")
Back to Journals » Drug Design, Development and Therapy » Volume 18
In vitro and in vivo Biological Evaluation of Newly Tacrine-Selegiline Hybrids as Multi-Target Inhibitors of Cholinesterases and Monoamine Oxidases for Alzheimer’s Disease
Authors Huang ST , Luo JC, Zhong GH , Teng LP, Yang CY, Tang CL , Jing L , Zhou ZB , Liu J, Jiang N
Received 21 August 2023
Accepted for publication 15 January 2024
Published 24 January 2024 Volume 2024:18 Pages 133—159
DOI https://doi.org/10.2147/DDDT.S432170
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Georgios Panos
Shu-Tong Huang,1,2,* Jin-Chong Luo,1,3,* Guo-Hui Zhong,1,2 Li-Ping Teng,4 Cai-Yan Yang,4 Chun-Li Tang,1 Lin Jing,1 Zhong-Bo Zhou,4 Jing Liu,1,3 Neng Jiang1,2
1Department of Pharmacy, Guangxi Medical University Cancer Hospital, Nanning, Guangxi, People’s Republic of China; 2Pharmaceutical College, Guangxi Medical University, Nanning, Guangxi, People’s Republic of China; 3School of Pharmacy, Jiangxi University of Chinese Medicine, Nanchang, People’s Republic of China; 4School of Pharmacy, Youjiang Medical University for Nationalities, Baise, Guangxi, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Jing Liu, School of Pharmacy, Jiangxi University of Chinese Medicine, No. 56, Yangming Road, Donghu District, Nanchang, Jiangxi, 330006, People’s Republic of China, Email [email protected] Neng Jiang, Department of Pharmacy, Guangxi Medical University Cancer Hospital, 71 Hedi Road, Qingxiu District, Nanning, Guangxi, 530021, People’s Republic of China, Tel/Fax +86-17307711726, Email [email protected]
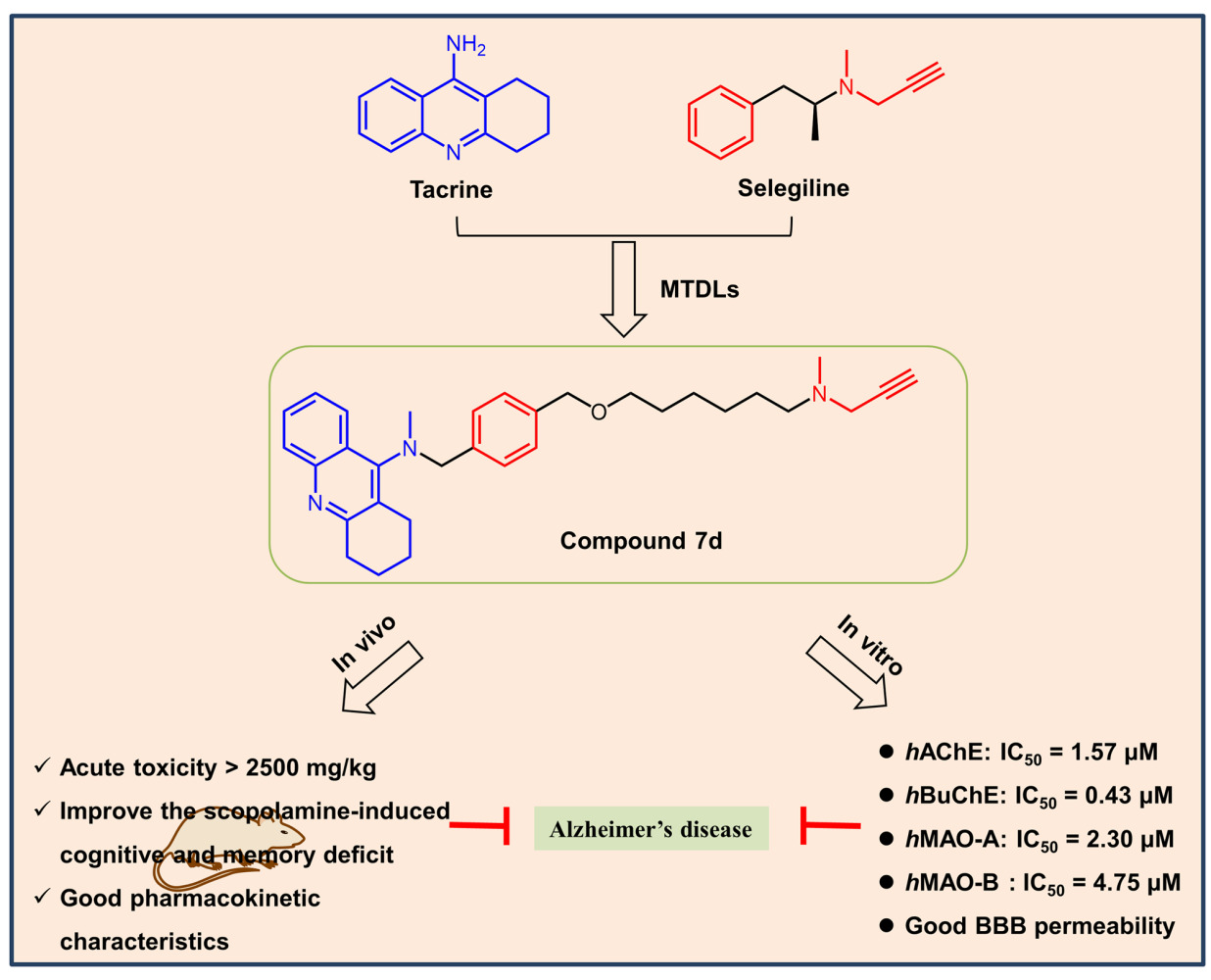
Purpose: Alzheimer’s disease (AD) is the most common neurodegenerative disease, and its multifactorial nature increases the difficulty of medical research. To explore an effective treatment for AD, a series of novel tacrine-selegiline hybrids with ChEs and MAOs inhibitory activities were designed and synthesized as multifunctional drugs.
Methods: All designed compounds were evaluated in vitro for their inhibition of cholinesterases (AChE/BuChE) and monoamine oxidases (MAO-A/B) along with their blood-brain barrier permeability. Then, further biological activities of the optimizing compound 7d were determined, including molecular model analysis, in vitro cytotoxicity, acute toxicity studies in vivo, and pharmacokinetic and pharmacodynamic property studies in vivo.
Results: Most synthesized compounds demonstrated potent inhibitory activity against ChEs/MAOs. Particularly, compound 7d exhibited good and well-balanced activity against ChEs (hAChE: IC50 = 1.57 μM, hBuChE: IC50 = 0.43 μM) and MAOs (hMAO-A: IC50 = 2.30 μM, hMAO-B: IC50 = 4.75 μM). Molecular modeling analysis demonstrated that 7d could interact simultaneously with both the catalytic active site (CAS) and peripheral anionic site (PAS) of AChE in a mixed-type manner and also exhibits binding affinity towards BuChE and MAO-B. Additionally, 7d displayed excellent permeability of the blood-brain barrier, and under the experimental conditions, it elicited low or no toxicity toward PC12 and BV-2 cells. Furthermore, 7d was not acutely toxic in mice at doses up to 2500 mg/kg and could improve the cognitive function of mice with scopolamine-induced memory impairment. Lastly, 7d possessed well pharmacokinetic characteristics.
Conclusion: In light of these results, it is clear that 7d could potentially serve as a promising multi-functional drug for the treatment of AD.
Keywords: Alzheimer’s disease, cholinesterase, monoamine oxidase, multi-target, designed
Graphical Abstract:
Introduction
Alzheimer’s disease (AD) is an age-related, chronic, primary brain nervous system degenerative disorder, characterized by memory loss, cognitive dysfunction and behavioral abnormalities that gradually worsen over time and ultimately result in death.1,2 The global number of AD cases had increased by 117% between 1990 and 2016 due to the aging of the population, with over 50 million people living with dementia today and a projected 152 million by 2050 according to the latest available data.3 AD, being the leading cause of dementia and the only one of the top 10 lethal diseases worldwide without an effective treatment, with care costs of around $1 trillion annually, is fast becoming one of the most expensive, deadly, and burdensome diseases of this century.2,4,5 Despite the urgent need, AD drug development has experienced obstacles. AD is a multifactorial disease whose exact pathological causes are not yet fully understood. However, several factors, such as a deficit of acetylcholine (ACh)6 and oxidative stress caused by increased activity of monoamine oxidase, play important roles in the pathogenesis of AD.7–9 Although strong efforts are underway to elucidate the underlying mechanisms of the disease, translating these findings into novel therapeutic approaches remains challenging.
To date, the FDA has approved three types of drugs for the treatment of AD: three central AChE inhibitors (Donepezil, Rivastigmine and Galantamine), a partial antagonist of the N-methyl-D-aspartate (NMDA) receptor (Memantine),10 and two anti-beta-amyloid (Aβ) monoclonal antibodies (Aducanumab and Lecanemab).11 However, the approval of these two anti-amyloid antibodies is controversial, and further data are needed to confirm their effectiveness.11 Thus, in deed, drug therapy for AD is primarily based on central AChE inhibitors to compensate for cholinergic defects in the brain.12 A significant decrease of endogenous ACh in the brain can result in the typical pathological features of AD, such as cognitive decline.13 AChE, mainly present in blood and synapses, is the principal enzyme responsible for the hydrolysis of acetylcholine (~90%) and is the most feasible therapeutic target for improving AD symptoms.9,14 In addition to AChE, the liver also contains butyl cholinesterase (BuChE).9 These two enzymes share almost 65% homologous amino acid sequences, and both possess a catalytic active site (CAS), a deep gorge and a peripheral anionic site (PAS).15 BuChE serves as a crucial compensatory enzyme in progressed AD. When neurons undergo severe damage in patients with AD, the AChE level in the brain declines to 90% of the normal value, while the BuChE level can increase up to 120% of the physiological state.5,15 Moreover, studies have suggested that the accumulation of BuChE in senile plaques is closely related to the progressive buildup of Aβ.16 Therefore, BuChE can also be used as a drug target to treat AD. Tacrine (9-amino-1,2,3,4-tetrahydroacridine) was the first FDA-approved cholinesterase inhibitor for AD treatment but limited by hepatotoxicity and low bioavailability.12,17 Nonetheless, it is widely used in medicinal chemistry since it has good blood-brain barrier permeability, low molecular weight, synthesizability, and easy modification, serving as a scaffold for the development of new multifunctional drugs with minimal side effects.18,19 In recent years, structural modifications of tacrine have mainly focused on substituting the benzene ring with a heterocycle and molecular hybridization based on tacrine.12 Studies have revealed that homologous and heterodimers improve and extend the function of tacrine, while reducing side effects.20,21
Monoamine oxidases (MAOs) are flavoproteins that catalyze the oxidative deamination of monoamines (endogenous or exogenous). They are localized as dimers on the outer mitochondrial membrane and are classified into two subtypes, MAO-A and MAO-B, based on their acting substrates and selective inhibitors.22 In the central nervous system (CNS), MAO-A is predominantly located in neuron axons and responsible for the oxidative deamination of a broad range of monoamine neurotransmitters, such as serotonin, norepinephrine, and epinephrine, while MAO-B is mainly expressed in glial cells and decomposes approximately 70% of dopamine.22–24 Inhibitions of MAO-A have been approved for the treatment of neurasthenia, depression, and anxiety, while MAO-B inhibitors hold promise for the management of AD and Parkinson’s disease (PD). With age, the expression level of MAO-B in the brain increases by up to fourfold, generating a massive number of free radicals that contribute to oxidative stress, which is identified as one of the pathogenic factors for AD.25 Moreover, AD patients exhibit reduced concentrations of several monoamine neurotransmitters, including serotonin, dopamine, and norepinephrine.26 These studies demonstrated that elevated MAO activity is closely related to the onset of AD. Therefore, MAO inhibitors, which slow the metabolism of monoamine neurotransmitters and exert indirect antioxidant effects by inhibiting MAO activity, have been identified as candidates for AD treatment. For instance, selegiline is an irreversible selective MAO-B inhibitor that shows neuroprotective effects in both in vitro and in vivo models of AD.27
Given the multifactorial nature of AD, single-target drugs often face difficulties in interfering with the complex network regulation of the disease and may produce high levels of toxicity. As a result, the development of multi-target directed ligands (MTDLs) has emerged as an important direction in current research, and multi-target drugs may be critical and effective in regulating the progression of AD. One of the most common approaches for the design of multi-target AD drugs involves pharmacophore conjugation, which combines two or more pharmacophores with known activities to form hybrid compounds. These hybrids not only inherit or amplify the anti-AD bioactivity of the parent compound but also offer the potential for lower side effects.28 As ChEs and MAOs are two key targets for the treatment of AD, numerous multi-targeted drugs have been discovered and developed that target both enzymes.21,28,29 One such drug is Ladostigil, a bifunctional agent that combines the carbamate moiety of rivastigmine and the indoleamine moiety of rasagiline. This drug has inhibitory activities on AChE, BuChE, MAO-A and MAO-B and has entered a phase IIb clinical trial,30 indicating that the combination of ChEs and MAOs inhibitors is a promising method for the development of new multifunctional anti-AD drugs.
Inspired by the above concept of drug molecular design, a series of novel tacrine-selegiline hybrids with ChE and MAO inhibitory activities were designed and synthesized as multifunctional drugs against AD, based on tacrine (AChE and BuChE inhibitory activities) and selegiline (MAO-B inhibitory activity) (Figure 1). All designed compounds were synthesized and evaluated for their in vitro cholinesterase and monoamine oxidase inhibitory activities as well as their blood-brain barrier permeability. Furthermore, we performed molecular simulation studies, acute toxicity and neurotoxicity assessments, and pharmacokinetic and pharmacodynamic property evaluations for the optimizing compound.
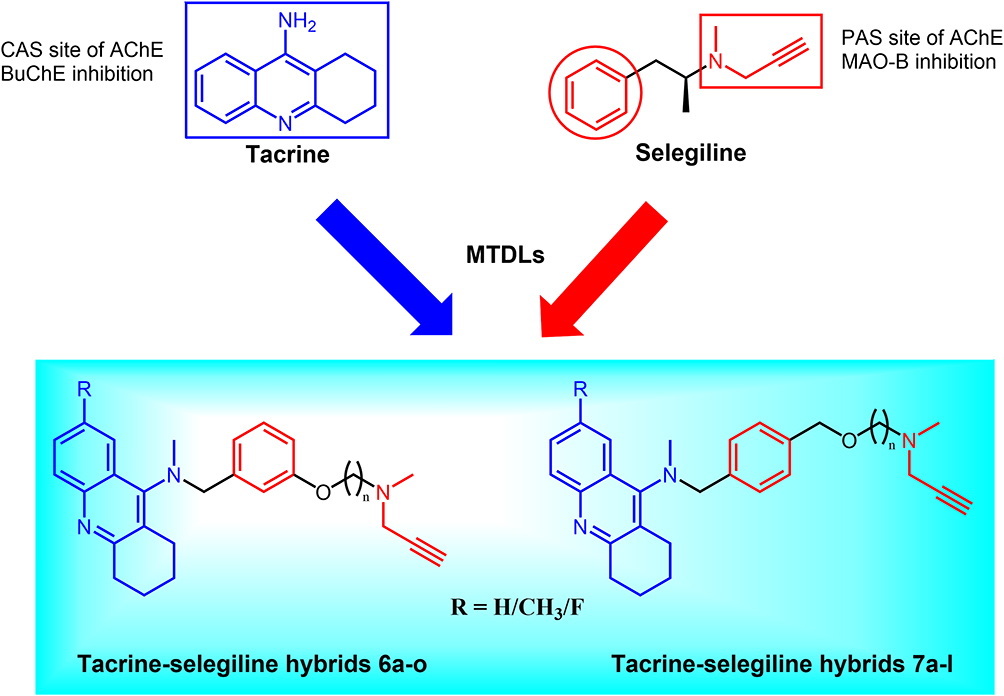 |
Figure 1 Design strategy toward tacrine-selegiline hybrids 6a-o and 7a-l. |
Materials and Methods
Chemistry
Reagents and solvents were purchased from commercial suppliers (China) and used without additional purification. All reaction processes were monitored by analytical thin layer chromatography (TLC) on glass-packed pre-coated silica gel GF254 (Qingdao Haiyang Chemical Co., Ltd., China) plates under 254 nm UV light. Compounds were purified through column chromatography using silica gel (90–150 mm, Qingdao Marine Chemical Inc.) as the stationary phase and a mixture of PE/EA or DCM/MeOH as the elution system. The 1H- (600 MHz) and 13C- (150 MHz) NMR spectra at room temperature were recorded on a Bruker AVANCE-NEO 600 MHz spectrometer using CD2Cl2, CDCl3, or DMSO-d6 solvents. Chemical shifts (δ) are expressed in parts per million (ppm), and coupling constants (J) in hertz (Hz). High-resolution mass spectra (HRMS) were obtained with a Mariner ESI-TOF spectrometer (HRESIMS). After being characterized by 1H NMR and HRMS, the intermediate undergoes the next reaction directly, while the target product was characterized using 1H NMR, 13C NMR, and HRMS. The samples of the tacrine-selegiline hybrids, which underwent pharmacological evaluation, possessed a purity > 95% as indicated by their high-performance liquid chromatography (HPLC) analyses.
Preparation of Intermediates 3a-3e
To a stirred solution of compound 1a-c (6.89 mmol, 1.0 equiv.) and KI (0.34 mmol, 0.05 equiv.) in 1-pentanol (20 mL), compound 2a 3-hydroxy benzylamine or 2b 4-hydroxymethyl benzylamine (13.78mmol, 2.0 equiv.) was added and refluxed (130 °C) for 8–10 h.31 After completion, it was concentrated to dryness, diluted with water, and extracted with EA (30 mL × 1, 20 mL × 3). The organic phase was washed with saturated salt water, dried with anhydrous sodium sulfate, filtered, and evaporated under reduced pressure. The crude product was purified by silica gel column chromatography (DCM/MeOH: 20/1~10/1, v/v) to give compounds 3a-3e.
3-(((1,2,3,4-tetrahydroacridin-9-yl)amino)methyl)phenol (3a). Yield 51%; yellow solid; 1H NMR (600 MHz, DMSO-d6) δ 8.10 (d, J = 8.6 Hz, 1H), 7.70 (d, J = 8.5 Hz, 1H), 7.51 (q, J = 6.8 Hz, 1H), 7.28 (q, J = 6.7 Hz, 1H), 7.08 (q, J = 6.6, 5.8 Hz, 1H), 6.75 (d, J = 9.1 Hz, 2H), 6.61 (d, J = 8.1 Hz, 1H), 6.00 (d, J = 10.2 Hz, 1H), 4.56 (t, J = 5.5 Hz, 2H), 2.89 (q, J = 5.8 Hz, 2H), 2.72 (d, J = 6.1 Hz, 2H), 1.78 (dt, J = 20.4, 6.5 Hz, 4H) (Figure S1 in the Supporting Information); HRMS (ESI): m/z calcd for C20H21N2O [M+H]+: 305.1648; found: 305.1691(Figure S2 in the Supporting Information).
3-(((7-methyl-1,2,3,4-tetrahydroacridin-9-yl)amino)methyl)phenol (3b). Yield 54%; yellow solid; 1H NMR (600 MHz, DMSO-d6) δ 9.36 (s, 1H), 7.90 (s, 1H), 7.60 (d, J = 8.5 Hz, 1H), 7.39–7.32 (m, 1H), 7.08 (t, J = 7.9 Hz, 1H), 6.75 (d, J = 7.2 Hz, 2H), 6.60 (dd, J = 8.1, 2.3 Hz, 1H), 6.11 (d, J = 7.6 Hz, 1H), 4.54 (d, J = 7.1 Hz, 2H), 2.86 (t, J = 6.3 Hz, 2H), 2.69 (t, J = 6.2 Hz, 2H), 2.38 (s, 3H), 1.82–1.70 (m, 4H) (Figure S3 in the Supporting Information); HRMS (ESI): m/z calcd for C21H23N2O [M+H]+: 319.1805; found: 319.1860 (Figure S4 in the Supporting Information).
3-(((7-fluoro-1,2,3,4-tetrahydroacridin-9-yl)amino)methyl)phenol (3c). Yield 72%; yellow solid; 1H NMR (600 MHz, DMSO-d6) δ 9.33 (s, 1H), 7.86 (dd, J = 11.4, 2.8 Hz, 1H), 7.75 (dd, J = 9.2, 5.8 Hz, 1H), 7.42 (td, J = 8.6, 2.7 Hz, 1H), 7.08 (t, J = 8.0 Hz, 1H), 6.73 (dd, J = 4.4, 2.3 Hz, 2H), 6.59 (dd, J = 8.1, 2.3 Hz, 1H), 6.15 (s, 1H), 4.51 (d, J = 7.1 Hz, 2H), 2.88 (t, J = 6.4 Hz, 2H), 2.72 (t, J = 6.3 Hz, 2H), 1.82–1.70 (m, 4H) (Figure S5 in the Supporting Information); HRMS (ESI): m/z calcd for C20H20FN2O[M+H]+: 323.1554; found: 323.1588 (Figure S6 in the Supporting Information).
(4-(((1,2,3,4-tetrahydroacridin-9-yl)amino)methyl)phenyl)methanol (3d). Yield 77%; yellow solid; 1H NMR (600 MHz, Methylene Chloride-d2) δ 7.99 (dd, J = 8.5, 1.4 Hz, 1H), 7.83 (dd, J = 8.5, 1.3 Hz, 1H), 7.54 (ddd, J = 8.3, 6.8, 1.4 Hz, 1H), 7.37–7.30 (m, 5H), 4.68 (s, 2H), 4.64–4.57 (m, 2H), 4.24 (s, 1H), 3.00 (t, J = 6.2 Hz, 2H), 2.66 (t, J = 6.2 Hz, 2H), 1.90–1.83 (m, 4H) (Figure S7 in the Supporting Information); HRMS (ESI): m/z calcd for C21H23N2O [M+H]+: 319.1805; found: 319.1858 (Figure S8 in the Supporting Information).
(4-(((7-methyl-1,2,3,4-tetrahydroacridin-9-yl)amino)methyl)phenyl)methanol (3e). Yield 62%; yellow solid; 1H NMR (600 MHz, DMSO-d6) δ 8.25 (s, 1H), 8.12 (s, 1H), 7.84 (d, J = 8.6 Hz, 1H), 7.60 (d, J = 8.6 Hz, 1H), 7.33 (d, J = 7.9 Hz, 2H), 7.29 (d, J = 7.9 Hz, 2H), 5.02 (d, J = 6.7 Hz, 2H), 4.46 (s, 2H), 2.98 (d, J = 5.2 Hz, 2H), 2.72 (d, J = 5.1 Hz, 2H), 2.34 (s, 3H), 1.79 (t, J = 3.6 Hz, 4H) (Figure S9 in the Supporting Information); HRMS (ESI): m/z calcd for C22H25N2O [M+H]+: 333.1961; found: 333.2015 (Figure S10 in the Supporting Information).
General Procedure for the Preparation of Compounds 4a-4e
A mixture of intermediate 3a-3e (4.24 mmol, 1.0 equiv.), 40% formaldehyde solution (14.84mmol, 3.5 equiv.), and glacial acetic acid (4.24 mmol, 1.0 equiv.) in DMF (30 mL) was stirred at room temperature for 60 min, then sodium cyanoborohydride (12.72 mmol, 3.0 equiv.) was added and stirred at room temperature for 8 h.32 After completion, the mixture was poured into appropriate water and extracted with EA (20 mL × 3, 10 mL × 1). The organic phase was washed with saturated salt water, dried with anhydrous Na2SO4, filtered, and concentrated in a vacuum. The residue was purified by silica gel chromatography (PE/EA: 1/1~1/3, v/v), and compounds 4a-4e were obtained.
3-((methyl(1,2,3,4-tetrahydroacridin-9-yl)amino)methyl)phenol (4a). Yield 78%; yellow solid; 1H NMR (600 MHz, DMSO-d6) δ 9.31 (s, 1H), 8.09 (d, J = 8.4 Hz, 1H), 7.89 (d, J = 8.4 Hz, 1H), 7.67 (s, 1H), 7.52 (s, 1H), 7.13 (t, J = 8.0 Hz, 1H), 6.75–6.70 (m, 2H), 6.67 (dd, J = 8.1, 2.4 Hz, 1H), 4.38 (s, 2H), 3.04 (t, J = 6.7 Hz, 2H), 2.92 (s, 3H), 2.83 (t, J = 6.3 Hz, 2H), 1.90–1.85 (m, 2H), 1.80–1.74 (m, 2 H) (Figure S11 in the Supporting Information); HRMS (ESI): m/z calcd for C21H23N2O [M+H]+: 319.1805; found: 319.1813 (Figure S12 in the Supporting Information).
3-((methyl(7-methyl-1,2,3,4-tetrahydroacridin-9-yl)amino)methyl)phenol (4b). Yield 80%; yellow solid; 1H NMR (600 MHz, Chloroform-d) δ 8.11 (d, J = 8.6 Hz, 1H), 7.74 (s, 1H), 7.45 (dd, J = 8.6, 1.8 Hz, 1H), 7.12 (t, J = 7.8 Hz, 1H), 6.83 (dd, J = 8.0, 2.5 Hz, 1H), 6.77 (t, J = 2.0 Hz, 1H), 6.69 (d, J = 7.5 Hz, 1H), 4.44 (s, 2H), 3.22 (t, J = 6.6 Hz, 2H), 3.01 (s, 3H), 2.67 (t, J = 6.2 Hz, 2H), 2.49 (s, 3H), 1.84 (m, 2H), 1.77–1.69 (m, 2H) (Figure S13 in the Supporting Information); HRMS (ESI): m/z calcd for C22H25N2O [M+H]+: 333.1961; found: 333.1991 (Figure S14 in the Supporting Information).
3-(((7-fluoro-1,2,3,4-tetrahydroacridin-9-yl)(methyl)amino)methyl)phenol (4c). Yield 72%; pale yellow solid; 1H NMR (600 MHz, DMSO-d6) δ 9.37 (s, 1H), 7.91 (dd, J = 9.2, 5.6 Hz, 1H), 7.70 (dd, J = 10.7, 2.9 Hz, 1H), 7.52 (td, J = 8.7, 2.8 Hz, 1H), 7.13 (t, J = 8.0 Hz, 1H), 6.76–6.70 (m, 2H), 6.68–6.63 (m, 1H), 4.27 (s, 2H), 2.99 (t, J = 6.7 Hz, 2H), 2.83 (s, 3H), 2.81 (d, J = 6.4 Hz, 2H), 1.85 (p, J = 6.4 Hz, 2H), 1.75 (p, J = 6.0 Hz, 2H) (Figure S15 in the Supporting Information); HRMS (ESI): m/z calcd for C21H22FN2O [M+H]+: 337.1711; found: 337.1746 (Figure S16 in the Supporting Information).
(4-((methyl(1,2,3,4-tetrahydroacridin-9-yl)amino)methyl)phenyl)methanol (4d). Yield 69%; yellow solid; 1H NMR (600 MHz, Methylene Chloride-d2) δ 8.09 (dd, J = 8.5, 1.4 Hz, 1H), 7.89 (dt, J = 8.4, 0.9 Hz, 1H), 7.57 (ddd, J = 8.3, 6.8, 1.4 Hz, 1H), 7.43 (ddd, J = 8.2, 6.7, 1.3 Hz, 1H), 7.35–7.30 (m, 4H), 4.67 (s, 2H), 4.40 (s, 2H), 3.07 (t, J = 6.7 Hz, 2H), 2.90 (s, 3H), 2.86 (t, J = 6.4 Hz, 2H), 1.96–1.90 (m, 2H), 1.84–1.79 (m, 2H) (Figure S17 in the Supporting Information); HRMS (ESI): m/z calcd for C22H25N2O [M+H]+: 333.1961; found: 333.2016 (Figure S18 in the Supporting Information).
(4-((methyl(7-methyl-1,2,3,4-tetrahydroacridin-9-yl)amino)methyl)phenyl)methanol (4e). Yield 76%; yellow solid; 1H NMR (600 MHz, Chloroform-d) δ 8.36 (d, J = 8.6 Hz, 1H), 7.73 (s, 1H), 7.57–7.48 (m, 1H), 7.35 (d, J = 8.1 Hz, 2H), 7.20 (dd, J = 8.1, 2.8 Hz, 2H), 4.72 (d, J = 1.9 Hz, 2H), 4.52 (s, 2H), 3.37 (t, J = 6.6 Hz, 2H), 3.00 (d, J = 2.0 Hz, 3H), 2.77–2.67 (m, 2H), 2.51 (d, J = 2.2 Hz, 3H), 1.91 (dp, J = 10.3, 3.9 Hz, 2H), 1.78 (qd, J = 5.8, 2.4 Hz, 2H) (Figure S19 in the Supporting Information); HRMS (ESI): m/z calcd for C23H27N2O [M+H]+: 347.2118; found: 347.2170 (Figure S20 in the Supporting Information).
General Procedure for the Preparation of Compounds 6a-o and 7a-l
To a solution of compound 4a-4e (3.10 mmol, 1.0 equiv.) in dry DMF (20mL), NaH (6.20 mmol, 2.0 equiv., 60% mineral oil) was added under the protection of nitrogen atmosphere and stirred at 0 °C for 30 min. Next, dibromide (18.6 mmol, 6.0 equiv.) was added and stirred at room temperature for 12 h. After completion, the reaction was quenched by ice water. The reaction mixture was extracted with EA (30 mL × 3), and the organic phase was washed with saturated salt water as well as dried with anhydrous Na2SO4. The crude product without excess solvent was purified by column chromatography (PE /EA: 4/1~1/1, v/v) to give compounds 5a-5e as pale yellow or yellow oil.33 A mixture of compound 5a-e (2.32 mmol, 1.0 equiv.) and K2CO3 (11.60 mmol, 5.0 equiv.) in acetonitrile (23 mL), N-methylpropargylamine (4.64 mmol, 2.0 equiv.) was added. The reaction was stirred for 12 h at 70 °C, the solvent was removed by vacuum, and appropriate water was added and extracted with DCM (30 mL × 3). The combined organic phases were washed with saturated salt water, dried with anhydrous Na2SO4, filtered, and concentrated under a vacuum. The crude product was purified by silica gel chromatography (PE/EA: 1/1~1/3 to DCM/MeOH: 20/1~10/1, v/v) to get the target compounds 6a-6o and 7a-7l.21
N-methyl-N-(3-(2-(methyl(prop-2-yn-1-yl)amino)ethoxy)benzyl)-1,2,3,4-tetrahydroacridin-9-amine (6a). Yield 62%; pale yellow oil; 1H NMR (600 MHz, Methylene Chloride-d2) δ 8.08 (dd, J = 8.4, 1.4 Hz, 1H), 8.00 (d, J = 8.4 Hz, 1H), 7.60 (ddd, J = 8.3, 6.7, 1.4 Hz, 1H), 7.45 (ddd, J = 8.2, 6.7, 1.3 Hz, 1H), 7.24 (t, J = 7.8 Hz, 1H), 6.91 (d, J = 7.5 Hz, 1H), 6.87 (t, J = 2.1 Hz, 1H), 6.84–6.79 (m, 1H), 4.41 (s, 2H), 3.99 (t, J = 5.7 Hz, 2H), 3.41 (d, J = 2.4 Hz, 2H), 3.13 (t, J = 6.7 Hz, 2H), 2.95 (s, 3H), 2.85 (t, J = 6.3 Hz, 2H), 2.82 (t, J = 5.7 Hz, 2H), 2.37 (s, 3H), 2.28 (t, J = 2.4 Hz, 1H), 1.96–1.91 (m, 2H), 1.82 (ddt, J = 9.5, 6.2, 3.5 Hz, 2H) (Figure S21 in the Supporting Information). 13C NMR (150 MHz, Chloroform-d) δ 160.59, 159.08, 154.51, 148.09, 141.12, 129.48, 128.98, 128.41, 127.96, 126.04, 125.01, 124.47, 121.19, 114.73, 113.65, 78.46, 73.53, 66.03, 60.43, 54.47, 46.10, 42.35, 40.50, 34.11, 27.03, 23.13, 23.03 (Figure S22 in the Supporting Information). HRMS (ESI): m/z calcd for C27H32N3O [M+H]+: 414.2540; found: 414.2603 (Figure S23 in the Supporting Information).
N-methyl-N-(3-(3-(methyl(prop-2-yn-1-yl)amino)propoxy)benzyl)-1,2,3,4-tetrahydroacridin-9-amine (6b). Yield 57%; pale yellow oil; 1H NMR (600 MHz, DMSO-d6) δ 8.07 (d, J = 8.4 Hz, 1H), 7.85 (d, J = 8.3 Hz, 1H), 7.62–7.57 (m, 1H), 7.49–7.44 (m, 1H), 7.22 (t, J = 7.8 Hz, 1H), 6.89 (d, J = 7.5 Hz, 1H), 6.86 (s, 1H), 6.80 (dd, J = 8.2, 2.4 Hz, 1H), 4.36 (s, 2H), 3.92 (t, J = 6.4 Hz, 2H), 3.25 (d, J = 2.5 Hz, 2H), 3.07–3.03 (m, 1H), 3.00 (t, J = 6.7 Hz, 2H), 2.88 (s, 3H), 2.84 (t, J = 6.4 Hz, 2H), 2.46 (t, J = 7.1 Hz, 2H), 2.20 (s, 3H), 1.89–1.83 (m, 2H), 1.81 (q, J = 6.8 Hz, 2H), 1.76 (q, J = 6.2 Hz, 2H) (Figure S24 in the Supporting Information). 13C NMR (150 MHz, DMSO-d6) δ 159.83, 158.63, 153.33, 147.42, 140.81, 129.19, 128.57, 127.95, 127.64, 125.37, 124.73, 123.99, 120.39, 114.09, 113.40, 78.93, 75.40, 65.57, 59.28, 51.65, 44.95, 41.14, 40.20, 33.34, 26.69, 25.92, 22.36, 22.22 (Figure S25 in the Supporting Information). HRMS (ESI): m/z calcd for C28H34N3O [M+H]+: 428.2696; found: 428.2763 (Figure S26 in the Supporting Information).
N-methyl-N-(3-(4-(methyl(prop-2-yn-1-yl)amino)butoxy)benzyl)-1,2,3,4-tetrahydroacridin-9-amine (6c). Yield 80%; pale yellow oil; 1H NMR (600 MHz, DMSO-d6) δ 8.08 (d, J = 8.4 Hz, 1H), 7.85 (d, J = 8.3 Hz, 1H), 7.60 (ddd, J = 8.3, 6.7, 1.4 Hz, 1H), 7.50–7.44 (m, 1H), 7.23 (t, J = 7.8 Hz, 1H), 6.89 (d, J = 7.5 Hz, 1H), 6.86 (t, J = 2.0 Hz, 1H), 6.81 (dd, J = 8.2, 2.5 Hz, 1H), 4.36 (s, 2H), 3.91 (t, J = 6.5 Hz, 2H), 3.27 (s, 2H), 3.04 (d, J = 2.4 Hz, 1H), 3.00 (t, J = 6.7 Hz, 2H), 2.88 (s, 3H), 2.84 (t, J = 6.3 Hz, 2H), 2.37 (t, J = 7.2 Hz, 2H), 2.19 (s, 3H), 1.87 (p, J = 6.6 Hz, 2H), 1.76 (p, J = 6.3 Hz, 2H), 1.71–1.65 (m, 2H), 1.50 (p, J = 7.3 Hz, 2H) (Figure S27 in the Supporting Information). 13C NMR (150 MHz, DMSO-d6) δ 162.86, 160.22, 158.93, 154.01, 147.09, 141.12, 129.69, 128.63, 127.93, 125.62, 125.29, 124.45, 120.73, 114.36, 113.70, 79.25, 75.99, 67.38, 59.56, 54.74, 45.07, 41.43, 40.59, 33.55, 26.68, 26.40, 23.58, 22.73, 22.54 (Figure S28 in the Supporting Information). HRMS (ESI): m/z calcd for C29H36N3O [M+H]+: 442.2853; found: 442.2919 (Figure S29 in the Supporting Information).
N-methyl-N-(3-((5-(methyl(prop-2-yn-1-yl)amino)pentyl)oxy)benzyl)-1,2,3,4-tetrahydroacridin-9-amine (6d). Yield 86%; pale yellow oil; 1H NMR (600 MHz, DMSO-d6) δ 8.07 (d, J = 8.4 Hz, 1H), 7.85 (d, J = 8.3 Hz, 1H), 7.63–7.55 (m, 1H), 7.47 (dd, J = 8.4, 6.7 Hz, 1H), 7.22 (t, J = 8.0 Hz, 1H), 6.89 (d, J = 7.5 Hz, 1H), 6.85 (d, J = 2.4 Hz, 1H), 6.80 (dd, J = 8.2, 2.4 Hz, 1H), 4.36 (s, 2H), 3.88 (t, J = 6.6 Hz, 2H), 3.26 (s, 2H), 3.03 (t, J = 2.1 Hz, 1H), 3.00 (t, J = 6.7 Hz, 2H), 2.88 (d, J = 1.5 Hz, 3H), 2.84 (t, J = 6.4 Hz, 2H), 2.33 (t, J = 6.9 Hz, 2H), 2.18 (s, 3H), 1.86 (p, J = 6.8 Hz, 2H), 1.76 (p, J = 6.2 Hz, 2H), 1.67 (dp, J = 14.6, 7.1 Hz, 2H), 1.41 (dp, J = 22.3, 8.0 Hz, 4H) (Figure S30 in the Supporting Information). 13C NMR (150 MHz, DMSO-d6) δ 159.84, 158.66, 153.33, 147.43, 140.78, 129.20, 128.58, 127.96, 127.67, 125.38, 124.72, 123.99, 120.34, 114.10, 113.38, 79.09, 75.27, 67.24, 59.28, 54.83, 44.88, 41.19, 40.22, 33.34, 28.41, 26.48, 25.92, 23.28, 22.37, 22.22 (Figure S31 in the Supporting Information). HRMS (ESI): m/z calcd for C30H38N3O [M+H]+: 456.3009; found: 456.3077 (Figure S32 in the Supporting Information).
N-methyl-N-(3-((6-(methyl(prop-2-yn-1-yl)amino)hexyl)oxy)benzyl)-1,2,3,4-tetrahydroacridin-9-amine (6e). Yield 83%; pale yellow oil; 1H NMR (600 MHz, DMSO-d6) δ 8.07 (d, J = 8.4 Hz, 1H), 7.85 (d, J = 8.3 Hz, 1H), 7.60 (t, J = 7.6 Hz, 1H), 7.47 (t, J = 7.6 Hz, 1H), 7.22 (t, J = 7.8 Hz, 1H), 6.89 (d, J = 7.5 Hz, 1H), 6.85 (d, J = 2.6 Hz, 1H), 6.80 (dd, J = 8.2, 2.6 Hz, 1H), 4.36 (s, 2H), 3.88 (t, J = 6.5 Hz, 2H), 3.26 (d, J = 2.4 Hz, 2H), 3.03 (d, J = 2.4 Hz, 1H), 3.00 (t, J = 6.7 Hz, 2H), 2.88 (s, 3H), 2.84 (t, J = 6.4 Hz, 2H), 2.32 (t, J = 7.2 Hz, 2H), 2.17 (s, 3H), 1.86 (p, J = 6.5 Hz, 2H), 1.76 (p, J = 6.3 Hz, 2H), 1.66 (q, J = 6.9 Hz, 2H), 1.38 (tdd, J = 14.7, 9.4, 5.1 Hz, 4H), 1.30 (q, J = 7.7, 7.1 Hz, 2H) (Figure S33 in the Supporting Information). 13C NMR (151 MHz, DMSO) δ 159.83, 158.66, 153.33, 147.42, 140.78, 129.19, 128.58, 127.95, 127.66, 125.38, 124.71, 123.99, 120.33, 114.06, 113.41, 79.07, 75.26, 67.22, 59.28, 54.85, 44.87, 41.19, 40.22, 33.34, 28.52, 26.75, 26.43, 25.92, 25.29, 22.37, 22.22 (Figure S34 in the Supporting Information). HRMS (ESI): m/z calcd for C31H40N3O [M+H]+: 470.3166; found: 470.3234 (Figure S35 in the Supporting Information).
N-methyl-N-(3-((7-(methyl(prop-2-yn-1-yl)amino)heptyl)oxy)benzyl)-1,2,3,4-tetrahydroacridin-9-amine (6f). Yield 75%; pale yellow oil; 1H NMR (600 MHz, DMSO-d6) δ 8.07 (d, J = 8.4 Hz, 1H), 7.85 (d, J = 8.4 Hz, 1H), 7.60 (t, J = 7.5 Hz, 1H), 7.47 (t, J = 7.6 Hz, 1H), 7.22 (t, J = 7.8 Hz, 1H), 6.89 (d, J = 7.5 Hz, 1H), 6.84 (d, J = 2.4 Hz, 1H), 6.80 (dd, J = 8.2, 2.6 Hz, 1H), 4.36 (s, 2H), 3.88 (t, J = 6.6 Hz, 2H), 3.25 (d, J = 2.5 Hz, 2H), 3.02 (d, J = 2.4 Hz, 1H), 3.00 (t, J = 6.7 Hz, 2H), 2.88 (s, 3H), 2.84 (t, J = 6.4 Hz, 2H), 2.31 (t, J = 7.2 Hz, 2H), 2.16 (s, 3H), 1.86 (p, J = 6.6 Hz, 2H), 1.76 (q, J = 6.2 Hz, 2H), 1.66 (q, J = 7.0 Hz, 2H), 1.37 (dt, J = 10.6, 5.2 Hz, 4H), 1.32–1.26 (m, 4H) (Figure S36 in the Supporting Information). 13C NMR (150 MHz, DMSO-d6) δ 159.84, 158.67, 153.33, 147.43, 140.78, 129.19, 128.58, 127.95, 127.66, 125.38, 124.72, 124.00, 120.33, 114.07, 113.41, 79.09, 75.25, 67.23, 59.29, 54.90, 44.86, 41.19, 40.23, 40.06, 33.34, 28.48, 26.71, 26.66, 25.92, 25.36, 22.38, 22.23 (Figure S37 in the Supporting Information). HRMS (ESI): m/z calcd for C32H42N3O [M+H]+: 484.3322; found: 484.3390 (Figure S38 in the Supporting Information).
N-methyl-N-(3-((8-(methyl(prop-2-yn-1-yl)amino)octyl)oxy)benzyl)-1,2,3,4-tetrahydroacridin-9-amine (6g). Yield 66%; pale yellow oil; 1H NMR (600 MHz, DMSO-d6) δ 8.08 (d, J = 8.4 Hz, 1H), 7.85 (dd, J = 8.4, 1.2 Hz, 1H), 7.60 (dd, J = 8.4, 6.8 Hz, 1H), 7.47 (dd, J = 8.4, 6.9 Hz, 1H), 7.22 (t, J = 7.8 Hz, 1H), 6.89 (d, J = 7.5 Hz, 1H), 6.85 (d, J = 2.6 Hz, 1H), 6.80 (dd, J = 8.2, 2.6 Hz, 1H), 4.36 (s, 2H), 3.88 (td, J = 6.6, 1.6 Hz, 2H), 3.25 (d, J = 2.4 Hz, 2H), 3.02 (t, J = 2.3 Hz, 1H), 3.00 (t, J = 6.7 Hz, 2H), 2.88 (s, 3H), 2.84 (t, J = 6.4 Hz, 2H), 2.31 (t, J = 7.3 Hz, 2H), 2.17 (s, 3H), 1.86 (p, J = 6.6 Hz, 2H), 1.76 (p, J = 6.3 Hz, 2H), 1.66 (q, J = 7.1 Hz, 2H), 1.41–1.35 (m, 4H), 1.32–1.25 (m, 6H) (Figure S39 in the Supporting Information). 13C NMR (150 MHz, DMSO-d6) δ 162.84, 160.19, 158.96, 153.98, 147.52, 141.09, 129.66, 128.57, 127.90, 125.61, 125.25, 124.43, 120.70, 114.23, 113.74, 79.18, 75.98, 67.49, 59.57, 55.16, 45.06, 41.50, 40.58, 33.56, 29.13, 28.94, 28.81, 26.99, 26.39, 25.72, 22.73, 22.54, 22.38 (Figure S40 in the Supporting Information). HRMS (ESI): m/z calcd for C33H44N3O [M+H]+: 498.3479; found: 498.3546 (Figure S41 in the Supporting Information).
N,7-dimethyl-N-(3-(3-(methyl(prop-2-yn-1-yl)amino)propoxy)benzyl)-1,2,3,4-tetrahydroacridin-9-amine (6h). Yield 85%; yellow oil; 1H NMR (600 MHz, Methylene Chloride-d2) δ 7.88–7.85 (m, 1H), 7.80 (d, J = 8.5 Hz, 1H), 7.42 (dd, J = 8.5, 1.9 Hz, 1H), 7.23 (t, J = 7.8 Hz, 1H), 6.94–6.88 (m, 2H), 6.81 (ddd, J = 8.2, 2.6, 1.0 Hz, 1H), 4.36 (s, 2H), 3.95 (t, J = 6.4 Hz, 2H), 3.33 (d, J = 2.4 Hz, 2H), 3.05 (t, J = 6.7 Hz, 2H), 2.91 (s, 3H), 2.84 (t, J = 6.3 Hz, 2H), 2.56 (t, J = 7.1 Hz, 2H), 2.53 (s, 3H), 2.29 (s, 3H), 2.24 (t, J = 2.4 Hz, 1H), 1.94–1.87 (m, 4H), 1.83–1.78 (m, 2H) (Figure S42 in the Supporting Information). 13C NMR (150 MHz, Chloroform-d) δ 159.28, 158.90, 154.83, 145.46, 140.85, 134.91, 131.03, 129.41, 127.80, 127.63, 125.69, 123.37, 120.87, 114.41, 113.73, 78.48, 73.28, 65.97, 60.47, 52.39, 45.72, 41.75, 40.49, 33.27, 27.48, 26.93, 22.99, 22.73, 22.12 (Figure S43 in the Supporting Information). HRMS (ESI): m/z calcd for C29H36N3O [M+H]+: 442.2853; found: 442.2918 (Figure S44 in the Supporting Information).
N,7-dimethyl-N-(3-(4-(methyl(prop-2-yn-1-yl)amino)butoxy)benzyl)-1,2,3,4-tetrahydroacridin-9-amine (6i). Yield 86%; yellow oil; 1H NMR (600 MHz, Methylene Chloride-d2) δ 7.85 (d, J = 9.6 Hz, 2H), 7.43 (dd, J = 8.7, 1.9 Hz, 1H), 7.22 (q, J = 6.7, 5.7 Hz, 1H), 6.93–6.85 (m, 2H), 6.80 (dt, J = 9.4, 4.7 Hz, 1H), 4.38 (s, 2H), 3.91 (q, J = 6.7 Hz, 2H), 3.32 (d, J = 2.4 Hz, 2H), 3.07 (q, J = 6.6 Hz, 2H), 2.92 (s, 3H), 2.83 (q, J = 6.5 Hz, 2H), 2.53 (s, 3H), 2.47–2.38 (m, 2H), 2.27 (s, 3H), 2.24 (t, J = 2.4 Hz, 1H), 1.94–1.88 (m, 2H), 1.83–1.74 (m, 4H), 1.63–1.56 (m, 2H) (Figure S45 in the Supporting Information). 13C NMR (150 MHz, Chloroform-d) δ 159.53, 159.36, 153.89, 146.73, 141.25, 134.57, 130.54, 129.39, 128.81, 128.09, 126.02, 123.32, 120.92, 114.44, 113.72, 78.74, 73.14, 67.68, 60.44, 55.39, 45.66, 41.83, 40.39, 34.04, 27.26, 26.98, 24.29, 23.16, 23.08, 22.18 (Figure S46 in the Supporting Information). HRMS (ESI): m/z calcd for C30H38N3O[M+H]+: 456.3009; found: 456.3075 (Figure S47 in the Supporting Information).
N,7-dimethyl-N-(3-((5-(methyl(prop-2-yn-1-yl)amino)pentyl)oxy)benzyl)-1,2,3,4-tetrahydroacridin-9-amine (6j). Yield 87%; yellow oil; 1H NMR (600 MHz, Methylene Chloride-d2) δ 7.86 (s, 1H), 7.80 (d, J = 8.5 Hz, 1H), 7.42 (dd, J = 8.5, 1.9 Hz, 1H), 7.22 (t, J = 7.8 Hz, 1H), 6.94–6.87 (m, 2H), 6.80 (dd, J = 8.2, 2.6 Hz, 1H), 4.36 (s, 2H), 3.90 (t, J = 6.5 Hz, 2H), 3.31 (d, J = 2.4 Hz, 2H), 3.05 (t, J = 6.7 Hz, 2H), 2.91 (s, 3H), 2.84 (t, J = 6.3 Hz, 2H), 2.52 (s, 3H), 2.40 (t, J = 7.0 Hz, 2H), 2.26 (s, 3H), 2.24 (t, J = 2.4 Hz, 1H), 1.91 (d, J = 6.3 Hz, 2H), 1.82–1.75 (m, 4H), 1.53–1.43 (m, 4H) (Figure S48 in the Supporting Information). 13C NMR (150 MHz, DMSO-d6) δ 158.78, 158.26, 154.09, 144.87, 140.83, 134.45, 130.85, 130.30, 129.40, 127.36, 125.12, 123.21, 120.51, 113.94, 113.62, 77.17, 69.84, 67.16, 59.41, 54.67, 44.70, 40.77, 40.38, 32.72, 28.48, 26.17, 25.74, 23.24, 22.47, 22.19, 21.63 (Figure S49 in the Supporting Information). HRMS (ESI): m/z calcd for C31H40N3O [M+H]+: 470.3166; found: 470.3232 (Figure S50 in the Supporting Information).
N,7-dimethyl-N-(3-((6-(methyl(prop-2-yn-1-yl)amino)hexyl)oxy)benzyl)-1,2,3,4-tetrahydroacridin-9-amine (6k). Yield 78%; yellow oil; 1H NMR (600 MHz, Methylene Chloride-d2) δ 7.86 (dt, J = 1.9, 0.9 Hz, 1H), 7.83 (d, J = 8.5 Hz, 1H), 7.43 (dd, J = 8.5, 2.0 Hz, 1H), 7.22 (d, J = 0.8 Hz, 1H), 6.91–6.87 (m, 2H), 6.80 (ddd, J = 8.2, 2.6, 1.0 Hz, 1H), 4.37 (s, 2H), 3.89 (t, J = 6.5 Hz, 2H), 3.31 (d, J = 2.4 Hz, 2H), 3.07 (t, J = 6.7 Hz, 2H), 2.91 (s, 3H), 2.84 (t, J = 6.3 Hz, 2H), 2.52 (s, 3H), 2.41–2.36 (m, 2H), 2.26 (s, 3H), 2.24 (t, J = 2.4 Hz, 1H), 1.91 (d, J = 6.4 Hz, 2H), 1.83–1.78 (m, 2H), 1.78–1.72 (m, 2H), 1.49–1.43 (m, 4H), 1.36 (d, J = 5.4 Hz, 2H) (Figure S51 in the Supporting Information). 13C NMR (150 MHz, Chloroform-d) δ 159.40, 158.08, 156.30, 143.75, 140.35, 135.47, 131.81, 129.54, 127.03, 126.46, 125.30, 123.54, 120.75, 114.43, 113.79, 78.27, 73.46, 67.78, 60.64, 55.53, 45.50, 41.73, 40.74, 32.32, 29.76, 29.22, 27.25, 26.96, 23.99, 22.85, 22.32, 22.13 (Figure S52 in the Supporting Information). HRMS (ESI): m/z calcd for C32H42N3O [M+H]+: 484.3322; found: 484.3386 (Figure S53 in the Supporting Information).
N,7-dimethyl-N-(3-((7-(methyl(prop-2-yn-1-yl)amino)heptyl)oxy)benzyl)-1,2,3,4-tetrahydroacridin-9-amine (6L). Yield 78%; yellow oil; 1H NMR (600 MHz, Methylene Chloride-d2) δ 7.89–7.85 (m, 1H), 7.78 (d, J = 8.5 Hz, 1H), 7.41 (dd, J = 8.5, 1.9 Hz, 1H), 7.22 (t, J = 7.7 Hz, 1H), 6.93–6.88 (m, 2H), 6.82–6.77 (m, 1H), 4.35 (s, 2H), 3.89 (t, J = 6.5 Hz, 2H), 3.30 (d, J = 2.4 Hz, 2H), 3.04 (t, J = 6.7 Hz, 2H), 2.90 (s, 3H), 2.85 (t, J = 6.4 Hz, 2H), 2.52 (s, 3H), 2.40–2.34 (m, 2H), 2.25 (s, 3H), 2.23 (t, J = 2.4 Hz, 1H), 1.94–1.89 (m, 2H), 1.84–1.71 (m, 6H), 1.48–1.40 (m, 4H), 1.35 (dt, J = 9.9, 6.5 Hz, 2H) (Figure S54 in the Supporting Information). 13C NMR (150 MHz, Methylene Chloride-d2) δ 159.83, 159.77, 153.90, 147.00, 141.73, 134.78, 130.52, 129.54, 128.98, 128.51, 126.30, 123.65, 121.03, 114.54, 113.93, 79.43, 72.81, 68.24, 60.59, 56.06, 45.84, 41.84, 40.49, 34.28, 29.69, 29.66, 27.90, 27.69, 27.21, 26.42, 23.44, 23.35, 22.11 (Figure S55 in the Supporting Information). HRMS (ESI): m/z calcd for C33H44N3O [M+H]+: 498.3479; found: 498.3545 (Figure S56 in the Supporting Information).
N,7-dimethyl-N-(3-((8-(methyl(prop-2-yn-1-yl)amino)octyl)oxy)benzyl)-1,2,3,4-tetrahydroacridin-9-amine (6m). Yield 83%; yellow oil; 1H NMR (600 MHz, Methylene Chloride-d2) δ 7.87 (s, 1H), 7.78 (d, J = 8.5 Hz, 1H), 7.41 (dd, J = 8.6, 1.9 Hz, 1H), 7.22 (t, J = 7.8 Hz, 1H), 6.92–6.89 (m, 2H), 6.81–6.77 (m, 1H), 4.35 (s, 2H), 3.89 (t, J = 6.6 Hz, 2H), 3.30 (d, J = 2.4 Hz, 2H), 3.04 (t, J = 6.7 Hz, 2H), 2.90 (s, 3H), 2.85 (t, J = 6.3 Hz, 2H), 2.52 (s, 3H), 2.40–2.33 (m, 2H), 2.25 (s, 3H), 2.23 (t, J = 2.4 Hz, 1H), 1.94–1.88 (m, 2H), 1.81 (t, J = 6.1 Hz, 2H), 1.77–1.72 (m, 2H), 1.44 (dt, J = 10.9, 7.2 Hz, 4H), 1.37–1.30 (m, 6H) (Figure S57 in the Supporting Information). 13C NMR (151 MHz, Methylene Chloride-d2) δ 159.83, 159.78, 153.89, 147.01, 141.74, 134.78, 130.51, 129.54, 129.00, 128.51, 126.30, 123.65, 121.03, 114.54, 113.93, 79.45, 72.78, 68.26, 60.59, 56.09, 45.85, 41.84, 40.49, 34.30, 29.89, 29.76, 29.68, 27.96, 27.70, 27.21, 26.41, 23.45, 23.36, 22.11 (Figure S58 in the Supporting Information). HRMS (ESI): m/z calcd for C34H46N3O [M+H]+: 512.3635; found: 512.3701 (Figure S59 in the Supporting Information).
7-fluoro-N-methyl-N-(3-((6-(methyl(prop-2-yn-1-yl)amino)hexyl)oxy)benzyl)-1,2,3,4-tetrahydroacridin-9-amine (6n). Yield 77%; yellow oil; 1H NMR (600 MHz, Chloroform-d) δ 7.97 (dd, J = 9.2, 5.5 Hz, 1H), 7.70 (dd, J = 10.5, 2.8 Hz, 1H), 7.39–7.33 (m, 1H), 7.25–7.22 (m, 1H), 6.89 (d, J = 7.5 Hz, 1H), 6.85 (t, J = 2.0 Hz, 1H), 6.82 (dd, J = 8.2, 2.4 Hz, 1H), 4.33 (s, 2H), 3.90 (t, J = 6.5 Hz, 2H), 3.36 (d, J = 2.3 Hz, 2H), 3.11 (t, J = 6.7 Hz, 2H), 2.90 (s, 3H), 2.85 (t, J = 6.4 Hz, 2H), 2.43 (t, J = 7.5 Hz, 2H), 2.32 (s, 3H), 2.22 (d, J = 2.2 Hz, 1H), 1.96–1.92 (m, 2H), 1.81 (d, J = 12.6 Hz, 2H), 1.77 (q, J = 7.1 Hz, 2H), 1.49 (dt, J = 14.5, 7.2 Hz, 4H), 1.38 (q, J = 7.8, 7.4 Hz, 2H) (Figure S60 in the Supporting Information). 13C NMR (150 MHz, Chloroform-d) δ 160.01 (d, JC-F = 243 Hz), 159.89 (d, JC-F = 1.5 Hz), 159.43, 153.91 (d, JC-F = 4.5 Hz), 145.15, 140.79, 131.33 (d, JC-F = 9 Hz), 129.50, 128.87, 126.91 (d, JC-F = 9 Hz), 120.81, 118.44 (d, JC-F = 25.5 Hz), 115.88, 113.80, 107.81 (d, JC-F = 22.5 Hz), 78.74, 73.12, 67.93, 60.20, 55.74, 45.62, 41.88, 40.18, 33.95, 29.36, 27.65, 27.30, 27.01, 26.13, 22.98, 22.94 (Figure S61 in the Supporting Information). HRMS (ESI): m/z calcd for C31H39FN3O [M+H]+: 488.3072; found: 488.3133 (Figure S62 in the Supporting Information).
7-fluoro-N-methyl-N-(3-((8-(methyl(prop-2-yn-1-yl)amino)octyl)oxy)benzyl)-1,2,3,4-tetrahydroacridin-9-amine (6o). Yield 77%; yellow oil; 1H NMR (600 MHz, Chloroform-d) δ 7.96 (dd, J = 9.2, 5.4 Hz, 1H), 7.70 (dd, J = 10.5, 2.8 Hz, 1H), 7.36 (td, J = 8.6, 2.8 Hz, 1H), 7.24 (t, J = 7.9 Hz, 1H), 6.88 (d, J = 7.5 Hz, 1H), 6.86 (d, J = 2.5 Hz, 1H), 6.82 (dd, J = 8.1, 2.5 Hz, 1H), 4.32 (s, 2H), 3.90 (t, J = 6.6 Hz, 2H), 3.34 (d, J = 2.5 Hz, 2H), 3.11 (t, J = 6.7 Hz, 2H), 2.90 (s, 3H), 2.84 (t, J = 6.4 Hz, 2H), 2.44–2.37 (m, 2H), 2.30 (s, 3H), 2.21 (t, J = 2.4 Hz, 1H), 1.94 (p, J = 6.5 Hz, 2H), 1.83–1.73 (m, 4H), 1.45 (q, J = 9.5 Hz, 4H), 1.34 (d, J = 9.7 Hz, 6H) (Figure S63 in the Supporting Information). 13C NMR (150 MHz, Chloroform-d) δ 160.02 (d, JC-F = 244.5 Hz), 159.9 (d, JC-F = 3 Hz), 159.46, 153.91 (d, JC-F = 6 Hz), 145.16, 140.78, 131.33 (d, JC-F = 9 Hz), 129.50, 128.88, 126.92 (d, JC-F = 9 Hz), 120.79, 118.44 (d, JC-F = 25.5 Hz), 114.46, 113.80, 107.81 (d, JC-F = 24 Hz), 78.78, 73.08, 68.02, 60.22, 55.86, 45.61, 41.89, 40.19, 33.96, 29.61, 29.44, 29.38, 27.71, 27.48, 27.01, 26.12, 22.98, 22.94 (Figure S64 in the Supporting Information). HRMS (ESI): m/z calcd for C33H43FN3O [M+H]+: 516.3385; found: 516.3446 (Figure S65 in the Supporting Information).
N-methyl-N-(4-((3-(methyl(prop-2-yn-1-yl)amino)propoxy)methyl)benzyl)-1,2,3,4-tetrahydroacridin-9-amine (7a). Yield 37%; yellow oil; 1H NMR (600 MHz, Methylene Chloride-d2) δ 8.09 (dd, J = 8.4, 1.4 Hz, 1H), 7.89 (d, J = 8.4 Hz, 1H), 7.57 (ddd, J = 8.5, 6.8, 1.5 Hz, 1H), 7.43 (ddd, J = 8.2, 6.7, 1.3 Hz, 1H), 7.34–7.28 (m, 4H), 4.47 (s, 2H), 4.40 (s, 2H), 3.51 (t, J = 6.5 Hz, 2H), 3.31 (d, J = 2.4 Hz, 2H), 3.10–3.04 (m, 2H), 2.90 (s, 3H), 2.86 (t, J = 6.3 Hz, 2H), 2.50–2.46 (m, 2H), 2.27 (s, 3H), 2.24 (t, J = 2.4 Hz, 1H), 1.93 (p, J = 6.6 Hz, 2H), 1.84–1.79 (m, 2H), 1.77–1.73 (m, 2H) (Figure S66 in the Supporting Information). 13C NMR (150 MHz, Chloroform-d) δ 158.20, 154.44, 150.89, 138.41, 137.20, 130.08, 129.52, 128.68, 128.17, 127.25, 126.47, 125.92, 125.04, 79.07, 72.72, 70.70, 68.60, 60.77, 52.69, 45.46, 41.47, 41.33, 32.06, 29.84, 27.38, 27.16, 22.67. HRMS (ESI): m/z calcd for C29H36N3O [M+H]+: 442.2853; found: 442.2902 (Figure S67 in the Supporting Information).
N-methyl-N-(4-((4-(methyl(prop-2-yn-1-yl)amino)butoxy)methyl)benzyl)-1,2,3,4-tetrahydroacridin-9-amine (7b). Yield 89%; yellow oil; 1H NMR (600 MHz, Methylene Chloride-d2) δ 8.09 (dd, J = 8.3, 1.4 Hz, 1H), 7.89 (d, J = 8.4 Hz, 1H), 7.57 (ddd, J = 8.3, 6.7, 1.4 Hz, 1H), 7.43 (ddd, J = 8.1, 6.7, 1.3 Hz, 1H), 7.34–7.27 (m, 4H), 4.47 (s, 2H), 4.40 (s, 2H), 3.49 (t, J = 6.4 Hz, 2H), 3.30 (d, J = 2.4 Hz, 2H), 3.07 (t, J = 6.7 Hz, 2H), 2.90 (s, 3H), 2.86 (t, J = 6.3 Hz, 2H), 2.42–2.37 (m, 2H), 2.26 (s, 3H), 2.23 (t, J = 2.4 Hz, 1H), 1.93 (p, J = 6.6 Hz, 2H), 1.84–1.79 (m, 2H), 1.63–1.60 (m, 2H), 1.54–1.49 (m, 2H) (Figure S68 in the Supporting Information). 13C NMR (150 MHz, Chloroform-d) δ 159.44, 157.88, 142.44, 138.46, 137.19, 131.09, 129.34, 128.66, 128.14, 126.41, 125.87, 125.03, 124.55, 74.84, 72.68, 70.68, 70.34, 60.74, 55.27, 45.20, 41.31, 32.04, 29.82, 27.50, 27.14, 23.76, 22.66, 21.67 (Figure S69 in the Supporting Information). HRMS (ESI): m/z calcd for C30H38N3O [M+H]+: 456.3009; found: 456.3069 (Figure S70 in the Supporting Information).
N-methyl-N-(4-(((5-(methyl(prop-2-yn-1-yl)amino)pentyl)oxy)methyl)benzyl)-1,2,3,4-tetrahydroacridin-9-amine (7c). Yield 80%; yellow oil; 1H NMR (600 MHz, Methylene Chloride-d2) δ 8.11–8.08 (m, 1H), 7.89 (dd, J = 8.4, 1.3 Hz, 1H), 7.57 (ddd, J = 8.4, 6.8, 1.5 Hz, 1H), 7.43 (ddd, J = 8.2, 6.7, 1.3 Hz, 1H), 7.34–7.28 (m, 4H), 4.46 (s, 2H), 4.40 (s, 2H), 3.47 (t, J = 6.6 Hz, 2H), 3.30 (d, J = 2.4 Hz, 2H), 3.07 (t, J = 6.7 Hz, 2H), 2.90 (s, 3H), 2.86 (t, J = 6.3 Hz, 2H), 2.40–2.34 (m, 2H), 2.25 (s, 3H), 2.23 (t, J = 2.4 Hz, 1H), 1.93 (p, J = 6.6 Hz, 2H), 1.82 (ddt, J = 12.1, 9.1, 4.4 Hz, 2H), 1.63–1.61 (m, 2H), 1.45 (tt, J = 7.4, 5.9 Hz, 2H), 1.41–1.36 (m, 2H) (Figure S71 in the Supporting Information). 13C NMR (150 MHz, Methylene Chloride-d2) δ 160.95, 154.52, 148.41, 139.15, 138.33, 129.18, 128.96, 128.47, 128.36, 128.06, 126.38, 125.05, 124.81, 79.40, 72.96, 72.83, 70.86, 60.37, 55.98, 45.85, 41.84, 40.47, 34.40, 30.08, 27.77, 27.22, 24.40, 23.40, 23.30 (Figure S72 in the Supporting Information). HRMS (ESI): m/z calcd for C31H40N3O [M+H]+: 470.3166; found: 470.3224 (Figure S73 in the Supporting Information).
N-methyl-N-(4-(((6-(methyl(prop-2-yn-1-yl)amino)hexyl)oxy)methyl)benzyl)-1,2,3,4-tetrahydroacridin-9-amine (7d). Yield 64%; yellow oil; 1H NMR (600 MHz, Methylene Chloride-d2) δ 8.09 (dd, J = 8.4, 1.4 Hz, 1H), 7.92–7.86 (m, 1H), 7.57 (ddd, J = 8.3, 6.7, 1.4 Hz, 1H), 7.43 (ddd, J = 8.2, 6.7, 1.3 Hz, 1H), 7.35–7.26 (m, 4H), 4.46 (s, 2H), 4.39 (s, 2H), 3.46 (t, J = 6.6 Hz, 2H), 3.29 (d, J = 2.4 Hz, 2H), 3.07 (t, J = 6.7 Hz, 2H), 2.90 (s, 3H), 2.86 (t, J = 6.3 Hz, 2H), 2.40–2.33 (m, 2H), 2.25 (s, 3H), 2.24–2.22 (m, 2H), 1.93 (p, J = 6.6 Hz, 2H), 1.82 (q, J = 6.1 Hz, 2H), 1.70–1.68 (m, 2H), 1.61–1.59 (m, 2H), 1.44–1.42 (m, 2H), 1.41–1.37 (m, 2H) (Figure S74 in the Supporting Information). 13C NMR (150 MHz, Chloroform-d) δ 158.82, 157.25, 144.62, 138.10, 137.64, 129.82, 128.55, 127.88, 126.60, 126.15, 125.68, 125.02, 124.68, 74.04, 72.56, 70.57, 60.37, 55.44, 45.19, 41.42, 40.76, 32.70, 32.06, 29.64, 27.14, 26.92, 26.05, 25.65, 22.69, 22.07 (Figure S75 in the Supporting Information). HRMS (ESI): m/z calcd for C32H42N3O [M+H]+: 484.3322; found: 484.3378 (Figure S76 in the Supporting Information).
N-methyl-N-(4-(((7-(methyl(prop-2-yn-1-yl)amino)heptyl)oxy)methyl)benzyl)-1,2,3,4-tetrahydroacridin-9-amine (7e). Yield 37%; yellow oil; 1H NMR (600 MHz, Methylene Chloride-d2) δ 8.11–8.07 (m, 1H), 7.89 (d, J = 8.4 Hz, 1H), 7.57 (ddd, J = 8.3, 6.7, 1.4 Hz, 1H), 7.43 (ddd, J = 8.3, 6.8, 1.3 Hz, 1H), 7.33–7.27 (m, 4H), 4.46 (s, 2H), 4.39 (s, 2H), 3.46 (t, J = 6.6 Hz, 2H), 3.29 (d, J = 2.4 Hz, 2H), 3.07 (t, J = 6.7 Hz, 2H), 2.90 (s, 3H), 2.86 (t, J = 6.3 Hz, 2H), 2.36 (dd, J = 8.2, 6.7 Hz, 2H), 2.24 (s, 3H), 2.22 (t, J = 2.4 Hz, 1H), 1.93 (p, J = 6.5 Hz, 2H), 1.82 (q, J = 6.1 Hz, 2H), 1.62–1.58 (m, 6H), 1.44–1.41 (m, 2H), 1.32–1.30 (m, 2H) (Figure S77 in the Supporting Information). 13C NMR (150 MHz, Chloroform-d) δ 159.47, 154.42, 146.56, 138.17, 137.69, 128.64, 128.36, 128.24, 127.57, 127.36, 126.58, 125.17, 124.36, 77.48, 71.66, 69.62, 62.74, 59.22, 54.70, 44.60, 40.62, 40.31, 32.99, 29.20, 28.65, 26.55, 26.17, 25.69, 22.46, 22.19, 22.18 (Figure S78 in the Supporting Information). HRMS (ESI): m/z calcd for C33H44N3O [M+H]+: 498.3479; found: 498.3526 (Figure S79 in the Supporting Information).
N-methyl-N-(4-(((8-(methyl(prop-2-yn-1-yl)amino)octyl)oxy)methyl)benzyl)-1,2,3,4-tetrahydroacridin-9-amine (7f). Yield 51%; yellow oil; 1H NMR (600 MHz, Methylene Chloride-d2) δ 8.09 (dd, J = 8.5, 1.4 Hz, 1H), 7.89 (dd, J = 8.5, 1.3 Hz, 1H), 7.57 (ddd, J = 8.4, 6.7, 1.5 Hz, 1H), 7.43 (ddd, J = 8.2, 6.7, 1.3 Hz, 1H), 7.34–7.27 (m, 4H), 4.46 (s, 2H), 4.39 (s, 2H), 3.46 (t, J = 6.6 Hz, 2H), 3.29 (d, J = 2.5 Hz, 2H), 3.07 (t, J = 6.7 Hz, 2H), 2.90 (s, 3H), 2.86 (t, J = 6.4 Hz, 2H), 2.39–2.32 (m, 2H), 2.24 (s, 3H), 2.22 (t, J = 2.4 Hz, 1H), 1.93 (p, J = 6.6 Hz, 2H), 1.85–1.78 (m, 2H), 1.73–1.65 (m, 4H), 1.62–1.57 (m, 2H), 1.45–1.39 (m, 2H), 1.39–1.32 (m, 4H) (Figure S80 in the Supporting Information). 13C NMR (150 MHz, Chloroform-d) δ 160.14, 155.39, 147.12, 138.48, 137.95, 128.81, 128.75, 128.27, 127.93, 127.63, 125.80, 125.23, 124.55, 78.51, 73.31, 72.78, 70.80, 60.31, 55.82, 45.54, 41.82, 40.48, 29.88, 29.83, 29.61, 29.55, 27.60, 27.48, 27.02, 26.29, 23.04, 22.81 (Figure S81 in the Supporting Information). HRMS (ESI): m/z calcd for C34H46N3O [M+H]+: 512.3635; found: 512.3688 (Figure S82 in the Supporting Information).
N,7-dimethyl-N-(4-((3-(methyl(prop-2-yn-1-yl)amino)propoxy)methyl)benzyl)-1,2,3,4-tetrahydroacridin-9-amine (7g). Yield 29%; pale yellow oil; 1H NMR (600 MHz, Chloroform-d) δ 8.01 (d, J = 8.6 Hz, 1H), 7.79 (s, 1H), 7.46 (d, J = 8.5 Hz, 1H), 7.32 (d, J = 7.8 Hz, 2H), 7.28 (d, J = 7.9 Hz, 2H), 4.51 (s, 2H), 4.42 (s, 2H), 3.55 (t, J = 6.5 Hz, 2H), 3.36 (d, J = 2.9 Hz, 2H), 3.19 (t, J = 6.7 Hz, 2H), 2.92 (s, 3H), 2.80 (t, J = 6.3 Hz, 2H), 2.55 (t, J = 7.4 Hz, 2H), 2.53 (s, 3H), 2.32 (s, 3H), 2.22 (q, J = 2.0 Hz, 1H), 1.92 (q, J = 6.7, 6.2 Hz, 2H), 1.84–1.77 (m, 4H) (Figure S83 in the Supporting Information). 13C NMR (150 MHz, Chloroform-d) δ 158.72, 157.73, 145.05, 138.46, 137.82, 135.21, 131.37, 128.77, 127.95, 127.92, 127.51, 125.60, 123.43, 78.54, 73.34, 72.84, 68.80, 60.36, 52.74, 45.71, 41.80, 40.52, 29.84, 27.94, 26.99, 23.00, 22.66, 22.21. HRMS (ESI): m/z calcd for C30H38N3O [M+H]+: 456.3009; found: 456.3047 (Figure S84 in the Supporting Information).
N,7-dimethyl-N-(4-((4-(methyl(prop-2-yn-1-yl)amino)butoxy)methyl)benzyl)-1,2,3,4-tetrahydroacridin-9-amine (7h). Yield 42%; pale yellow oil; 1H NMR (600 MHz, Chloroform-d) δ 8.11 (d, J = 8.7 Hz, 1H), 7.79 (s, 1H), 7.49 (dd, J = 8.7, 1.9 Hz, 1H), 7.32 (d, J = 7.8 Hz, 2H), 7.27 (d, J = 5.1 Hz, 2H), 4.51 (s, 2H), 4.45 (s, 2H), 3.52 (t, J = 6.4 Hz, 2H), 3.36 (d, J = 2.4 Hz, 2H), 3.24 (t, J = 6.7 Hz, 2H), 2.95 (s, 3H), 2.79 (t, J = 6.3 Hz, 2H), 2.53 (s, 3H), 2.50–2.45 (m, 2H), 2.33 (s, 3H), 2.22 (t, J = 2.4 Hz, 1H), 1.93 (dt, J = 13.1, 6.7 Hz, 2H), 1.80 (p, J = 6.2 Hz, 2H), 1.69–1.64 (m, 2H), 1.61–1.56 (m, 2H) (Figure S85 in the Supporting Information). 13C NMR (150 MHz, Chloroform-d) δ 159.09, 156.22, 144.03, 138.12, 138.07, 135.56, 131.86, 128.74, 127.96, 127.14, 126.58, 125.38, 123.52, 78.40, 73.42, 72.75, 70.48, 60.48, 55.53, 45.56, 41.75, 40.69, 29.83, 27.65, 27.00, 24.32, 22.91, 22.40, 22.20 (Figure S86 in the Supporting Information). HRMS (ESI): m/z calcd for C31H40N3O [M+H]+: 470.3166; found: 470.3222 (Figure S87 in the Supporting Information).
N,7-dimethyl-N-(4-(((5-(methyl(prop-2-yn-1-yl)amino)pentyl)oxy)methyl)benzyl)-1,2,3,4-tetrahydroacridin-9-amine (7i). Yield 61%; pale yellow oil; 1H NMR (600 MHz, Chloroform-d) δ 7.92 (d, J = 8.5 Hz, 1H), 7.80 (s, 1H), 7.42 (dd, J = 8.5, 1.9 Hz, 1H), 7.33–7.26 (m, 4H), 4.49 (s, 2H), 4.38 (s, 2H), 3.48 (t, J = 6.6 Hz, 2H), 3.33 (d, J = 2.4 Hz, 2H), 3.13 (t, J = 6.7 Hz, 2H), 2.89 (s, 3H), 2.80 (t, J = 6.4 Hz, 2H), 2.51 (s, 3H), 2.44–2.38 (m, 2H), 2.29 (s, 3H), 2.20 (t, J = 2.4 Hz, 1H), 1.92 (p, J = 6.5 Hz, 2H), 1.79 (p, J = 6.2 Hz, 2H), 1.65 (p, J = 6.8 Hz, 2H), 1.48 (p, J = 7.2 Hz, 2H), 1.44–1.36 (m, 2H) (Figure S88 in the Supporting Information). 13C NMR (150 MHz, Chloroform-d) δ 159.14, 154.42, 146.00, 138.64, 137.73, 134.77, 130.80, 128.74, 128.21, 127.85, 127.83, 125.81, 123.29, 78.61, 73.16, 72.81, 70.56, 60.22, 55.67, 45.55, 41.83, 40.33, 33.59, 29.73, 27.46, 26.91, 24.12, 23.04, 22.86, 22.14 (Figure S89 in the Supporting Information). HRMS (ESI): m/z calcd for C32H42N3O [M+H]+: 484.3322; found: 484.3375 (Figure S90 in the Supporting Information).
N,7-dimethyl-N-(4-(((6-(methyl(prop-2-yn-1-yl)amino)hexyl)oxy)methyl)benzyl)-1,2,3,4-tetrahydroacridin-9-amine (7j). Yield 82%; pale yellow oil; 1H NMR (600 MHz, Chloroform-d) δ 8.01 (d, J = 8.6 Hz, 1H), 7.78 (s, 1H), 7.45 (dd, J = 8.6, 1.9 Hz, 1H), 7.33–7.25 (m, 4H), 4.49 (s, 2H), 4.41 (s, 2H), 3.48 (t, J = 6.7 Hz, 2H), 3.34 (d, J = 2.4 Hz, 2H), 3.18 (t, J = 6.7 Hz, 2H), 2.92 (s, 3H), 2.79 (t, J = 6.4 Hz, 2H), 2.52 (s, 3H), 2.43–2.38 (m, 2H), 2.30 (s, 3H), 2.20 (t, J = 2.4 Hz, 1H), 1.92 (p, J = 6.6 Hz, 2H), 1.79 (p, J = 6.0 Hz, 2H), 1.63 (p, J = 6.8 Hz, 2H), 1.46 (p, J = 7.5 Hz, 2H), 1.39 (p, J = 7.0 Hz, 2H), 1.33 (q, J = 8.4, 7.8 Hz, 2H) (Figure S91 in the Supporting Information). 13C NMR (150 MHz, Chloroform-d) δ 158.63, 155.33, 144.93, 138.34, 137.92, 135.14, 131.31, 128.72, 127.92, 127.89, 127.43, 125.54, 123.40, 78.47, 73.28, 72.74, 70.67, 60.33, 55.71, 45.52, 41.80, 40.49, 33.02, 29.80, 27.56, 27.35, 26.94, 26.24, 22.96, 22.61, 22.16 (Figure S92 in the Supporting Information). HRMS (ESI): m/z calcd for C33H44N3O [M+H]+: 498.3479; found: 498.3529 (Figure S93 in the Supporting Information).
N,7-dimethyl-N-(4-(((7-(methyl(prop-2-yn-1-yl)amino)heptyl)oxy)methyl)benzyl)-1,2,3,4-tetrahydroacridin-9-amine (7k). Yield 61%; pale yellow oil; 1H NMR (600 MHz, Chloroform-d) δ 8.31 (d, J = 8.6 Hz, 1H), 7.75 (s, 1H), 7.53 (d, J = 8.6 Hz, 1H), 7.32 (d, J = 7.7 Hz, 2H), 7.22 (d, J = 7.6 Hz, 2H), 4.50 (s, 2H), 4.49 (s, 2H), 3.51–3.45 (m, 2H), 3.42 (d, J = 2.5 Hz, 2H), 3.35 (t, J = 6.7 Hz, 2H), 2.99 (s, 3H), 2.74 (t, J = 6.3 Hz, 2H), 2.52 (s, 3H), 2.49 (t, J = 7.7 Hz, 2H), 2.37 (s, 3H), 2.27 (t, J = 2.4 Hz, 1H), 1.92 (p, J = 6.6 Hz, 2H), 1.79 (p, J = 6.0 Hz, 2H), 1.62 (p, J = 6.7 Hz, 2H), 1.50 (q, J = 7.0, 6.5 Hz, 2H), 1.38–1.31 (m, 6H) (Figure S94 in the Supporting Information). 13C NMR (150 MHz, Chloroform-d) δ 157.93, 156.65, 143.34, 138.22, 137.91, 135.73, 132.12, 128.72, 127.98, 126.95, 126.10, 125.25, 123.57, 77.80, 73.89, 72.72, 70.81, 60.52, 55.60, 45.29, 41.54, 40.75, 32.03, 29.81, 29.46, 27.42, 27.20, 27.00, 26.25, 22.84, 22.25, 22.19 (Figure S95 in the Supporting Information). HRMS (ESI): m/z calcd for C34H46N3O [M+H]+: 512.3635; found: 512.3678 (Figure S96 in the Supporting Information).
N,7-dimethyl-N-(4-(((8-(methyl(prop-2-yn-1-yl)amino)octyl)oxy)methyl)benzyl)-1,2,3,4-tetrahydroacridin-9-amine (7l). Yield 65%; pale yellow oil; 1H NMR (600 MHz, Chloroform-d) δ 8.03 (s, 1H), 7.79 (s, 1H), 7.51–7.41 (m, 1H), 7.32 (d, J = 8.0 Hz, 2H), 7.28 (d, J = 7.9 Hz, 2H), 4.50 (s, 2H), 4.42 (s, 2H), 3.48 (t, J = 6.7 Hz, 2H), 3.35 (d, J = 2.4 Hz, 2H), 3.20 (s, 2H), 2.93 (s, 3H), 2.80 (t, J = 6.3 Hz, 2H), 2.53 (s, 3H), 2.45–2.38 (m, 2H), 2.31 (s, 3H), 2.22 (t, J = 2.4 Hz, 1H), 1.93 (p, J = 6.6 Hz, 2H), 1.82–1.77 (m, 2H), 1.62 (p, J = 6.8 Hz, 2H), 1.46 (q, J = 7.5 Hz, 2H), 1.37 (t, J = 7.3 Hz, 2H), 1.34–1.30 (m, 6H) (Figure S97 in the Supporting Information). 13C NMR (150 MHz, Chloroform-d) δ 158.05, 156.59, 143.62, 138.19, 137.97, 135.64, 131.99, 128.72, 127.97, 127.03, 126.32, 125.31, 123.55, 77.89, 73.81, 72.72, 70.85, 60.49, 55.65, 45.32, 41.58, 40.71, 32.19, 29.85, 29.55, 29.51, 27.42, 27.28, 26.99, 26.26, 22.86, 22.31, 22.19 (Figure S98 in the Supporting Information). HRMS (ESI): m/z calcd for C35H48N3O [M+H]+: 526.3792; found: 526.3851 (Figure S99 in the Supporting Information).
In vitro Inhibition Experiments of AChE/BuChE
To assess the inhibitory activity of compounds against AChE or BuChE, we followed the Ellman spectrophotometry method.34 AChE (E.C. 3.1.1.7, eeAChE and human recombinant AChE from electric eel and human serum, respectively), BuChE (E.C. 3.1.1.8, eqBuChE and human recombinant BuChE from equine serum and human serum, respectively), 5.5 -dithiobis-(2-nitrobenzoic acid) (Ellman Reagent, DTNB), S-butylthiocholine iodide (BTCI), acetylthiocholine iodide (ATCI), and donepezil were obtained from Sigma-Aldrich. The tested compound was first dissolved in DMSO and then diluted with Tris-HCl buffer (pH = 8.0) to the required concentration (DMSO < 1%). Subsequently, 160 µL DTNB (1.5 mM), 50 µL AChE (0.25 U/mL eeAChE or hAChE) or 50 µL BuChE (0.12 U/mL eqBuChE or hBuChE), and 10 µL of test drug were successively added into the 96-well plate and incubated for 6 min at 37 °C. Then, 30 µL of the substrate (initial concentration of 3 mM, ATCI or BTCI) was added, and the absorbance values of 0, 60, 120, and 180 s were detected at 405nm in a microplate reader (SpectraMax Plus 384, Molecular Devices, CA, USA). The percent inhibition due to the presence of test compounds was calculated by the following expression: [1-(absorbance change of the experimental group/absorbance change of the blank group)] × 100%, where the absorbance changes in the experimental and blank groups were obtained in the presence and absence of the inhibitor, respectively. The concentration of the compound at 50% inhibition was calculated by Prism software (GraphPad, United States), and this value was considered as the IC50 of the compound. Each experiment was repeated three times, and the experimental results are expressed as mean ± SD.
In vitro Inhibition Experiments of hMAO-A and hMAO-B
The inhibitory effects of all synthetic compounds on MAOs were determined using an Amplex Red fluorescence assay with rasagiline and iproniazid as control drugs.35 Human recombinant Monoamine oxidases (EC 1.4.3.4, hMAO-A and hMAO-B), Amplex Red reagent, horseradish peroxidase (HRP), rasagiline, and iproniazid were purchased from Sigma-Aldrich. The detailed experimental steps were as follows. The test compounds were dissolved in DMSO and diluted with PBS buffers (0.05 M, pH = 7.4) to prepare different final concentrations (DMSO < 1%). Various concentrations of test compounds (20 μL) were incubated with PBS buffer (80 μL) of hMAO-A or hMAO-B for 15 min at 37 °C in the dark. Next, the reaction was initiated by adding 100 µL of a mixture containing 200 μM of Amplex Red reagent, 2 U/mL of HRP, and 2 mM of tyramine (final concentration 1mM) to the system. The fluorescence changes of each solution within 20 min were recorded by a multi-detection microplate fluorescence reader (SpectraMax Plus 384, Molecular Devices, CA, USA). The detection conditions were an excitation wavelength of 545 nm and an emission wavelength of 590 nm. The percent inhibition due to the presence of test compounds was calculated by the following expression: [1-(change of fluorescence value of the experimental group/change of fluorescence value of the blank group)] × 100%, where the changes in fluorescence values of the experimental and blank groups were obtained in the presence and absence of the inhibitor, respectively. The concentration of the compound at 50% inhibition was calculated by Prism software (GraphPad, United States), and this value was considered as the IC50 of the compound. Experimental results are expressed as the mean ± SD of three independent experiments.
Docking Studies
Molecular simulations were performed using the Molecular Operating Environment (MOE) 2015.10 software (Chemical Computing Group, Montreal, Canada), following previously reported method.36 The X-ray crystal structures of hAChE, hBuChE and hMAO-B (PDB codes: 4EY7, 7QHD and 2V61, respectively) downloaded from the Protein Data Bank (PDB) were processed by removing water molecules and adding charges. Compound 7d was constructed through MOE’s structure preparation module and subjected to energy minimization by the Merck Molecular force field (MMFF94x, RMSD gradient: 0.05 kcal mol−1 Å−1), followed by docking to the active site of the corresponding protein via the Triangle Matcher method. Scoring and refinement were performed using the ASE scoring function and Force field, respectively. From the output, the optimal 10 conformations were retained. Finally, MOE’s pose viewer utility was employed to analyze the geometry of the docking complex.
PAMPA-BBB Penetration Assay
Based on the previous work of Di et al, parallel artificial membrane permeation analysis (PAMPA) with the blood-brain barrier was used to predict the brain permeation capacity of the tested compounds.37 Commercially available drugs, as well as DMSO and dodecane, were purchased from Sigma and Aladdin, while porcine brain phospholipids (PBL) were provided by Avanti Polar Lipids. Donor 96-well filter plates (PVDF membrane, pore size 0.45 μM) and acceptor indented 96-well microplates were obtained from Corning Inc., the 96-well UV plate was sourced from Millipore. The commercially available drug and the tested compound were dissolved in DMSO and diluted with PBS/EtOH (70/30, v/v) buffer to achieve a concentration of 25 μg/mL in the tested well. A 20 mg/mL dodecane solution of 4 μL PBL was evenly coated on the lipophilic filter membrane of the upper layer of a 96-well filter plate and left to stand for 5 min. Subsequently, 200 μL diluted solution of the tested drug and 300 μL PBS/EtOH (70/30, v/v) buffer were added to the donor and acceptor wells, respectively. The 96-well filter plate was placed over the acceptor plate to form a “sandwich” assembly, ensuring that the filter membrane could contact the buffer solution. Following incubation at 25 °C for 18 h, the donor plate was carefully removed, and the drug concentration was calculated by measuring the absorbance of the test compound in the donor, acceptor, and reference wells with a UV plate reader (SpectraMax Plus 384, Molecular Devices, CA, USA). Each sample was analyzed at five wavelengths, in four wells, and in at least three independent runs. The results are expressed as the means ± SD.
Cell Culture and Toxicity
For cell treatment, PC12 and BV-2 cells provided by the Stem Cell Bank of the Chinese Academy of Sciences were seeded in DMEM high glucose medium containing 10% fetal bovine serum, 100 U/mL penicillin, and 100 μg/mL streptomycin, and cultured under a humidified atmosphere with 5% CO2 in air at 37 °C.
For cytotoxicity experiments, the CCK-8 assay was performed with donepezil as the reference compound.38 The cells were planted in 96-well plates at a density of 10,000 per well and incubated in 1% fetal bovine serum medium for 24 h with six concentrations of compound 7d or donepezil (0.3125, 0.625, 1.25, 2.5, 5, and 10 μM) after the cell growth met the requirements. After the incubation period, we removed the medium and added 100 μL of medium containing 10% CCK-8 to each well in the dark and incubated for 4 h, and the absorbance was detected at 450 nm to calculate the cell viability.
In vivo Acute Toxicity
After 5 days of adaptive feeding under standard conditions, 40 Kunming mice (20–24 g, half male and half female) were randomly divided into four groups: control group, high-, medium-, and low-dose groups (2500 mg/kg, 1250 mg/kg, and 625 mg/kg, respectively), with 10 mice in each group.36,38,39 Compound 7d was uniformly dispersed in 0.5% sodium carboxymethyl cellulose (CMC-Na) salt solution and administered orally according to the divided groups. The mice were closely monitored for death, dietary and behavioral abnormalities within 4 h after administration, and were then observed for 14 days and recorded in detail. On the 14th day after administration, all the mice were sacrificed after being anesthetized with ether. The heart, liver, kidney, lung and brain tissues of the high-dose and control mice were collected, and the tissues’ pathological damage was observed by HE staining.
In vivo Step-Down Passive Avoidance Test
Scopolamine hydrobromide injection and donepezil hydrochloride were obtained from Zhengzhou Suicheng Pharmaceutical Co., Ltd and Annaiji Chemical Co., Ltd, respectively. Clean-grade male Kunming mice (18–22 g) from Hunan SJA Laboratory Animal Co., Ltd were housed under standard control conditions (22 ± 2 °C, 55 ± 10% humidity, 12 h: 12 h light:dark cycle) with free access to water and food. The step-down test, consisting of a training trial and a recall trial, was performed in a passive avoidance chamber with a steel grated floor and a plastic platform in the bottom right corner.38,40,41
During the training trial, each mouse was allowed to acclimatize to the chamber’s environment for 5 min before the power supply was turned on and the current stimulation intensity was set to 0.5 mA (24 V). When the mice stepped down onto the steel grated floor, they received an electric shock that prompted them to return to the platform to avoid injury from the stimulus. For the passive avoidance test, 48 mice were randomly divided into 6 groups with 8 mice in each group. Compound 7d and donepezil were suspended in a 0.5% CMC-Na solution to prepare the required concentrations. One hour before the training trial, the mice were given oral doses of compound 7d (20, 10 and 5 mg/kg) or donepezil (10 mg/kg, positive control). After 30 min, mice were injected with scopolamine hydrobromide (3 mg/kg) intraperitoneally to induce memory impairment.40,41 The recall trial was conducted 24 hours after training. During this trial, the mice were placed once again on the platform, and the latency to step down onto the grid for the first time and errors that caused an electric shock within 5 min were recorded. Latency and the number of errors were used to evaluate learning and memory performance.
Pharmacokinetics Assay in Sprague-Dawley Rats
The pharmacokinetic parameters of compound 7d were determined in male SD rats (n = 3, 220 ± 20 g).42,43 Samples were collected at appropriate time points (0.033, 0.083, 0.25, 0.5, 1, 2, 4, 6, 8, and 24 h) after administering 7d to the rats by intravenous (1 mg/kg) or intraperitoneal (10 mg/kg) injection, followed by centrifugation at 10,000 rpm for 2 min and storage at −20 °C. Plasma proteins were precipitated with 0.5% formic acid/acetonitrile (v/v), and the supernatants obtained by centrifugation (14,000 rpm, 14 min) were analyzed using LC-MS/MS (Waters, ACQUITY-TQ-XS). The PK parameters were calculated by DAS2.0 software (China).
Statistical Analysis
The sample size of the step-down passive avoidance test was determined using Power Analysis and Sample Size (PASS) 15 software based on previous studies.38,44 A two-sided significance level (α) of 0.05 and statistical power of 90% were selected, with equal group allocation ratios. By calculation, a minimum total of 48 mice should be included in the experiment. Thus, we included a total of 48 mice in this experiment, with 8 mice in each group. All data were tested for normality using Q-Q plots. For normally distributed data, the Brown-Forsythe test was used for homogeneity of variance, and results are presented as mean ± standard error of mean (SEM) (n = 8 per group). Statistical differences between groups were assessed using one-way analysis of variance (ANOVA) followed by Tukey’s Multiple Comparison Test for data with homogeneous variance. In cases where data did not meet the normal distribution, differences were evaluated using a Kruskal–Wallis test followed by Dunn’s Multiple Comparison Test, and the results are expressed as boxplots, illustrating medians within boxes from the first quartile (25th percentile) to the third quartile (75th percentile) and whiskers ranging from minimum to maximum (n = 8 per group). A p-value of less than 0.05 was considered to indicate statistically significant differences with a 95% confidence interval (CI). Statistical analyses were performed using Prism software (GraphPad, United States).
Results
Chemistry
The target compounds were synthesized according to the procedure shown in Scheme 1. The initial compounds 1a-1c and 2a-2b were commercially available. Firstly, compounds 1a-1c were reacted with the corresponding benzylamines 2a-2b to obtain intermediates 3a-3e in the presence of KI,31 then a selective N-methylation was employed to afford compounds 4a-4e with high yield by treatment with formaldehyde/acetic acid and the reductant NaBH3CN,32 followed by O-alkylation with the corresponding dibromide under NaH and DMF conditions to yield the key intermediates 5a-5e.33 Finally, the target compounds 6a-6o and 7a-7l were obtained via another N-alkylation of compounds 5a-5e with N-methylpropargylamine under the alkaline condition at 70 °C.21 The target compounds were characterized by proton NMR carbon, NMR and high resolution mass spectrometry (HRMS). However, it is noteworthy that, for the intermediates, only proton NMR and HRMS were utilized for characterization, excluding13C NMR.
 |
Scheme 1 Synthesis of tacrine-selegiline hybrids 6a-6o and 7a-7l. Reagents and conditions: (i) KI, 1-pentanol, reflux at 130 °C for 10 h; (ii) HCHO, AcOH, DMF, r.t. for 60 min, followed by NaBH3CN, 8 h; (iii) Br(CH2)nBr, NaH, DMF, 0 °C to r.t., 12 h; (iv) N-methylpropargylamine, anhydrous K2CO3, MeCN, reflux at 70 °C for 12 h. |
In vitro Inhibition of AChE and BuChE
The inhibitory effects of all target compounds on electric eel acetylcholinesterase (eeAChE) and equine serum butyrylcholinesterase (eqBuChE) were evaluated using classical spectrophotometry as described by Ellman et al,34 with donepezil, a well-known cholinesterase inhibitor, used as a reference compound. Additionally, in view of the high cost of human cholinesterases, only 6a-g, 7b-d and 7h-j hybrids, which demonstrated relatively good and balanced inhibitory activities against both eeAChE and eqBuChE, were selected for further testing on human recombinant cholinesterases from human serum (hAChE/hBuChE). The results of these experiments were expressed as IC50 (μM) and are summarized in Table 1.
 |
Table 1 Inhibition of AChE and BuChE by Tacrine-Selegiline Hybrids 6a-o and 7a-l |
The assays demonstrated that the majority of the compounds potently inhibited AChE and BuChE. Compounds with alkyl chains of different lengths between tacrine and N-methylpropargylamine were synthesized to evaluate the inhibitory activities against ChEs, as the length of the alkyl chain can influence the inhibitory ability of both AChE and BuChE. Compound 6h, with a 3-carbon spacer, exhibited the strongest inhibitory activity against eeAChE (IC50 = 5.36 μM). For 6a-6g, compounds containing two- and three-carbon spacers showed better eeAChE inhibition. For 6h-6m, compounds with short linkers (n = 3–4) demonstrated stronger eeAChE inhibitory activity. Among compounds 7a-7f and 7g-7l, 7b (n = 4) and 7d (n = 6), as well as 7h-7j (n = 4–6), exhibited better eeAChE inhibitory activity. Furthermore, we investigated the effect of introducing different substituents at position 7 of tacrine on ChEs inhibition. As shown in Table 1, compounds with a methyl group introduced (6i, 7h, 7i) exhibited about 2-fold more potent eeAChE inhibitory activity than their corresponding unsubstituted compounds (6c, 7b, 7c), whereas compounds (6h, 6j, 7g, 7j) with the same substituent showed similar eeAChE inhibition as their unsubstituted counterparts (6b, 6d, 7a, 7d). Introduction of a fluorine atom as an electron-absorbing group at site 7 of tacrine significantly reduced the eeAChE inhibitory potency of the compounds (6n-6o). Furthermore, we explored the structure-activity relationships (SARs) of the benzene rings connecting tacrine and N-methylpropargylamine by comparing target compounds (6b-6g vs 7a-7f and 6h-6m vs 7g-7l). The results suggested that compounds (7b-7d, 7h-7j) containing benzyl ether had higher inhibitory activity against eeAChE than compounds (6c-6e, 6i-6k) containing phenol ether. Interestingly, most compounds showed superior inhibitory activity against hAChE compared to eeAChE. However, it should be noted that the effects of different chain lengths, substituent groups at position 7 of tacrine and linked benzene ring on the inhibitory activity of hAChE and eeAChE were not completely consistent. Compounds (6d, 6e) containing phenol ether inhibited hAChE more strongly than the corresponding compounds (7c, 7d) containing benzyl ether. Among 6a-6g, the long linker compounds 6e and 6f (n = 6–7) demonstrated relatively strong hAChE inhibition. The unsubstituted compound (7b-7d) exhibited better hAChE inhibitory activity than the corresponding compounds (7h-7j) with an introduced methyl group. In general, compounds with 4 to 8 carbon spacers demonstrated stronger eqBuChE inhibitory activity, and this SAR was found in compounds 6a-6g, 7a-7f and 7g-7l. Compound 7e was the best eqBuChE inhibitor in this series of hybrids with an IC50 value of 0.19 μM, approximately 18-fold more potent than the reference compound donepezil (eqBuChE: IC50 = 3.50 μM). Introducing a methyl or fluorine atom at position 7 of tacrine significantly reduced the inhibitory activity of compounds toward eqBuChE (6b-6g vs 6h-6m vs 6n-6o and 7a-7f vs 7g-7l). When the chain length between tacrine and N-methylpropargylamine was consistent, the change of the linked benzene ring had little effect on the inhibitory activity of eqBuChE for the compounds without substitution at site 7 of tacrine. However, for the methyl-substituted compounds, compounds (7h-7l) containing benzyl ether exhibited stronger eqBuChE inhibitory activity than the corresponding compounds (6i-6m) containing phenol ether. The SARs of the effects of changes in alkyl chain length, substituent at position 7 of tacrine moiety, and the linked benzene ring on the inhibitory activity of hBuChE were similar to those of eqBuChE. Among them, compound 6f (hBuChE: IC50 = 0.31 μM) showed the best inhibitory activity against hBuChE activity, approximately 7-fold stronger than donepezil (hBuChE: IC50 = 2.25 μM).
Considering the results of both human and eel cholinesterase assays, several of the hybrids demonstrated a favorable balance of AChE/BuChE inhibitory activities. Notably, 7d showed the best balance inhibition of the two esterases, with IC50 values of 8.76 μM for eeAChE, 1.57 μM for hAChE, 0.46 μM for eqBuChE, and 0.43 μM for hBuChE. Therefore, this hybrid may have greater potential for the treatment of AD.
In vitro Inhibition of hMAO-A and hMAO-B
To study the multifunctional biological properties of the hybrids, we evaluated their inhibitory activities against both human recombinant MAO-A and MAO-B from human serum (hMAO-A and hMAO-B) by an Amplex Red fluorescence assay.35 Iproniazid, an irreversible non-selective MAO-A inhibitor, and rasagiline, an irreversible selective MAO-B inhibitor, served as control drugs. AD patients often exhibit symptoms of depression, which are even considered a risk factor for the onset of AD.33 Moreover, MAO-A can also impact the regulation of neurotransmitters in the brain; hence, dual inhibition of MAO-A and MAO-B, rather than just inhibiting MAO-B alone, may hold therapeutic value for AD.22 Table 2 showed that most of the synthesized compounds exhibited potent inhibitory effects against both MAO-A and MAO-B. Importantly, all compounds except for 6n and 6o (IC50 values of 65.61 and 71.39 μM for hMAO-B, respectively) showed stronger anti-MAO-B activity (IC50 ranging from 1.12 to 5.78 μM) than iproniazid (IC50 = 7.54 μM), though weaker than rasagiline (IC50 = 0.23 μM). Of all the synthetic compounds, 7l (IC50 = 0.22 μM for hMAO-A) and 6m (IC50 = 1.12 μM for hMAO-B), with eight alkyl chains between tacrine and N-methylpropargylamine, exhibited the strongest inhibitory potency against MAO-A and MAO-B, respectively. Analysis of the MAO-A inhibitory activities of 6a-6g, 6h-6m, 7a-7f and 7g-7l showed that compounds with a linker length of 5 or 6 carbon atoms, such as 6e, 6j-k, 7c-d and 7i-j were potent MAO-A inhibitors, with some demonstrating sub-micromolar activity (eg, 7i-7j). The introduction of a methyl group at position 7 of tacrine had little impact on the MAO-A inhibition characteristics of compounds (6b-g vs 6h-m) containing phenol ether; however, it greatly increased the inhibitory effect of MAO-A of compounds (7a-f vs.7g-l) containing benzyl ether. Notably, a fluorine substitution at position 7 of tacrine significantly reduced the inhibitory properties of MAO-A (eg, 6n: IC50 = 50.67 μM, 6o: IC50 = 61.33 μM). Additionally, compounds (7c-7f, 7g-7l) containing benzyl ether showed stronger inhibitory activity against MAO-A than the corresponding compounds (6d-6g, 6h-6m) containing phenol ether. Furthermore, we discussed the SARs of changes in chain length, substituent at position 7 of tacrine moiety, and linked benzene ring on the inhibitory activity of MAO-B. The extension of the carbon chain did not significantly alter the IC50 value of MAO-B inhibition for compounds 6a-6g (n = 2–8), and similar SARs were observed for 6h-6m, 7a-7f, and 7g-7l. With a few exceptions, introducing a methyl group at site 7 of tacrine enhanced the inhibitory effect on MAO-B. However, the introduction of a fluorine atom significantly decreased the MAO-B inhibitory activity, similar to that of MAO-A. Overall, compounds containing phenol ether exhibited higher inhibitory effects on MAO-B than the corresponding compounds containing benzyl ether, which is the opposite trend compared to that of MAO-A.
 |
Table 2 Inhibition of hMAO-A and hMAO-B by Tacrine-Selegiline Hybrids 6a-o and 7a-l |
Considering the above findings, 7d was selected as the representative compound in this series of hybrids for further study due to its well-balanced and excellent inhibitory effect on ChEs and MAOs.
Molecular Docking of 7d with hAChE and hBuChE
Based on the results obtained from the in vitro inhibition of cholinesterase, it was observed that, except for compound 7h-7j, the inhibitory activities of the other compounds (6a-6g and 7b-7d) on electric eel cholinesterases were similar to those observed on human recombinant cholinesterases. Consequently, selecting the less expensive cholinesterases from electric eel for the preliminary screening of compound activity was representative, with the final results being determined based on inhibitory activities on human cholinesterases. Thus, to determine the capacity of compound 7d targeting cholinesterase at the Molecular level, we selected the crystal structure of hAChE and hBuChE for conducting molecular docking studies using the Molecular Operating Environment (MOE) 2015.10 version software from the Canadian Chemical Computing Group. Based on the results in Figure 2A and C, compound 7d occupies the CAS, the mid-gorge site, and the PAS of the entire hAChE enzyme. Specifically, the N-methylpropargylamine moiety of 7d can act on the PAS of AChE and binds to Trp286 through hydrophobic interaction. The benzene ring of the benzylamine unit establishes π-π stacking interaction with Trp341, and the alkyl chain shows a hydrophobic interaction with Phe338 at the aromatic gorge. Moreover, the tacrine ring binds to the CAS through π-π stacking formed by its benzene ring with Trp86. The above docking results indicated that compound 7d is a two-site inhibitor of the AChE, capable of binding to the CAS and PAS simultaneously. From the docking results of compound 7d and hBuChE (Figure 2B and D), it can be seen that the cavity of BuChE can also be occupied by compound 7d. In the hBuChE-7d complex, the tacrine ring establishes two π-π stacking interactions with Trp82 and binds to Ala328 and His438 through hydrophobic interaction. Additionally, the benzyl ether unit forms hydrophobic interactions with Tyr332, Thr120, and Leu286.
 |
Figure 2 Molecule docking results: (A and C) were 3D and 2D docking models of compound 7d with hAChE; (B and D) were 3D and 2D docking models of compound 7d with hBuChE. Atom colors: yellow-carbon atoms of 7d, grey-carbon atoms of residues of hAChE or hBuChE, dark blue-nitrogen atoms, and red-oxygen atoms. The figure was prepared using the ligand interactions application in MOE. |
Molecular Docking of 7d with hMAO-B
To clearly explain the binding mode of compound 7d and hMAO-B, we performed molecular docking studies using MOE software. As shown in Figure 3A and B, compound 7d can enter the substrate cavity of MAO-B, and the tacrine ring establishes a π-H interaction with Tyr398 of the flavin adenine dinucleotide (FAD) cofactor and forms a hydrophobic interaction with Phe343, Tyr60, and Tyr435. The N-methylpropargylamine unit occupies the entrance cavity of the enzyme and interacts with Pro102, Pro104, Ile316, Phe168, Leu167, Leu164, Trp199 and Phe103 via van der Waals and hydrophobic forces. The results also suggested that the inhibition of MAO-B by compound 7d may be affected by the length of the alkylene chain.
 |
Figure 3 Molecule docking results: (A) 3D docking model of compound 7d with hMAO-B. Atom colors: yellow-carbon atoms of 7d, grey-carbon atoms of residues of hMAO-B, green-carbon atoms of FAD, dark blue-nitrogen atoms, and red-oxygen atoms; (B) 2D schematic diagram of docking model of compound 7d with hMAO-B. The figure was prepared using the ligand interactions application in MOE. |
In vitro Blood–Brain Barrier Permeation
One of the main requirements for anti-AD drugs is good brain permeability to reach the target site of AD located in the CNS. Therefore, screening the ability of target compounds to infiltrate the brain is of great significance. We used the parallel artificial membrane permeability assay (PAMPA) described by Di et al, which has the advantages of simplicity, speed, and high accuracy, to investigate the blood-brain barrier (BBB) permeability of tacrine-selegiline hybrids 6a-o and 7a-l.37 10 commercially available drugs with reported values were used as reference compounds (Table 3). From the measured results, the plot of experimental data and bibliographic values for the 10 control drugs provided a good linear correlation: Pe (exp.) = 1.0654 Pe (bibl.) + 0.1625 (R2 = 0.9967) (Figure 4). Based on the equation described above and followed the criteria established by Di et al for BBB permeability, we classified compounds as follows: Pe (× 10−6 cm/s) > 4.42 represented high BBB permeation predicted (CNS+), Pe (× 10−6 cm/s) < 2.29 represented low BBB permeation predicted (CNS-), 2.29 < Pe (× 10−6 cm/s) < 4.42 represented uncertain BBB permeation (CNS±). The permeability and predicted CNS penetration for all target hybrids are shown in Table 4. Notably, with the exception of compounds 6e and 7l, the Pe values of the remaining compounds were greater than 4.42 (× 10−6 cm/s), indicating that they were able to cross the BBB via passive diffusion and potentially exert biological effects in the central nervous system.
 |
Table 3 Permeability Pe (× 10−6 Cm/s) in the PAMPA-BBB Assay for 10 Commercial Drugs in the Experiment Validation |
 |
Table 4 Permeability Pe (× 10−6 Cm/s) in the PAMPA-BBB Assay for the Target Compounds and Their Predicted Penetration into CNS |
 |
Figure 4 Lineal correlation between experimental and reported permeability of commercial drugs using the PAMPA-BBB assay. Pe (exp.) = 1.0654 Pe (bibl.) + 0.1625 (R2 = 0.9967). |
In vitro Cytotoxicity and in vivo Acute Toxicity Evaluation
The cytotoxicity of compound 7d was evaluated in PC12 and BV-2 cells using donepezil as a positive control. The cells were exposed to different concentrations of compound 7d for 24 hours, and cell viability was assessed by the Cell Counting Kit-8 (CCK-8) assay (Figure 5A and B).38 Our results showed that, within a concentration range of 0.325 to 2.5 μM, compound 7d had no significant effect on the viability of either cell line, which was comparable to the toxicity of donepezil. However, at higher concentrations (above 2.5 μM), we observed a decrease in cell viability, with some variability between the two cell lines. Notably, at a concentration of 10 μM, the cytotoxicity of 7d to BV-2 cells surpassed 50%. Therefore, compound 7d has moderate in vitro cellular safety.
 |
Figure 5 Cytotoxicity of compound 7d and donepezil on (A) PC12 and (B) BV-2 cells. PC12 and BV-2 cells were incubated with different concentrations of compound 7d or donepezil (0.3125–10 μM) for 24 h. The results were shown as cell viability after treated with compound 7d or donepezil vs untreated control cells. Data are shown as mean ± SD of three independent experiments. |
To assess the safety of compound 7d in vivo, an acute toxicity study was performed using KM mice (20–24 g, 10 mice per group, half male and half female). We administered compound 7d orally at doses of 625, 1250 and 2500 mg/kg36,38,39 and monitored the mice for 4 h for any signs of death, abnormal behavior, or dietary changes. No such effects were observed in any of the animals. We also recorded the body weights of all mice in the control and treatment groups for 14 days after a single-dose administration. Our results showed that the mice in all groups exhibited a gradual increase in body weight, and the average weight change in the low, middle, and high dose groups was comparable to that in the control group, indicating no obvious acute toxicity (Figure 6). To further evaluate the potential of compound 7d to cause tissue damage, we observed paraffin sections of tissues (heart, liver, kidney, lung and brain) from mice that had been treated with vehicle (control group) or compound 7d (2500 mg/kg) for 14 days. Hematoxylin-eosin (HE) staining revealed no obvious signs of tissue damage in the treated mice, indicating that compound 7d was well tolerated at the dosage of 2500 mg/kg (Figure 7).
 |
Figure 6 The change on body weight of mice after oral administration of different concentrations of compound 7d. Data are presented as the average weight ± SD of mice (n = 10). |
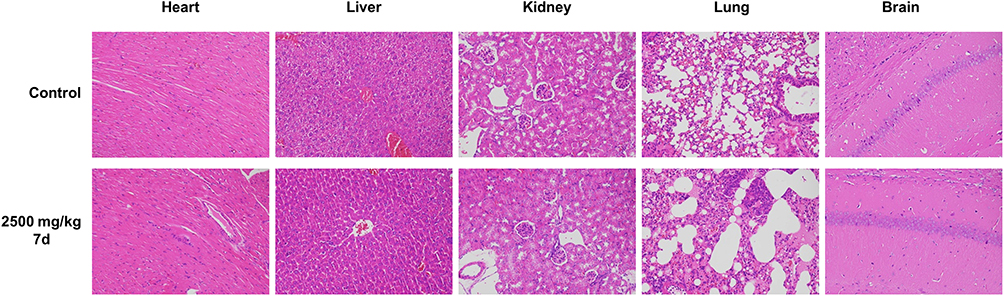 |
Figure 7 Histological analysis of heart, liver, kidney, lung and brain for the acute toxicity studies of compound 7d at a dosage of 2500 mg/kg in mice. Representative images of HE-stained are shown. Bar = 200 mm. |
In vivo Efficacy Study
Given the promising in vitro pharmacological activity and in vitro and in vivo safety of compound 7d, we next selected the scopolamine-induced memory impairment mouse model as the AD animal model for the step-down passive avoidance assay. Using donepezil as a positive control to determine whether 7d could improve the cognitive deficit in vivo.38,40,41 The experimental results (Figure 8A and B) showed that compared to the control group, the model group had significantly shorter latency (Tukey’s post hoc test, p < 0.0001) and a higher number of errors (Kruskal–Wallis test, p < 0.0001), indicating the successful establishment and reliability of the AD model. Administration of compound 7d could prolong the latency and reduce the number of errors in mice in a dose-dependent manner. Compared with donepezil, the high-dose group of compound 7d (20 mg/kg) exhibited longer latency and fewer errors, while the medium-dose group (10 mg/kg) showed the similar number of errors and latency. Additionally, although the latency (Tukey’s post hoc test, p = 0.01476) and the number of errors (Kruskal–Wallis test, p = 0.2219) in the low-dose group (5 mg/kg) were not statistically significant compared to the model group, improvements were observed in both. In summary, consistent with the expected results, compound 7d could cross the BBB and act on targets in the CNS, leading to the reversal of memory impairment.
 |
Figure 8 The effects of compound 7d on scopolamine-induced learning and memory impairment in the step-down test. (A) Latency. Data were analyzed by one-way analysis of variance (ANOVA) followed by Tukey’s Multiple Comparison Test, and the results are expressed as mean ± SEM (n = 8 per group). (B) Number of errors. Data were analyzed by the Kruskal–Wallis test followed by Dunn’s Multiple Comparison Test, and the results are expressed as boxplots illustrating medians within boxes from the first quartile (25th percentile) to the third quartile (75th percentile) and whiskers ranging from minimum to maximum (n = 8 per group). A p < 0.05 was considered the threshold for statistical significance with 95% CI. ####p < 0.0001 vs control group, *p < 0.05, **p < 0.01, ***p < 0.001 vs model group. |
Pharmacokinetic Evaluation of Compound 7d
A promising candidate compound for new drugs should exhibit not only high pharmacological activity and low toxicity but also desirable pharmacokinetic (PK) properties. Initial screening of the pharmacokinetics of candidate compounds is therefore essential in the early stages of development. Taking compound 7d as a representative, we performed pharmacokinetic studies in SD rats following intravenous (i.v.) and intraperitoneal (i.p.) administration to obtain basic pharmacokinetic parameters of compound 7d for comprehensive evaluation. The main pharmacokinetic parameters of compound 7d are shown in Table 5. When dosed with i.v. at 1 mg/kg, compound 7d exhibited a reasonable peak plasma concentration (Cmax = 465.33 μg/L) and area under the curve (AUC = 170.06 μg/L*h), with a half-life (t1/2) of 0.82 h and a plasma clearance (CL) of 5.92 L/h/kg. Furthermore, when dosed with i.p. at 10 mg/kg, compound 7d displayed a favorable Cmax (364.67 μg/L) and AUC (1530.61 μg/L*h), with a suitable t1/2 (3.16 h) and CL (6.54 L/h/kg). Additionally, the bioavailability of 7d was up to 90.04%. These results suggested that compound 7d possesses favorable pharmacokinetic properties.
 |
Table 5 Pharmacokinetic Parameters of Compound 7d After I.v. and I.p. Administration |
Discussion
To develop effective treatments for AD, a class of novel multifunctional hybrids, 6a-o and 7a-l, were designed and synthesized based on a multi-target design strategy by linking to tacrine and N-methylpropargylamine moiety. The ability of all target compounds to inhibit cholinesterase (ChEs) and monoamine oxidase (MAOs) was investigated. Results suggested that most of the compounds exhibited potent inhibitory activities against both enzymes in vitro. Compound 7d showed suitable and best-balanced inhibitory activities against hAChE (IC50 = 1.57 μM), hBuChE (IC50 = 0.43 μM), hMAO-A (IC50 = 2.30 μM) and hMAO-B (IC50 = 4.75 μM). Molecular docking studies revealed that 7d was a dual-site inhibitor of hAChE, while also displaying binding affinity towards hBuChE and hMAO-B. Compound 7d presented low neurotoxicity and good in vivo safety with tolerated doses up to 2500 mg/kg while being able to cross the BBB and exert central activity. In the step-down passive avoidance test, compound 7d significantly ameliorated memory impairment in a scopolamine-induced mouse model in a dose-dependent manner. Further pharmacokinetic studies showed that compound 7d had acceptable pharmacokinetic properties after intravenous and intraperitoneal administration.
However, it should be noted that this study has a limitation in that although tacrine-selegiline hybrids 6a-o and 7a-l were characterized by 1H NMR, 13C NMR and HRMS, the intermediates were only characterized by 1H NMR and HRMS, excluding 13C NMR. In fact, from a rigorous standpoint, it would be advantageous to have carbon NMR characterization for the new intermediates.
Conclusion
Based on the above findings, compound 7d could be a promising multi-target agent for further investigation as a potential treatment for AD.
Supporting Information
The 1H NMR and HRMS spectra of intermediates 3a-e, 4a-4e, as well as the 1H NMR, 13C NMR, HRMS spectra and HPLC analysis of the target compounds 6a-o, 7a-l, were available as Supporting Information.
Ethical Approval
All animal work was carried out in accordance with the institutional guidelines for animal care and use and was approved by the Ethical Committee at Guangxi Medical University Cancer Hospital (Approval LW2023114, date 10/01/2023).
Acknowledgments
This work is supported by the Guangxi Science Fund for Distinguished Young Scholars (Grant No. 2022GXNSFFA035030), the Special Funds for Science and Technology Development under the Guidance of the Central Government (Grant No. ZY20198020), the National Natural Science Foundation of China (Grant Nos. 31970371, 31600265), the Open Project Program of Guangxi Key Laboratory of Brain and Cognitive Neuroscience (Guilin Medical University, GKLBCN-202301-02), the Guangxi Medical University Training Program for Distinguished Young Scholars, the Guangxi Postdoctoral Research Fund.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conceptualization, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Alzheimer’s A. 2016 Alzheimer’s disease facts and figures. Alzheimers Dement. 2016;12:459–509. doi:10.1016/j.jalz.2016.03.001
2. Scheltens P, De Strooper B, Kivipelto M, et al. Alzheimer’s disease. Lancet. 2021;397:1577–1590. doi:10.1016/S0140-6736(20)32205-4
3. Collaborators GDF. Estimation of the global prevalence of dementia in 2019 and forecasted prevalence in 2050: an analysis for the global burden of disease study 2019. Lancet Public Health. 2022;7:105–125. doi:10.1016/s2468-2667(21)00249-8
4. Levey AI. Progress with Treatments for Alzheimer’s Disease. N Engl J Med. 2021;384:1762–1763. doi:10.1056/NEJMe2103722
5. Li Q, Xing S, Chen Y, et al. Discovery and biological evaluation of a novel highly potent selective butyrylcholinsterase inhibitor. J Med Chem. 2020;63:10030–10044. doi:10.1021/acs.jmedchem.0c01129
6. Scarpini E, Scheltens P, Feldman H. Treatment of Alzheimer’s disease: current status and new perspectives. Lancet Neurol. 2003;2:539–547. doi:10.1016/s1474-4422(03)00502-7
7. Ballard C, Gauthier S, Corbett A, Brayne C, Aarsland D, Jones E. Alzheimer’s disease. Lancet. 2011;377:1019–1031. doi:10.1016/s0140-6736(10)61349-9
8. Chun H, Im H, Kang YJ, et al. Severe reactive astrocytes precipitate pathological hallmarks of Alzheimer’s disease via H(2)O(2)(-) production. Nat Neurosci. 2020;23:1555–1566. doi:10.1038/s41593-020-00735-y
9. Simunkova M, Alwasel SH, Alhazza IM, et al. Management of oxidative stress and other pathologies in Alzheimer’s disease. Arch Toxicol. 2019;93:2491–2513. doi:10.1007/s00204-019-02538-y
10. Vaz M, Silvestre S. Alzheimer’s disease: recent treatment strategies. Eur J Pharmacol. 2020;887:173554. doi:10.1016/j.ejphar.2020.173554
11. Høilund-Carlsen PF, Revheim ME, Costa T, et al. Passive Alzheimer’s immunotherapy: a promising or uncertain option? Ageing Res Rev. 2023;90:101996. doi:10.1016/j.arr.2023.101996
12. Przybyłowska M, Kowalski S, Dzierzbicka K, Inkielewicz-Stepniak I. Therapeutic Potential of Multifunctional Tacrine Analogues. Curr Neuropharmacol. 2019;17:472–490. doi:10.2174/1570159x16666180412091908
13. Perry EK, Tomlinson BE, Blessed G, Bergmann K, Gibson PH, Perry RH. Correlation of cholinergic abnormalities with senile plaques and mental test scores in senile dementia. Br Med J. 1978;2:1457–1459. doi:10.1136/bmj.2.6150.1457
14. Kucukoglu K, Gul HI, Taslimi P, Gulcin I, Supuran CT. Investigation of inhibitory properties of some hydrazone compounds on hCA I, hCA II and AChE enzymes. Bioorg Chem. 2019;86:316–321. doi:10.1016/j.bioorg.2019.02.008
15. Li Q, Yang H, Chen Y, Sun H. Recent progress in the identification of selective butyrylcholinesterase inhibitors for Alzheimer’s disease. Eur J Med Chem. 2017;132:294–309. doi:10.1016/j.ejmech.2017.03.062
16. Gómez-Ramos P, Morán MA. Ultrastructural localization of butyrylcholinesterase in senile plaques in the brains of aged and Alzheimer disease patients. Mol Chem Neuropathol. 1997;30:161–173. doi:10.1007/bf02815095
17. Dogterom P, Nagelkerke JF, Mulder GJ. Hepatotoxicity of tetrahydroaminoacridine in isolated rat hepatocytes: effect of glutathione and vitamin E. Biochem Pharmacol. 1988;37:2311–2313. doi:10.1016/0006-2952(88)90356-5
18. Zemek F, Drtinova L, Nepovimova E, et al. Outcomes of Alzheimer’s disease therapy with acetylcholinesterase inhibitors and memantine. Expert Opin Drug Saf. 2014;13:759–774. doi:10.1517/14740338.2014.914168
19. Nepovimova E, Korabecny J, Dolezal R, et al. Tacrine-trolox hybrids: a novel class of centrally active, nonhepatotoxic multi-target-directed ligands exerting anticholinesterase and antioxidant activities with low in vivo toxicity. J Med Chem. 2015;58:8985–9003. doi:10.1021/acs.jmedchem.5b01325
20. Tumiatti V, Minarini A, Bolognesi ML, Milelli A, Rosini M, Melchiorre C. Tacrine derivatives and Alzheimer’s disease. Curr Med Chem. 2010;17:1825–1838. doi:10.2174/092986710791111206
21. Lu C, Zhou Q, Yan J, Du Z, Huang L, Li X. A novel series of tacrine-selegiline hybrids with cholinesterase and monoamine oxidase inhibition activities for the treatment of Alzheimer’s disease. Eur J Med Chem. 2013;62:745–753. doi:10.1016/j.ejmech.2013.01.039
22. Finberg JP. Update on the pharmacology of selective inhibitors of MAO-A and MAO-B: focus on modulation of CNS monoamine neurotransmitter release. Pharmacol Ther. 2014;143:133–152. doi:10.1016/j.pharmthera.2014.02.010
23. Chen JJ, Wilkinson JR. The monoamine oxidase type B inhibitor rasagiline in the treatment of Parkinson disease: is tyramine a challenge? J Clin Pharmacol. 2012;52:620–628. doi:10.1177/0091270011406279
24. Kalgutkar AS, Dalvie DK, Castagnoli N, Taylor TJ. Interactions of nitrogen-containing xenobiotics with monoamine oxidase (MAO) isozymes A and B: SAR studies on MAO substrates and inhibitors. Chem Res Toxicol. 2001;14:1139–1162. doi:10.1021/tx010073b
25. Boppana K, Dubey PK, Jagarlapudi SA, Vadivelan S, Rambabu G. Knowledge based identification of MAO-B selective inhibitors using pharmacophore and structure based virtual screening models. Eur J Med Chem. 2009;44:3584–3590. doi:10.1016/j.ejmech.2009.02.031
26. Gottfries CG. Alzheimer’s disease and senile dementia: biochemical characteristics and aspects of treatment. Psychopharmacology. 1985;86:245–252. doi:10.1007/bf00432208
27. Naoi M, Maruyama W. Monoamine oxidase inhibitors as neuroprotective agents in age-dependent neurodegenerative disorders. Curr Pharm Des. 2010;16:2799–2817. doi:10.2174/138161210793176527
28. Zhang P, Xu S, Zhu Z, Xu J. Multi-target design strategies for the improved treatment of Alzheimer’s disease. Eur J Med Chem. 2019;176:228–247. doi:10.1016/j.ejmech.2019.05.020
29. Farina R, Pisani L, Catto M, et al. Structure-based design and optimization of multitarget-directed 2H-chromen-2-one derivatives as potent inhibitors of monoamine oxidase B and cholinesterases. J Med Chem. 2015;58:5561–5578. doi:10.1021/acs.jmedchem.5b00599
30. Schneider LS, Geffen Y, Rabinowitz J, et al. Low-dose ladostigil for mild cognitive impairment: a Phase 2 placebo-controlled clinical trial. Neurology. 2019;93:1474–1484. doi:10.1212/wnl.0000000000008239
31. Hiremathad A, Keri RS, Esteves AR, Cardoso SM, Chaves S, Santos MA. Novel tacrine-hydroxyphenylbenzimidazole hybrids as potential multitarget drug candidates for Alzheimer’s disease. Eur J Med Chem. 2018;148:255–267. doi:10.1016/j.ejmech.2018.02.023
32. Frantz MC, Pellissier LP, Pflimlin E, et al. LIT-001, the first nonpeptide oxytocin receptor agonist that improves social interaction in a mouse model of autism. J Med Chem. 2018;61:8670–8692. doi:10.1021/acs.jmedchem.8b00697
33. Bolea I, Juárez-Jiménez J, de Los Ríos C, et al. Synthesis, biological evaluation, and molecular modeling of donepezil and N-[(5-(benzyloxy)-1-methyl-1H-indol-2-yl)methyl]-N-methylprop-2-yn-1-amine hybrids as new multipotent cholinesterase/monoamine oxidase inhibitors for the treatment of Alzheimer’s disease. J Med Chem. 2011;54:8251–8270. doi:10.1021/jm200853t
34. Ellman GL, Courtney KD, Andres V, Feather-Stone RM. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem Pharmacol. 1961;7:88–95. doi:10.1016/0006-2952(61)90145-9
35. Matos MJ, Terán C, Pérez-Castillo Y, Uriarte E, Santana L, Viña D. Synthesis and study of a series of 3-arylcoumarins as potent and selective monoamine oxidase B inhibitors. J Med Chem. 2011;54:7127–7137. doi:10.1021/jm200716y
36. Jiang N, Huang Q, Liu J, et al. Design, synthesis and biological evaluation of new coumarin-dithiocarbamate hybrids as multifunctional agents for the treatment of Alzheimer’s disease. Eur J Med Chem. 2018;146:287–298. doi:10.1016/j.ejmech.2018.01.055
37. Di L, Kerns EH, Fan K, McConnell OJ, Carter GT. High throughput artificial membrane permeability assay for blood-brain barrier. Eur J Med Chem. 2003;38:223–232. doi:10.1016/s0223-5234(03)00012-6
38. Zhong GH, Guo J, Pang CY, et al. Novel AP2238-clorgiline hybrids as multi-target agents for the treatment of Alzheimer’s disease: design, synthesis, and biological evaluation. Bioorg Chem. 2023;130:106224. doi:10.1016/j.bioorg.2022.106224
39. He Q, Liu J, Lan JS, et al. Coumarin-dithiocarbamate hybrids as novel multitarget AChE and MAO-B inhibitors against Alzheimer’s disease: design, synthesis and biological evaluation. Bioorg Chem. 2018;81:512–528. doi:10.1016/j.bioorg.2018.09.010
40. Ukai M, Kobayashi T, Shinkai N, Shan-Wu X, Kameyama T. Dynorphin A-(1-13) potently improves scopolamine-induced impairment of passive avoidance response in mice. Eur J Pharmacol. 1995;274:89–93. doi:10.1016/0014-2999(94)00710-o
41. Sang Z, Pan W, Wang K, et al. Design, synthesis and evaluation of novel ferulic acid-O-alkylamine derivatives as potential multifunctional agents for the treatment of Alzheimer’s disease. Eur J Med Chem. 2017;130:379–392. doi:10.1016/j.ejmech.2017.02.039
42. Li Q, Meng L, Zhou S, et al. Rapid generation of novel benzoic acid-based xanthine derivatives as highly potent, selective and long acting DPP-4 inhibitors: scaffold-hopping and prodrug study. Eur J Med Chem. 2019;180:509–523. doi:10.1016/j.ejmech.2019.07.045
43. Xie SS, Liu J, Tang C, et al. Design, synthesis and biological evaluation of rasagiline-clorgyline hybrids as novel dual inhibitors of monoamine oxidase-B and amyloid-β aggregation against Alzheimer’s disease. Eur J Med Chem. 2020;202:112475. doi:10.1016/j.ejmech.2020.112475
44. Jiang N, Ding J, Liu J, et al. Novel chromanone-dithiocarbamate hybrids as multifunctional AChE inhibitors with β-amyloid anti-aggregation properties for the treatment of Alzheimer’s disease. Bioorg Chem. 2019;89:103027. doi:10.1016/j.bioorg.2019.103027
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.