")
Back to Journals » Therapeutics and Clinical Risk Management » Volume 12
In vitro and in vivo postmarketing surveillance of valsartan, alone or in combination with amlodipine or hydrochlorthiazide, among Palestinian hypertensive patients
Authors Zaid AN , Ghanem M, Shweiki D, Shtewi H, Shaheen R, Al Helaly S, Khayyat Z, Al Ramahi R , Zyoud S
Received 16 April 2016
Accepted for publication 10 June 2016
Published 19 September 2016 Volume 2016:12 Pages 1425—1432
DOI https://doi.org/10.2147/TCRM.S110727
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Garry Walsh
Abdel Naser Zaid,1 Masshour Ghanem,2 Dua’a Shweiki,1 Hala Shtewi,1 Raja’ Shaheen,1 Sondos Al Helaly,1 Zeina Khayyat,1 Rowa’a Al Ramahi,1 Sa’ed H Zyoud1
1Department of Pharmacy, Faculty of Medicine & Health Sciences, An-Najah National University, Nablus, 2Pharmacare Ltd, Ramallah, Palestine
Objectives: The objectives of this study were to evaluate the general quality of the most prescribed products of valsartan (VL; alone or in combination) and to evaluate their efficacy and safety among Palestinian population through in vivo postmarketing surveillance.
Patients and methods: The first part was pharmacopeial quality control assay, including dissolution, disintegration, friability, and weight uniformity for VL. The second part was a 3-month cardiology clinics, observational, postmarketing surveillance pilot study that included 103 hypertensive patients who were prescribed 80 mg or 160 mg of VL as monotherapy or combination therapy. The end points were reduction in blood pressure (BP) and the rate of incidence of adverse effects (AEs) at weeks 4 and 8.
Results: According to our quality control tests, all VL products showed high-quality standards according to the international guidelines. A reduction in BP was observed at weeks 4 and 8, and no significant difference was observed between the strengths of 80 mg and 160 mg. Higher BP reduction was observed after the use of combination therapy. Moreover, VL was well tolerated; most of the AEs were of mild-to-moderate intensity. In general, the most frequently reported AEs included headache (17.5%), dizziness (11.75%), and weakness (11.7%). No serious AEs or death cases were reported during the study period.
Conclusion: High quality of VL tablet products was used; hence, the observed efficacy and safety results should be related to patient’s factors and not due to any product defects or substandard quality. Moreover, VL is an effective treatment for essential hypertension.
Keywords: valsartan, quality control, postmarketing, surveillance, Palestine
Introduction
Valsartan (VL) is a nonpeptide angiotensin receptor blocker (ARB) used orally for the treatment of high blood pressure (BP).1 It inhibits the angiotensin type 2 receptor that is responsible for the decrease in aldosterone secretion. This causes an arteriolar and venous dilation, resulting in a decrease in BP.1 Accordingly, this drug has been approved for the initial treatment of primary BP worldwide.2,3
In fact, the European Society of Hypertension/European Society of Cardiology has included this medication as a first-line therapy in patients with high BP.4 VL is available in several strengths, 40 mg, 80 mg, 160 mg, and 320 mg, in order to meet the needs of patients.5
However, 80 mg and 160 mg are the only strengths available in Palestine. These two strengths are available as monotherapy or in combination therapy with other antihypertensive agents such as hydrochlorothiazide (HCT) and amlodipine (AML; Table 1).6
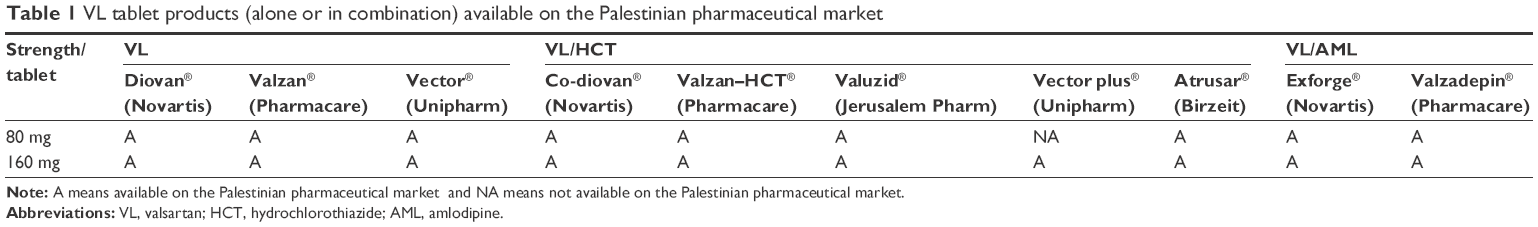 | Table 1 VL tablet products (alone or in combination) available on the Palestinian pharmaceutical market |
The efficacy of VL is independent of age and sex, but may depend to a certain extent on ethnic origin, since Afro-American patients are less sensitive to ARBs than Caucasians.7
Unlike angiotensin-converting enzyme inhibitors, ARBs are relatively well tolerated and do not induce cough. ARBs are indicated in mild, moderate, and severe essential high BP, where their efficacy has been proven in many studies.7–9
Moreover, VL has a very low incidence of side effects, approximately similar to that of placebo, with an acceptable overall safety profile.4,6 Biswas et al10 have assessed several adverse effects (AEs), including dizziness, diarrhea, headache, palpitation, nausea/vomiting, rash, sweating, cough, dyspepsia, dyspnea, epistaxis, and malaise, as suspected adverse drug reactions among patients under VL treatment. Dizziness, headache, and fatigue were commonly reported in patients who used this drug; the most common reason that led to stop the use of VL was dizziness.
In fact, in another study conducted on Taiwanese population, the incidence rate of dizziness as AE was ~3.58% and 4.33% using VL alone and in combination with other antihypertensive drugs, respectively.11 It was also found that the incidence of dizziness increased by increasing the dose.12 Therefore, it was suggested that ARBs should be initially started in low doses and then increased according to their efficacy and safety in order to minimize dizziness.13 Other AEs have been also reported such as headache (1.04% in monotherapy vs 1.44% in combination), cough (1.04% in monotherapy vs 0.76% in combination), and constipation (1.04% in monotherapy vs 0.76% in combination).11
Several in vivo postmarketing surveillance studies are usually conducted on marketed drugs in order to assess the efficacy and safety of these drugs.11,14,15
However, these studies are costly and time consuming since they need years and involve the contribution of several physicians, clinical institutions, and patients. Therefore, in vitro postmarketing surveillance, which involves pharmacopeial and technological quality control (QC) assessment of drug products, may be used as a quick and cheap alternative to the in vivo surveillance studies.16,17
To the best of our knowledge, there are neither in vitro nor in vivo postmarketing studies about VL in Arab countries. Accordingly, the objectives of this study were to evaluate the pharmacopeial QC of the most prescribed products of VL alone or in combination with other drugs. Moreover, a pilot study was used in order to evaluate the efficacy and safety of VL alone or in combination with other antihypertensive agents among Palestinian population through in vivo postmarketing surveillance.
Patients and methods
Postmarketing surveillance study
In vivo postmarketing surveillance study
Study design
This was a 3-month cardiology clinics, observational, post- marketing surveillance pilot study designed and approved by the local ethics committee (Institutional Review Board [IRB] of An-Najah National University). In this contest, the primary objective was to evaluate the efficacy of VL as mono- or combination therapy among Palestinian patients with high BP. The secondary objective was to assess the incidence rate of side effects that appeared during the use of VL as monotherapy or combination with other antihypertensive drugs.
Patients
Participants aged >20 years and diagnosed with high BP (sitting diastolic BP [DBP] >90 mmHg and/or systolic BP [SBP] >140 mmHg) were included in the study. Patients with severe medical conditions, using any other investigational drugs at the time of study, or with known hypersensitivity to VL or any component in the formulation; breastfeeding or pregnant women; or patients with a history of any malignancy within the previous 5 years were excluded from the study. All participants signed a written informed consent, and the protocol was in accordance with the ethical principles laid down in the Declaration of Helsinki.
Treatment and assessment
At the first visit (week 0), several information were recorded from the patients, including their sociodemographic data, medical history, and used medications. BP was measured by the physician according to his discernment; the patients were prescribed VL as a monotherapy or in combination. At the following visit (week 4), the patient’s BP, medication changes, and AEs were recorded. Similar evaluation was done at the final visit (week 8).
The incidence rate of side effects of VL alone or in combination therapy was assessed according to the patient’s answer to our standard question: “Did you feel or sense any of the following side effects?”
Moreover, the efficacy of VL on BP was assessed by measuring the mean change between weeks 0, 4, and 8, and the reduction in BP was calculated.
Statistical analyses
All statistical analyses were performed using the SPSS 16 software package (SPSS Inc., Chicago, IL, USA). Categorical variables were expressed as frequencies and percentages. Numerical variables that did not follow a normal distribution were represented as median and interquartile range. Categorical variables were compared by Fisher’s exact test when the expected frequency was ≤5. Two-sample comparisons were carried out by the Mann–Whitney U-test for nonnormally distributed variables. Numerical variables were tested for normal distribution with the Kolmogorov–Smirnov test. P-values <0.05 were considered statistically significant.
In vitro postmarketing QC
Materials and methods
Formulations
Ten different brands of VL (alone and in combination) were available in the Palestinian market as shown in Table 1; however, actually only six brands were studied because these were the most prescribed for high BP in the clinic during the study period. These brands were subjected to visual and pharmacopeial inspection according to the United States Pharmacopoeia 38 (USP 38)/National Formulary 33.18
Chemicals and reagents
USP VL, HCT, and AML besylate reference standards (RIs; Holland Moran, Holon, Israel) were used. Valzan 160 mg tablets, valzan–HCT (160/25), and valzadepin (10/160) were obtained from Pharmacare PLC (Ramallah, Palestine).
High-performance liquid chromatography (HPLC)-grade solvents such as acetonitrile and methanol were purchased from EMD Millipore (Billerica, MA, USA) and were used as received. Potassium dihydrogen phosphate, sodium hydroxide pellets, trifluoroacetic acid, triethylamine, phosphoric acid, and glacial acetic acid were also purchased from EMD Millipore. High purified water was prepared by using a EMD Millipore Milli-Q plus water purification system. All other reagents were of analytical grade.
Instruments and chromatographic conditions
The HPLC system consisted of LaChrom (Merck–Hitachi, Kent, England) equipped with model L-7100 pump, L-7200 autosampler, L-7300 column oven, DAD L-7450 photodiode array detector, and D-7000 software HSM Version 3.1 (Merck–Hitachi).
The HPLC experimental conditions were optimized on octadecyl silane C18 chemically bonded column (125×3.0 mm id, 5 mcm particles) that was purchased from ACE (London, UK). Weights were measured using Ohaus balance (Model DV215CD; Shekel Ltd, Israel); pH was identified using Toledo GmbH pH meter (Model S47-K; Agentek, Mettler Toledo, Switzerland).
Method
All VL tablet products available in the Palestinian pharmaceutical market were reported and assessed for their price. However, only products that were prescribed in the postmarketing surveillance were subjected to complete in vitro QC. Accordingly, visual examination was used to assess the general look and appearance of these tablet products. The assay of VL alone or in combination with other drugs was assessed according to the procedure reported in the related reference (Table 2).
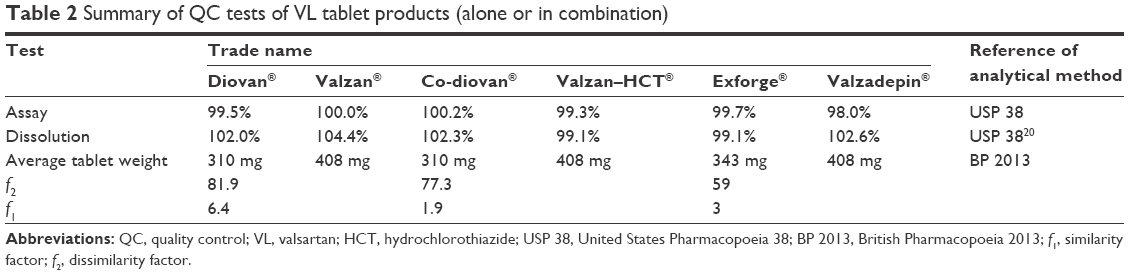 | Table 2 Summary of QC tests of VL tablet products (alone or in combination) |
For weight uniformity test, 20 tablets from each of the six products of VL were weighed individually using an analytical weighing balance. The percentage relative standard deviation and the average weights for each product were calculated according to the USP for weight uniformity test.18
Regarding the crushing strength of tablet products, it was determined using tablet hardness tester (Monsanto, Cambridge, UK). For this purpose, ten tablets were randomly selected from each product and force of crushing was recorded. Moreover, friability test was used to assess the resistance of tablet products to any stress that may cause powder loss >1% of the initial weight. Exactly, 20 tablets were taken and weighed from each brand. The selected tablets were subjected to abrasion by using a Roche friabilator (ERWEKA GmbH, Heusenstamm, Germany) at 100 revolutions in 4 minutes (25 revolutions/min). At the end of this procedure, the loss of powder was calculated using the following equation:
|
|
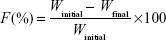
where F is the friability, Winitial is the initial weight, and Wfinal is the final weight.
Concerning the disintegration test,19 six tablets from each product were tested in a freshly prepared medium that contained 0.1 N HCl at 37°C by using ERWEKA disintegration apparatus (ERWEKA GmbH). The disintegration time was the time when no particle remained in the basket of the system. The disintegration time for all products was <10 minutes (accepted limit <15 minutes).
Regarding the release profile of VL from tablet products, 12 tablets of each VL product (alone or in combination) were selected for the dissolution profile tests. The dissolution profile test was carried out using USP Apparatus 2 (paddle) using the recommended US Food and Drug Administration dissolution media. Details of dissolution profile procedure and time intervals of sampling for each product are reported in the related dissolution profile procedure as per the USP 38 and US Food and Drug Administration.
Sample aliquots (10 mL) were withdrawn and replaced with an equal volume of fresh medium to maintain a constant total volume. The samples were filtered through an EMD Millipore filter 0.45 μm. The amount of VL in the samples was determined using the validated HPLC analytical method referenced in Table 1.
The dissolution profile of VL tablet (alone or in combination) was generated from the graph of the percent of VL released versus time using Microsoft Office Excel. The amount of VL in each sample was plotted against time, and similarity (f1) and dissimilarity factors (f2) were calculated according to Equations 2 and 3. The f2 factor measures the closeness between two profiles, and f1 measures the difference between two profiles:
|
|
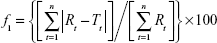
|
|
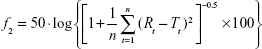
where Rt and Tt are the percentages of drug dissolved at each time point for the reference and test products, respectively. An f1 value >15 indicates significant dissimilarity, and an f2 value >50 indicates significant similarity.20–23
Results
In vitro postmarketing surveillance
The six commercial tablet products containing VL included two monotherapy and four combinations (two with HCT and two with AML; Table 1).
Bioequivalence studies were conducted on all these products by the corresponding manufacturers as a requirement of product registration prior to commercialization of premarketing assessment. All these products passed successfully these bioequivalence studies as a precondition of drug registration protocol of the Palestinian Ministry of Health.
Regarding the in vitro postmarketing study, all VL tablet products (brands and the mostly prescribed generic products) were subjected to several QC analyses, including visual and instrumental analyses. There were no visible signs of defects or abnormalities in the shape and color or any sign of spots in any of the tested product. Accordingly, further in vitro investigations were conducted, such as on weight uniformity. All products were in compliance with the USP weight uniformity test, and no single tablet was out of the range as summarized in Table 2.
Regarding hardness and friability, all products showed sufficient resistance strength and did not lose >1% of their powder content in the friability tests (Table 2).
As summarized in Table 2, the assay of VL in the monotherapy tablet products and in combination also was within the USP requirements. In fact, the assay of our analyzed products containing VL as mono therapy or combination was always close to 100%.
Concerning tablet dissolution, all products showed complete release of their VL content within 30 minutes (ranging from 99.1% to 102%) as reported in Table 2.
Moreover, the release profile of VL from the generic tablet products was comparable with the related reference listed drug since it showed f2 >50 and f1 <15 (Table 2).
In vivo postmarketing surveillance
Demographic analysis
A total of 103 patients were studied in terms of baseline demographic and clinical characteristics as shown in Table 3. About half of the patients were males (53.4%) with an average age between 60 years and 69 years. Less than half of them were of secondary education level (41.7%). The majority of them were nonsmokers (70.9%) and had no other diseases (56.3%).
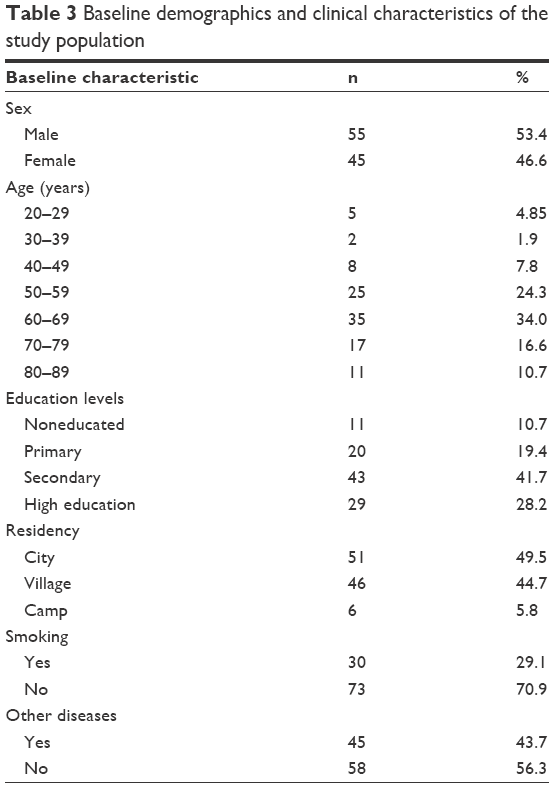 | Table 3 Baseline demographics and clinical characteristics of the study population |
All patients were included in the study, and most of them (80.6%) were on combination therapy. Over the period of the study, 19.4% of patients received monotherapy, and 80.6% patients received the combination therapy (VL in combination with another antihypertensive drug).
Moreover, VL treatment showed that at baseline, 15.5% and 84.5% of patients were prescribed VL at a dose of 80 mg and 160 mg, respectively, either as monotherapy or as combination therapy.
Assessment of the efficacy profile
Regarding the efficacy, it was found that the use of the lower dose of VL (80 mg/tablet) was sufficient to produce reduction in both SBP and DBP. In fact, the means were close to each other, and the P-value was not significant as reported in Tables 4 and 5, respectively.
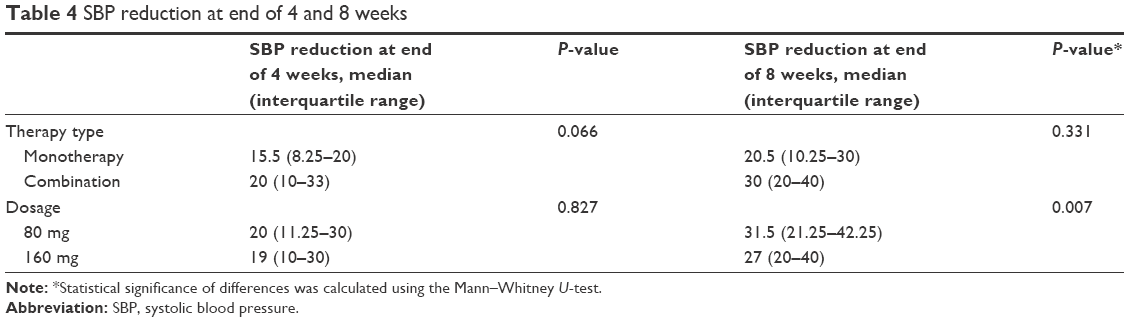 | Table 4 SBP reduction at end of 4 and 8 weeks |
 | Table 5 DBP reduction at end of 4 and 8 weeks |
Moreover, even the same results were observed when the lower dose in combination therapy was compared to the higher dose.
The efficacy of the use of VL alone was compared with that in combination using the same strengths of VL. It was found that the combination was more effective than the monotherapy, resulting in a reduction in both SBP and DBP (Tables 4 and 5, respectively). After 8 weeks, the number of patients who reached BP <140/90 was 70 (68%).
Assessment of the safety profile
In general, our pilot study showed that VL was well tolerated. In fact, only ~6.06% of patients reported AEs. Most of these AEs were of mild-to-moderate intensity. In general, the most frequently reported AEs included headache (17.5%), dizziness (11.75%), and weakness (11.7%) as clarified in Table 6. No serious AEs or death cases were reported during the study period.
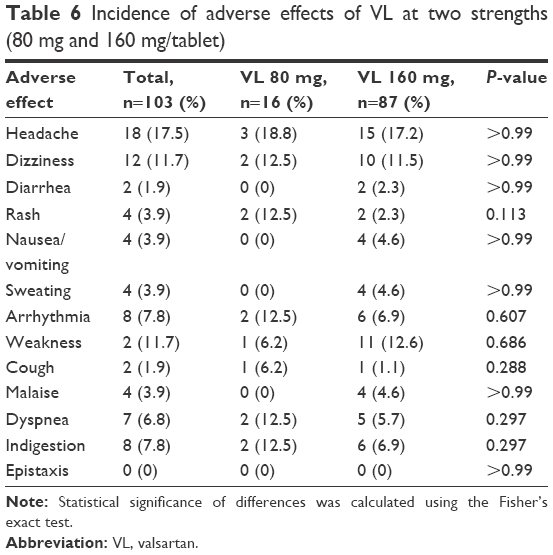 | Table 6 Incidence of adverse effects of VL at two strengths (80 mg and 160 mg/tablet) |
Despite the existence of differences in the rate of incidence of some AEs between the two investigated VL strengths as seen in cough, dyspnea, and rash, these differences were not significant since the related P-values were >0.05. These AEs may be shown less in 80 mg of VL than in 160 mg of VL as shown in Table 6. However, there are no significant differences in AEs between the uses of combination therapy or monotherapy of VL except for weakness, where the P-value was 0.011 as shown in Table 7.
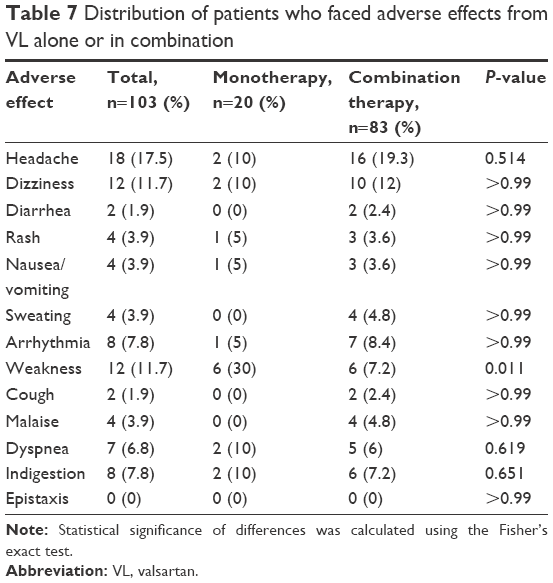 | Table 7 Distribution of patients who faced adverse effects from VL alone or in combination |
Discussion
In vitro postmarketing surveillance
Our in vitro postmarketing surveillance assessment demonstrated a high overall quality of all prescribed VL products as reported in Table 2.
In fact, all products containing VL drug were close to 100%, and the dissolution of the drug was within the recommended international guideline. This indicates that these products have suitable clinical and safety profiles, and any results in the terms of efficacy and safety should be closely related to patients’ factors and not to any defect in the manufacturing of the used product.
In vivo postmarketing surveillance
Several postmarketing studies were conducted on VL alone or in combination worldwide, in order to investigate the efficacy and safety of these products.10,11,24
However, similar studies on Palestinian or Arab patients are not available. Our pilot study showed that a reduction in BP at weeks 4 and 8 was observed similar to other previous studies. However, our studies showed no difference in the reduction between 80 mg and 160 mg strengths in VL alone or in combination. This result was unexpected when compared with other studies where 160 mg dose showed greater BP reduction.11 Regarding the effect of combination on BP reduction, our results were comparable with similar studies. In fact, the combination showed better BP reduction than VL alone.
Many postmarketing studies were conducted in order to investigate the safety and efficacy of ARBs.7–9 These studies proved that VL either alone or in combination is well tolerated. However, a Taiwanese study showed that the most frequent AEs were dizziness, headache, and constipation,11 while a study conducted in England showed that the most frequent AEs were malaise, dizziness, and headache.10 Moreover, dizziness, headache, and upper respiratory tract infection were the most frequently occurring AEs according to a Chinese postmarketing study.24
Mainly, the AEs that were obtained in our study were less than the AEs that occurred in other studies as seen in Figure 1. The most frequent AE was headache, which was close to the study conducted in the UK that also showed epistaxis, a rare AE that was not observed in our patients or in Chinese and Taiwanese patients as shown in Figure 1.
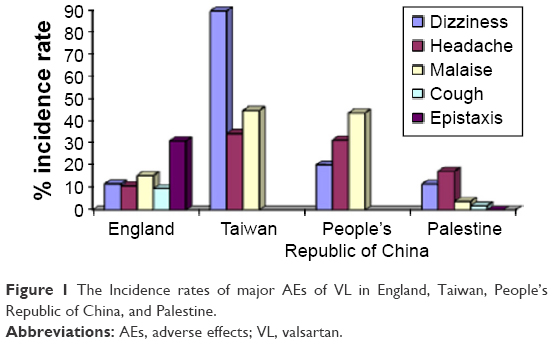 | Figure 1 The Incidence rates of major AEs of VL in England, Taiwan, People’s Republic of China, and Palestine. |
We assessed the efficacy and safety of VL as monotherapy and combination therapy in the Palestinian population. The incidence rates of AEs with VL 160 mg were higher than with VL 80 mg. The most frequently occurring AE was headache. There was no difference in the occurrence of AEs in patients receiving monotherapy or combination therapy.
BP reduction was greater at week 8 than week 4. In addition, the mean SBP reduction at week 8 was significantly greater in patients receiving combination therapy than in those receiving monotherapy. There was no significant difference in the occurrence of AEs between the use of monotherapy and combination therapy, and this proved that the main cause of AEs was VL drug rather than the combined drug, such as HCT or AML.
The main AE in this study was headache (17.5%), which was different from the main AE in the Taiwanese study that was dizziness,11 and this may be attributed to many reasons. The first reason was using of pilot study with small number of participants that could give different results. Also, VL is known to be one of the medications that is genetic dependent,7 and there is a race difference between the Palestinian and Taiwanese patients. Moreover, a study that focused on the bioavailability found that it may be different from one country to another.25 Our study showed that despite the existence of differences in the incidence of certain AEs, these differences are not significant since the P-value is >0.05 as seen in cough, dyspnea, indigestion, and rash (Table 6). In a Taiwanese study, the safety profile was independent of dose.11 Moreover, there is no significant difference in AEs between the uses of combination therapy or monotherapy of VL as clarified in Table 7.
Conclusion
The in vitro postmarketing surveillance revealed high quality of the used VL tablet products, and accordingly, any result in efficacy or safety should be related to patient’s factors and not product defect or substandard quality. The in vivo pilot postmarketing surveillance conducted on our Palestinian patients showed interesting efficacy profile, since there was no significant difference in BP reduction between both doses (80 mg or 160 mg, alone or in combination). However, a higher reduction in BP was observed when the combination therapy was used despite the strength of VL in the used tablets. Regarding the safety profile of VL, no serious SEs were observed and most of the minor AEs were lower than those observed in patients from other populations. However, future studies with higher number of patients and a wider geographical area are recommended in order to confirm or deny these interesting clinical observations.
Acknowledgment
Many thanks go to Doctor Jamal Alol and Doctor Yaser Yaseen who allowed us to meet their patients and provided us with the required information and help for the success of this study.
Disclosure
The authors report no conflicts of interest in this work.
References
Harvey RA, Clark M, Finkel R, Rey J, Whalen K. Lippincott’s Illustrated Reviews: Pharmacology. Philadelphia, PA: Wolters Kluwer; 2012. | ||
Chazova IE, Dongre N, Vigdorchik AV. Real-life safety and effectiveness of amlodipine/valsartan combination in the treatment of hypertension. Adv Ther. 2011;28(2):134–149. | ||
Black HR, Bailey J, Zappe D, Samuel R. Valsartan: more than a decade of experience. Drugs. 2009;69(17):2393–2414. | ||
Verdecchia P, Gentile G, Angeli F, Mazzotta G, Mancia G, Reboldi G. Influence of blood pressure reduction on composite cardiovascular endpoints in clinical trials. J Hypertens. 2010;28(7):1356–1365. | ||
Drugs [webpage on the Internet]. Valsartan Dosage. 2016. Available from: http://www.drugs.com/dosage/valsartan.html. Accessed April 10, 2016. | ||
Kjeldsen SE, Brunner HR, McInnes GT, Stolt P. Valsartan in the treatment of hypertension. Aging Health. 2005;1(1):27–36. | ||
Corvol P, Plouin PF. Angiotensin II receptor blockers: current status and future prospects. Drugs. 2002;62(Spec No 1):53–64. | ||
Akioyamen L, Levine M, Sherifali D, et al. Cardiovascular and cerebrovascular outcomes of long-term angiotensin receptor blockade: meta-analyses of trials in essential hypertension. J Am Soc Hypertens. 2016;10(1):55.e–69.e. | ||
Matchar DB, McCrory DC, Orlando LA, et al. Systematic review: comparative effectiveness of angiotensin-converting enzyme inhibitors and angiotensin II receptor blockers for treating essential hypertension. Ann Intern Med. 2008;148(1):16–29. | ||
Biswas PN, Wilton LV, Shakir SW. The safety of valsartan: results of a postmarketing surveillance study on 12881 patients in England. J Hum Hypertens. 2002;16(11):795–803. | ||
Liou CW, Yeh TC, Chen IC, et al. Efficacy and safety of valsartan in hypertensive Taiwanese patients: post-marketing surveillance study. Blood Press Suppl. 2011;2(sup 2):13–21. | ||
Oparil S, Dyke S, Harris F, et al. The efficacy and safety of valsartan compared with placebo in the treatment of patients with essential hypertension. Clin Ther. 1996;18(5):797–810. | ||
Kirk JK. Angiotensin-II receptor antagonists: their place in therapy. Am Fam Physician. 1999;59(11):3140–3148. | ||
Karpov Y, Dongre N, Vigdorchik A, Sastravaha K. Amlodipine/valsartan single-pill combination: a prospective, observational evaluation of the real-life safety and effectiveness in the routine treatment of hypertension. Adv Ther. 2012;29(2):134–147. | ||
Ganzoni E, Santoni JP, Chevillard V, Sebille M, Mathy B. Zolpidem in insomnia: a 3-year post-marketing surveillance study in Switzerland. J Int Med Res. 1995;23(1):61–73. | ||
Zaid AN, Cortesi R, Qaddomi A, Khammash S. Formulation and bioequivalence of two valsartan tablets after a single oral administration. Sci Pharm. 2011;79(1):123–135. | ||
Zaid A, Rinno T, Jaradat N, Jodeh S, Khammash S. Interchangeability between paracetamol tablets marketed in Palestine. Is there a quality reason for a higher price? East Mediterr Health J. 2013;19(6):542–546. | ||
United States Pharmacopeial Convention. The United States Pharmacopia 38 – National Formulary 33 (USP 38-NF 33). USP 38 – NF 33 ed. Rockville, MD: United States Pharmacopeial Convention; 2014. | ||
The United Stade Pharmacopoeial Inc. Pharmacopoeia UP. x II ed. Rockville: The United Stade Pharmacopoeial Inc; 2008. | ||
Zaid AN, Qaddomi A, Ghanem M, et al. Development of a dissolution method to compare tablet formulations containing Valsartan/Amlodipine. Dissolution Technologies. 2015;22(3):32–38. | ||
Emami J. In vitro – in vivo correlation: from theory to applications. J Pharm Pharm Sci. 2006;9(2):169–189. | ||
Fortunato D. Dissolution method development for immediate release solid oral dosage forms. Dissolut Technol. 2005;12(3):12–14. | ||
Gupta AG, Ram S, Ganga S. Development of discriminating dissolution method for an insoluble drug: nisoldipine. Int J Pharmtech Res. 2010;2(1):931–939. | ||
Hu D, Liu L, Li W. Efficacy and safety of valsartan/amlodipine single-pill combination in 11,422 Chinese patients with hypertension: an observational study. Adv Ther. 2014;31(7):762–775. | ||
Wagner JG. Drug bioavailability studies. Hosp Pract. 1977;12(1):119–127. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.