Back to Journals » International Journal of Nanomedicine » Volume 20
In vitro Evaluation of Mitochondrial-Targeted Andrographolide Nanoparticles Against 4T1 Breast Cancer Cells
Authors Yuan X, Bi Y, Liu J, Xu W, Li M, Li K, Zou Y, Wang X, Chen Y, Yang Q
Received 28 February 2025
Accepted for publication 5 December 2025
Published 20 December 2025 Volume 2025:20 Pages 15395—15414
DOI https://doi.org/10.2147/IJN.S525568
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Dong Wang
Xiaoyan Yuan,1,* Yunfeng Bi,2,* Ji Liu,1 Wenhao Xu,1 Mingjuan Li,3 Kexin Li,1 Yan Zou,1 Xing Wang,4 Yanqing Chen,1 Qiming Yang1
1Department of Pharmacy, Panzhihua Central Hospital, Panzhihua, People’s Republic of China; 2Department of Pharmacy, Pingbian Maternal and Child Health Hospital, Honghe, People’s Republic of China; 3Department of Pharmacy, Chuantou Xichang Hospital, Xichang, People’s Republic of China; 4College of Medicine, Southwest Jiaotong University, Chengdu, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Xiaoyan Yuan, Email [email protected] Qiming Yang, Email [email protected]
Objective: Andrographolide (AG) demonstrated promising anticancer efficacy against the initiation and progression of breast cancer by triggering the mitochondria-mediated intrinsic apoptotic pathway. However, its clinical translation is still hindered by drawbacks such as poor bioavailability and off-target effects; therefore, an optimized drug-delivery system that minimizes these effects is urgently needed. To address these issues, we successfully developed a mitochondria-targeting nanocarrier (TPP-PEG-PCL) with high drug-loading capacity and excellent biocompatibility.
Methods: The mitochondria-targeting copolymer (TPP-PEG-PCL) was synthesized chemically and used to prepare AG-loaded polymeric micelles (TPP-PEG-PCL@AG) by solvent-evaporation method. In vitro, the blank micelles were first evaluated for biocompatibility with mouse breast-cancer cells (4T1) and endothelial cells (EC). Subsequently, a panel of cellular assays was performed on 4T1 cells to compare the antitumor activity of free AG, PEG-PCL@AG, and TPP-PEG-PCL@AG, confirming the enhanced cancer-cell killing achieved through mitochondria-targeted delivery of AG.
Results: The results showed that TPP-PEG-PCL micelles were readily taken up by 4T1 cells and selectively accumulated in mitochondria with a Pearson’s correlation (Rr) 0.47 compared to 0.25 in PEG-PCL micelles group, leading to a pronounced inhibition of proliferation and migration. By elevating intracellular ROS, decreasing mitochondrial membrane potential, and activating the caspase cascade, the micelles induced apoptosis and thereby achieved mitochondria-targeted potentiation of TPP-PEG-PCL@AG. However, this study is limited to in vitro validation using the 4T1 murine model, and further in vivo investigations are warranted to assess translational efficacy and potential systemic toxicity..
Conclusion: PCL-PEG nanoparticles decorated with TPP combine pronounced mitochondria-targeting specificity, high drug-loading capacity, excellent biocompatibility and readily tunable architecture, making them an ideal platform for constructing a precise mitochondrial-intervention system for AG. This strategy is particularly attractive for tumor-targeted delivery of AG and opens a new avenue for its clinical translation.
Keywords: andrographolide, nanoparticles, mitochondrial targeting, anti-breast cancer
Introduction
As of now, breast cancer remains the most prevalent malignant tumor among women globally, accounting for approximately 16% of newly diagnosed cancer cases in females and resulting in a mortality rate of nearly 10%, which poses a serious threat to women’s health.1,2 Currently, common clinical treatment modalities for breast cancer include surgical intervention, neoadjuvant chemotherapy, targeted therapy, endocrine therapy, and radiotherapy.3–5 In the early stages of breast cancer, surgical resection of the tumor along with surrounding normal tissue and lymph nodes is often performed to achieve a curative outcome. However, the presence of undetected and incompletely eradicated malignant cells, coupled with technical challenges, contributes to a high rate of recurrence following surgical treatment.6 Consequently, postoperative pharmacotherapy has become a primary method for extending survival in breast cancer patients.7 Although significant advancements have been made in tumor immunotherapy, offering hope for curing cancer, the low response and efficacy rates limit its benefits to only a small subset of patients.8 Therefore, chemotherapy remains the mainstay of breast cancer treatment in clinical practice.9 While chemotherapy is effective in providing broad-spectrum therapeutic effects, the emergence of drug resistance poses a significant challenge as treatment progresses.10 The discovery of new chemotherapeutic agents for breast cancer is crucial for advancing its treatment. Over 60% of anticancer drugs are derived from natural products, such as plants and microorganisms, which exhibit potent antitumor activity by inhibiting tumor cell metastasis, inducing apoptosis, and reversing multidrug resistance in cancer cells. Thus, the exploration of natural products for the development of breast cancer therapeutics represents an important direction in current research.
Andrographolide (AG), a principal active compound of the plant Andrographis paniculata from the Acanthaceae family, is a diterpenoid lactone commonly employed in clinical settings to treat inflammatory diseases and upper respiratory infections. Recent studies have shown that AG exhibits promising anticancer properties against breast cancer,11,12 offering new possibilities for novel breast cancer therapies. The antitumor mechanisms of AG may involve its ability to inhibit tumor angiogenesis,13–18 suppress tumor cell migration,19–22 impede tumor cell proliferation,23,24 induce cell cycle arrest,25–27 and trigger apoptosis in tumor cells.28–31 Despite its favorable anticancer effects, poor water solubility of AG leads to its conversion into less effective sulfated metabolites through first-pass metabolism. Furthermore, AG is rapidly eliminated via the intestinal tract due to the action of P-glycoprotein (P-gp), significantly reducing its bioavailability.32 These factors considerably limit the development of AG for breast cancer treatment.
With the continuous advancement of nanomedicine, nanoparticle carriers for drug delivery have shown promising applications in cancer treatment. Compared to conventional drug therapies, nanoparticle carriers, such as polymer micelles, nanocrystals, vesicles, mesoporous materials, gold nanoparticles, and nanoemulsions, could enhance the pharmacokinetic properties of drugs in vivo,33 prolong the half-life and effective circulation time of drugs in the bloodstream, facilitate drug delivery to tumor tissues, minimize toxic side effects, and improve antitumor efficacy.34 The application of nanotechnology in drug delivery enables poorly soluble drugs, highly toxic chemotherapeutic agents, and drugs with low bioavailability to meet clinical needs. Furthermore, due to their customizable chemical structures and adjustable particle sizes, nanodrugs can acquire novel functionalities, such as targeted delivery to specific cells or organelles, stealth properties during circulation, and rapid release at action sites. Among these functionalities, organelle targeting presents a significant challenge for conventional chemotherapeutic drugs.35 The primary organelles influencing apoptosis in cells are mitochondria and nucleus. Mitochondria are essential for regulating cellular energy and metabolism, playing a crucial role in tumor growth and survival, while also being closely associated with tumor cell apoptosis.36 Depolarization of the mitochondrial membrane potential (MMP) can lead to the release of cytochrome C and Smac proteins from the mitochondrial membrane, subsequently activating caspase-related apoptotic pathways, ultimately resulting in tumor cell death.36 The notable differences in MMP between tumor and normal cells provide a foundation for mitochondrial-targeted cancer therapy.37
Among reported mitochondrial targeting strategies, besides TPP, approaches include mitochondrial penetrating peptides (MPPs), membrane-fusion liposomes, and physical methods like electroporation38–41 However, most existing systems still face limitations, including complex synthesis procedures, insufficient in vivo stability, and limited targeting efficiency. In contrast, TPP has emerged as one of the most widely used mitochondrial targeting moieties due to its high chemical stability, ease of modification, and strong membrane penetration capability.42–44 By focusing on mitochondria, which are essential for meeting the high metabolic demands of cancer cells, interventions can trigger apoptosis through the disruption of cellular energy pathways.45–47 Triphenylphosphonium (TPP) is a polar cationic molecule widely utilized for mitochondrial targeting.48 The incorporation of TPP onto micelles imparts a positive charge, enabling them to interact with negatively charged cell membranes and enhancing cellular uptake. Additionally, TPP functions as a “proton sponge” increasing lysosomal pH, promoting the influx of extracellular chloride ions and water, resulting in osmotic expansion, lysosomal membrane rupture, and the release of nanoparticles into the cytoplasm.49 Nanoparticle carriers bearing TPP, due to the cationic nature and lipophilicity of TPP, can penetrate mitochondrial membranes, driving the accumulation of positively charged and lipophilic molecules within mitochondria, thereby enhancing the drug’s anti-apoptotic effects.48 In this study, we developed a TPP-PEG-PCL carrier that combines TPP’s targeting ability, PEG’s hydrophilicity and long circulation, and PCL’s biodegradability and sustained release50,51 This system achieves efficient mitochondrial targeting, good biocompatibility, and controlled drug release, significantly enhancing drug accumulation in mitochondria to induce dysfunction and promote apoptosis more effectively than non-targeted or other organelle-targeted systems.
This study aims to address the poor water solubility and low bioavailability of AG, as well as its capacity to enhance reactive oxygen species (ROS) accumulation, induce mitochondrial membrane potential (MMP) loss, facilitate phosphatidylserine externalization, and activate Caspase-9 and Caspase-3. These mechanisms collectively contribute to the induction of apoptosis.52 We propose the development of a naturally biodegradable, mitochondria-targeted nanoparticle drug delivery system, TPP-PEG-PCL@AG, to facilitate substantial accumulation of AG within mitochondria (Figure 1). Compared with previous studies, the innovation of this design lies in integrating the natural active compound AG with a mitochondria-targeted delivery strategy. By employing a TPP-modified nanocarrier to enhance drug-targeted accumulation, the system simultaneously elevates ROS levels, decreases mitochondrial membrane potential (MMP), and activates the caspase cascade, thereby promoting tumor cell apoptosis. This approach not only improves the pharmacokinetic profile of AG but also enhances its antitumor efficacy through precise organelle-level intervention, offering a novel strategy for suppressing breast cancer growth and metastasis.
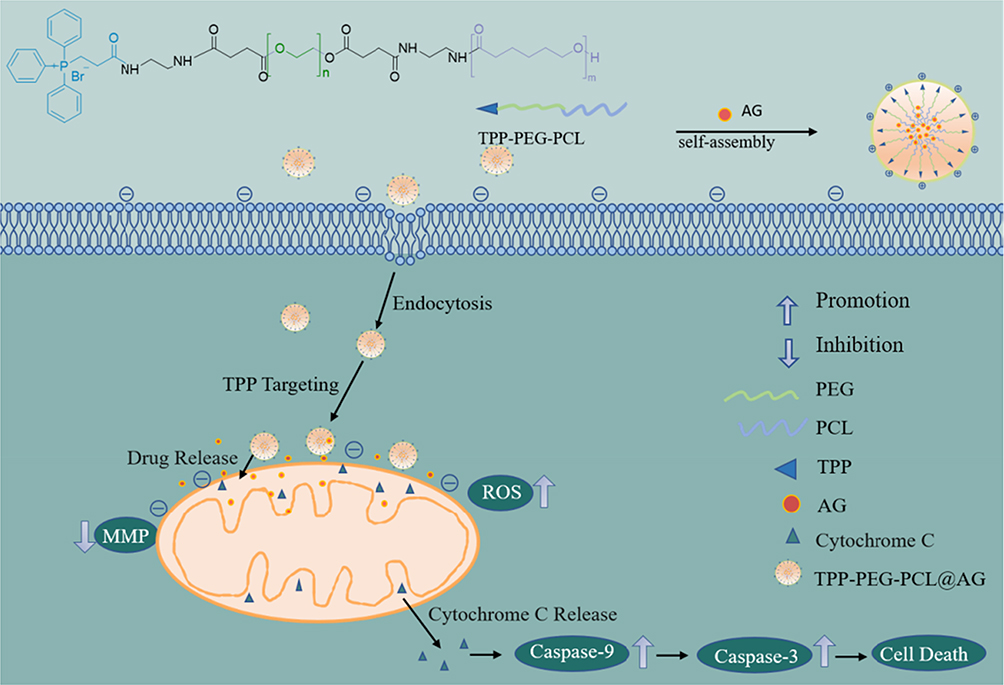 |
Figure 1 The composition of nanoparticles and the process of mitochondrial targeting. |
Materials and Methods
Materials
Andrographolide (AG) and triphenyl phosphine (TPP) were supplied by Aladdin Scientific Co., Ltd. (Shanghai, China). PEG2000, ε-caprolactone, stannous chloride, succinic anhydride, 4-dimethylaminopyridine (DMAP), and dicyclohexylcarbodiimide (DCC) were procured from Kelon Chemical Co., Ltd. (Shanghai, China). Cell Counting Kit-8 (CCK-8), Live/Dead Cell Staining Kit, DAPI, Annexin V-PI Kit, BCA Protein Concentration Assay Kit, DCFH-DA fluorescent probe, Caspase-3 and Caspase-9 Detection Kits were acquired from Beyotime Biotechnology Co., Ltd. (Shanghai, China). JC-1 fluorescent probe was obtained from Yeasen Biotechnology Co., Ltd. (Shanghai, China). Rhodamine B was purchased from Aladdin Scientific Co., Ltd. (Shanghai, China). Fetal bovine serum was sourced from Biological Industries Co., Ltd. (Beit-Haemek, Israel). 4T1 and HUVECs cell lines were purchased from National Collection of Authenticated Cell Cultures (NCACC, China). All other reagents were purchased from Adamas-Beta Co., Ltd., (Shanghai, China).
Synthesis of PEG-PCL Copolymer
As previously reported,53 PEG-PCL copolymer was synthesized in a round-bottom flask where PCL (0.8 g) dissolved in 5 mL dichloromethane (DCM). The reaction mixture’s pH was adjusted to 2–3 using trifluoroacetic acid (TFA), and the reaction was carried out under a nitrogen atmosphere at room temperature for 2 h. Triethylamine was added to adjust the pH to slightly alkaline, followed by solvent evaporation under reduced pressure to obtain white residues. Subsequently, COOH-PEG-COOH (1.06 g), DCC (0.219 g) and DMAP (0.13 g) were added into the residues dissolved in DCM, and the reacted under nitrogen at room temperature for 48 hours. The mixture was then filtered to remove the white solid by-product (dicyclohexylurea) and concentrated under vacuum. Then the residues were redissolved in ethyl acetate stored at 0 °C for 12 h, and filtered to remove any precipitates. The filtrates were evaporated under vacuum and redissolved in a small amount of DCM. The solution was precipitated with ice-cold ethanol to get white precipitates, and product was yield by freeze drying.
Synthesis of N-Tert-Butyloxycarbonyl Triphenyl Phosphate (Boc-NH-TPP)
A mixture of TPP-COOH (5 g), Boc-NH-NH2 (1.3 g), EDC (2.38 g), and NHS (2.78 g) was dissolved in 20 mL of dichloromethane and reacted under nitrogen at room temperature for 72 h, monitored by thin-layer chromatography (TLC). The reaction was quenched by adding 5 mL of deionized water, and the mixture was then washed with slightly acidic water to remove EDC and NHS residues. The organic layer was separated, dried over anhydrous sodium sulfate, and concentrated under reduced pressure to yield a pale-yellow solid.
Synthesis of TPP-PEG-PCL
TPP-PEG-PCL was synthesized by dissolving PEG-PCL (0.7 g), Boc-TPP (0.55 g), EDC (0.049 g), and NHS (0.029 g) in 5 mL of DCM and reacted under nitrogen at room temperature for 48 h. The solution was further dissolved in 30 mL of DCM and washed with 10 mL of water each for three times. The organic layer was dried over anhydrous sodium sulfate. The solution was precipitated with ice-cold ethanol, filtered, and dried to yield a pale-yellow powder.
Fabrication of Micelles
Drug-loaded polymer micelles were synthesized via the solvent evaporation technique.54 TPP-PEG-PCL (10 mg) and AG (1 mg) were dissolved in 6 mL of tetrahydrofuran (THF). The solution was incrementally added into 10 mL deionized water with continuous stirring (300 rpm). Following complete evaporation of THF, the drug-loaded polymer micelle solution TPP-PEG-PCL@AG was formed. Micelles containing Rhodamine B (PEG-PCL@RB and TPP-PEG-PCL@RB) were prepared by same methods.
Characterization
The structure of polymers was identified using Fourier-transform infrared (FT-IR) spectroscopy (Thermo Nicolet 5700) and proton nuclear magnetic resonance (1H-NMR) spectroscopy (Bruker). The size and zeta potential of the micelles were determined using dynamic light scattering (DLS; Malvern Zetasizer Nano ZS90), while their morphology was examined via transmission electron microscopy (TEM; JEL2100F). The critical micelle concentrations (CMC) of micelles were determined using a fluorescence spectrophotometer (Hitachi F-7000). Drug loading capacity (LC) and encapsulation efficiency (EE) was determined by high-performance liquid chromatography (HPLC; Agilent 1260, methanol: water = 60: 40, flow rate 0.4 mL/min, 254nm). The cumulative drug release rates of TPP-PEG-PCL@AG under different pH conditions were evaluated by HPLC.
In vitro Cell Uptake Assays
The efficiency of cellular uptake for the micelles was evaluated using inverted fluorescence microscope (IFM; Olympus CKX41) and flow cytometry (FCM; BD). Breast cancer cells (4T1) were seeded at a density of 5×104 per well in a 12-well plate and incubated with 5% CO2 until full adherence. Cells were treated for 6 hours with fresh medium containing equivalent amounts of free Rhodamine B, Rhodamine B-labeled PEG-PCL@RB, and TPP-PEG-PCL@RB. The cells were then fixed with 4% paraformaldehyde, washed with PBS, and incubated with DAPI for 10 min. Cellular uptake of micelles was observed under IFM and detected by FCM.
In vitro Cytotoxicity Assays
All cytotoxicity evaluations of both blank (TPP-PEG-PCL) and AG-loaded micelles against 4T1 cells and endothelial cells (ECs) were carried out through the CCK-8 assay and live/dead cell staining. In brief, 4T1 cells and ECs were seeded into a 96 - well plate at a density of 5 × 103 cells per well and incubated in an environment with 5% CO2 until they reached full adherence. Then, the original culture medium was substituted with TPP - PEG - PCL micelles at concentrations spanning from 10 to 200 µg/mL, and the cells were further incubated for 24 h. After this incubation period, 100 µL of medium containing 10% CCK-8 solution was added to each well, followed by an additional 30 - minute incubation, and subsequently, the optical density (OD) was measured at 450 nm using a microplate reader (Thermo). For the live/dead cell staining, acridine orange (AO) and ethidium bromide (EB) were introduced to the cells treated with blank micelles after 24-hour treatment, and then the cells were incubated for another 30 minutes. Finally, cell viability was observed with an inverted fluorescence microscope (IFM; Olympus CKX41).
Mitochondria-Targeting Analysis
To assess the mitochondria-targeting ability endowed by TPP groups, the nile red (NR) loaded TPP-PEG-PCL and PEG-PCL micelles were incubated with 4T1. The intracellular distribution of NR was observed by confocal laser scanning microscopic (CLSM). In brief, 4T1 cells were seeded in glass bottom confocal dishes (15mm, Biosharp, China) at a density of 5×104 cells per dish, after incubated for 1 h the pure NR, NR loaded TPP-PEG-PCL and PEG-PCL micelles were added. After 1 h’s incubation, cells were washed with PBS for three times, and then stained with Mitotracker Green (MTG, Beyotime Biotech, China) for 30 minutes, followed by washing with PBS for three times. Mitochondrion were visualized via CLSM (A1R+, Nikon, Japan), and the co-localization of nile red and mitochondrion was analyzed using Image J.
Scratch Test
To evaluate the effects of free AG and AG-loaded micelles (TPP-PEG-PCL@AG) on the migratory ability of 4T1 cells, a scratch wound healing assay was performed. Briefly, 4T1 cells were seeded in 6-well plates at a density of 2 × 105 cells per well and cultured until reaching 80–90% confluency. A uniform scratch was created in the cell monolayer using a sterile pipette tip, and the initial wound edges were clearly marked under a microscope. The cells were then washed with PBS to remove detached cells, and fresh serum-free medium containing equivalent concentrations of free AG or TPP-PEG-PCL@AG was added. Wound closure was monitored by capturing microscopic images at the marked sites at 0, 24, and 36 h. The migration rate was assessed based on the reduction in the scratch area over time.
Reactive Oxygen Species (ROS) Assays
Intracellular reactive oxygen species (ROS) levels in 4T1 cells were measured using the 2’,7’-dichlorodihydrofluorescein diacetate (DCFH-DA) fluorescence probe. Briefly, 4T1 cells were seeded in 12-well plates at a density of 5 × 104 cells per well and cultured until adherent. Subsequently, the cells were treated with fresh medium containing equivalent concentrations of free AG, PEG-PCL@AG, or TPP-PEG-PCL@AG for 48 h. After treatment, the cells were incubated with DCFH-DA (diluted in serum-free medium) in the dark at 37°C for 30 min to allow ROS-dependent fluorescence generation. The cells were then gently washed three times with PBS to remove excess probe. To visualize cell nuclei, DAPI staining was performed for 10 min, followed by another PBS wash to eliminate residual dye. Fluorescence imaging was conducted using an IFM, capturing DCFH-DA (green, ROS indicator) and DAPI (blue, nuclear counterstain) signals to assess ROS production.
Mitochondrial Membrane Potential (MMP) Assays
The effects of free AG and AG-loaded micelles (PEG-PCL@AG and TPP-PEG-PCL@AG) on mitochondrial membrane potential (MMP) in 4T1 cells were evaluated using inverted fluorescence microscopy (IFM) and flow cytometry (FCM). 4T1 cells were seeded in 12-well plates at a density of 5 × 104 cells per well. After 24 hours of treatment with fresh medium containing equivalent concentrations of free AG, PEG-PCL@AG, or TPP-PEG-PCL@AG, the cells were gently washed with PBS to remove residual medium. For MMP assessment, an appropriate volume of JC-1 staining solution (prepared according to the manufacturer’s protocol) was added to each well. The cells were then incubated in the dark at 37°C for 20 minutes to allow mitochondrial staining. Following incubation, the cells were thoroughly washed with PBS to remove excess dye. MMP changes were visualized by capturing fluorescence images under IFM (detecting both J-aggregate [red] and monomer [green] forms of JC-1), and quantitatively analyzed by FCM to determine the MMP status.
In vitro Cell Apoptosis Assays
Apoptotic cell death in 4T1 cells was quantitatively assessed using annexin V-FITC/PI double staining followed by FCM analysis. 4T1 cells were seeded in 12-well plates at a density of 5 × 104 cells per well. After 24 hours of treatment with fresh culture medium containing equivalent concentrations of free AG, PEG-PCL@AG, or TPP-PEG-PCL@AG, the cells were harvested for analysis. For apoptosis detection, the collected cells were gently washed with cold PBS and resuspended in binding buffer. The cell suspension was then incubated in the dark at room temperature for 20 minutes with a mixture of annexin V-FITC (a phosphatidylserine-binding protein that specifically labels apoptotic cells) and propidium iodide (PI, a DNA intercalating dye that stains necrotic and late apoptotic cells with compromised membranes). The stained cells were immediately analyzed by flow cytometry to distinguish between viable (annexin V−/PI−), early apoptotic (annexin V⁺/PI−), late apoptotic (annexin V⁺/PI⁺), and necrotic (annexin V−/PI⁺) cell populations. Apoptosis-related protein Caspase-3 and Caspase-9 were detected using BCA protein assay.
Statistical Analysis
All raw data were derived from at least three independent biological replicates (n ≥ 3) and presented as mean ± standard deviation (SD) to reflect both central tendency and variability. Data processing and statistical analyses were performed using GraphPad Prism software (version 9.0 or later). For comparisons across multiple treatment groups, one-way ANOVA was employed to assess overall differences between group means. Statistical significance was defined as p < 0.05 (*), p < 0.01 (**), and p < 0.001 (***).
Results and Discussion
Preparation and Characterization of AG-Loaded Micelles
The diblock polymers TPP-PEG-PCL was precisely synthesized step by step as shown in Figure 2. The amide condensation reaction between COOH-PEG-COOH and Boc-NH-PCL was carried out under catalyzing of EDC and DCC in DCM after getting rid of Boc protecting groups. The resulting solution was precipitated and purified by ice-cold ethanol to reduce unreacted COOH-PEG-COOH. In Figure 3a, the peaks of PEG, PCL and linker ethane diamine were presented in the 1H-NMR spectrum, which indicated the successful synthesis of the amphiphilic polymers PEG-PCL.
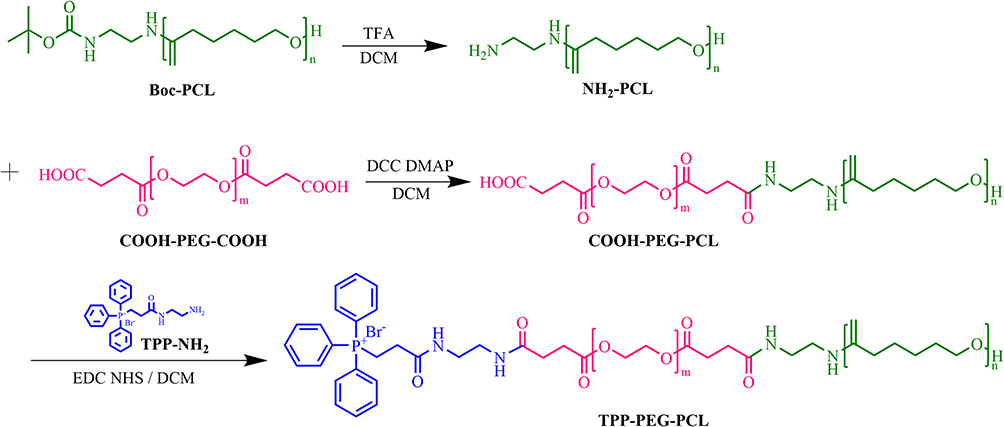 |
Figure 2 The step-by-step synthesis pathway of TPP-PEG-PCL. |
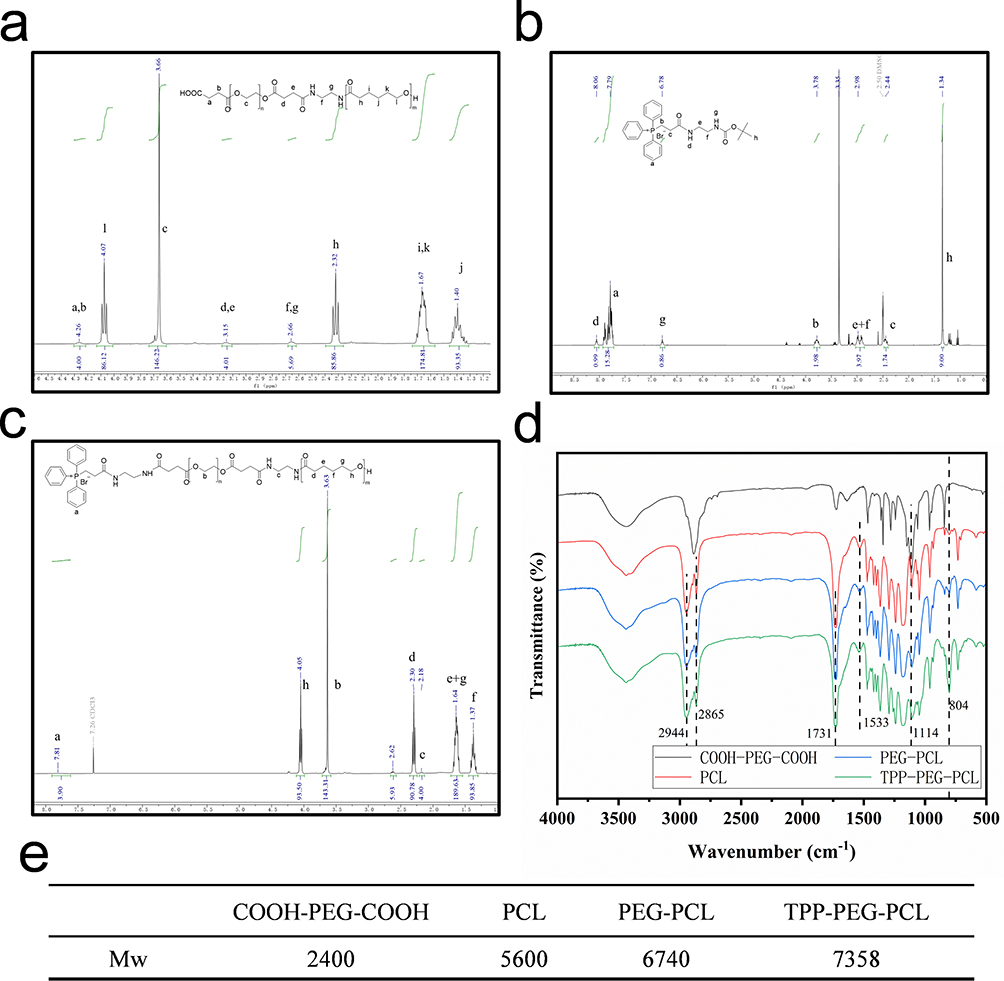 |
Figure 3 1H-NMR spectra of COOH-PEG-PCL (a), Boc-TPP (b), TPP-PEG-PCL (c), FT-IR spectra of COOH-PEG-COOH, PCL, PEG-PCL, and TPP-PEG-PCL (d), and molecular weights of COOH-PEG-COOH, PCL, PEG-PCL, and TPP-PEG-PCL (e). Statistical significance was defined as p < 0.05 (*), p < 0.01 (**), and p < 0.001 (***). |
Boc-NH-TPP was synthesis based on amide condensation between TPP-COOH and Boc-NH-NH2, as shown in Figure 3b, characteristic peaks of TPP (δ 2.44, 3.78 and 7.79) and Boc-NH-NH2 (δ 1.34, 2.98 and 8.06) were observed, indicating the successful synthesis of Boc-NH-TPP. Then, TPP-PEG-PCL was synthesized by grafting Boc-NH-TPP to the remaining carboxyl groups of COOH-PEG-PCL. In Figure 3c, characteristic peaks of TPP (δ 7.81), PEG-PCL (δ 1.37, 1.64, 2.18, 2.3, 2.62, 3.63 and 4.05) were presented. These results confirmed the successful synthesis of TPP-PEG-PCL. As shown in Figure 3d, the infrared spectrum of COOH-PEG-COOH displayed a stretching vibration peak for -CH2- at 2865 cm-1, a C-O-C peak at 1114 cm−1, and a carbonyl peak at 1731 cm−1. The PCL spectrum exhibited a typical amide I peak at 1533 cm−1,53,55 The spectrum of TPP-PEG-PCL showed a characteristic aromatic ring peak at 804 cm−1. These results also verified the successful synthesis of TPP-PEG-PCL. By comparing the integrated areas of characteristic peaks in COOH-PEG-COOH, PCL, PEG-PCL, and TPP-PEG-PCL, the actual molecular weights of the three polymers were calculated, as shown in Figure 3e. The grafting rate of TPP was calculated to be 26% using 1H-NMR.Meanwhile, the structures of COOH-PEG-COOH, PCL, PEG-PCL, and TPP-PEG-PCL were confirmed using FT-IR.
Drug-loaded micelles were prepared through the self-assembly of TPP-PEG-PCL copolymers in aqueous solution. Size and zeta potential were measured with a Malvern particle size analyzer. The particle size of PEG-PCL@AG was approximately 155.53 nm, and its zeta potential was about 2.97 mV (Figure 4a). The positive charge inherent in TPP resulted in a significant increase in zeta potential (from 2.97 mV to 22.77 mV) when TPP-targeting groups were grafted onto the micelles, along with a slight increase in particle size. The particle size and morphology of TPP-PEG-PCL@AG drug-loaded polymer micelles were examined using DLS and TEM. According to Figure 4b, the average particle size of the TPP-PEG-PCL@AG micelles was 168.6 nm, following a normal distribution. Figure 4b and c indicates that the micelles were uniformly spherical, with consistent size and good dispersion.
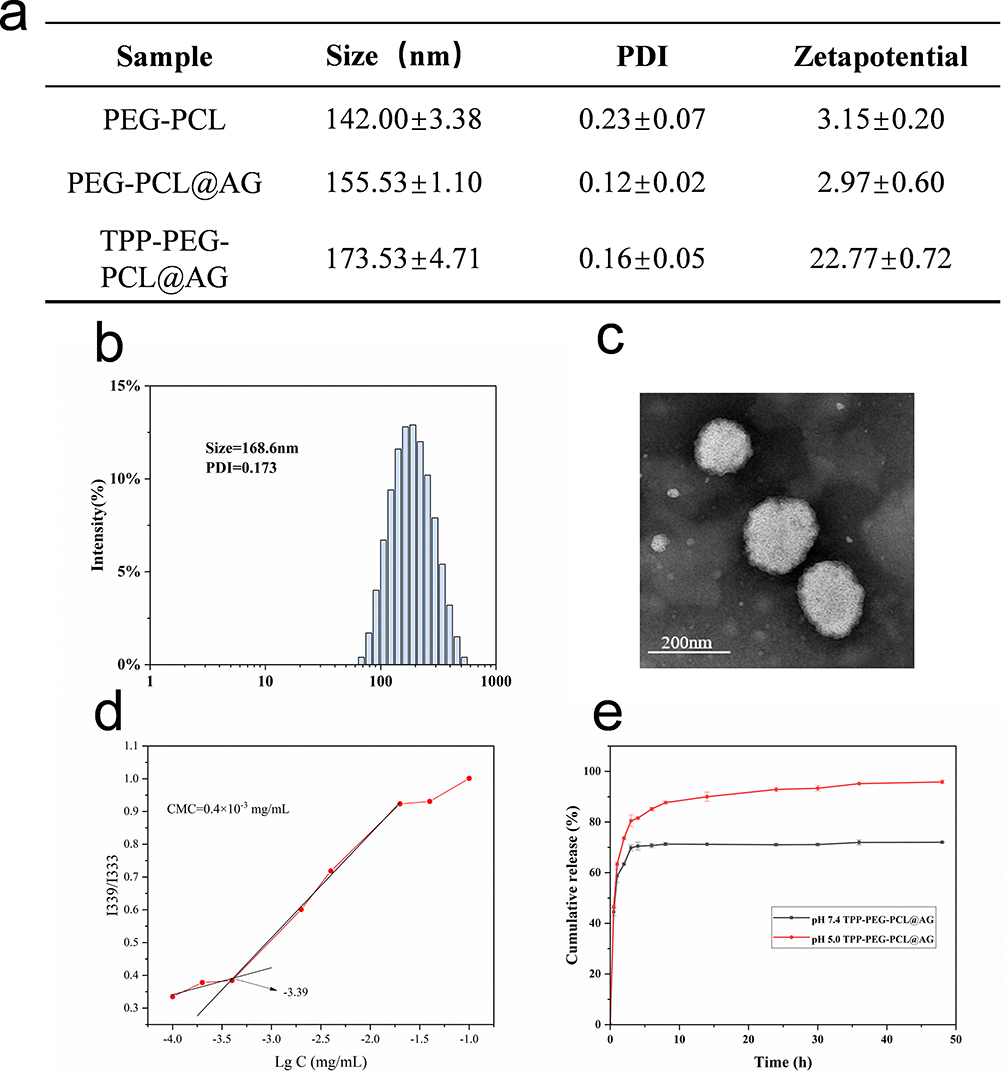 |
Figure 4 The nanoparticle was fabricated using solvent evaporation method. Particle size and potential of polymeric micelles were summarized in the table (a), the particle size distribution of TPP-PEG-PCL@AG by DLS (b), TEM of TPP-PEG-PCL (c), the CMC value of TPP-PEG-PCL (d), and in vitro drug release curves of TPP-PEG-PCL@AG micelles (e). Statistical significance was defined as p < 0.05 (*), p < 0.01 (**), and p < 0.001 (***). |
The CMC value represents the stability of micelle formation. A lower CMC value indicates that a lower polymer concentration is required to form micelles, which could endow micelles a good stability in blood.56 To measure the CMC of polymeric micelles, we used pyrene as a fluorescent probe. The results, shown in Figure 4d, revealed that the CMC of polymeric micelles was 0.4×10−3 mg/mL, which is quite low. This suggested that the polymeric micelles can form at low concentrations and are resistant to dilution. HPLC was used to measure the content of AG in drug-loaded micelles. The drug loading capacity of TPP-PEG-PCL@AG micelles was calculated to be 4.23±0.22%, with a drug encapsulation efficiency of 46.55±2.47%. As shown in Figure 4e, the cumulative drug release from TPP-PEG-PCL@AG micelles at pH 7.4 over a 72-hour period was approximately 75%, with more complete drug release at pH 5.0.
In vitro Cellular Uptake and Cytotoxicity Analysis
Due to AG’s lack of fluorescence, RB was used as a fluorescent model drug to study the uptake efficiency of drug-loaded micelles by 4T1 cells. The micelles containing RB were prepared using the same method as TPP-PEG-PCL@AG micelles, resulting in either unmodified (PEG-PCL@RB) or TPP-modified (TPP-PEG-PCL@RB) micelles. Fluorescence microscopy was employed to visualize the cells, with DAPI-emitted blue fluorescence and RB-emitted red fluorescence. As shown in Figure 5a, the red fluorescence in PEG-PCL@RB was significantly weaker than that in free RB and TPP-PEG-PCL@RB. Flow cytometry was used for quantitative analysis, as shown in Figure 5b and c and supplementary table 1, which corroborated the fluorescence results. The free RB group exhibited the strongest fluorescence, the TPP-modified group had slightly weaker fluorescence, and the non-targeted group had the weakest fluorescence. This is likely because free RB, as a small-molecule drug, can freely diffuse into cells, thereby resulting in a stronger red fluorescence. The TPP-PEG-PCL@RB group, with positive surface charge, enhanced the cellular uptake by interacting with the negatively charged cell membrane of 4T1. In contrast, the non-targeted PEG-PCL@RB group exhibited relatively weaker red fluorescence compared to the TPP-PEG-PCL@RB group. These results suggested that TPP modification can enhance the cellular uptake of drug-loaded micelles.
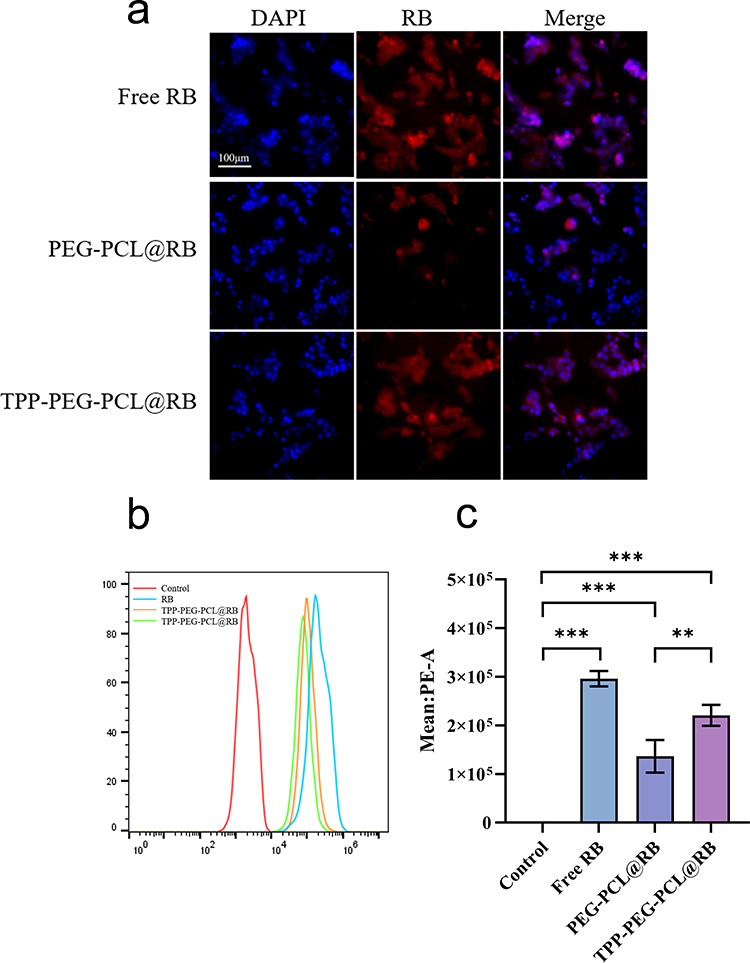 |
Figure 5 Cellular uptake of AG-loaded micelles. Fluorescence images of cellular uptake (a). The flow cytometry analyses (b) and quantitative analysis (c) after incubated 4T1 with AG and AG-loaded micelles. Statistical significance was defined as p < 0.05 (*), p < 0.01 (**), and p < 0.001 (***). Scale bar = 100 um. |
An ideal nanocarrier should possess not only suitable physicochemical properties but also high biocompatibility. To evaluate the effect of blank materials on cells, various concentrations of the blank material were co-cultured with 4T1 cells and EC cells for 24 hours. As shown in Figure 6a and b, no significant cell death was observed, indicating that the blank material exhibits good cellular compatibility. The results shown in Figure 6c indicate that cell viability for both cell types remained above 85% within the given concentration range of 0–200 μg/mL, with negligible toxicity observed at any concentration within this range.
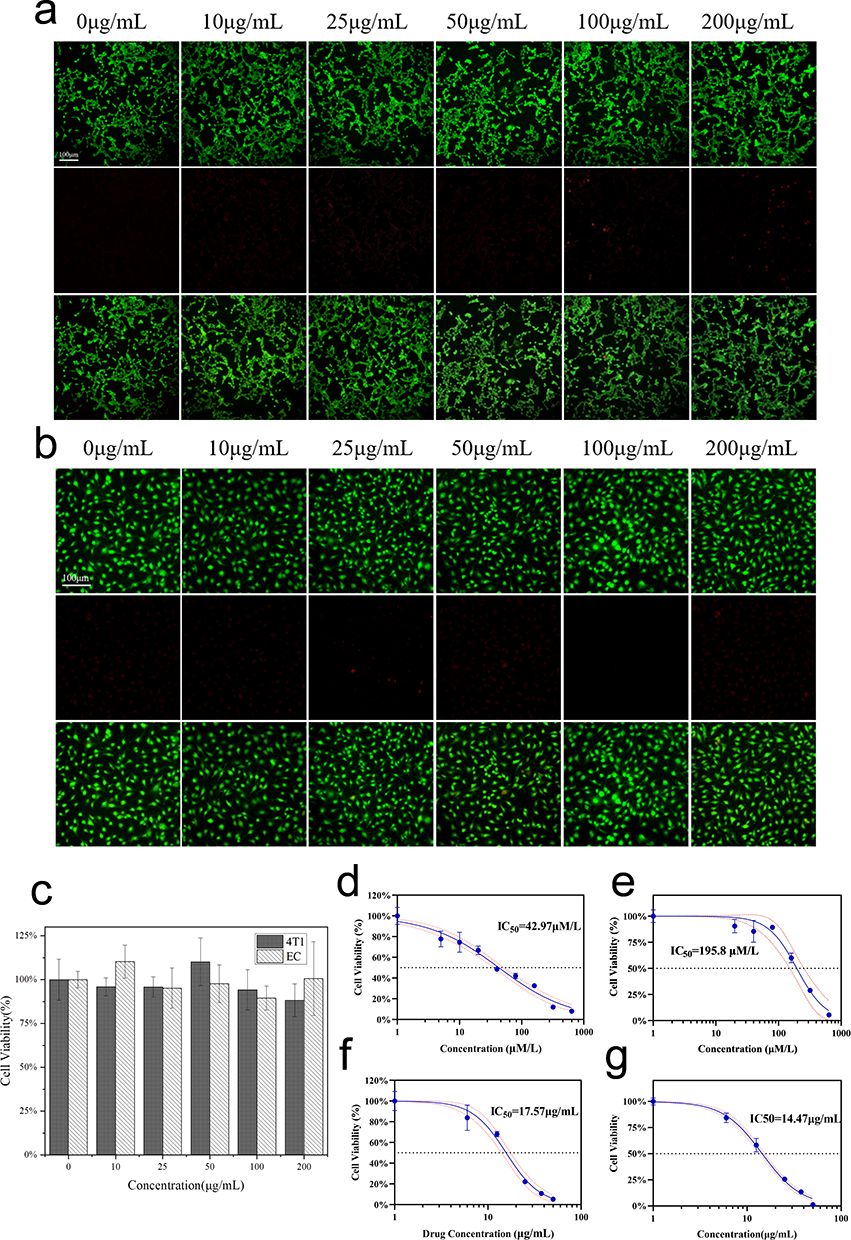 |
Figure 6 Fluorescence images of live/dead staining of blank micelles against 4T1 (a) and ECs (b). Cell viabilities of blank micelles against 4T1 and ECs (c). Cytotoxicity of AG against 4T1 (d) and EC (e), PEG-PCL@AG (f) and TPP-PEG-PCL@AG (g) against 4T1. Scale bar = 100 um. |
The inhibitory effect of drug-loaded micelles on 4T1 proliferation was evaluated using the CCK-8 assay. As illustrated in Figure 6d–g the IC50 values of free AG co-incubated with 4T1 and ECs for 24 hours were 42.9 μM/L (15.06 μg/mL) and 195.8 μM/L (68.63 μg/mL), respectively. These findings suggested a notable inhibition of 4T1 proliferation within the experimental concentration range by free AG, while its impact on ECs was comparatively minimal. The IC50 values for PEG-PCL@AG and TPP-PEG-PCL@AG on 4T1 were 17.57 and 14.47 μg/mL, respectively. The results indicated a similar inhibitory effect of free AG and TPP-PEG-PCL@AG on 4T1 proliferation, with PEG-PCL@AG demonstrating weaker inhibition. This disparity can be attributed to the small molecular nature of free AG, enabling it to freely diffuse into cells, whereas drug-loaded micelles require endocytosis and drug release. TPP-PEG-PCL@AG can enhance the cellular uptake and mitochondrial damage, consequently reinforced the inhibitory effect on 4T1 cell proliferation.
In vitro Mitochondria Targeting Ability Assessment
To assess the mitochondria targeting ability of TPP-PEG-PCL micelles, we compare the mitochondria enrichment of free NR, NR-loaded PEG-PCL and TPP-PEG-PCL micelles using CLSM. As shown in Figure 7a, mitochondrion of 4T1 cells were stained with Mito-tracker Green (MTG) dye, and NR was excited to emit red fluorescence. The yellow fluorescence appeared when the red and green fluorescence overlapped and co-localized. In the free NR group, the red fluorescence of free dye and green fluorescence of mitochondria were nearly separated, suggesting that free NR did not possess a mitochondria affinity. In TPP-PEG-PCL micelles group, yellow fluorescence was observed in the merged image, which indicating a good co-localization between NR loaded TPP-PEG-PCL micelles and mitochondrion of 4T1 cells. To quantitatively evaluate the intracellular NR distribution, Pearson’s correlation (Rr) was applied to evaluate the co-localization of two types of fluorescence. When the Rr value is closer to 1, it indicates a stronger co-localization of two channels of fluorescence, reflecting a better mitochondria-targeting ability. As shown in Figure 7b and supplementary table 2, compare to control group (Rr = 0.25) and NR loaded PEG-PCL group (Rr = 0.25), TTP modified group exhibited increased Rr (0.47), indicating that TPP modification can bring about a significant mitochondrial targeting ability.
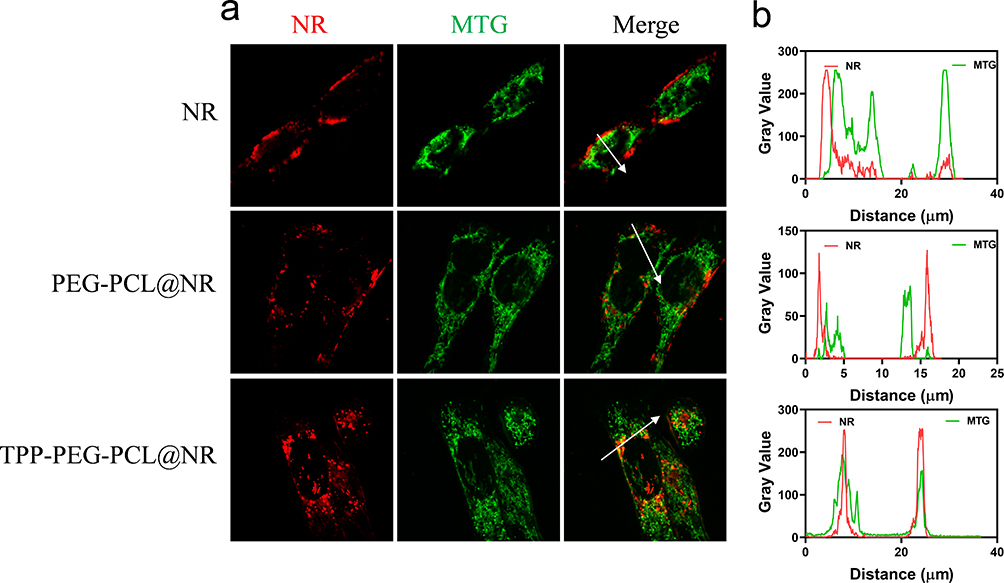 |
Figure 7 Mitochondria-targeting analysis of NR, NR loaded PEG-PCL and TPP-PEG-PCL micelles on 4T1 cells. (a) CLSM images and (b) quantitative analysis between NR and mitochondria co-localization. |
In vitro Antitumor Activity Analysis
Migration Inhibition Effects on 4T1
The in vitro biological effects evaluations further demonstrated the good antitumor activity of TPP-PEG-PCL@AG. A scratch assay was performed to assess the inhibitory effect of drug-loaded micelles on the migration of 4T1 cells. Upon reaching 80–90% confluence, cells were scratched using a pipette tip, followed by co-incubation with drug-loaded micelles for a designated time interval before image capture for analysis. Figure 8a illustrated that the control group showed no inhibition of cell migration, whereas both the free drug AG and TPP-PEG-PCL@AG group exhibited differing levels of inhibition. Nevertheless, statistical analysis (Figure 8b and c and supplementary table 3) showed that the free drug AG group demonstrated superior efficacy compared to the TPP-PEG-PCL@AG group.
 |
Figure 8 Scratch tests of 4T1 incubated with PBS, AG and TPP-PEG-PCL@AG (a). Statistical analysis of scratch tests of 4T1 in 24 h (b) and 36 h (c). Statistical significance was defined as p < 0.05 (*), p < 0.01 (**), and p < 0.001 (***). Scale bar = 100 um. |
In vitro ROS Generation and Apoptosis Induction Effects on 4T1
Reactive oxygen species (ROS) play a critical role in cellular signal transduction, with slightly elevated ROS levels in cancer cells essential for promoting cell proliferation and activating survival pathways. However, excessive ROS generation can cause oxidative stress, leading to damage in cancer cells. To investigate the effects of free AG and AG-loaded micelles on ROS levels in 4T1, intracellular ROS levels were measured using the DCFH-DA probe after a 24-hour incubation. As shown in Figure 9 and supplementary table 4, all drug-treated groups exhibited elevated ROS levels compared to the control group. Among these, the PEG-PCL@AG group displayed the lowest increase in intracellular ROS levels, while the TPP-PEG-PCL@AG group significantly increased ROS levels (p < 0.05). This suggested that TPP-PEG-PCL@AG micelles enhanced AG-induced ROS generation likely due to their mitochondrial targeting capability.
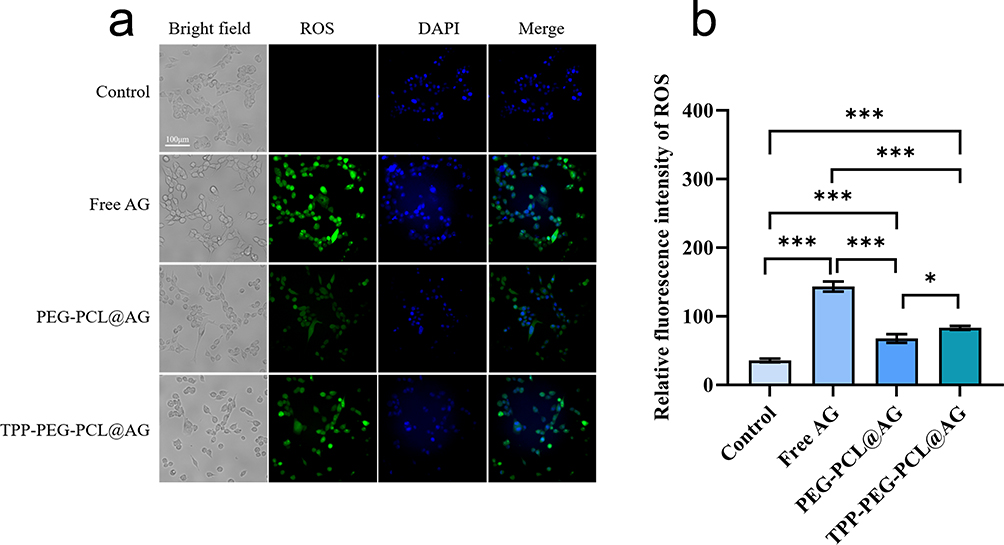 |
Figure 9 Fluorescence images (a) and mean fluorescence intensity analysis (b) of DCFH-DA probe representing the ROS level of 4T1 after incubation with PBS, AG, PEG-PCL@AG and TPP-PEG-PCL@AG. Statistical significance was defined as p < 0.05 (*), p < 0.01 (**), and p < 0.001 (***). Scale bar = 100 um. |
Excessive ROS generation were believed to induce apoptosis, resulting in antitumor effects;57 thus, Annexin V-PI kit was applied to assess the effects of AG-loaded micelles on apoptosis against 4T1. The results, depicted in Figure 10, show that the percentage of apoptotic cells was approximately 6.98% in the control group, 13.97% in the free AG group, and around 8.87% and 18.07% in the PEG-PCL@AG and TPP-PEG-PCL@AG groups, respectively. Compared to the control group, free AG significantly increased the apoptosis rate of 4T1 (p < 0.001), indicating that AG can induce apoptosis in these cells. The TPP-PEG-PCL@AG group induced apoptosis more effectively than the PEG-PCL@AG group (p < 0.001). This finding suggested that TPP-PEG-PCL@AG micelles can promote greater accumulation of the drug in tumor cell mitochondria, thus furthering the apoptotic process.
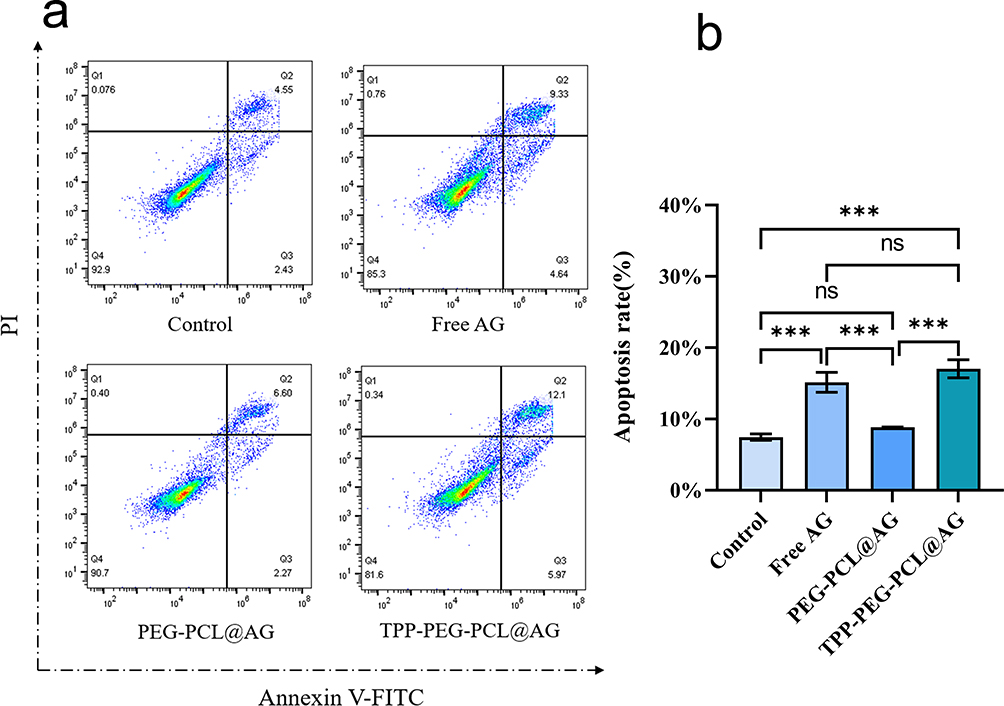 |
Figure 10 Flow cytometry analyses (a) and quantitative analyses (b) of apoptosis in 4T1 after incubation with PBS, AG, PEG-PCL@AG and TPP-PEG-PCL@AG. |
MMP Disruption and Apoptosis-Related Protein Expression
Disruption of MMP is an early indicator of apoptosis.58 To further investigate mitochondrial dysfunction in 4T1 cells treated with TPP-PEG-PCL@AG, different drug-loaded micelles were co-incubated with 4T1 cells for 24 hours, followed by staining with the JC-1 kit. In normal cells with intact mitochondrial membranes and properly functioning mitochondria, JC-1 primarily exists as a polymer, emitting red fluorescence due to high potential. When mitochondrial potential is low, JC-1 cannot aggregate and exist as a monomer, emitting green fluorescence, allowing changes in mitochondrial membrane potential to be detected through a shift in fluorescence color. The results, as shown in Figure 11a, indicate that in the control group, which received no drug treatment, mitochondrial membrane potential was normal, with bright red fluorescence and faint green fluorescence. In the free AG-treated group, the red fluorescence was noticeably dimmer, and the green fluorescence was significantly enhanced compared to the control group, suggesting that free AG can reduce mitochondrial potential.
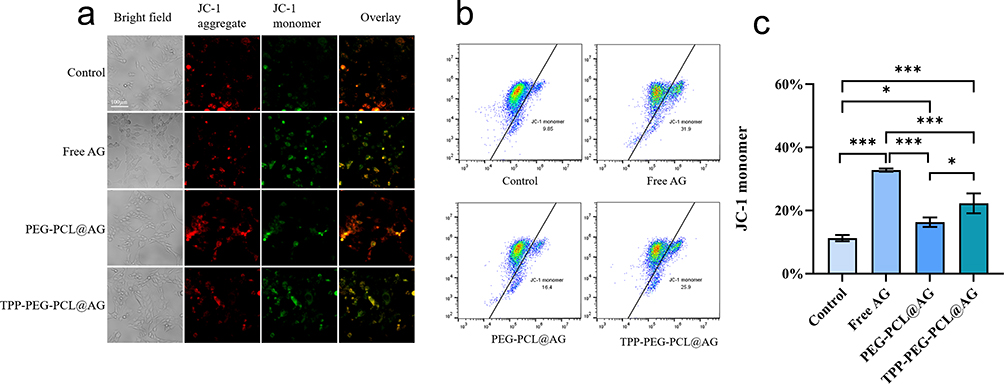 |
Figure 11 Fluorescence images (a) and flow cytometry analyses (b and c) of JI-C probe representing the changes in mitochondrial membrane of 4T1 after incubation with PBS, AG, PEG-PCL@AG and TPP-PEG-PCL@AG. Statistical significance was defined as p < 0.05 (*), p < 0.01 (**), and p < 0.001 (***). |
The FCM results (Figure 11b and c, and supplementary table 5) are consistent with the fluorescence findings, demonstrating a significant difference between the proportion of JC-1 monomers in 4T1 cells treated with free AG for 24 hours, which was 31.9%, and that in the control group, which was 9.85% (p < 0.001). The proportions for the PEG-PCL@AG and TPP-PEG-PCL@AG groups were 16.4% and 25.9%, respectively, with a statistically significant difference between these two groups (p < 0.05). Based on these results, we conclude that AG can cause mitochondrial membrane damage in 4T1 cells, reducing mitochondrial membrane potential. The TPP-PEG-PCL@AG micelles, with TPP targeting for mitochondria, can lead to AG accumulation in the mitochondria, further reducing mitochondrial membrane potential.
Damage to the MMP can lead to the release of cytochrome c from the mitochondria, which, in turn, activates the caspase-related apoptosis pathway, ultimately resulting in tumor cell death.59,60 Therefore, we further investigated the effects of AG-loaded micelles on the expression of apoptosis-related proteins, caspase-3 and caspase-9. As shown in Figure 12 and supplementary tables 6–7, the expression of caspase-3 and caspase-9 significantly increased in the AG-treated group compared to the control group (p < 0.001). This finding suggests that AG can induce the expression of caspase-3 and caspase-9 in 4T1. In contrast to the PEG-PCL@AG group, the TPP-PEG-PCL@AG group induced greater expression of caspase-3 in 4T1 (p < 0.01). Although no significant difference was observed in caspase-9 expression, these results collectively indicated that TPP-PEG-PCL@AG micelles can enhance AG-induced expression of caspase-3 and caspase-9, ultimately leading to cell apoptosis due to their mitochondrial targeting capability.
 |
Figure 12 Caspase-3 (a) and Caspase-9 (b) protein contents of within 4T1 after incubation with PBS, AG, PEG-PCL@AG and TPP-PEG-PCL@AG. Statistical significance was defined as p < 0.05 (*), p < 0.01 (**), and p < 0.001 (***). Scale bar = 100 um. |
Discussion
AG induces apoptosis in breast cancer cells by activating caspase-3 and caspase-9, but its efficacy is limited by the drug’s effective intracellular concentration and targeting capacity.58 This study successfully constructed a mitochondrial-targeted nanocarrier system, TPP-PEG-PCL, by introducing a triphenylphosphine (TPP) cationic group. This endows the nanoparticles with mitochondrial targeting capability, significantly enhancing AG’s enrichment efficiency within tumor cell mitochondria. As a core organelle regulating cellular energy metabolism and apoptosis, mitochondria represent a critical therapeutic target in cancer treatment.45–47 The incorporation of TPP not only enhances the nanoparticle’s cell membrane penetration but also promotes drug release from lysosomes into the cytoplasm via its “proton sponge” effect, thereby boosting mitochondrial targeting efficiency.
Compared to unmodified PEG-PCL@AG, TPP-PEG-PCL@AG demonstrated stronger 4T1 cytotoxicity, greater elevation of intracellular ROS levels, more pronounced MMP reduction, and more efficient apoptosis induction in vitro experiments. These results align with previous studies, indicating that AG-induced apoptosis relies on mitochondrial dysfunction and caspase activation. Furthermore, TPP-PEG-PCL@AG demonstrated favorable effects in migration inhibition assays, suggesting potential for suppressing tumor metastasis.
Notably, the TPP-PEG-PCL blank carrier maintained excellent biocompatibility even at high concentrations without significant toxicity to normal cells, indicating robust safety. This stands in stark contrast to the dose-limiting toxicities (eg, cardiotoxicity, neurotoxicity) commonly associated with many conventional chemotherapy regimens, highlighting a key potential advantage of our nano-platform. This advantage is crucial for the clinical translation of nanomedicines. Although the in vitro results of this study are encouraging, several limitations exist. First, the current research is confined to the 4T1 mouse breast cancer cell line and has not yet validated its broad applicability across multiple breast cancer subtypes, such as triple-negative or hormone receptor-positive. A critical next step is to benchmark its performance against standard-of-care chemotherapies and targeted agents in a panel of cell lines representing these subtypes. Second, in vivo animal studies to evaluate pharmacokinetic properties, tissue distribution, and long-term toxicity have not been conducted. Such studies are essential to determine if the improved therapeutic index observed in vitro translates in vivo, which would be a key differentiator from the often-narrow therapeutic window of existing drugs Furthermore, while TPP modification significantly enhances mitochondrial targeting, its potential impact on mitochondria within normal tissues requires further assessment.
Conclusion
In this study, we developed a mitochondrial-targeted polymer nanoparticle designed to improve the intracellular uptake and controlled release of AG. The amphiphilic polymer was synthesized via amide condensation, incorporating a high density of TPP groups to enhance mitochondrial targeting, while ester bonds within the polymer backbone enabled enzymatic hydrolysis-mediated drug release. In vitro evaluations indicated that the AG-loaded TPP-PEG-PCL nanoparticles exhibited promising antitumor activity on 4T1 cell line in vitro and acceptable biocompatibility. These findings suggest that the polymer nanoparticle system has potential as a targeted drug delivery platform for cancer therapy, though further in vivo studies are needed to validate its therapeutic efficacy and safety.
Funding
This work was supported by Sichuan Provincial Administration of Traditional Chinese Medicine Research Project (Technology Projects, 2023MS207).
Disclosure
The authors declare that they have no competing financial interests or personal relationships that could have influenced the work reported in this paper.
Xiaoyan Yuan and Yunfeng Bi share first authorship.
References
1. Henderson JT, Webber EM, Weyrich MS, Miller M, Melnikow J. Screening for breast cancer: evidence report and systematic review for the US preventive services task force. JAMA. 2024;331(22):1931–1946. doi:10.1001/jama.2023.25844
2. Loibl S, Poortmans P, Morrow M, Denkert C, Curigliano G. Breast cancer. Lancet. 2021;397(10286):1750–1769. doi:10.1016/S0140-6736(20)32381-3
3. Britt KL, Cuzick J, Phillips KA. Key steps for effective breast cancer prevention. Nat Rev Cancer. 2020;20(8):417–436. doi:10.1038/s41568-020-0266-x
4. Trayes KP, Cokenakes SEH. Breast cancer treatment. Am Fam Physician. 2021;104(2):171–178.
5. Pondé NF, Zardavas D, Piccart M. Progress in adjuvant systemic therapy for breast cancer. Nat Rev Clin Oncol. 2019;16(1):27–44. doi:10.1038/s41571-018-0089-9
6. Franzoi MA, Romano E, Piccart M. Immunotherapy for early breast cancer: too soon, too superficial, or just right? Ann Oncol. 2021;32(3):323–336. doi:10.1016/j.annonc.2020.11.022
7. Dumas E, Grandal Rejo B, Gougis P, et al. Concomitant medication, comorbidity and survival in patients with breast cancer. Nat Commun. 2024;15(1):2966. doi:10.1038/s41467-024-47002-3
8. Hegde PS, Chen DS. Top 10 challenges in cancer immunotherapy. Immunity. 2020;52(1):17–35. doi:10.1016/j.immuni.2019.12.011
9. Wei G, Wang Y, Yang G, Wang Y, Ju R. Recent progress in nanomedicine for enhanced cancer chemotherapy. Theranostics. 2021;11(13):6370–6392. doi:10.7150/thno.57828
10. Wang Y, Wu X, Ren Z, et al. Overcoming cancer chemotherapy resistance by the induction of ferroptosis. Drug Resist Updat. 2023;66:100916. doi:10.1016/j.drup.2022.100916
11. Tohkayomatee R, Reabroi S, Tungmunnithum D, Parichatikanond W, Pinthong D. Andrographolide exhibits anticancer activity against breast cancer cells (MCF-7 and MDA-MB-231 cells) through suppressing cell proliferation and inducing cell apoptosis via inactivation of ER-α receptor and PI3K/AKT/mTOR signaling. Molecules. 2022;27(11):3544. doi:10.3390/molecules27113544
12. Xu T, Jiang Y, Yuan S, et al. Andrographolide inhibits ER-positive breast cancer growth and enhances fulvestrant efficacy via ROS-FOXM1-ER-α axis. Front Oncol. 2022;12:899402. doi:10.3389/fonc.2022.899402
13. Shi L, Zhang G, Zheng Z, Lu B, Ji L. Andrographolide reduced VEGFA expression in hepatoma cancer cells by inactivating HIF-1α: the involvement of JNK and MTA1/HDCA. Chem Biol Interact. 2017;273:228–236. doi:10.1016/j.cbi.2017.06.024
14. Li J, Zhang C, Jiang H, Cheng J. Andrographolide inhibits hypoxia-inducible factor-1 through phosphatidylinositol 3-kinase/AKT pathway and suppresses breast cancer growth. Onco Targets Ther. 2015;8:427–435. doi:10.2147/OTT.S76116
15. Blanchard TG, Lapidus R, Banerjee V, et al. Upregulation of RASSF1A in colon cancer by suppression of angiogenesis signaling and Akt activation. Cell Physiol Biochem. 2018;48(3):1259–1273. doi:10.1159/000492012
16. Lu CY, Yang YC, Li CC, Liu KL, Lii CK, Chen HW. Andrographolide inhibits TNFα-induced ICAM-1 expression via suppression of NADPH oxidase activation and induction of HO-1 and GCLM expression through the PI3K/Akt/Nrf2 and PI3K/Akt/AP-1 pathways in human endothelial cells. Biochem Pharmacol. 2014;91(1):40–50. doi:10.1016/j.bcp.2014.06.024
17. Yuan M, Meng W, Liao W, Lian S. Andrographolide antagonizes TNF-α-induced IL-8 via inhibition of NADPH oxidase/ROS/NF-κB and Src/MAPKs/AP-1 axis in human colorectal cancer HCT116 cells. J Agric Food Chem. 2018;66(20):5139–5148. doi:10.1021/acs.jafc.8b00810
18. Li L, Yang LL, Yang SL, et al. Andrographolide suppresses breast cancer progression by modulating tumor-associated macrophage polarization through the Wnt/β-catenin pathway. Phytother Res. 2022;36(12):4587–4603. doi:10.1002/ptr.7578
19. Lai YH, Yu SL, Chen HY, Wang CC, Chen HW, Chen JJ. The HLJ1-targeting drug screening identified Chinese herb andrographolide that can suppress tumour growth and invasion in non-small-cell lung cancer. Carcinogenesis. 2013;34(5):1069–1080. doi:10.1093/carcin/bgt005
20. Chao CY, Lii CK, Hsu YT, et al. Induction of heme oxygenase-1 and inhibition of TPA-induced matrix metalloproteinase-9 expression by andrographolide in MCF-7 human breast cancer cells. Carcinogenesis. 2013;34(8):1843–1851. doi:10.1093/carcin/bgt131
21. Liu W, Fan T, Li M, et al. Andrographolide potentiates PD-1 blockade immunotherapy by inhibiting COX2-mediated PGE2 release. Int Immunopharmacol. 2020;81:106206. doi:10.1016/j.intimp.2020.106206
22. Dai L, Wang G, Pan W. Andrographolide inhibits proliferation and metastasis of SGC7901 gastric cancer cells. Biomed Res Int. 2017;2017:6242103. doi:10.1155/2017/6242103
23. Yang ES, Do Y, Cheon SY, et al. Andrographolide suppresses aerobic glycolysis and induces apoptotic cell death by inhibiting pyruvate dehydrogenase kinase 1 expression. Oncol Rep. 2023;49(4). doi:10.3892/or.2023.8509
24. Chen Z, Tang WJ, Zhou YH, Chen ZM, Liu K. Andrographolide inhibits non-small cell lung cancer cell proliferation through the activation of the mitochondrial apoptosis pathway and by reprogramming host glucose metabolism. Ann Transl Med. 2021;9(22):1701. doi:10.21037/atm-21-5975
25. Wang S, Li H, Chen S, et al. Andrographolide induces apoptosis in human osteosarcoma cells via the ROS/JNK pathway. Int J Oncol. 2020;56(6):1417–1428. doi:10.3892/ijo.2020.5032
26. Othman NS, Mohd Azman DK. Andrographolide induces G2/M cell cycle arrest and apoptosis in human glioblastoma DBTRG-05MG cell line via ERK1/2 /c-Myc/p53 signaling pathway. Molecules. 2022;27(19):6686. doi:10.3390/molecules27196686
27. Zhang HT, Yang J, Liang GH, et al. Andrographolide induces cell cycle arrest and apoptosis of chondrosarcoma by targeting TCF-1/SOX9 axis. J Cell Biochem. 2017;118(12):4575–4586. doi:10.1002/jcb.26122
28. Yang T, Yao S, Zhang X, Guo Y. Andrographolide inhibits growth of human T-cell acute lymphoblastic leukemia Jurkat cells by downregulation of PI3K/AKT and upregulation of p38 MAPK pathways. Drug Des Devel Ther. 2016;10:1389–1397. doi:10.2147/DDDT.S94983
29. Xuan L, Hu JH, Bi R, Liu SQ, Wang CX. Andrographolide inhibits proliferation and promotes apoptosis in bladder cancer cells by interfering with NF- κ B and PI3K/AKT signaling in vitro and in vivo. Chin J Integr Med. 2022;28(4):349–356. doi:10.1007/s11655-022-3464-4
30. Bao GQ, Shen BY, Pan CP, Zhang YJ, Shi MM, Peng CH. Andrographolide causes apoptosis via inactivation of STAT3 and Akt and potentiates antitumor activity of gemcitabine in pancreatic cancer. Toxicol Lett. 2013;222(1):23–35. doi:10.1016/j.toxlet.2013.06.241
31. Lim SC, Jeon HJ, Kee KH, Lee MJ, Hong R, Han SI. Andrographolide induces apoptotic and non-apoptotic death and enhances tumor necrosis factor-related apoptosis-inducing ligand-mediated apoptosis in gastric cancer cells. Oncol Lett. 2017;13(5):3837–3844. doi:10.3892/ol.2017.5923
32. Ye L, Wang T, Tang L, et al. Poor oral bioavailability of a promising anticancer agent andrographolide is due to extensive metabolism and efflux by P-glycoprotein. J Pharm Sci. 2011;100(11):5007–5017. doi:10.1002/jps.22693
33. Fan D, Cao Y, Cao M, Wang Y, Cao Y, Gong T. Nanomedicine in cancer therapy. Signal Transduct Target Ther. 2023;8(1):293. doi:10.1038/s41392-023-01536-y
34. Nam J, Son S, Park KS, Zou WP, Shea LD, Moon JJ. Cancer nanomedicine for combination cancer immunotherapy. Nature Rev Mater. 2019;4(6):398–414. doi:10.1038/s41578-019-0108-1
35. Wei X, Yang M. Cell- and subcellular organelle-targeting nanoparticle-mediated breast cancer therapy. Front Pharmacol. 2023;14:1180794. doi:10.3389/fphar.2023.1180794
36. Momcilovic M, Jones A, Bailey ST, et al. In vivo imaging of mitochondrial membrane potential in non-small-cell lung cancer. Nature. 2019;575(7782):380–384.
37. Jeena MT, Palanikumar L, Go EM, et al. Mitochondria localization induced self-assembly of peptide amphiphiles for cellular dysfunction. Nat Commun. 2017;8(1):26. doi:10.1038/s41467-017-00047-z
38. Faria R, Vivés E, Boisguerin P, Sousa A, Costa D. Development of peptide-based nanoparticles for mitochondrial plasmid DNA delivery. Polymers. 2021;13(11):19. doi:10.3390/polym13111836
39. Pegoraro C, Sanchis E, Dordevic S, et al. Multifunctional polypeptide-based nanoconjugates for targeted mitochondrial delivery and nonviral gene therapy. Chem Mat. 2025;37(4):1457–1467. doi:10.1021/acs.chemmater.4c02742
40. Yamada Y, Hibino M, Sasaki D, Abe J, Harashima H, Harashima H. Power of mitochondrial drug delivery systems to produce innovative nanomedicines. Adv Drug Delivery Rev. 2020;154:187–209. doi:10.1016/j.addr.2020.09.010
41. Yoshinaga N, Numata K. Rational designs at the forefront of mitochondria-targeted gene delivery: recent progress and future perspectives. ACS Biomater Sci Eng. 2022;8(2):348–359. doi:10.1021/acsbiomaterials.1c01114
42. Batheja S, Gupta S, Tejavath K, Gupta U. TPP-based conjugates: potential targeting ligands. Drug Discov Today. 2024;29(6):15. doi:10.1016/j.drudis.2024.103983
43. Yin-Hua Y, Qi G, Shan-Shan Z, Mi T. Preparation of curcumin TPP-PEG-PE nanomicelles with mitochondrial targeting and lysosomal escape functions and its effect on promoting breast cancer cell apoptosis. Zhongguo Zhong Yao Za Zhi. 2020;45(22):5495–5503. doi:10.19540/j.cnki.cjcmm.20200819.303
44. Zhou L, Du Y, Shang Y, Xiang D, Xia X. A novel triptolide nano-liposome with mitochondrial targeting for treatment of hepatocellular carcinoma. Int J Nanomed. 2024;19:12975–12998. doi:10.2147/IJN.S498099
45. Guo X, Yang N, Ji W, et al. Mito-bomb: targeting mitochondria for cancer therapy. Adv Mater. 2021;33(43).
46. Cho H, Cho YY, Shim MS, Lee JY, Lee HS, Kang HC. Mitochondria-targeted drug delivery in cancers. Biochimica Et Biophysica. 2020;1866(8). doi:10.1016/j.bbadis.2020.165808
47. Tao J, Ning W, Shi X, et al. Self-delivery nanomedicine for selective mitochondrial copper depletion and oxidative stress amplification. Acs Nano. 2025;19(37):33587–33600. doi:10.1021/acsnano.5c11565
48. Li WQ, Wu JY, Xiang DX, et al. Micelles loaded with puerarin and modified with triphenylphosphonium cation possess mitochondrial targeting and demonstrate enhanced protective effect against isoprenaline-induced H9c2 cells apoptosis. Int J Nanomed. 2019;14:8345–8360. doi:10.2147/IJN.S219670
49. Marrache S, Dhar S. Engineering of blended nanoparticle platform for delivery of mitochondria-acting therapeutics. Proc Natl Acad Sci USA. 2012;109(40):16288–16293. doi:10.1073/pnas.1210096109
50. Gao Z, Liu P. Improving drug release performance of phosphazene-based crosslinked polyprodrug nanoparticles for tumor chemotherapy by enhancing hydrophilicity with polyethylene glycol. Eur Polym J. 2025;237:8. doi:10.1016/j.eurpolymj.2025.114196
51. Li P, Li J, Cheng J, et al. Hypoxia-responsive liposome enhances intracellular delivery of photosensitizer for effective photodynamic therapy. J Control Release. 2025;377:277–287. doi:10.1016/j.jconrel.2024.11.032
52. Gao J, Wang Z, Guo Q, et al. Mitochondrion-targeted supramolecular “nano-boat” simultaneously inhibiting dual energy metabolism for tumor selective and synergistic chemo-radiotherapy. Theranostics. 2022;12(3):1286–1302. doi:10.7150/thno.67543
53. Wang Y, Zhang L, Zhang X, Wei X, Tang Z, Zhou S. Precise polymerization of a highly tumor microenvironment-responsive nanoplatform for strongly enhanced intracellular drug release. ACS Appl Mater Interfaces. 2016;8(9):5833–5846. doi:10.1021/acsami.5b11569
54. Zhang X, Wang Y, Wei G, Zhao J, Yang G, Zhou S. Stepwise dual targeting and dual responsive polymer micelles for mitochondrion therapy. J Control Release. 2020;322:157–169. doi:10.1016/j.jconrel.2020.03.011
55. Behl A, Solanki S, Paswan SK, et al. Biodegradable PEG-PCL nanoparticles for co-delivery of MUC1 inhibitor and doxorubicin for the confinement of triple-negative breast cancer. J Polym Environ. 2023;31(3):999–1018. doi:10.1007/s10924-022-02654-4
56. Kahana A, Lancet D. Self-reproducing catalytic micelles as nanoscopic protocell precursors. Nat Rev Chem. 2021;5(12):870–878. doi:10.1038/s41570-021-00329-7
57. Zeng Z, Luo Y, Xu X, et al. A mitochondria-targeting ROS-activated nanoprodrug for self-augmented antitumor oxidation therapy. J Control Release. 2023;359:415–427. doi:10.1016/j.jconrel.2023.06.004
58. Troiano L, Ferraresi R, Lugli E, et al. Multiparametric analysis of cells with different mitochondrial membrane potential during apoptosis by polychromatic flow cytometry. Nat Protoc. 2007;2(11):2719–2727. doi:10.1038/nprot.2007.405
59. Huang Q, Li F, Liu X, et al. Caspase 3-mediated stimulation of tumor cell repopulation during cancer radiotherapy. Nat Med. 2011;17(7):860–866. doi:10.1038/nm.2385
60. Olsson M, Zhivotovsky B. Caspases and cancer. Cell Death Differ. 2011;18(9):1441–1449. doi:10.1038/cdd.2011.30
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Triphenylphosphine-Based Mitochondrial Targeting Nanocarriers: Advancing Cancer Therapy

Ali MS, Gowda BHJ, Shukla R, Kesharwani P
Clinical Pharmacology: Advances and Applications 2025, 17:119-141
Published Date: 10 June 2025