Back to Journals » International Journal of Nanomedicine » Volume 15
In vitro Characterization and Release Studies of Combined Nonionic Surfactant-Based Vesicles for the Prolonged Delivery of an Immunosuppressant Model Drug
Authors Rasul A, Imran Khan M, Rehman MU ![]() , Abbas G
, Abbas G ![]() , Aslam N, Ahmad S, Abbas K
, Aslam N, Ahmad S, Abbas K ![]() , Akhtar Shah P, Iqbal M, Ahmed Al Subari AM, Shaheer T, Shah S
, Akhtar Shah P, Iqbal M, Ahmed Al Subari AM, Shaheer T, Shah S ![]()
Received 1 July 2020
Accepted for publication 12 September 2020
Published 14 October 2020 Volume 2020:15 Pages 7937—7949
DOI https://doi.org/10.2147/IJN.S268846
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Anderson Oliveira Lobo
Akhtar Rasul,1,* Muhammad Imran Khan,2,* Mujeeb Ur Rehman,1,* Ghulam Abbas,1,* Nosheen Aslam,3,* Shabbir Ahmad,4,* Khizar Abbas,5,* Pervaiz Akhtar Shah,6,* Muhammad Iqbal,7,* Ali Mohammad Ahmed Al Subari,6,* Talal Shaheer,8,* Shahid Shah9,*
1Department of Pharmaceutics, Faculty of Pharmaceutical Sciences, Government College University, Faisalabad, Pakistan; 2Riphah Institute of Pharmaceutical Sciences, Riphah International University, Lahore Campus, Lahore, Pakistan; 3Department of Biochemistry, Government College University, Faisalabad, Pakistan; 4Cancer Center, Faculty of Health Sciences, University of Macau, Taipa, Macau, People’s Republic of China; 5Department of Pharmacognosy, Faculty of Pharmacy, Bahauddin Zakariya University, Multan, Pakistan; 6University College of Pharmacy, University of the Punjab, Lahore, Pakistan; 7Faculty of Pharmacy, Gomal University, Dera Ismail Khan, Pakistan; 8Department of Pharmacognosy, Xian Jiaotong University, Xian, People’s Republic of China; 9Department of Pharmacy Practice, Faculty of Pharmaceutical Sciences, Government College University, Faisalabad, Pakistan
*All authors contributed equally to this work
Correspondence: Akhtar Rasul Email [email protected]
Background: Cyclosporine A (CsA) is an exceptional immunosuppressant used for the treatment of immune disorders. Niosomal vesicles are promising drug carriers that are formed by self-association of nonionic surfactants and cholesterol in an aqueous phase. The objective of the study was to formulate combined nonionic surfactant based vesicles and to evaluate their in vitro characterization, release studies and in vivo studies.
Materials and Methods: Five niosomal formulations (F7 to F11) were prepared using the thin film hydration method. The molar ratio of cholesterol and non-ionic surfactant taken was 1:1. In formulation F10, the combination of surfactants Span 20 and Brij 35 was used. The niosomes were characterized by zeta sizer and SEM for particle size analysis, in vitro drug release and stability studies. The pharmacokinetic studies were conducted on healthy albino rabbits.
Results: The size of niosome was found in the range of 427.1 nm to 972.3 nm. SEM image of optimized formulations F10 exhibit the spherical nature of niosomal vesicles. DSC thermograms of niosomal formulations exhibited a broadened endothermic peak. The stability study exhibited that all formulations are stable and negligible change of vesicle size and entrapment was observed with time. The percentage drug release was significantly higher as compared to CsA plain dispersion for all niosomal formulations at pH 1.2 and 7.4. The release kinetic behavior showed that all preparations were best described by zero order and can release active ingredient in a sustained manner. The pharmacokinetic data showed the test formulation (F10) possessed greater bioavailability as compared to the reference formulation (CsA aqueous dispersion).
Conclusion: The formulation F10 demonstrated a comparatively more delayed rate of release with enhanced dissolution as compared to a single surfactant scheme. The F10 formulation can be a remarkable nanotechnology for prolonged delivery of CsA orally with improved dissolution profile and bioavailability.
Keywords: in vitro study, cyclosporine A, niosomes, nonionic surfactants
Introduction
An ideal drug delivery system must transport the drug to the site of action and effectively release it over a predetermined amount of time.1 Colloidal particulate carriers like niosomes and liposomes act as drug delivery systems having distinctive advantages over conventional dosage forms.2 Niosomes can act as drug reservoirs from which the active pharmaceutical ingredient can be released at the targeted site and the release of drug can be adjusted by changing the composition of the niosomes.3
Cyclosporine A (CsA) has revolutionized transplant medicine. Initially discovered while searching for novel antifungal agents, it was established to have many immunologic properties that made it a peculiar agent for immunosuppression following renal and other solid organ transplants.4 The data from United Network for Organ Sharing (UNOS) from 1998 to 2007 reveals current one-year survival rates to be 96.6. CsA has nephrotoxic effects but it was found that CsA nephrotoxicity may be dose dependent and reversible upon dose reductions or discontinuation of the drug.5 CsA is an immunosuppressive drug that acts selectively on T-cells by inhibiting calcineurin phosphorylase. It is also used in psoriasis and for the treatment of various inflammatory skin conditions, including atopic dermatitis, blistering disorders, and connective tissue diseases.6
Niosomes reduce the nephrotoxicity of the drugs as exhibited in the study of niosomes of nystatin.7 Niosomes behave in vivo like liposomes, prolonging the circulation of the entrapped drug and altering its organ distribution and metabolic stability. Furthermore they are more stable and economical than liposomes.8
In this study, niosomes were developed to improve the solubility profile of CsA and to enhance the oral bioavailability. The nonionic surfactants were used to enhance the solubility and bioavailability of CsA. Five niosomal formulations were developed, ie, F7, F8, F9, F10 and F11. In formulations F7, F8 and F9 nonionic surfactants span 20, brij 35 and span 60 were used respectively. The formulation F10 contained a combination of span 20 and brij 35 along with an equimolar concentration of cholesterol and in formulation F11 span 60 and brij 35 were used along with an equimolar concentration of cholesterol (1:1). These nonionic surfactants were also used previously in research for development of niosomes, eg, in a study by Yoshioka et al span 60 and span 80 were used successfully to prepare niosomes.9 Span 20 and brij 35 were also used for niosomal formulation of salbutamol along with cholesterol.10 In the current study these nonionic surfactants were used for the first time to prepare niosomes of CsA. Then, in vitro characterization, drug release and bioavailability studies of these developed formulations were investigated.
Materials and Methods
Materials
Polyoxyl (23) lauryl ether (brij 35) was purchased from Avonchem Ltd, Macclesfield, Cheshire SK11 6PJ, United Kingdom. Sorbitan monostearate (span 60), and sorbitan monododecanoate (span 20) were procured from Daejung Chemicals & Metals Co., Ltd. CsA was obtained from Xi’an Lyphar Biotech Co., Ltd, China. Cholesterol and 1-hexadecyl pyridinium chloride monohydrate were acquired from Alfa Aesar GmbH & Co., KG. Chloroform and methyl alcohol were taken from Daejung Chemicals and Metals Co., Ltd, Korea. Distilled water was prepared in the pharmaceutics laboratory, Department of Pharmaceutics, Government College University, Faisalabad, Pakistan.
Solubility Studies
The solubility studies were carried out in phosphate buffer saline pH 1.2 and 7.4. Fixed amount of drug was added to the phosphate buffer saline (pH 1.2 and 7.4) containing various concentrations of surfactant (span 20, brij 35 and span 60) and cholesterol. These samples were placed on an orbit shaker for 24 h at 37 ºC. Samples were collected and filtered through a membrane filter (0.45 µm). The concentration of CsA was determined using a UV-Visible spectrophotometer (PerkinElmer, USA) at the wavelength of 210 nm with a proper dilution.
Preparation of Niosomal Formulations
Thin film hydration method was used for the preparation of niosomal formulations of immunosuppressant drug (CsA).11 In a 250 mL round-bottomed flask appropriate quantities of surfactant and cholesterol in different molar ratios as given in Table 1 were dissolved in 15 mL of chloroform/methanol mixture (2:1, v/v). In each formulation 25 mg (21 µmoles) of CsA and 2.5% of charge inducer 1-Hexadecyl pyridinium chloride monohydrate was incorporated. This mixture was allowed to rotate in a rotary evaporator at 60–65 °C under vacuum until a thin film was formed on the wall of the flask. This thin film was then hydrated with 20 mL phosphate buffer saline (pH 7.4), in a water bath along with gentle stirring at 60 °C for 1 h. Then this niosomal system was allowed to mature overnight at 4 °C.12
 |
Table 1 Composition of Different Niosomal Formulations |
Preparation of Physical Mixtures
For the preparation of physical mixtures equimolar quantities of CsA, cholesterol and nonionic surfactants were mixed in a pestle and mortar. Physical mixture 3 (PM3) was prepared by span 20, brij 35, cholesterol and CsA, and physical mixture 4 (PM4) was prepared by span 60, brij 35, cholesterol and CsA in ratio (1:1:1:1). In mortar the constituents were mixed by pestle for about 15 min to prepare a homogenous physical mixtures.13
Thermal Analysis
Thermal properties of CsA, niosomal formulations, physical mixtures and other ingredients were investigated with differential scanning calorimetry (DSC) (TA Instruments SDT-Q600 Simultaneous TGA/DSC). Samples were placed in an aluminum pan and sealed. The rate of purging was taken at 50 mL min−1. Thermograms were taken at a scan rate of 10 ºC min−1 by heating the samples from 10 ºC to 250 ºC under nitrogen atmosphere.14
Measurement of Size and Zeta Potential of Vesicles
The mean size of niosomal vesicles, polydispersity index (PDI) and zeta potential were evaluated using zetasizer (Malvern zetasizer version 7.11, UK) at 24 °C. The niosomal formulations were diluted with double distilled water for analysis. All measurements were conducted in triplicate and then results were presented as a mean along with standard deviation.14
Study of Morphology of Niosomes
To study the shape and morphology of niosomes, scanning electron microscopy was used. A drop of optimized niosomal formulation F10 was taken and put on an aluminum stub with adhesive silver tape. Under vacuum, aluminum stubs were stored overnight. Then, by utilizing gold film of thickness 0.20 µm, sputter coated. At suitable magnification photographs were taken.15,16 Energy-dispersive X-ray analysis (EDX) of selected niosomal formulation (F10) was also done for elemental analysis. In this study the standards used were CaCO3 (C), SiO2 (O), Albite (Na) and KCl (Cl).17
Drug Entrapment Studies
The Agilentz1200 series HPLC technique was employed to analyze CsA. The system was furnished with autosampler, quaternary pump and heated column compartment.18 In it G1315B Diode-array detector (DAD) was employed. For drug entrapment studies of niosomes ultracentrifugation method was utilized at 12,000 × g. Ultracentrifugation was done at 4 °C for 30 minutes. To disrupt the niosomes methanol was used.19 The mobile phase used was water-acetonitrile in 3:7 (v/v). The column used was nucleosil C18 (25 cm × 3.2 mm, 5 µm particle size) and operated at 70 °C. The flow rate was taken at 1.2 mL/min. Injections using 20 µL fixed loop were made and chromatograms were recorded at 210 nm.20
Stability Studies
The stability of CsA loaded niosomes was examined at different storage conditions. The stability was determined at two different conditions as mentioned in ICH stability guidelines. The niosomal formulations were stored in a refrigerator at temperature (4–8 °C) and a climatic chamber at 24±2 ºC in transparent vessels. Specimens were taken at appropriate time intervals for up to 3 months. Vesicle size, zeta potential and entrapment was evaluated to characterize the stability profile. Visual examination was also done to evaluate any color changes in formulation.15,21
Drug Release Studies
In vitro release of niosomal formulations (F7-F11) and drug aqueous suspension was studied using a dialysis bag. Release studies were performed at gastric pH (1.2) and intestinal pH (7.4) by using phosphate buffer saline (PBS). Over night a dialysis membrane was soaked in distilled water. One end was sealed with a clip then 1 mL of formulation or aqueous drug suspension was pipetted into the dialysis membrane and the bag was sealed with another closure clip to prevent leakage. The dialysis bag was placed in 100 mL of PBS of pH 1.2 or pH 7.4 at 37 ± 2 °C. The medium acted as receptor compartment and was stirred at 100 rpm. Samples of medium (2 mL) were withdrawn at 0, 0.25, 0.50, 0.75, 1, 1.25, 1.75, 2.5, 4, 5.5, 8, 12, 16, 20 and 24 h. The same amount of fresh PBS was added when sample was collected.22 The samples were analyzed by utilizing HPLC.18 Results were the mean value of three runs. The mechanism of release of CsA was determined by applying mathematical models like zero order kinetics, first order kinetics, higuchi kinetics and korsmeyer-Peppas models.
Pharmacokinetic Studies
The niosomal preparations (F10) were studied for in vivo performance of niosomal vesicles in twelve healthy albino rabbits. The animals received care in compliance to the principles of Laboratory animal care and the guide for the care and use of laboratory animals. The study complies with the ‘3Rs’ and ARRIVE guidelines for use of animals in research. The F10 and CsA aqueous dispersion were respectively administered to rabbits by oral route at an equivalent dose of 10 mg/kg CsA.23,24 Single dose (non-crossover design) was used for in vivo study of optimized niosomal preparations. Twelve healthy male albino rabbits weighing 1.2–1.6 Kg (average weight of 1.5 Kg) were chosen. Each carrier of CsA was administered to a group with 6 rabbits (n= 6). Rabbits were retained in a light controlled room. Its average temperature was maintained at (22±2 °C) and humidity at (50–60%). The rabbits were not allowed to eat for twelve hours, before the administration of dose with sufficiently available water ad libitum.25 On the day of experiment, rabbits were engaged in restrainers of metal. Dose of CsA (10 mg/Kg) of two carriers of CsA were administered by oral route to each group of rabbits by utilizing a flexible catheter. After administration of the dose, samples of blood (1 mL) were drawn from the jugular vein of the rabbits at 0, 0.5, 0.75, 1, 1.5, 2, 2.5, 3, 4, 6, 8, 12 and 24 hours. The samples of blood were collected in EDTA tubes. After collecting blood samples the rabbits were reverted to their appropriate cages.23 A simple and rapid HPLC method was used for estimation of CsA in rabbit plasma. The Agilentz1200 series HPLC technique was employed to analyze CsA. The system was furnished with autosampler, quaternary pump, and heated column compartment. In it a G1315B Diode-array detector (DAD) was employed and the column used was Nucleosil C18 (25 cm ×z3.2 mm, 5 µm particle size). The effluents were evaluated at a wavelength of 210 nm. To prepare the sample for HPLC analysis the protein precipitation method was used. Plasma level time profiles of niosomal preparations and aqueous dispersion were evaluated for twenty-four hours. Microsoft Excel based Pk solver was used to determine pharmacokinetic parameters.
Data Analysis and Statistics
Data are expressed as mean ± SD. Analysis of variance (ANOVA) was used to evaluate results of mean values of size of niosomes, entrapment efficiency of niosomal preparations. ANOVA was conducted at 95% confidence interval by GraphPad Prism 6 software. The significance was defined at p values < 0.05.
Results and Discussion
Solubility Studies
The effect of different concentration surfactants on the solubility of CsA were studied at pH 1.2 and 7.4. The combination of span 60, cholesterol and brij 35 enhance the solubility of the drug up to 7180% at pH 1.2 and 7.4 respectively as shown in Figure 1. The use of span 20, cholesterol and brij 35 in combined form significantly enhance the solubility of CsA by more than 9096% at pH 1.2 and 7.4 respectively as shown in Figure 1. The maximum solubility was observed at pH 7.4. The similar findings of solubility studies of carvedilol using surfactant were previously reported by Incecayir, in 2015.26
 |
Figure 1 Solubility profile of CsA with nonionic surfactants at pH 1.2 and 7.4. |
Preparation of Niosomes
Niosomes of CsA were successfully prepared by thin film hydration method as reported earlier with slight modifications. The different ratios of surfactants and cholesterol used in niosomal formulation are given in Table 1. In formulation F7, F8 and F9, span 20, Brij 35 and span 60 were used along with cholesterol in molar ratio 1:1. In formulation F10 a combination of span 20 and Brij 35 was used along with cholesterol, and in formulation F11 a combination of span 60 and Brij 35 was used along with cholesterol. 1-Hexadecyl pyridinium chloride monohydrate was added to the formulations that act as a positive charge inducer. It provides more effective drug delivery and keeps the niosomal formulation stable for a long period of time.
Thermal Analysis
Pure raw ingredients, physical mixtures and formulations of niosomes were analyzed through thermal analysis. In Figure 2, DSC thermograms of span 20, brij 35, physical mixture 3 (PM3), physical mixture 4 (PM4) and F7, F8, F9, F10 and F11 are demonstrated. DSC thermograms of brij 35 and span 20 depicted the peculiar endothermic melting peaks. The DSC thermograms of physical mixtures and F7-F11 showed the melting range of ingredients and CsA.27,28 DSC thermograms of niosomal formulations exhibited a broadened endothermic peak which showed improved solubility, dissolution and the sustained release nature of formulations.29 The outcomes of DSC demonstrated that among the active and inactive ingredients used in this niosomal formulation there is no significant interaction.30
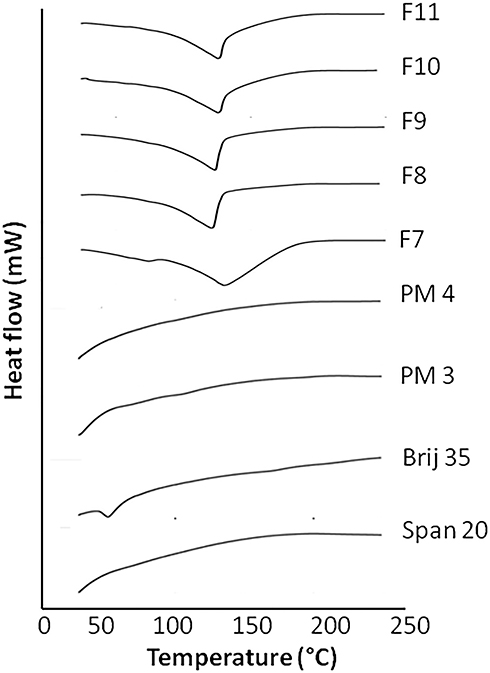 |
Figure 2 DSC thermograms of Span 20, Brij 35, physical mixture 3 (PM3), physical mixture 4 (PM4), F7, F8, F9, F10 and F11 formulations. |
Size and Zeta Potential of Niosomal Vesicles
The mean size, polydispersity index (PDI) and zeta potential of niosomal preparations is given in Table 2. All niosomes have a size range of 427.1 nm to 972.3 nm. The graphical representation is given in Figure 3A. Cholesterol is one of the important constituents of niosomes that can influence their physicochemical characteristics and stability. The molar ratio of surfactant and cholesterol was taken as 1:1. The addition of cholesterol can enhance the bilayer hydrophobicity, leading to a decrease in the surface free energy and therefore a decrease of particle size and enhanced entrapment efficiency.31
 |
Table 2 Mean Size, Polydispersity Index and Zeta Potential of Niosomal Formulations |
 |
Figure 3 Size (A) and zeta potential (B) of niosomal formulations. |
It is well established that the size of niosomes is dependent on the length of the alkyl chain of the surfactants. Surfactants with longer alkyl chains generally give larger vesicles. This might be the reason for the smaller particle size of span 20 and larger particle size of span 60 niosome. Span 20 showed comparatively a lesser degree of polydispersity. The size of niosomes depends upon the chain length of surfactant. As the length carbon increases, the size of niosomes also increases. In this study the size of niosomes of formulation F9 and F11 containing span 60 are higher than other formulations. It was due to the fact that Span 60 has a longer saturated alkyl chain as compared to span 20 and it was reported that surfactants with longer alkyl chains generally give larger vesicles.32
The range of polydispersity index found was 0.259 to 0.497. This value was considered to be within the range sufficient for attaining stable and aggregation resistant systems.33 The range of zeta potential of niosomes was from 26.2 to 35.3 mV. The graphical representation of zeta potential is given in Figure 3B. These values are sufficient to provide acceptable repulsion between vesicles to prevent the aggregation and provide stable niosomes.21
Morphology of Niosomes
Scanning electron microscopy (SEM) was employed for morphological study. The SEM image of optimized formulation F10 is presented in Figure 4. In the current study the SEM image of optimized formulation F10 exhibits the spherical nature of niosomal vesicles, which was also alike to the niosomes of naltrexone prepared by using sorbitan monostearate.34 Furthermore the mean niosome size was in good agreement with the size that was obtained by Malvern zetasizer. SEM of formulation F10 showed the smooth surface of niosomes formed. Some unevenness of vesicles that were observed under the study may be due to the drying process. Similar results were exhibited in niosomes of cefdinir.16
 |
Figure 4 SEM appearance of optimized formulation F10. |
EDX spectra were used for the elemental analysis or chemical characterization of a sample. EDX spectra of selected niosomal formulation F10 is shown in Figure 5 and in Table 3 atomic % and weight % of elements in formulation F10 are demonstrated. The atomic % of C K, Na K, Cl K and Na K was 77.95, 10.60, 8.57 and 2.87 respectively.
 |
Table 3 Atomic % and Weight % of Elements in EDX Spectra of Formulation F10 |
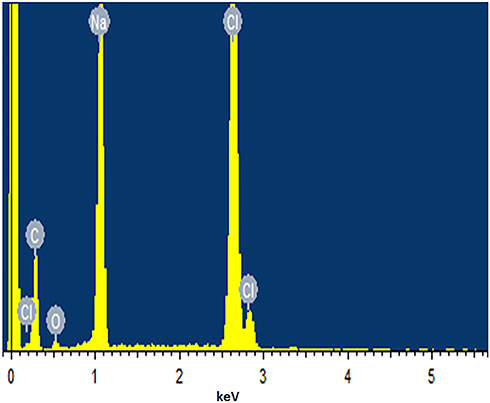 |
Figure 5 EDX spectra of selected niosomal formulation F10. |
Drug Entrapment Studies
Niosomes were separated using ultracentrifugation, and the percentage entrapment efficiency of CsA in different formulations determined by HPLC is given below in Table 4. The niosomal formulation F10 demonstrated the highest entrapment efficiency of 89.31%. In formulations F7, F8 and F9 nonionic surfactants span 20, brij 35 and span 60 were used along with cholesterol in ratio (1:1) respectively as shown in Table 4. In formulation F10 a combination of span 20 and brij 35 was used along with cholesterol and in formulation F11 span 60 and brij 35 were used along with cholesterol in equimolar ratio (1:1). Formulation F10 showed maximum entrapment efficiency (89.31 ± 0.37).
 |
Table 4 Mean Entrapment Efficiency of Niosomal Formulations |
The explanation for this fact was due to a combination of brij 35 and span 20 surfactants in niosomes, which result in more stable and less leaky niosomal vesicles. Brij 35 has a lengthy lauryl (c12) chain, so the long chain effects the HLB of surfactants and also results in a raised drug entrapment and improved stability. Cholesterol was also a very significant additive added in the niosomal preparation to formulate stable niosomes. Cholesterol prevents leakiness, stabilizes bilayer, and reduces pervasion of solutes encircled in the aqueous core of niosomes. The molar ratio of cholesterol and non-ionic surfactant was 1:1, and this exhibited better results in the case of F10. Similarly, in a study of proniosomal gel of flurbiprofen, the surfactant brij 35 was utilized which resulted in high entrapment efficiency.35 In a combination of surfactants, span 20 gives high entrapment efficiency, as shown in a study of niosomes of diacerein, a combination of sorbitan monolaurate and poloxamer 184 results in high entrapment efficiency.36
Stability Studies of Niosomal Formulations
For stability studies at the end of each month the vesicle size and zeta potential of CsA niosomes was evaluated for the duration of 3 months. The variation in vesicle size, PDI and zeta potential of niosomal formulations F7–F11 at temperature 4–8 °C and 25±2 °C over three months is presented in Tables 5 and 6 respectively. For stability studies the entrapment efficiency was also evaluated at the termination of each month for the duration of three months. For stability studies the % entrapment efficiency of niosomal formulations F7, F8, F9, F10, and F11 were determined at temperature 4–8 °C and at 25±2 °C at 0, 1, 2 and 3 month intervals and presented in Table 7.
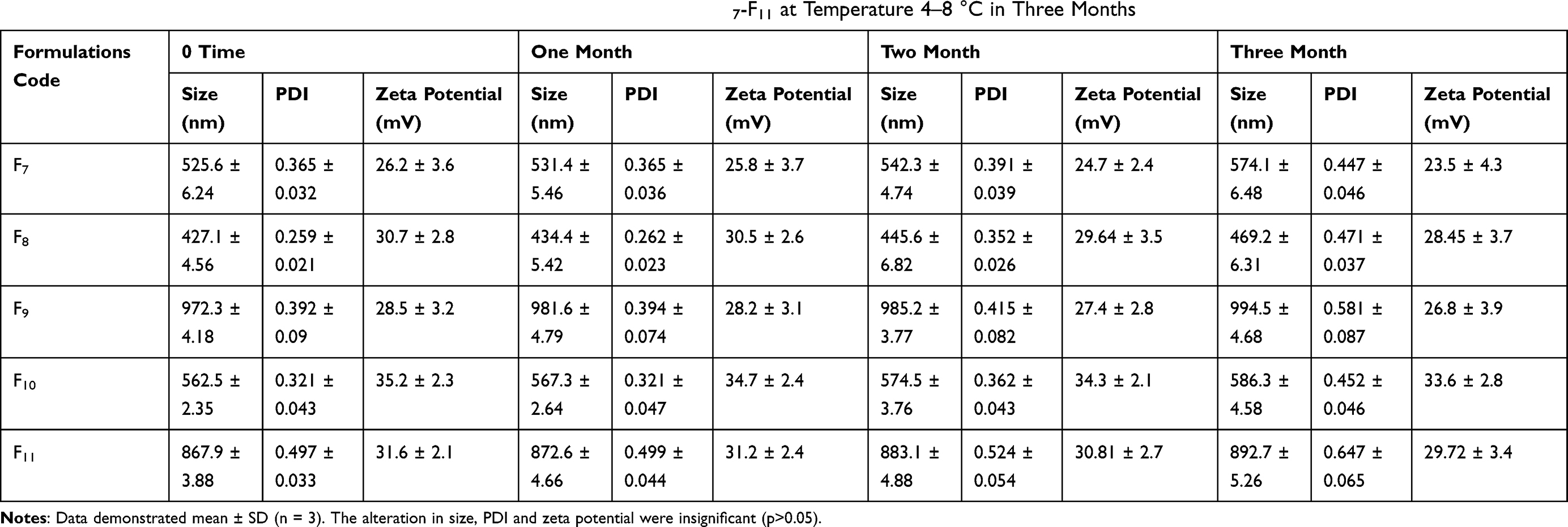 |
Table 5 Change in Vesicle Size, PDI and Zeta Potential of Niosomal Formulations F7-F11 at Temperature 4–8 °C in Three Months |
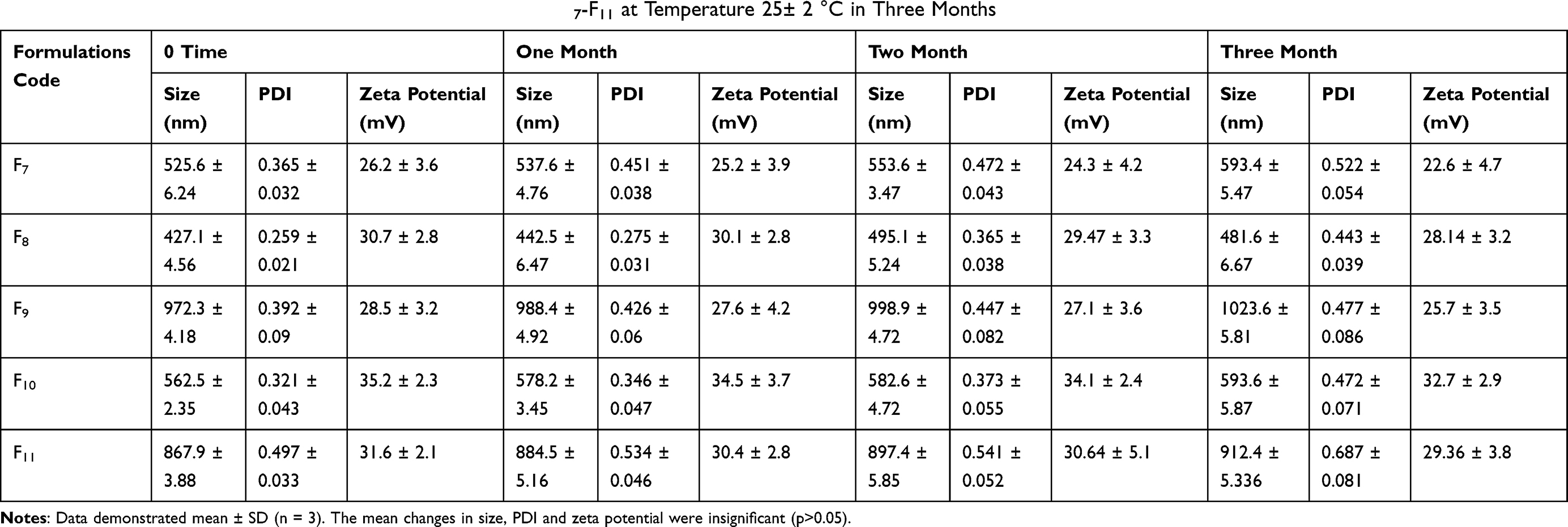 |
Table 6 Change in Size, PDI and Zeta Potential of Preparations F7-F11 at Temperature 25± 2 °C in Three Months |
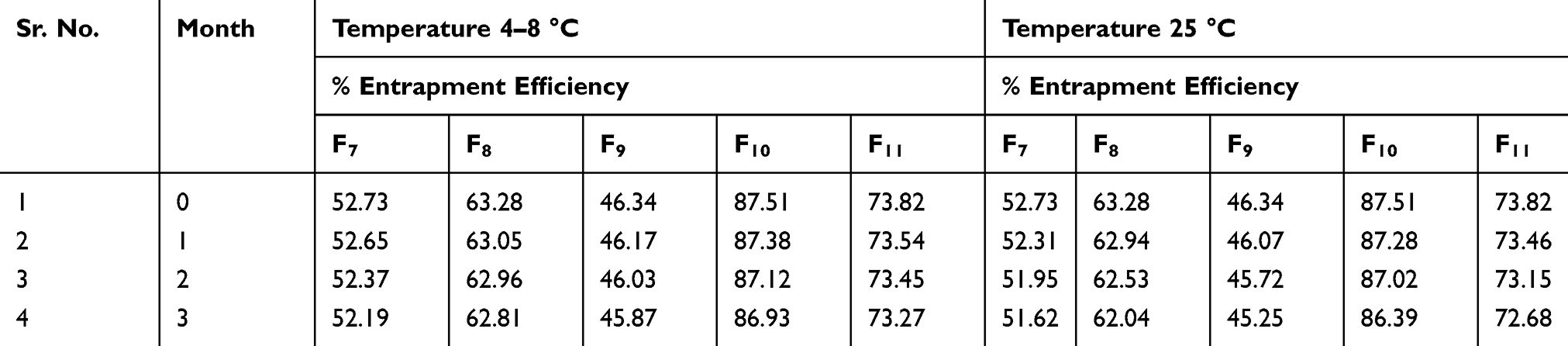 |
Table 7 Stability Studies of Cyclosporine a Niosomes at Different Temperatures |
The results demonstrated that with the passage of time there is a slight change in vesicle size and zeta potential. The variation in size of niosomes was comparatively greater at 25 °C than 4 °C as shown in Tables 5 and 6. This is due to the temperature effect on niosomal preparations. This might be due to the merging of vesicles, which occur owing to molecular movements with the passage of time.37 Cholesterol has been used in niosomes to enhance stability. In general all preparations were found stable.
The percentage entrapment of selected niosomal formulation F10 was decreased from 87.51 to 86.93% at 4°C. Similarly, at 25°C, it was decreased from 87.51 to 86.39%. In the current study the competency of niosomal vesicles to maintain the entrapment efficacy is in good agreement with preceding studies.38 It was further concluded that at refrigerated temperature 4–8 °C the amount of drug retained in niosomes was greater than at 25 °C. Visual examination shows no significant color change after three months in formulations at 4–8 °C, but at 25 °C the color of formulation slightly fades or becomes a bit light yellow. So these niosomes can be an effective formulation with good stability but it is better to store them at 4–8 °C. In stability studies, CsA leakage at high temperature was maybe due to the high flexibility of lipid bilayers.39
Drug Release Studies
The release data of CsA, which was developed by formulations F7, F8, F9, F10, and F11 and drug aqueous suspension in phosphate buffer saline solutions at two dissimilar pH (1.2 and 7.4), are presented in Figures 6 and 7 respectively. The percentage drug release was significantly higher as compared to CsA plain dispersion for all niosomal formulations at pH 1.2 and 7.4 as presented in Figures 6 and 7. This showed the solubilization effect of CsA in the surfactant vesicles, which resulted in augmented release of the drug. The dissolution profile was also improved in all the formulations. At pH 1.2 the percentage of drug released in 24 hours by F7-F11 was 82%, 87%, 91%, 71% and 76% respectively. And percentage drug released at pH 7.4 in 24 hours by F7-F11 was 63%, 67%, 73%, 53% and 60% respectively.
 |
Figure 6 Drug release profiles of formulations F7-F11 and drug aqueous suspension at pH 1.2. |
 |
Figure 7 Drug release profiles of formulations F7-F11 and drug aqueous suspension at pH 7.4. |
In vitro release studies showed sustained release behavior in all formulations (F7–F11). At low pH 1.2 the cumulative drug release was greater as compared to at pH 7.4, this may be due to the effect of pH on release of CsA. Similar results were achieved in the in vitro release of diacerein niosomal vesicles36 and niosomal formulations for prolonged delivery of clarithromycin.40 So it was established that formulated niosomal vesicles are competitive candidates for increasing the solubility of CsA and can result in improved bioavailability.
Release Kinetic Behaviour
The in vitro release data was applied to different kinetic models to determine the mechanism of release of drug from niosomal formulations. The constant of release and regression coefficient (r2) was evaluated from the slope of suitable plots. The data of release studies of niosomal preparations at pH 1.2 and 7.4 was fitted into zero order, first order, Higuchi model and Korsemeyer Peppas models. Kinetic modeling of formulations F7 to F11 at pH 1.2 and pH 7.4 is presented in Table 8.
 |
Table 8 Drug Release Data and Kinetic Modeling of Formulations F7-F11 at pH 1.2 and 7.4 |
All formulations exhibited raised R2 values for zero order kinetic as associated to first order. The results established that all preparations were best described by zero order, and can release active ingredients in a sustained manner. The dosage forms which follow zero order kinetics release equal quantity of active pharmaceutical agent by unit of time and it is the best technique of drug release to accomplish a delayed effect.41 The data of in vitro release demonstrated that the release of CsA from all niosomal preparations was most fitted to the Higuchi model which is a diffusion-controlled mechanism. So high R2 values were detected in the Higuchi plots, and the drug release was very close to Higuchi kinetics in all preparations. So the drug diffuses at a slower rate as the distance for diffusion increases, referred to the square root kinetics. These findings are consistent with niosomes of metoprolol. In which the data of in vitro release showed that the release of metoprolol from niosomal dispersion and film was best fitted to the Higuchi model.42
The model of Korsemeyer-Peppas was applied to all preparations and n value was determined. It indicated a good linearity for all the preparations. For niosomal formulations F7 to F11 the values of “n” were in the range of 0.531 to 0.821. In these formulations the release exponent (n) suggests the drug transport mechanism is non-fickian (anomalous). The R2 value for selected niosomal formulation F10 is comparatively high for the Higuchi equation. The n value of selected niosomal formulation F10 at pH 1.2 and 7.4 is 0.531 and 0.562 respectively, so the drug transport mechanism is through anomalous transport which depicts a coupling of erosion and diffusion mechanisms.41 These findings were parallel to release kinetics modeling of niosomes of tenofovir disoproxil fumarate. The Korsmeyer-Peppas model exhibited good linearity for all preparations.19 The Korsmeyer-Peppas models values of release exponent (n) established in our research were similar with the niosomal preparation of aceclofenac. In this niosomal formulation of aceclofenac follows model of Korsmeyer-Peppas. The “n” value was found in the range of 0.60 to 0.79 which also indicates that release is non fickian (anomalous), ie, a combination of erosion and diffusion.43 Similarly, in the proniosomes of risperidone the n value was found to be 0.7751 which also exhibited an anomalous release of the drug.44
Pharmacokinetic Studies
In the bioavailability study, F10 (test formulation) and aqueous drug dispersion (reference formulation) were used. The concentration of CsA in whole blood was determined after a single dosage administration to healthy albino rabbits. The graphical presentation of blood concentration verses time plots of optimized niosomal formulations F10 and aqueous drug dispersion is presented in Figure 8. The pharmacokinetic parameters like Cmax, Tmax, AUC0-t, and MRT0-inf_obs, etc are demonstrated in Table 9. Niosomal formulation F10 exhibited a higher blood concentration of CsA than the aqueous dispersion of CsA. At all data points the difference was significant (p<0.05) for both niosomal preparations F10. The high values of blood drug concentrations may be due to the increased retention of niosomes in the gastrointestinal tract and due to lipids and nonionic surfactants.45 It may also be due to lipophilicity of the components of the formulation and the small size of the vesicles.46 It was observed that niosomal formulation F10 exhibited relatively greater blood concentrations as compared to the aqueous dispersion. The increased bioavailability of these formulations was maybe the result of well-packed bimolecular film formation by mixed nonionic surfactants, uniform vesicle size, and higher lipid concentration was incorporated in niosomes to establish a positive charge, for the reason that charged vesicles are described to be additionally proficient for drug delivery.47
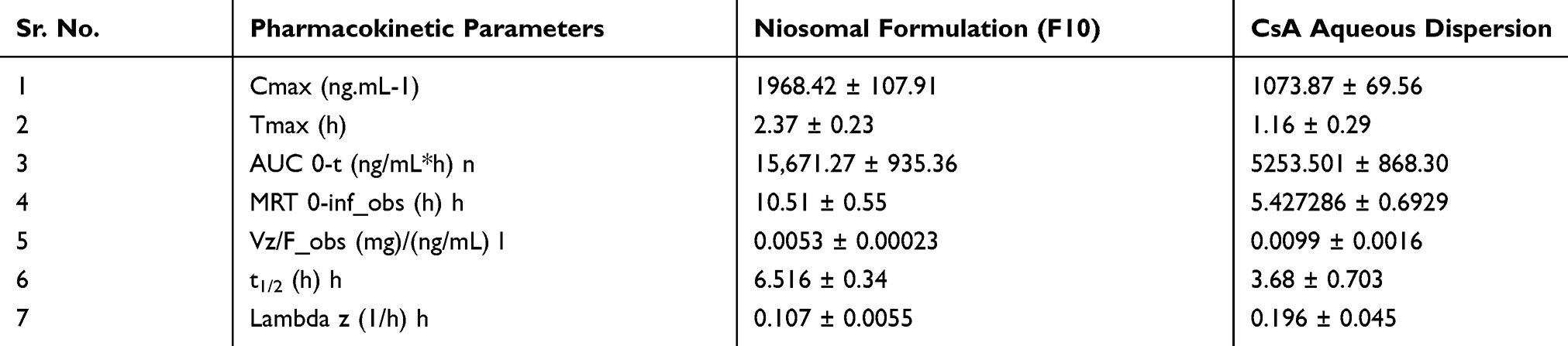 |
Table 9 Mean Pharmacokinetic Parameters of Niosomal Formulations F10 and CsA Aqueous Dispersion Administered in Dose (10 Mg/Kg) Orally to Albino Rabbits (n=6) |
 |
Figure 8 Pharmacokinetic profile of F10 niosomal formulation and aqueous dispersion of CsA in healthy albino rabbits. |
The mean ± SD values of Cmax for the F10 formulation was calculated as 1968.419 ± 107.91 ng/mL and it was 1073.87 ± 69.56 ng/mL for FTS and aqueous drug dispersion (reference formulation). ANOVA results showed a significant difference in the Cmax values (P<0.05) for niosomal formulation and Cmax of the CsA aqueous dispersion. The greater value of Cmax of niosomal formulations as compared to aqueous drug dispersion clearly indicates the enhanced bioavailability of CsA. These outcomes might be the result of the impact of nonionic surfactants and cholesterol. Generally, colloidal systems have a tendency to improve the bioavailability of BCS class II drugs orally like CsA, which exhibit low aqueous solubility and high permeability. In the absorption of BCS class II drugs, dissolution is the rate-limiting step. So a slight increase in dissolution rate owing to a raised surface area can result in substantial improvement in oral absorption.46 Raised blood drug concentrations of niosomal formulations might be due to the nonionic surfactants, which act as penetration enhancers. Other factors include improved penetration of mucosa which results from the lipophilic nature of niosomal vesicles, small size of vesicles and higher rate of dissolution and solubility in contrast to aqueous suspensions. Furthermore, the surface charge of the F10 formulation was 35.2 mV. This may also enhance vesicle uptake and transcytosis through M-cells of Peyer’s patches in the small intestine.48
In the current investigation, the mean ± SD values of AUC0-t for niosomal formulation F10 were 15,671.27 ± 935.36 ng.h.mL−1. Whereas it was 5253.501 ± 868.30 ng.h.mL−1 for the control plain dispersion. The results of ANOVA demonstrated that values of AUC0-t of both the niosomal formulation was significantly higher (p<0.05) as compared to the control plain dispersion. This may be due to the presence of hydrophobic surfactants, ie, span 20 and brij 35, in the formulation of F10. MRT is the mean residence time of the unchanged drug in the systemic circulation.49 The value of MRT0-inf of F10 niosomal formulations were calculated as 10.50651 ± 0.5519 hrs and the values of MRT 0-inf for control plain dispersion of the CsA was 5.427286 ± 0.6929 hrs. The values of half-life (t1/2) determined in the current study were 3.683 ± 0.703 hrs and 6.515 ± 0.3380 hrs for control plain dispersion and F10 formulation respectively. The results of ANOVA demonstrated a significant difference between the t1/2 of niosomal formulations and plain. The improved half-life of niosomal formulations can be explained by the effect of nonionic surfactants.
Conclusion
The niosomal formulations of CsA were successfully developed by using span 20, brij 35 and span 60. The mixed surfactant system of span 20 and brij 35 gives maximum entrapment of 89.31% along with cholesterol in formulation F10. DSC thermograms of niosomal formulations exhibited a broadened endothermic peak which showed improved solubility, dissolution and the sustained release nature of formulations. In vitro release studies showed that the dissolution profile was improved. Furthermore all formulations exhibit sustain release behavior. These formulations follow zero order kinetic models. The Korsemeyer-Peppas model showed that release of the drug is non fickian (anomalous). So it was concluded that CsA can be encapsulated in niosomes by using nonionic surfactants, ie, span 20 and brij 35, with a high dissolution profile. The bioavailability studies showed the bioavailability of F10 niosomal formulation was greater as compared to the aqueous dispersion of CsA. The bioavailability of this developed niosomal formulation can be conducted on human volunteers. In future, the topical delivery of CsA through the niosomal formulation will be conducted on animal as well as on human volunteers.
Disclosure
The authors report no conflicts of interest for this work.
References
1. Sezgin-Bayindir Z, Yuksel N. Investigation of formulation variables and excipient interaction on the production of niosomes. Aaps Pharmscitech. 2012;13(3):826–835. doi:10.1208/s12249-012-9805-4
2. Shatalebi M, Mostafavi S, Moghaddas A. Niosome as a drug carrier for topical delivery of N-acetyl glucosamine. Res Pharm Sci. 2010;5(2):107.
3. Hamishehkar H, Rahimpour Y, Kouhsoltani M. Niosomes as a propitious carrier for topical drug delivery. Expert Opin Drug Deliv. 2013;10(2):261–272. doi:10.1517/17425247.2013.746310
4. Calne R, et al. Cyclosporin A in patients receiving renal allografts from cadaver donors. 1978. J Am Society Nephrol. 1998;9(9):1751–1756.
5. Tedesco D, Haragsim L. Cyclosporine: a review. J Transplant. 2012;2012. doi:10.1155/2012/230386
6. Dehesa L, et al. The use of cyclosporine in dermatology. JDD. 2012;11(8):979–987.
7. El-Ridy MS, et al. Niosomes as a potential drug delivery system for increasing the efficacy and safety of nystatin. Drug Dev Ind Pharm. 2011;37(12):1491–1508. doi:10.3109/03639045.2011.587431
8. Kazi KM, et al. Niosome: a future of targeted drug delivery systems. J Adv Pharm Technol Res. 2010;1(4):374. doi:10.4103/0110-5558.76435
9. Yoshioka T, Sternberg B, Florence AT. Preparation and properties of vesicles (niosomes) of sorbitan monoesters (Span 20, 40, 60 and 80) and a sorbitan triester (Span 85). Int J Pharm. 1994;105(1):1–6. doi:10.1016/0378-5173(94)90228-3
10. Bhaskaran S, Panigrahi L. Formulation and evaluation of niosomes using different non-ionic surfactants. Indian J Pharm Sci. 2002;64(1):63.
11. Ravalika V, Sailaja A. Formulation and evaluation of etoricoxib niosomes by thin film hydration technique and ether injection method. Nano Biomed Eng. 2017;9(3):242–248. doi:10.5101/nbe.v9i3.p242-248
12. Yeo L, et al. Brief effect of a small hydrophobic drug (cinnarizine) on the physicochemical characterisation of niosomes produced by thin-film hydration and microfluidic methods. Pharmaceutics. 2018;10(4):185. doi:10.3390/pharmaceutics10040185
13. Maruthapillai A, Palanisamy K, Sunkara M. Preparation and characterization of rilpivirine solid dispersions with the application of enhanced solubility and dissolution rate. Beni-Suef University J Basic Applied Sci. 2015;4(1):71–79. doi:10.1016/j.bjbas.2015.02.010
14. Sezgin-Bayindir Z, Antep MN, Yuksel N. Development and characterization of mixed niosomes for oral delivery using candesartan cilexetil as a model poorly water-soluble drug. AAPS PharmSciTech. 2015;16(1):108–117. doi:10.1208/s12249-014-0213-9
15. Khan MI, et al. Formulation design and characterization of a non-ionic surfactant based vesicular system for the sustained delivery of a new chondroprotective agent. Brazilian J Pharm Sci. 2015;51(3):607–615. doi:10.1590/S1984-82502015000300012
16. Bansal S, et al. Design and development of cefdinir niosomes for oral delivery. J Pharm Bioallied Sci. 2013;5(4):318. doi:10.4103/0975-7406.120080
17. Fay F, et al. SEM and EDX analysis: two powerful techniques for the study of antifouling paints. Progress Organic Coatings. 2005;54(3):216–223. doi:10.1016/j.porgcoat.2005.05.005
18. Pecchio M, et al. Development and validation of a HPLC method for the determination of cyclosporine a in new bioadhesive nanoparticles for oral Administration. Indian J Pharm Sci. 2014;76(2):132.
19. Kamboj S, Saini V, Bala S. Formulation and characterization of drug loaded nonionic surfactant vesicles (niosomes) for oral bioavailability enhancement. Scientific World J. 2014;2014.
20. Aziz F, Gupta A, Khan M. Development and validation of a RP-HPLC method for determination of cyclosporine in capsule. Indian J Pharm Sci. 2010;72(2):252. doi:10.4103/0250-474X.65030
21. Ceren Ertekin Z, Sezgin Bayindir Z, Yuksel N. Stability studies on piroxicam encapsulated niosomes. Curr Drug Deliv. 2015;12(2):192–199. doi:10.2174/1567201811666140723115852
22. Ruckmani K, Sankar V. Formulation and optimization of zidovudine niosomes. Aaps Pharmscitech. 2010;11(3):1119–1127. doi:10.1208/s12249-010-9480-2
23. Wang K, et al. Enhancement of oral bioavailability of cyclosporine A: comparison of various nanoscale drug-delivery systems. Int J Nanomedicine. 2014;9:4991.
24. Al-Meshal MA, et al. Oral administration of liposomes containing cyclosporine: a pharmacokinetic study. Int J Pharm. 1998;168(2):163–168. doi:10.1016/S0378-5173(98)00066-0
25. Campos S, et al. In vivo study of hepatic oxidative stress and mitochondrial function in rabbits with severe hypotension after propofol prolonged infusion. SpringerPlus. 2016;5(1):1349. doi:10.1186/s40064-016-2970-2
26. Incecayir T. The effects of surfactants on the solubility and dissolution profiles of a poorly water-soluble basic drug, carvedilol. Die Pharmazie- Int J Pharm Sci. 2015;70(12):784–790.
27. Zaki RM, et al. Formulation and in vitro evaluation of diacerein loaded niosomes. Int J Pharm Pharm Sci. 2014;6(Suppl 2):515–521.
28. Jain S, Mittal A, Jain AK. Enhanced topical delivery of cyclosporin-A using PLGA nanoparticles as carrier. Curr Nanosci. 2011;7(4):524–530. doi:10.2174/157341311796196835
29. Deb TK, et al. In vitro-in vivo evaluation of xanthan gum and eudragit inter polyelectrolyte complex based sustained release tablets. Int J Pharm Investig. 2015;5(1):65. doi:10.4103/2230-973X.147236
30. Lakshmana Prabu S, et al. Formulation and evaluation of sustained release microspheres of rosin containing aceclofenac. Ars Pharm 2009;50(2):1–12.
31. Nowroozi F, et al. Effect of surfactant type, cholesterol content and various downsizing methods on the particle size of niosomes. Iranian j Pharm Res. 2018;17(Suppl2):1.
32. Abhinav K, et al. Review on niosomes as novel drug delivery system. Int Res j Pharm. 2011;2(5):61–65.
33. Chaw CS, Ah Kim KY. Effect of formulation compositions on niosomal preparations. Pharm Dev Technol. 2013;18(3):667–672. doi:10.3109/10837450.2012.672988
34. Abdelkader H, et al. Niosomes and discomes for ocular delivery of naltrexone hydrochloride: morphological, rheological, spreading properties and photo-protective effects. Int J Pharm. 2012;433(12):142–148. doi:10.1016/j.ijpharm.2012.05.011
35. Kumar S. Proniosomal gel of flurbiprofen: formulation and evaluation. J Drug Delivery Therapeutics. 2012;2:1. doi:10.22270/jddt.v2i1.31
36. Khan MI, Madni A, Peltonen L. Development and in-vitro characterization of sorbitan monolaurate and poloxamer 184 based niosomes for oral delivery of diacerein. European J Pharm Sci. 2016;95(p):88–95. doi:10.1016/j.ejps.2016.09.002
37. Biswal S, et al. Vesicles of non-ionic surfactants (niosomes) and drug delivery potential. Int J Pharm Sci Nanotechnol. 2008;1(1):1–8.
38. Jin Y, et al. Development of a novel niosomal system for oral delivery of Ginkgo biloba extract. Int J Nanomedicine. 2013;8(p):421. doi:10.2147/IJN.S37984
39. Nadzir MM, et al. Size and stability of curcumin niosomes from combinations of Tween 80 and Span 80. Sains Malaysiana. 2017;46(12):2455–2460. doi:10.17576/jsm-2017-4612-22
40. Shilakari Asthana G, Sharma PK, Asthana A. In vitro and in vivo evaluation of niosomal formulation for controlled delivery of clarithromycin. Scientifica. 2016;2016.
41. Costa P, Lobo JMS. Modeling and comparison of dissolution profiles. European j Pharm Sci. 2001;13(2):123–133. doi:10.1016/S0928-0987(01)00095-1
42. Allam A, Fetih G. Sublingual fast dissolving niosomal films for enhanced bioavailability and prolonged effect of metoprolol tartrate. Drug Des Devel Ther. 2016;10:2421. doi:10.2147/DDDT.S113775
43. Diva B, Ishwarya K, Naganjaneyulu R. In vitro drug release profile of aceclofenac niosomes formed with different ratios of cholestrol using sorbitan esters. Int J Chem Sci. 2014;12(1):237–247.
44. Sambhakar S, Paliwal S, Sharma S, et al. Formulation of risperidone loaded proniosomes for effective transdermal delivery: an in-vitro and in-vivo study. Bulletin Faculty Pharm Cairo University. 2017;55(2):239–247. doi:10.1016/j.bfopcu.2017.09.003
45. Aungst BJ. Intestinal permeation enhancers. J Pharm Sci. 2000;89(4):429–442. doi:10.1002/(SICI)1520-6017(200004)89:4<429::AID-JPS1>3.0.CO;2-J
46. Arzani G, et al. Niosomal carriers enhance oral bioavailability of carvedilol: effects of bile salt-enriched vesicles and carrier surface charge. Int J Nanomedicine. 2015;10:4797.
47. Jadon PS, et al. Enhanced oral bioavailability of griseofulvin via niosomes. AAPS PharmSciTech. 2009;10(4):1186. doi:10.1208/s12249-009-9325-z
48. Roger E, et al. Biopharmaceutical parameters to consider in order to alter the fate of nanocarriers after oral delivery. Nanomedicine. 2010;5(2):287–306. doi:10.2217/nnm.09.110
49. Gabrielsson J, Weiner D. Non-Compartmental Analysis, in Computational Toxicology. Springer. 2012;377–389.
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.