1007/BF02659652
18. Ahmed M, Pickova J, Ahmad T, Liaquat M, Farid A, Jahangir M. Oxidation of lipids in foods. Sarhad J Agric. 2016;32(3):230–238. doi:10.17582/journal.sja/2016.32.3.230.238
19. Orgeig S, Morrison JL, Daniels CB. Effect of environment and aging on the pulmonary surfactant system. In: Harding R, Pinkerton KE, editors. The Lung. New York: Elsevier; 2014:447–469.
20. Krpetić Ž, Anguissola S, Garry D, Kelly PM, Dawson KA. Nanomaterials: impact on cells and cell organelles. In: Capco DG, Chen Y, editors. Nanomaterial: Impacts on Cell Biology and Medicine. New York: Springer; 2014:135–156.
21. Liu X, Cui C, Zhao M, et al. Identification of phenolics in the fruit of emblica (Phyllanthus emblica L.) and their antioxidant activities. Food Chem. 2008;109(4):909–915.
22. Bangham A, Standish MM, Watkins JC. Diffusion of univalent ions across the lamellae of swollen phospholipids. J Mol Biol. 1965;13(1):238–252. doi:10.1016/S0022-2836(65)80093-6
23. Vandana V, Karuna MS, Vijayalakshmi P, Prasad RB. A simple method to enrich phospholipid content in commercial soybean lecithin. J Am Oil Chem Soc. 2001;78(5):555–556. doi:10.1007/s11746-001-0303-2
24. Stewart JCM. Colorimetric determination of phospholipids with ammonium ferrothiocyanate. Anal Biochem. 1980;104(1):10–14. doi:10.1016/0003-2697(80)90269-9
25. John B, Sulaiman C, George S, Reddy V. Total phenolics and flavonoids in selected medicinal plants from Kerala. Int J Pharm Pharm Sci. 2014;6(1):406–408.
26. Ardhammar M, Lincoln P, Nordén B. Nonlinear partial differential equations and applications: invisible liposomes: refractive index matching with sucrose enables flow dichroism assessment of peptide orientation in lipid vesicle membrane. Proc Natl Acad Sci. 2002;99(24):15313–15317. doi:10.1073/pnas.192583499
27. Blessing T, Remy J-S, Behr J-P. Monomolecular collapse of plasmid DNA into stable virus-like particles. Proc Natl Acad Sci. 1998;95(4):1427–1431. doi:10.1073/pnas.95.4.1427
28. Makra I, Terejánszky P, Gyurcsányi RE. A method based on light scattering to estimate the concentration of virus particles without the need for virus particle standards. MethodsX. 2015;2:91–99. doi:10.1016/j.mex.2015.02.003
29. Woff S. Ferrous ion oxidation in presence of ferric ion indicator xylenol orange for the measurement of hydroperoxide. Methods Enzymol. 1994;233:182–189.
30. Shimamura T, Tairabune T, Kogo T, et al. Investigation of the release test method for the topical application of pharmaceutical preparations: release test of cataplasm including nonsteroidal anti-inflammatory drugs using artificial sweat. Chem Pharm Bull. 2004;52(2):167–171. doi:10.1248/cpb.52.167
31. Zhang Y, Huo M, Zhou J, et al. DDSolver: an add-in program for modeling and comparison of drug dissolution profiles. AAPS J. 2010;12(3):263–271. doi:10.1208/s12248-010-9185-1
32. International Organization for Standardization. Biological Evaluation of Medical Devices. Part 5: Tests for in vitro Cytotoxicity. Geneva: International Organization for Standardization; 2009:10993–10995.
33. Sage D, Donati L, Soulez F, et al. DeconvolutionLab2: an open-source software for deconvolution microscopy. Methods. 2017;115:28–41. doi:10.1016/j.ymeth.2016.12.015
34. Lucy LB. An iterative technique for the rectification of observed distributions. Astron J. 1974;79:745–754. doi:10.1086/111605
35. Richardson WH. Bayesian-based iterative method of image restoration. J Opt Soc Am. 1972;62(1):55–59. doi:10.1364/JOSA.62.000055
36. Bolte S, Cordelières FP. A guided tour into subcellular colocalization analysis in light microscopy. J Microsc. 2006;224(3):213–232. doi:10.1111/j.1365-2818.2006.01706.x
37. Stauffer W, Sheng H, Lim HN. EzColocalization: an ImageJ plugin for visualizing and measuring colocalization in cells and organisms. Sci Rep. 2018;8(1):15764. doi:10.1038/s41598-018-33592-8
38. Li Q, Lau A, Morris TJ, Guo L, Fordyce CB, Stanley EF. A syntaxin 1, Gαo, and N-type calcium channel complex at a presynaptic nerve terminal: analysis by quantitative immunocolocalization. J Neurosci. 2004;24(16):4070–4081. doi:10.1523/JNEUROSCI.0346-04.2004
39. Sheng H, Stauffer W, Lim HN. Systematic and general method for quantifying localization in microscopy images. Biol Open. 2016;5(12):1882–1893. doi:10.1242/bio.019893
40. Spurr AR. A low-viscosity epoxy resin embedding medium for electron microscopy. J Ultrastruct Res. 1969;26(1–2):31–43. doi:10.1016/S0022-5320(69)90033-1
41. Conover WJ. Practical Nonparametric Statistical.
42. Wobbrock JO, Findlater L, Gergle D, Higgins JJ. The aligned rank transform for nonparametric factorial analyses using only ANOVA procedures. Proc SIGCHI Conf Hum Factor Comput Syst. 2011:143–146.
43. Mozafari M. Nanoliposomes: preparation and analysis. In: Weissig V, editor. Liposomes: Methods and Protocols, Volume 1: Pharmaceutical Nanocarriers. New York: Springer; 2010:29–50.
44. Gomes A, Fernandes E, Lima JL. Fluorescence probes used for detection of reactive oxygen species. J Biochem Biophys Methods. 2005;65(2–3):45–80. doi:10.1016/j.jbbm.2005.10.003
45. Sangster J, Databank LOGKOW. A databank of Evaluated Octanol-Water Partition Coefficients (Log P) on Microcomputer Diskette. Montreal, Quebec, Canada: Sangster Research Laboratories; 1993.
46. Riddick TM. Control of Colloid Stability Through Zeta Potential. Wynnewood, PA: Livingston Publishing Co; 1968.
47. Adeleye OA, Femi-Oyewo MN, Odeniyi MA. Effect of compression pressure on mechanical and release properties of tramadol matrix tablets. Curr Issues Pharm Med Sci. 2015;28(2):120–125. doi:10.1515/cipms-2015-0057
48. Lucio D, Martínez-Ohárriz MC, Gu Z, et al. Cyclodextrin-grafted poly (anhydride) nanoparticles for oral glibenclamide administration. In vivo evaluation using C. elegans. Int J Pharm. 2018;547(1–2):97–105. doi:10.1016/j.ijpharm.2018.05.064
49. Shain SA. A note on multicomponent diffusion. AiChE J. 1961;7(1):17–19.
50. Wang F, Gómez‐Sintes R, Boya P. Lysosomal membrane permeabilization and cell death. Traffic. 2018;19(12):918–931. doi:10.1111/tra.12613
51. Yang G, Tian J, Chen C, et al. An oxygen self-sufficient NIR-responsive nanosystem for enhanced PDT and chemotherapy against hypoxic tumors. Chem Sci. 2019;10(22):5766–5772.
52. Liu C-G, Han Y-H, Kankala RK, Wang S-B, Chen A-Z. Subcellular performance of nanoparticles in cancer therapy. Int J Nanomedicine. 2020;15:675–704. doi:10.2147/IJN.S226186
53. Luby-Phelps K. Preparation of fluorescently labeled dextrans and ficolls. In: Wang Y-L, Taylor DL, editors. Methods in Cell Biology: volume 29 Fluorescence Microscopy of Living Cells in Culture, Part A: Fluorescent Analogs, Labeling Cells and Basic Microscopy. California: Academic Press; 1989:59–73.
54. Scriven DR, Lynch RM, Moore ED. Image acquisition for colocalization using optical microscopy. Am J Physiol Cell Physiol. 2008;294(5):C1119–C1122. doi:10.1152/ajpcell.00133.2008
55. van Steensel B, van Binnendijk EP, Hornsby CD, et al. Partial colocalization of glucocorticoid and mineralocorticoid receptors in discrete compartments in nuclei of rat hippocampus neurons. J Cell Sci. 1996;109(4):787–792.
56. Pearson K
57. Manders E, Verbeek F, Aten J. Measurement of co-localization of objects in dual‐colour confocal images. J Microsc. 1993;169(3):375–382. doi:10.1111/j.1365-2818.1993.tb03313.x
58. Papadopoulos C, Kravic B, Meyer H. Repair or lysophagy: dealing with damaged lysosomes. J Mol Biol. 2020;432(1):231–239. doi:10.1016/j.jmb.2019.08.010
59. Eriksson I, Wäster P, Öllinger K. Restoration of lysosomal function after damage is accompanied by recycling of lysosomal membrane proteins. Cell Death Dis. 2020;11(5):1–16. doi:10.1038/s41419-020-2527-8
60. Zhao Y Quantum dots and doped nanocrystals: synthesis, optical properties and bio-applications [PhD Thesis]. Utrecht University; 2013.
61. Zana R, Yiv S, Kale K. Chemical relaxation and equilibrium studies of association in aqueous solutions of bolaform detergents. 3. Docosane-1, 22-bis (trimethylammonium bromide). J Colloid Interface Sci. 1980;77(2):456–465.
62. Lasic DD. Liposomes: From Physics to Applications. New York: Elsevier; 1993.
63. Ho S, Huang L. A novel cytochemical marker for liposome decomposition in lysosomes. J Histochem Cytochem. 1983;31(3):404–410. doi:10.1177/31.3.6827078
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.