Back to Journals » Open Access Rheumatology: Research and Reviews » Volume 13
Incidence and Clinical Pattern of Mixed Connective Tissue Disease in Sudanese Patients at Omdurman Military Hospital: Hospital-Based Study
Authors Abdelgalil Ali Ahmed S, Adam Essa ME ![]() , Ahmed AF, Elabib EM, Eltahir NIA, Awadallah H, Hassan A, khair ASM, Ebad MAB
, Ahmed AF, Elabib EM, Eltahir NIA, Awadallah H, Hassan A, khair ASM, Ebad MAB
Received 23 August 2021
Accepted for publication 30 November 2021
Published 9 December 2021 Volume 2021:13 Pages 333—341
DOI https://doi.org/10.2147/OARRR.S335206
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Chuan-Ju Liu
Sulafah Abdelgalil Ali Ahmed,1,2 Mohammed Elmujtba Adam Essa,3,4 Amar F Ahmed,5 Elnour Mohammed Elagib,2,6 Noha Ibrahim Ahmed Eltahir,2,6 Huyam Awadallah,7 Abubakr Hassan,3 Amna Sirag Mohammed khair,8 Mustafa Abdalla bakhit Ebad4
1Department of Medicine, Faculty of Medicine, Ahfad University, Khartoum, Sudan; 2Department of Rheumatology, Omdurman Military Hospital, Khartoum, Sudan; 3Department of Clinical Medicine, Medical and Cancer Research Institute, Nyala, Sudan; 4Faculty of Medicine, Al Fashir University, Al Fashir, Sudan; 5Department of Internal Medicine, Wayne State University, Detroit, MI, USA; 6Department of Medicine, Faculty of Medicine, Karrary University, Khartoum, Sudan; 7Department of Internal Medicine, Detroit Medical Centre, Detroit, MI, USA; 8Department of Medicine, Sudan Medical Specialization Board, Khartoum, Sudan
Correspondence: Mohammed Elmujtba Adam Essa
Department of Clinical Medicine, Medical and Cancer Research Institute, Nyala, Sudan
Tel +24990700938
Email [email protected]
Background: Mixed connective tissue disease (MCTD) is a rare autoimmune disease, characterized by the production of specific autoantibody anti-RNP, which presents with varied overlapping symptoms of different connective tissue disorders. The aim of this study is to identify the frequency and patterns of MCTD.
Methods: This is a descriptive cross-sectional hospital-based study conducted at the rheumatology clinic at Omdurman Military Hospital between February 2019 and July 2019. The study included 30 patients and data were collected using a designated questionnaire.
Results: The study showed that the majority of patients (96.7%) were females and only 3.3% was male. About 30% of the patients aged between 30 and 39 years were the most affected. As a first diagnosis, 10% of the patients had a MCTD fulfilling the Alarcon-Segovia criteria. The remaining 90% of the patients were diagnosed with other diseases before evolving into MCTD. The most common clinical presentation was arthralgia in 100% of the patients, 90% were symmetrically followed by myositis in 70% of the patients, arthritis in 63.3% of the patients, puffy fingers in 63.3% of the patients, and hand swelling in 60% as major musculoskeletal symptoms. Regarding the initial results in immunological profile, the most common positive autoantibodies among the patients were anti-RNP titer in 96.7% of the patients, ANA in 90%, anti-Sm in 50%, RF in 50%, anti-Ds DNA in 46.7%, and anti-Ro in 43.3%.
Conclusion: This study showed that MCTD is more common in females, only 10% of patients presented with a fulfilling criteria of the disease at diagnosis, and the rest of the patients presented with other rheumatologic diseases before evolving into MCTD.
Keywords: MCTD, females, arthralgia, anti-RNP titer
Introduction
Mixed connective tissue disease (MCTD), also known as Sharp´s syndrome, was originally defined in 1972 as a connective tissue disorder characterized by the presence of high titers of a distinctive autoantibody, now called anti-U1 RNP. MCTD is an autoimmune condition with overlapping of at least two connective tissue diseases, including Systemic Lupus erythematosus (SLE), Scleroderma and Polymyositis (PM), and rheumatoid arthritis.1 The distinctive overlap features commonly appear sequentially over time.2
MCTD occurs worldwide and in all races, with a peak incidence in adolescence and the 20s. About 80% of people who have the disease are women.3 The cause of MCTD is unknown.4 The natural history and outcome of MCTD patients are not well-characterized as preceding reports yielded inconsistent results. One study reported that more than 50% of patients with MCTD evolved to either SS or SLE, while a subsequent study found such evolution in only 13% of their cohort.5,6
There is very little information available regarding the prevalence and incidence of MCTD.7 In a 2011 nationwide study in Norway, the prevalence of MCTD was 3.8 per 100,000 adults, with an incidence of 2.1 million per year. Frequently the first manifestations resemble early SLE, Systemic scleroderma, Polymyositis, or even Rheumatoid arthritis, with many patients appearing to have an undifferentiated connective tissue disease initially.8 The early clinical features of MCTD are non-specific and may consist of general malaise, arthralgia, myalgia, and low-grade fever. Raynaud phenomenon may precede other manifestations by years. Swollen hands and puffy fingers are typical, skin findings include lupus or dermatomyositis-like rashes, and diffuse scleroderma-like skin changes may develop.9
Almost all patients have polyarthralgia and 75% have frank arthritis, with proximal muscle weakness being common. The absence of renal disease is a hallmark of MCTD1, although some degree of membranous nephropathy occurs in 25% of patients. Disorders motility in the upper gastrointestinal tract is the most common overlap feature with scleroderma. Interstitial lung disease is the most common lung manifestation; pulmonary hypertension is a major cause of death. A high titer speckled ANA whose fine specificity is anti-U1 RNP is the diagnostic serological finding in MCTD.10 The management of MCTD generally rests upon the known effectiveness of specific therapies for similar problems seen in SLE, Scleroderma, or Polymyositis.10,11
Among patients requiring long-term glucocorticoids, Hydroxychloroquine or Methotrexate is reasonable. Intravenous immunoglobulin (IVIG) may also have a role in patients with resistant thrombocytopenia or severe eruptive skin disease. The overall 10-year survival rate is about 80%, but the prognosis depends largely on which manifestations predominate.12,13 The aim of this study is to identify the frequency and patterns of mixed connective tissue disease to characterize his epidemiology in Sudanese patients.
Methodology
This is a descriptive cross-sectional hospital-based study, conducted at the rheumatology department at Omdurman Military hospital, Khartoum, Sudan, for 6 months periods between February to July 2019. The study included all adult patients diagnosed with MCTD who presented during the period of study at the hospital. The data was collected by using a designated questioner containing all adult patients who are confirmed to have MCTD fulfilling Alarcon-Segovia criteria which are as follow: A) Serological criteria; Anti-RNP antibodies with a hemagglutination titer of ≤1:1,600. B) Clinical criteria; swollen hands, Synovitis, Myositis, Raynaud’s phenomenon, acrosclerosis. Information also includes the received treatment, such as Prednisolone, HCQ, DMARD, Methotrexate, Azathioprine, Vitamin D, Warfarin, Lasix, Sildenafil, Osteocare, Omeprazole, Folic acid, and IVIG. Autoantibody tests and inflammatory markers such as ANA profile, RF, Anti-CCP, Anti-cardiolipin, ESR, CRP, hematological findings, associated complications (autoimmune thyroiditis, IHD, PAH, Sjogren’s syndrome, HTN and DM). Initial cutaneous symptoms were lung and musculoskeletal manifestations. Age was split into five groups (between 18–29 years, 30–39 years, 40–49 years, 50–59 years, and above 60 years), and geographical distributionswere the western, eastern, northern, and central states of Sudan.
MCTD is present if criterion A is accompanied by three or more clinical criteria, one of which must include synovitis or myositis. Total coverage of MCTD patients presented in the study area due to the rarity of the condition. The final sample-size obtained during the study period was 30 patients who fulfilled the inclusion criteria of the study. The questionnaire was completed directly with the patients by a clinician for obtaining basic information, and results of investigations were obtained from the files of the patients.
Statistical Analysis
Data was analyzed by the Statistical Packages for Social Sciences (SPSS) 21 software which was used to analyze all the collected data of MCTD as percentages, categories, and p-values for statistical significance testing. Evaluation data were analyzed by one-way analysis of variance (referred to as ANOVA) to test the relationship between the different variables and means. The level of statistical significance was set as p<0.05.
Ethical Considerations
Ethical clearance was obtained from SMSB Ethical Committee, and written consent was obtained from the hospital administration and from the patients.
Results
The majority of the patients (96.7%) were female and only 3.3% were male. The study shows that 30% were aged between 30–39 years, 26.6% were aged 40–49 years, 16.7% were aged 60 years and above, 16.7% were aged 18–29 years, and the remaining 10% were aged 30–39 years (Figure 1). Regarding the geographical distribution of the patients, 43.3% of them were from northern states, 26.7% from central states, 26.7% from western states, and only 3.3% from eastern states (Figure 2). The duration of illness was less than 5 years for 76.6% of the patients, while 16.7% were between 5–10 years, and the remaining 6.7% of the patients were more than 10 years. Arthralgia occurred in all patients as an initial symptom, followed by skin rash and fatigability in 13.3% and 10%, respectively; other symptoms are described in Table 1. Initially only 10% of the patients presented with MCTD fulfilling the Alarcon criteria, the remaining patients presented sequentially from other diseases as a first diagnosis, only 40% diagnosed as SLE, 26.7% diagnosed as RA, 2.7% diagnosed as SS, 2.7% diagnosed as SLE PM, 6.7% diagnosed as SS RA, and 3.3% diagnosed as PM (Table 2). Fatigue manifested as a constitutional symptom in all patients, fever in 63.3% and weight loss in 63.3% (Table 3). In musculoskeletal symptoms, joint pain manifested in all patients who were symmetrical in 90%, with joint stiffness in 73.3%, followed by arthritis in 63.3%, puffy fingers in 63.3%, and hand swelling in 60% as major symptoms; others are shown in Table 4. Myositis manifestations were muscle pain in 70%, muscle weakness in 60%, muscle tenderness in 56.7%, and muscle wasting in 10% (Table 5). The reported vascular symptoms were Raynaud’s phenomenon in 10% and digital gangrene in another 10% of the patients. Vascular symptoms did not manifestin 80% of the patients. The most common cutaneous symptoms were erythematous rash in half of the patients, skin tightness in 23.3%, and sclerodactyly in 20%. Other symptoms are shown in Table 6. The reported GIT symptoms were heartburn in 56.7% of all the patients and dysphagia in 10%. GIT symptoms were not reported in 33.3% of all patients. Lung manifestations that occurred among the studied patients were SOB 9 in 30%, dry cough in 30%, and accentuated P2 in 3.3%; with 36.7% of the patients not reporting (Figure 3).
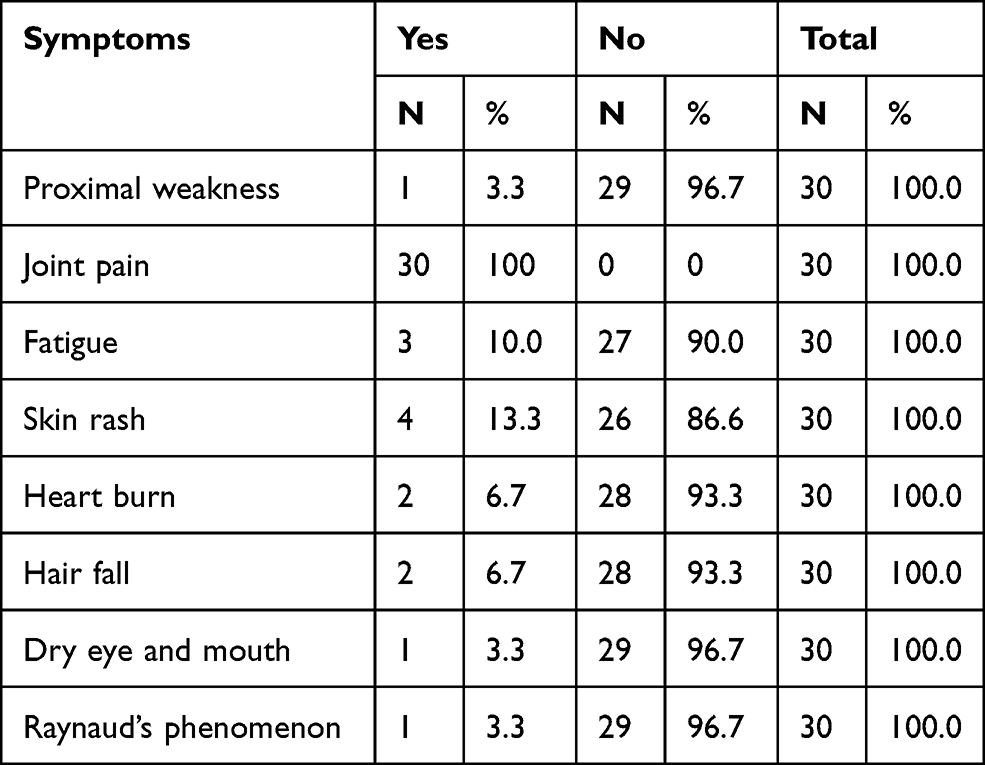 |
Table 1 The First Associated Symptoms which Occurred in the Patients |
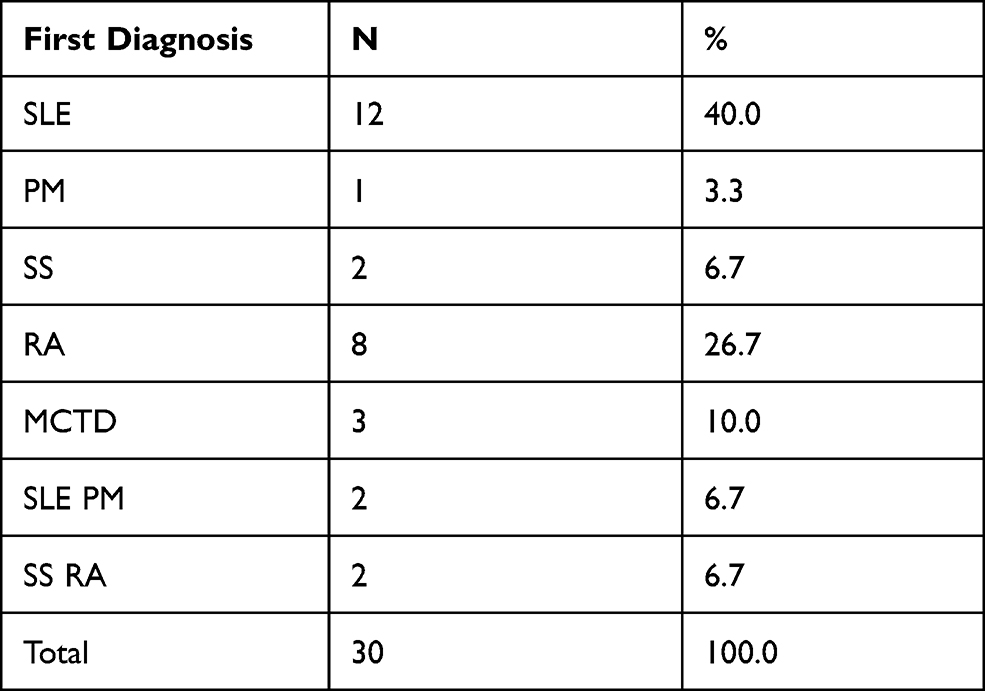 |
Table 2 The Patients Distribution According to First Diagnosis |
 |
Table 3 The Patients Constitutional Symptoms |
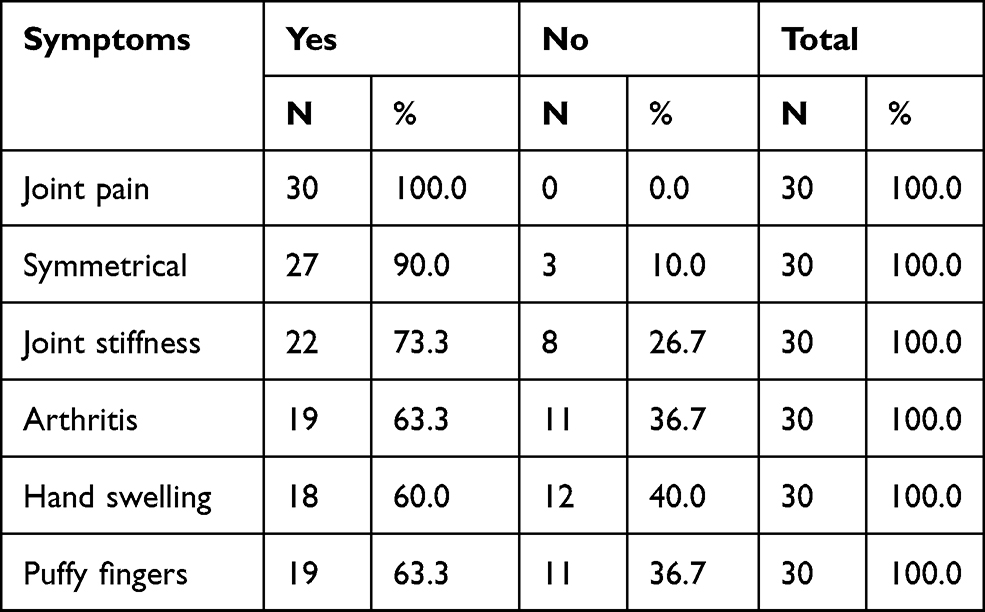 |
Table 4 The Musculoskeletal Symptoms on the Patients |
 |
Table 5 The Muscles Manifestations of the Patients |
 |
Table 6 The Cutaneous Symptoms Associated with MCTD in the Patients |
 |
Figure 1 Age groups of the patients. |
 |
Figure 2 Geographical distribution of the patients. |
 |
Figure 3 Distribution of the patients according to lung manifestations. |
Only two neurological symptoms were reported, which were a headache in 46.7% and peripheral neuropathy in 13.3%. In the remaining 40% of the patients, no neurological manifestations were reported. The associated complications were autoimmune thyroiditis, which was described in 16.7%, PAH in 10%, Sjogren syndrome in 10%, HTN in 10%, IHD in 6.7%, and DM in 3.3%. In 43.3% of all the patients, no associated complications were reported (Table 7). Forty percent of the patients reported a family history association. High TWBC was reported in only 3.3% of the patients, low Hb in 73.3%, and low platelet count in 13.3%.
 |
Table 7 The Associated Complications |
In 53.4% of the patients, ESR was raised, while positive CRP was reported in only 20% of the patients (Table 8). Urine analysis showed 76.7% were clear, while the remaining 23.3% indicated the opposite.
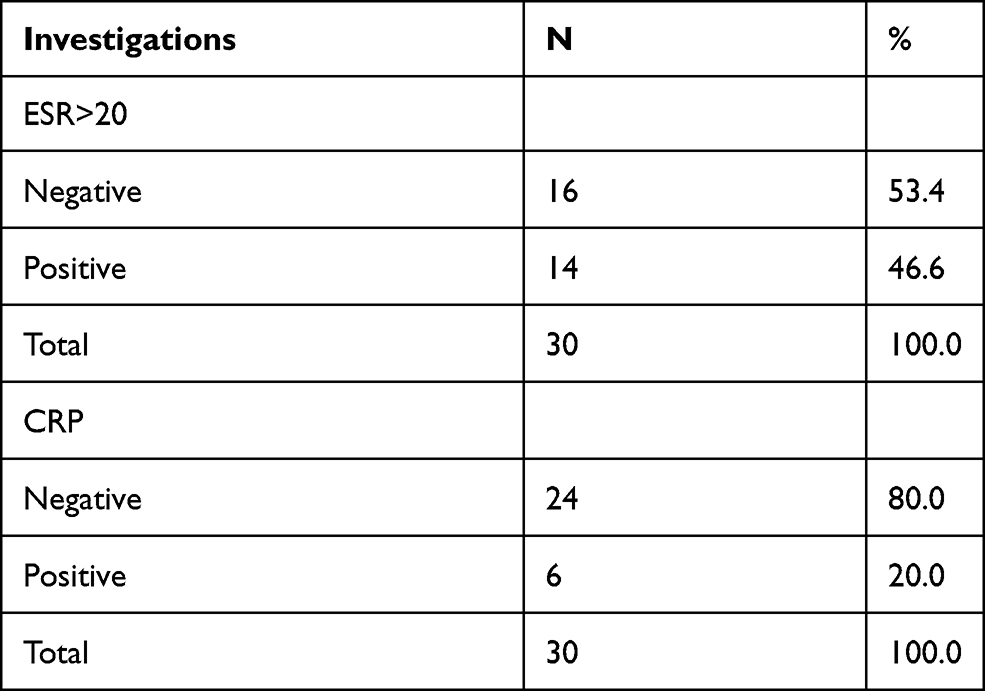 |
Table 8 The Percentage of Inflammatory Markers in the Patients |
The most common positive autoantibody was an Anti-RNP titer, which was seen in 96.7%, followed by an ANA in 90% of patients, Anti-SM and RF both in 50%, Anti-Ds DNA was reported in 46.7%, and Anti-Ro in 43.3%. Other antibodies are shown in Table 9. Regarding the treatment regimens, drugs given to treat patients were Prednisolone in 93.3%, Hydroxychloroquine (HCQ) in 80%, and DMARDS were given in 63.3%. Out of the DMARDS, azathioprine was prescribed to 43.3% of all patients and 20% were given Methotrexate. Other drugs are shown in Table 10.
 |
Table 9 The Autoantibody Tests Performed on the Patients |
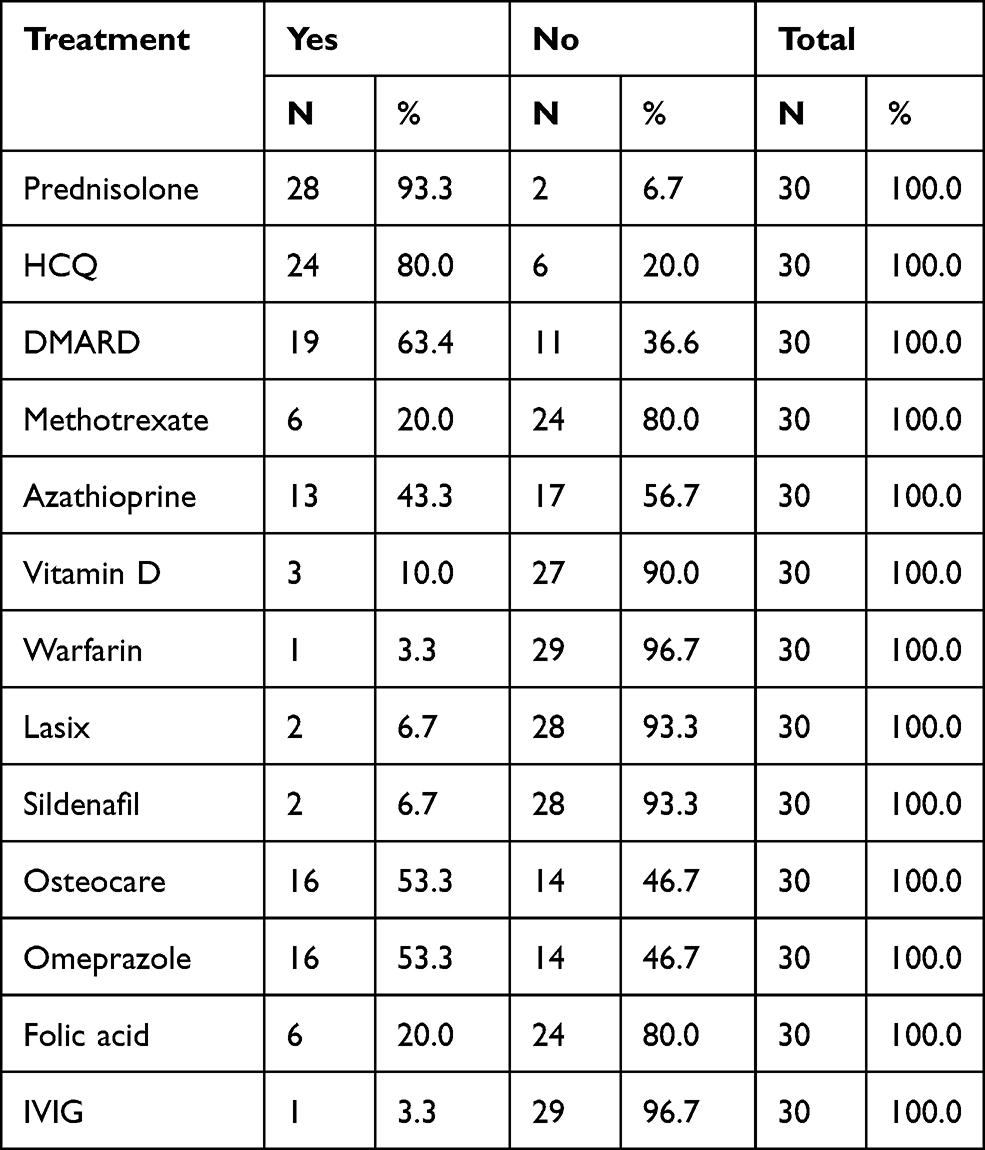 |
Table 10 Treatment given to the patients |
Discussion
MCTD is a rare disorder and presents with varied overlapping manifestations of different connective tissue disorders.14 Many patients evolve into other connective tissue disorders with the passage of time. In this study, MCTD has been investigated in 30 Sudanese patients at Omdurman Military Hospital during the period from February 2019 to July 2019 in the rheumatology clinic patients, according to Alarcon-Segovia criteria.
The majority of the patients (96.7%) were females and only 3.3% were male. Thirty percent of all the patients aged were between 30–39 years with a mean age of 34.5 years. This is comparable to previous studies, for example a study in Karachi, Pakistan reported that among patients with MCTD 80% were females and 20% of patients were males. The mean age was 30.5 years.15 A study in Gabon showed that seven patients with mixed connective tissue disease were women (100%), with an average age of 39.5 years.16 The most common clinical presentation was arthralgia in all the patients, which were symmetrical in 90% of the patients, followed by arthritis in 63.3%, puffy fingers in 63.3%, and hand swelling in 60% as major musculoskeletal symptoms. A previous study by Amigues et al reported that about 60% of patients complain of arthralgia.17 This is comparable to a study in the Philippines in which the chief complaint was most commonly joint pain, at 67%.18,19
The first diagnosis in 10% had a full blown picture of MCTD (SLE, SS, DM), is similar to a study in Minnesota, USA which reported that the annual incidence of MCTD was 1.9 per 100,000 population.20 Our study showed that 40% evolved from SLE, 26.7% evolved from RA, 6.7% from SS, and 3.3% evolved from PM to MCTD, similar to a study in Pakistan which found that, over the disease course of 6 years, 10% of patients evolved into SLE. Also this is comparable to the Minnesota study, in which evolution from other connective tissue diseases occurred at 8.5% and 6.3% for SLE and SS, respectively.7
Fatigue manifested as a constitutional symptom in all patients (100%) in our study followed by fever in 63.3% and weight loss in 63.3%. This is comparable to a study in Filipinos reporting that the majority had initial constitutional symptoms, with generalized weakness and fatigue being the most frequent, in 93%.21
Myositis manifestations were muscle pain (70%), muscle weakness (60%), muscle tenderness (56.7%), and muscle wasting (10%). This finding is similar to Aringer and Smolen which reported that two third of MCTD have overt myositis ranging from mild to severe.22
The study showed that the reported vascular symptoms were Raynaud’s phenomenon in 10% and digital gangrene in another 10% of the patients. This was in contrast to a study in Minnesota, USA which aimed to characterize the epidemiology of MCTD from 1985 to 2014, based on comprehensive individual medical records. Raynaud's phenomenon was the most common initial symptom, in 50%, followed by arthralgia in 30%, and swollen hands in 16%.23 This study is similar to a study in a hospital population in Gabon, reporting that articular manifestations included polyarthritis, myalgias, chubby fingers, and Raynaud’s.24
In this study, the most common cutaneous symptoms were erythematous rash (50%), skin tightness (23.3%), and sclerodactyly (20%). This is similar to a study in Filipinos which aimed to present the clinical pattern in 14 patients with MCTD. The chief complaint was most commonly joint pain in 67% followed by skin tightness in 13%.
The reported GIT symptoms in the study were heartburn (56.7%) and dysphagia (10%). A comparable study by Alarcon et al showed that dysphagia and dysfunction of esophageal motility resemble those occurring in systemic sclerosis. Other abnormalities of the gut include esophagitis, constipation, diarrhea, and malabsorption.25
Our study revealed that lung manifestations among the studied patients were SOB in 30%, dry cough in 30%, and accentuated P2 in 3.3%. As compared to a study in Gabon which described the clinical features of the disease over a period of 6 years, out of seven patients a case of death due to pulmonary arterial hypertension (PAH) was certified.26
The study shows regarding immunological profile, the most common positive autoantibody among the patients were Anti-RNP titer 29 (96.7%), ANA (90%), Anti-SM (50%), RF (50%), Anti-Ds DNA (46.7%), and Anti-Ro (43.3%), in contrast to other studies the study revealed that anti-RNP titer (96.7%) is higher than ANA (90%). A comparable study in Filipinos showed that, for serological studies, all patients with MCTD have speckled ANA and very high titers of anti-U1 RNP.27 Another study in Pakistan showed that patients had positive ANA and anti-RNP antibodies.28
Our study showed 50% of patients are positive for RF and anti-CCP positive in 33.3% of them. This is comparable to Aringer and Smolen, who reported that arthritis in MCTD has a possible association with RF and anti-CCP. Whereas RF was positive in 30–100% of MCTD patients, anti-CCP was found in only 9%. Anti-U1RNP may be a predictor of more aggressive erosive arthritis.29,30
The common drugs given for treatment were Prednisolone, Hydroxychloroquine (HCQ), Azathioprine, and Methotrexate, in 93.3%, 80%, 43.3%, and 20% of the patients, respectively. A similar study in Filipinos showed that most patients with MCTD were in remission on a low dose of prednisolone (79%), hydroxychoroquine (50%), nifedipine (36%), and methotrexate (21%).31
The geographical distributions of the patients showed that most of the patients come from the Northern states of Sudan (43% of all the patients), which contains six localities (Dongola, Merowe, Wadi Halfa, Al Dabbah, Delgo, Al Goled, and Al Burgaig). According to the last estimate in 2006 the population numbere 833,743. Central and western states both have an equal percentage of the total number of patients, which is 26.7%, followed by 3.3% from the eastern states. Central states of Sudan include Khartoum, which is the capital and the largest and most densely populated city in Sudan. Western states are the five main states known by Darfur regions, the eastern states are the region of Sudan lying to the east of Khartoum, along the Blue Nile.
Conclusion
The study showed that only 30 patients out of those attending the rheumatology clinic during the study period were fulfilling the Alarcon–Segovia’s criteria for MCTD, revealing that the disease is rare among Sudanese patients. The disease is most common in females than males, mainly among the age group 30–39 years. The most common positive auto-antibodies initially were anti-RNP titer and ANA. Only 10% of the patients presented with full blown manifestations of MCTD, the remaining patients sequentially presented with other rheumatological diseases initially before evolving to MCTD presenting as SLE in 40% of all the patients, RA in 26.7% of the patients, and PM in 3.3% in decreasing order of frequency. The common clinical presentations were arthralgia, followed by arthritis, puffy fingers, hand swelling, and myositis, presenting as muscle pain and weakness mainly.
Abbreviations
Anti-CCP, Anti-Cyclic Citrullinated Peptide; ANA, Anti-nuclear antigen; Anti-Sm, Anti-Smith antibody; Anti-Ds DNA, Anti-double stranded deoxyribonucleic acid; CBC, Complete Blood Count; CI, confidence index; CK, creatine kinase; CNS, Central Nervous System; CT Scan, Computerized Tomography Scan; CRP, C-reactive protein; DMARD, Disease Modifying Anti-Rheumatic Drugs; DM, Diabetes Mellitus; DM, Dermatomyositis; ECG, Electrocardiography; EDC, Education and Development Center; ESR, Erythrocyte Sedimentation Rate; EULAR, European League Against Rheumatism; FUO, fever of unknown origin; GIT, Gastro Intestinal Tract; HCQ, Hydroxychloroquine; Hb, Hemoglobin; HTN, Hypertension; HLA, Human Leukocyte Antigen; HR, hazard ratio; IgG, Immunoglobulin G; IVIG, Intravenous immunoglobulin; ILD, Interstitial Lung Disease; IHD, Ischemic Heart Disease; JPMC, Jinnah Postgraduate Medical Clinic; MCV, Mean Corpuscular Volume; MCH, Mean Corpuscular Hemoglobin; MI, Myocardial Infarction; MCTD, Mixed Connective Tissue Disease; P2, second pulmonary heart sound; PAH, pulmonary arterial hypertension; PM, Polymyositis; RNP, ribonucleoprotein; RF, Rheumatoid Factor; RFT, Renal Function Test; SSc, Systemic Sclerosis; SLE, Systemic Lupus Erythematosus; SPSS, Statistical Packages for Social Sciences; SS, Systemic sclerosis; TWBC, Total White Blood Count; UG, Urine General; USA, United States of America; GIT, Gastrointestinal Tract; SOB, shortness of the breath; PAH, Pulmonary arterial hypertension; HTN, hypertension; DM, Diabetes Mellitus; TWBC, total white blood cells.
Ethical Approval and Consent to Publish
Obtained from Sudan Federal ministry of health.
Written Consent from the Patients
Signed consent was obtained from all the patients for the publication of this study who was informed about the purpose of the study, which complies with the Declaration of Helsinki.
Disclosure
All authors declare no conflicts of interest in this work.
References
1. Marwa K, Anjum F. Undifferentiated Connective Tissue Disease. Treasure Island (FL): StatPearls; 2021.
2. Radin M, Rubini E, Cecchi I, et al. Disease evolution in a long-term follow-up of 104 undifferentiated connective tissue disease patients. Clin Exp Rheumatol. 2021. PubMed PMID: 34251309.
3. Yoo H, Hino T, Han J, et al. Connective tissue disease- related interstitial lung disease (CTD-ILD) and interstitial lung abnormality (ILA): evolving concept of CT findings, pathology and management. Eur J Radiol Open. 2021;8:100311. PubMed PMID: 33364263. Pubmed Central PMCID: PMC7750149. doi:10.1016/j.ejro.2020.100311
4. Miao C, Wang X, Zhou W, Huang J. The emerging roles of exosomes in autoimmune diseases, with special emphasis on microRNAs in exosomes. Pharmacol Res. 2021;169:105680. PubMed PMID: 34010670. doi:10.1016/j.phrs.2021.105680
5. Burdt MA, Hoffman RW, Deutscher SL, Wang GS, Johnson JC, Sharp GC. Long-term outcome in mixed connective tissue disease: longitudinal clinical and serologic findings. Arthritis Rheum. 1999;42(5):899–909. PubMed PMID: 10323445. doi:10.1002/1529-0131(199905)42:5<899::AID-ANR8>3.0.CO;2-L
6. Fernandez-Gutierrez B, Leon L, Madrid A, et al. Hospital admissions in inflammatory rheumatic diseases during the peak of COVID-19 pandemic: incidence and role of disease-modifying agents. Ther Adv Musculoskelet Dis. 2021;13:1759720X20962692. PubMed PMID: 33613703. Pubmed Central PMCID: PMC7869066. doi:10.1177/1759720X20962692
7. Ungprasert P, Crowson CS, Chowdhary VR, Ernste FC, Moder KG, Matteson EL. Epidemiology of mixed connective tissue disease, 1985–2014: a Population-Based Study. Arthritis Care Res. 2016;68(12):1843–1848. PubMed PMID: 26946215. Pubmed Central PMCID: PMC5426802. doi:10.1002/acr.22872
8. Hetlevik SO, Flato B, Rygg M, et al. Long-term outcome in juvenile-onset mixed connective tissue disease: a nationwide Norwegian study. Ann Rheum Dis. 2017;76(1):159–165. PubMed PMID: 27283334. doi:10.1136/annrheumdis-2016-209522
9. Latuskiewicz-Potemska J, Zygmunt A, Biernacka-Zielinska M, Stanczyk J, Smolewska E. Mixed connective tissue disease presenting with progressive scleroderma symptoms in a 10-year-old girl. Postepy Dermatol Alergol. 2013;30(5):329–336. PubMed PMID: 24353496. Pubmed Central PMCID: PMC3858664. doi:10.5114/pdia.2013.38365
10. Chen R, Wang J, Xie Q, Xue J, Hao C. Sjogren’s syndrome complicated with membranous nephropathy, a cause or coincidence? Int J Rheum Dis. 2021;24(8):1086–1347. PubMed PMID: 34223708. doi:10.1111/1756-185X.14168
11. Hanset N, Ronco P, Plaisier E. [Membranous nephropathy]. Rev Prat. 2021;71(1):85–89. French. PubMed PMID: 34160953.Glomerulonephrite extramembraneuse.
12. Nishizawa K, Yamashita T, Ogawa Y, Kobayashi H. Membranous nephropathy complicated by immune thrombocytopenia treated with low-density lipoprotein apheresis: a case report and literature review. CEN Case Rep. 2021. PubMed PMID: 34287815. doi:10.1007/s13730-021-00630-w
13. Koeks Z, Bladen CL, Salgado D, et al. Clinical outcomes in Duchenne muscular dystrophy: a study of 5345 patients from the TREAT-NMD DMD global database. J Neuromuscul Dis. 2017;4:
14. Ahsan T, Erum U, Dahani A, Khowaja D. Clinical and immunological profile in patients with mixed connective tissue disease. J Pak Med Assoc. 2018;68(6):959–962. PubMed 359 PMID: 30323370.
15. Nedumaran RCP, Porkodi R, Parthiban R. Mixed connective tissue disease– clinical and immunological profile. J Assoc Physicians India. 2001;49:412–414. PubMed 362 PMID: 11762609.
16. John KJ, Sadiq M, George T, et al. Clinical and immunological profile of mixed connective tissue disease and a comparison of four diagnostic criteria. Int J Rheumatol. 2020;2020:9692030. PubMed PMID: 32411251. Pubmed Central PMCID: PMC7204172. doi:10.1155/2020/9692030
17. Wiefel K, Aringer M. [Polyarthritis - from symptoms to diagnosis]. Polyarthritiden – vom Symptom zur Diagnose. Dtsch Med Wochenschr. 2021;146(9):582–590. German. PubMed PMID: 33931836. doi:10.1055/a-1294-1205
18. Michotte L. [The differential diagnosis of polyarthritis]. Le diagnostic differentiel des polyarthrites. Acta Physiother Rheumatol Belg. 1947;2(1):11–20. Swedish. PubMed PMID: 20247629.
19. Alpay-Kanitez N, Celik S, Bes C. Polyarthritis and its differential diagnosis. Eur J Rheumatol. 2019;6(4):167–173. PubMed PMID: 31657698. Pubmed Central PMCID: 375 PMC6.812894. doi:10.5152/eurjrheum.2019.19145
20. Kundi M, Assad S, Babar S, et al. Mixed connective tissue disorder complicated by polymyositis, sjogren’s syndrome, pleural effusion and pericarditis. Cureus. 2016;8(12):e906. PubMed PMID: 28083450. Pubmed Central PMCID: PMC5208556. doi:10.7759/cureus.906
21. Institue of Medicine (US) Committee on multiple sclerosis: current status and strategies for the the future. Joy JE, Johnston RB Jr, editors. Washington (DC): national academies press (us); 2001. PMID:25057543.
22. Lokesh L, Tony K, Raghupathy R, V S, Malepati B. A rare case of Mixed Connective Tissue Disease (MCTD) with intricate features of lupus, polymyositis and rheumatoid arthritis presenting with severe myositis. J Clin Diagn Res. 2015;9(3):OD05–OD07. PubMed PMID: 25954655. Pubmed Central PMCID: PMC4413104. doi:10.7860/JCDR/2015/11575.5695
23. Carnero-Montoro E, Barturen G, Povedano E, et al. Epigenome-Wide Comparative Study reveals key differences between mixed connective tissue disease and related systemic autoimmune diseases. Front Immunol. 2019;10:1880. PubMed PMID: 31440254. Pubmed Central PMCID: PMC6693476. doi:10.3389/fimmu.2019.01880
24. Missounga L, Ba JI, Nseng nseng ondo IR, et al. [Mixed connective tissue disease: prevalence and clinical characteristics in African black, study of 7 cases in Gabon and review of the literature]. La connectivite mixte: prevalence et caracteristiques cliniques chez le noir africain, etude de 7 cas au Gabon et revue de la litterature. Pan Afr Med J. 2017;27:162. French. PubMed PMID: 28904690. Pubmed Central PMCID: PMC5567953. doi:10.11604/pamj.2017.27.162.12572
25. Nagaraja V, McMahan ZH, Getzug T, Khanna D. Management of gastrointestinal involvement in scleroderma. Curr Treat Options Neurol. 2015;1(1):82–105. PubMed PMID: 26005632. Pubmed Central PMCID: PMC4437639. doi:10.1007/s40674-014-0005-0
26. Xu ZY, Li QQ, Zhang C, Zhang HS, Gu H. [Risk factors for death and the clinical features of different subtypes of patients with pulmonary arterial hypertension related to congenital heart disease]. Zhonghua Xin Xue Guan Bing Za Zhi. 2020;48(4):315–322. Chinese. PubMed PMID: 32370483. doi:10.3760/cma.j.cn112148-20190628-00364
27. Didier K, Bolko L, Giusti D, et al. Autoantibodies associated with connective tissue diseases: what meaning for clinicians? Front Immunol. 2018;9:541. PubMed PMID: 29632529. Pubmed Central PMCID: PMC5879136. doi:10.3389/fimmu.2018.00541
28. Cozzani E, Agnoletti AF, Pappalardo F, Schiavetti I, Torino A, Parodi A. The high incidence of anti-Ro/SSA and anti-p200 antibodies in female patients with connective tissue diseases confirms the importance of screening for congenital heart block-associated autoantibodies during pregnancy. Arch Dermatol Res. 2016;308(2):139–143. PubMed 410 PMID: 26830903. doi:10.1007/s00403-016-1622-2
29. Wilson H. Biomarkers in autoimmune rheumatic diseases. Biomark Med. 2015;9(6):497–498. PubMed PMID: 26079956. doi:10.2217/bmm.15.27
30. Giacomelli R, Afeltra A, Alunno A, et al. Guidelines for biomarkers in autoimmune rheumatic diseases - evidence based analysis. Autoimmun Rev. 2019;18(1):93–106. PubMed PMID: 30408582. doi:10.1016/j.autrev.2018.08.003
31. Amissah-Arthur MB, Gordon C. Contemporary treatment of systemic lupus erythematosus: an update for clinicians. Ther Adv Chronic Dis. 2010;1(4):163–175. PubMed PMID: 23251736. Pubmed Central PMCID: PMC3513867. doi:10.1177/2040622310380100
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.