Back to Journals » International Journal of Nanomedicine » Volume 14
Improving the anti-keloid outcomes through liposomes loading paclitaxel–cholesterol complexes
Authors Wang MJ, Chen LQ, Huang W, Jin MJ ![]() , Wang QM, Gao ZG
, Wang QM, Gao ZG ![]() , Jin ZH
, Jin ZH
Received 20 November 2018
Accepted for publication 12 January 2019
Published 21 February 2019 Volume 2019:14 Pages 1385—1400
DOI https://doi.org/10.2147/IJN.S195375
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Linlin Sun
Mengjiao Wang,1 Liqing Chen,2 Wei Huang,2 Mingji Jin,2 Qiming Wang,2 Zhonggao Gao,2 Zhehu Jin1
1Klebs Research Center, Department of Dermatology, Yanbian University Hospital, Yanji 133000, China; 2State Key Laboratory of Bioactive Substance and Function of Natural Medicines, Institute of Materia Medica, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing 100050, China
Background: Keloids represent benign fibroproliferative tumors which result from elevated expression of inflammation. Paclitaxel (PTX) was an effective chemotherapeutic agent and has been reported to have anti-fibrotic effects, but the strong hydrophobicity brings a challenge for its clinical application.
Purpose: The objective of this study was to improve the water solubility of PTX and investigate its anti-keloid effects.
Methods: We prepared a PTX-cholesterol-loaded liposomes (PTXL) by thin film evaporation fashion and characterized their physicochemical properties. We also investigated the effects of PTX on proliferation, invasion and fibrosis of keloid fibroblasts in vitro and in vivo.
Results: The prepared PTXL have a spherical appearance, a particle size of 101.43 nm and a zeta potential of -41.63 mV. PTXL possessed a high drug entrapment efficiency of 95.63% and exhibited a good stability within 30 days. The drugs in PTXL were released in a slow and sustained mode. The PTXL could be effectively uptaken into human keloids fibroblast (HKFs) in a time-dependent manner. In vitro, PTXL showed better ability on inhibiting cell proliferation, migration and invasion, and effectively on promoting apoptosis and arresting cell cycle in G2/M phase compared to PTX. Meanwhile, in vivo studies indicated that the PTXL had better performance on inhibiting the keloids growth compared to the PTX in keloid-bearing BALB/c nude mice model. Finally, we found PTX treatment suppressed the production of tumor necrosis factor alpah (TNF-α), interleukin 6 (IL-6) and transforming growth factor beta (TGF-β) and inhibited the expression of alpha smooth muscle actin (β-SMA) and collagen I in HKFs. The activation of protein kinase B (AKT)/glycogen synthase kinase 3 beta (GSK3β) signaling pathway also blocked by PTX in cultured HKFs and keloid tissues. LY294002, a PI3K (phosphatidylinositol 3-kinase)/AKT inhibitor, also suppressed the expression of TNF-α, IL-6 and TGF-β, and simultaneously, reduced the production of α-SMA and collagen I in HKFs. The inhibition of AKT/GSK3β signaling pathway contribute to inhibit the generation of fibrogenic cytokines by PTXL on ameliorating fibrosis progress in keloids.
Conclusion: Our results suggested that the developed PTXL would become a promising therapeutic agent in the field of anti-keloid therapy.
Keywords: keloids, paclitaxel, liposomes, AKT, GSK3β, fibrosis
Introduction
Fibroblast activation and fiber formation in place of damaged cells and tissues are essential for the wound healing process. Keloids are formed by abnormally sustained wound healing processes following dermal injury or inflammation in genetically susceptible individuals.1 Keloids occur more frequently at specific skin areas that are more susceptible to damage, and can result from ear piercings and folliculitis.2 Surgical and/or intralesional steroid injection remain the foremost and effective therapy of keloids. However, there is a high recurrence rate and exacerbation after recurrence is common.3–5 Keloids are symbolized by inordinate deposition of extracellular matrix (ECM)2 and gradually increasing size. The subsequent damage to physical appearance can lead to serious emotional stress in patients, necessitating efficacious therapy of keloids. The pathogenesis of keloid development has been incompletely clarified, and most etiologies regard keloids as a chronic inflammatory and fibrotic disease.6,7 The main effector cells are fibroblasts, which are closely related to the excess production of inflammation in reticular dermis and the deposition of ECM.6,8
Abnormal levels of inflammation and growth factors were recognized as fundamental factors in keloids.9 TNF-α can transmit inflammatory signals to cells and recruit T cells to the trauma position, which contributes to its migration and proliferation.6 IL-6 is a major factor involved in acute and chronic inflammation and has a crucial position in both humoral and cellular immunity.10 TGF-β serves as an essential point in the evolution of fibrosis.2,7 TGF-β can activate TGF-β receptors that have an intrinsic serine/threonine kinase property to stimulate the phosphorylation of Smad2 and Smad3 proteins. Subsequently, the active product forms a complex with Smad4 and this complex regulates gene transcription in the nucleus.11 The activated TGF-β promotes the transformation of fibroblasts into myofibroblasts, characterized by the production of α-SMA, and leading to the production and remodeling of ECM. It is demonstrated that the elevated TNF-α, IL-6 and TGF-β was the primary element in the acceleration of fibrosis.12
Paclitaxel (PTX), derived from the berry of Taxus brevifolia, is a diterpenoid compound with the ability to enhance microtubule assembly and stabilize polymerized microtubules. PTX can arrest the cell cycle in G0/G1 or G2/M phases and subsequently induce apoptosis of cancer cells.13,14 It exhibits significant therapeutic capability for various types of cancers, including human ovarian cancer,15,16 breast cancer,17 gastric cancer13 and other malignancies.14 As well as its anti-cancer activity, PTX has been shown to adjust fibrogenic signaling and inflammatory responses. For instance, PTX can block TGF-β/Smad signaling through suppressing the action of STAT3,18–20 and inhibit nuclear factor-κB activation and the expression of inflammation to attenuate fibrosis by intervening with toll-like receptor 4.21 Therefore PTX has been widely applied clinically owing to its anti-fibrosis function.22,23 However, the efficacy of PTX on keloids remains unclear.
Despite its efficacy in various diseases, PTX solvent used clinically contains a high concentration of Kolliphor EL (BSAF Company, Rheinland-Pfalz, German),24 which can result in severe allergic reactions in patients. New drug delivery systems have been exploited to avoid hypersensitivity, eg, micelles, hydrogel, cyclodextrin nanocarriers, and liposomes.25–29 Liposomes are a biocompatible and biodegradable drug delivery system which are capable of entrapping lipophilic drugs and offering a slow and sustained release of drugs within a tumor.30 Therefore, it could reduce the cytotoxicity to normal tissues and enhance the anti-tumor efficacy of chemotherapeutic agents.31 However, the aggregation and membrane fusion caused by PTX results in unstable liposomes for long-term application. It has been reported that stability of PTX nanoparticles could be extremely enhanced after encapsulating the complex formed by PTX and lipid components.32,33
In the present study, we choose cholesterol to form complexes with PTX, and stable PTX–cholesterol-loaded liposomes (PTXL) have been developed and well characterized. In addition, we investigated the anti-keloid and anti-fibrotic efficacy of PTXL aganist keloids. Also, we explored the related activation of AKT/GSK3β signaling in anti-keloid therapy using PTXL.
Materials and methods
Materials
PTX was provided by Meilun Biotech (Dalian, China). mPEG-DSPE2000 was bought from Laysan Bio, Inc. (Arab, AL, USA). Acetonitrile, methanol (HPLC grade) and cholesterol were all supplied by Sigma-Aldrich Co. (St Louis, MO, USA). Soybean phosphatidylcholine (SPC) and egg yolk phosphatidylcholine (EPC) were obtained from AVT (Shanghai) Pharmaceutical Co., Ltd (Shanghai, China). Coumarin-6 was purchased from Sigma-Aldrich Co. Cell Counting Kit-8 (CCK-8) was supplied by Dojindo Laboratories (Kumamoto, Japan). Annexin V-fluorescein isothiocyanate (FITC) apoptosis detection kit was provided by NeoBioscience (Shenzhen, China). DNA content quantitation assay (cell cycle) was supplied by Solarbio (Shanghai, China). ELISA kit for IL-6, TGF-β and TNF-α were purchased from USCN Kit, Inc. (Wuhan, China). The primary antibodies against the following proteins were obtained from Abcam (Cambridge, UK): AKT, phospho-AKT (p-AKT), glycogen synthase kinase 3β (GSK3β), p-GSK3β, α-SMA and collagen I. HPR-goat anti-rabbit secondary antibody IgG H&L was purchased from Abcam. The BCA protein assay kit was provided by CW Biotech (Beijing, China).
Preparation of PTX–cholesterol
PTX and cholesterol were dissolved in acetone at a ratio of 1:0.45 to adjust the PTX concentration to 2 mg/mL, and stirred at 45°C, 300 rpm/min for 2 hours under nitrogen protection. The acetone was evaporated through rotary evaporation and vacuum drying for 15 hours at 45°C. Finally, the PTX–cholesterol complex was attached to a flask, scraped and stored at −20°C.32
Preparation of PTXL
A film-hydration method was performed to prepare PTXL. PTX–cholesterol, EPC, SPC, cholesterol and mPEG-DSPE2000 were dissolved with chloroform. The mixture was transferred into round bottom flask and then chloroform was eliminated by rotary evaporation at 40°C to gain a uniform thin film. Subsequently, the film was hydrated with 5% glucose to obtain a liposome solution and then subjected to ultrasound (time: 15 minutes, power: 65 W, ultrasonic 2 seconds, intermittent 1 second) under a Ultrasonic cell pulverizer (Scientz, Wuhan, China). Finally, the obtained PTXL was filtered with a 0.22 μm filter (EMD Millipore, Billerica, MA, USA) three times. The blank liposomes (Blank-L) solution was identical to PTXL except for the absence of drug.
Morphology of PTXL
PTXL morphology was observed under a transmission electron microscope (TEM) (H-7650, Hitachi Ltd, Tokyo, Japan) at 200 kV. Briefly, the PTXL was rested on a carbon net for 5 minutes to be precipitated and sucked dry. Then the carbon net was incubated with 2% phosphotungstic acid solution for 5 minutes and dried out under an infrared lantern. Finally, the morphology of PTXL on carbon net was observed and imaged under a TEM.
Particle size and zeta potential of PTXL
The dynamic light scattering and electrophoretic light scattering are the most common methods for detecting the average size, polydispersity index (PDI) and zeta potential of nanoparticles. PTXL solution was diluted with distilled water 10 times and the average size, PDI and zeta potential were detected with a Zetasizer Nano ZS90 (Malvern, UK).
Entrapment efficiency (EE) and loading capacity (LC) of PTXL
EE and LC of PTXL were evaluated using a HPLC analysis system (Agilent 1200, Agilent Technologies, Santa Clara, CA, USA). A Grace Allsphere ODS-2 C18 column (250×4.6 mm, 5 μm; Grace, Columbia, MD, USA) was used for the separation. Flow phase was constituted of water-acetonitrile-methanol (41:36:23). The flow velocity and column temperature were determined at 1.0 mL/min and 25°C, separately. The UV absorbance was 227 nm. The sample injection volume was 10 μL.
Five mg of PTX powder was weighed accurately and a PTX solution at a concentration of 200 μg/mL was prepared with methanol. The PTX solution was diluted into the concentrations of 100, 50, 25, 12.5, 5, 2.5 μg/mL with flow phase. The above solutions were analyzed through HPLC based on the mentioned qualifications. Afterward, a standard curve was obtained between PTX concentration (C) and peak area (A). Further, the filtered and unfiltered PTXL solutions were diluted five times accurately with methanol. The lyophilized powder of PTXL was precisely weighed and dissolved with methanol. The solutions were subjected to vortexing for 5 minutes and ultrasound for 10 minutes to release the encapsulated PTX. Finally, the 0.22 μm membrane (Millipore) was used to remove large particles. PTX concentration was determined by HPLC and the weight of PTX was calculated separately. EE is the percentage of encapsulated drug in total drug, and LC is the percentage of the encapsulated drug in the total drug and material.
In vitro release
One mL PTXL solution was transferred into a dialysis bag (MWCO 20KD, MYM Technologies Ltd, Hyderabad, India) and immersed in 40 mL phosphate buffer solution (PBS) (pH 7.4, 0.5% (w/v) Tween-80). They were put in a Erlenmeyer flask and swirled at a speed of 100 rpm at 37°C. At the designated time, 0.5 mL solvent was sampled and substituted with 0.5 mL fresh solvent. At the end of the assay, the dialysis bag was broken and sampled. Afterward, the obtained samples were centrifuged at 10,000 rpm for 10 minutes and the supernatants collected for HPLC to obtain the PTX concentration in samples. Ultimately, the cumulative release of PTX was calculated and plotted.
Cell culture
HKFs (Shanghai Xin Yu Biotechnology, Shanghai, China) and human normal skin fibroblasts (NF) (American Type Culture Collection (ATCC), Manassas, VA, USA) were cultured with DMEM (HyClone Laboratories, Inc., South Logan, UT, USA) incorporation with 10% fetal bovine serum (Gibco®, Thermo Fisher Scientific, Waltham, MA, USA) at 37°C in a humidified atmosphere containing 5% CO2.
Cellular uptake and uptake mechanism assay
HKFs were inoculated in a plate with 6 wells, adjusted to 1.5×104 cells/well and cultured for 24 hours. Fresh medium containing coumarin-6 (C6) or coumarin-6-loaded liposomes (C6-L) (concentration of C6=1 μg/mL) was added to substitute the previous medium. After incubation for 15 or 30 minutes, excess C6 and C6-L were rinsed 3 times with PBS, and then cells in wells were collected and analyzed for fluorescence intensity using a FACSAria flow cytometry (BD, Franklin Lakes, NJ, USA).
Simultaneously, in order to observe the fluorescence intensity in the cells, HKFs were seeded in wells with coverslips, and were incubated with C6 or C6-L for 30 minutes after 24 hours. The excess C6 or C6-L were rinsed with cold PBS, and then 4% paraformaldehyde solution was used to fix the coverslips for 10 minutes. Afterward, antifade solution (Applygen, Beijing, China) was dropped onto slides and coverslips were placed on them. Finally, the slides were observed and photographed with 488 excitation wavelength under a confocal laser scanning microscope (TCS-SP2, Leica, Germany).
In order to determine the endocytosis mechanism of PTXL, methyl-β-cyclodextrin (M-β-CD) (caveolin inhibitor) and chlorpromazine (CPZ) (clathrin inhibitor) were used as endocytosis inhibitors to pretreat cells for one hour and removed before adding C6-L. The rest of the processing was the same as above.
Cell proliferation inhibition assay
A CCK-8 assay was performed to detect the cell viability. HKFs were inoculated in a plate with 96 wells and adjusted to 5.0×103 cells/well. After 24 hours’ culture, the primary medium was replaced with the fresh medium containing a series of different test samples for another 24 hours or 48 hours. Ultimately, cells in each well was incubated with 100 μL serum-free medium containing 10% CCK-8 for 2 hours. Subsequently, the optical density (OD) values were detected at 450 nm with 650 nm as reference by a Synergy H1 Microplate Reader (BioTek, Winooski, VT, USA). The below formula was applied for cell viability:
|
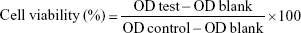
Apoptosis analysis of HKFs
The apoptosis proportion of HKFs was measured through Annexin V-FITC/propidium iodide (PI) staining. HKFs were inoculated in a plate with 6 wells and adjusted to 1.5×104 cells/well. After 24 hours, primary medium was replaced with fresh medium containing 0.1 μg/mL PTX or PTXL. Cells were harvested and washed with PBS twice, then dyed with Annexin V-FITC and PI according to the manufacturer’s instructions (Annexin V-FITC apoptosis detection kit, NeoBioscience, Shenzhen, China). Then cells were filtered through 300 mesh Nylon net and assayed using flow cytometry.
Cell cycle distribution
Cell cycle arrest was determined through PI staining. HKFs were cultured and treated in the same manner as apoptosis analysis. After treatment for 24 hours, cells were collected and fixed with cold 70% ethanol at −20°C overnight. Subsequently, the cells were washed twice and resuspended with PBS. Then RNaseA was added at the final concentration of 100 μg/mL and cell suspension was inoculated at 37°C for 30 minutes. Followed by dyeing with PI at the final concentration of 50 μg/mL for one hour at room temperature. Then cell cycle was analyzed through flow cytometry.
Wound healing assay
HKFs were inoculated in a plate with 12 wells and adjusted to 5.0×104 cells/well. After 24 hours’ incubation, scratches were made in the middle of each well artificially and rinsed with PBS for three times. Then the tested groups were treated with medium containing 0.1 μg/mL PTX or PTXL solution while the control group was replaced with fresh medium. The wound healing was observed and imaged at design time under a light microscope (Olympus Corporation, Tokyo, Japan).
Transwell invasion assay
HKFs were firstly seeded into a culture flask and cultured for 24 hours to adhere. After incubation with medium containing 0.1 μg/mL PTX or PTXL solution for 24 hours, cells were harvested and inoculated in the pre-treated upper chamber with matrigel (Thermo Fisher Scientific) in serum absent medium at a density of 1.0×105 cells/well. The upper chamber was put in medium possessing 10% FBS. After incubation for 24 hours, uninvaded cells from the upper side of the membrane were washed and removed. The invaded cells in the lower surface were fixed, stained with crystal violet and photographed under a microscope. For quantitative assay, the crystal violet staining cells were dissolved in 33% acetic acid and the OD value was measured at 570 nm using a Synergy H1 Microplate Reader.
Measurement of TNF-α, IL-6, and TGF-β
The standards of TNF-α, IL-6, and TGF-β were evaluated through ELISA kits (USCN Kit Inc.). Cytokine levels were normalized based on the protein concentration according to BCA protein assay kit (CW Biotech, Beijing, China).
Measurement of AKT/GSKW3β signaling, α-SMA and collagen I
Protein expression was determined by Western blot assay. The cell precipitate was lysed with 200 μL RIPA buffer (50 mM Tris-HCl (pH 7.4), 150 mM NaCl, 1% NP-40, 0.1% SDS) and replenished with 1× protease inhibitor cocktail and 1× phosphatase inhibitor cocktail (Hoffman-La Roche Ltd., Basel, Switzerland) on ice. The lysate was sonicated and centrifuged at 13,000 rpm at 4°C for 20 minutes to obtain the supernatant. Total protein content in supernatant was determined by BCA assay. The extracted protein was adjusted to a uniform concentration and denatured at 95°C for 5 minutes. Proteins were segregated on 10%–15% SDS-PAGE and migrated to PVDF membranes (Millipore). Then, membranes were incubated with 5% BSA-TBST Tris-buffered for blocking followed by primary antibodies overnight at 4°C: rabbit anti-AKT1/2, rabbit anti-AKT1 (phospho S473), rabbit anti-GSK3β, rabbit anti-GSK3β (phospho S9), rabbit anti-α-SMA, rabbit anti-collagen I. Three washes were performed for 10 minutes with TBST. The membranes were incubated with secondary antibodies for an hour at room temperature and washed as above. Blots were detected with ECL Western blot detection reagent (Millipore).
Nude mice subcutaneous keloid model
The 6-week-old BALB/c female nude mice were provided by Beijing Vital River Laboratory Animal Technology Co., Ltd (Beijing, China). All animal experiments were ratified by Animal Ethics Committee, abided by the Principles of Laboratory Animal Ethics Committee of the Institute of Materia Medica in Peking Union Medical College, and were conducted with necessary humane care. The keloid model was established through subcutaneously inoculating 5×106 HKFs cells in nude mice. The experiments were started when keloids grew to qualified volume.
Anti-keloid effect of PTXL in vivo
A digital caliper was used to measure the size of keloids and the short diameter and long diameter of the keloid were represented by a and b respectively. Keloid volume = a2 × b/2. When the keloid volume reached above 100 mm3, keloid-bearing nude mice were divided into three groups randomly (n=8), including 5% glucose solution group, PTX group and PTXL group. The intratumoral injection dose is 0.05 mL/100 mm3 according to the keloid volume (drug concentration =100 μg/mL). All groups were treated every seven days four times, while keloid volume was measured before each injection. After the experiment finished, nude mice were killed by cervical dislocation. The keloid, liver, heart, spleen, lung and kidney were taken out, fixed and preserved in 4% neutral formaldehyde. Keloids in each group were imaged and weighed for evaluation of efficacy. All organs taken out were embedded with paraffin and sliced. Hematoxylin and eosin (H&E) staining was performed on slides to observe the organizational structure. Additionally, the apoptosis in keloid tissue was detected by TUNEL assay.
Immunohistochemistry
The effect of PTX on keloid fibrosis and AKT/GSK3β signaling expression was evaluated by immunohistochemistry. Keloid slides were treated with the primary antibody overnight at 4°C and secondary antibody for 30 minutes at 37°C sequentially. Then they were incubated with diaminobenzidine (DAB) for 2 minutes to visualize the slides and neutralize them quickly. Finally, intensity was observed and evaluated in a blind manner.
Statistical analysis
Data are expressed as mean ± SD. Statistical analysis was executed through the SPSS Statistics 22 (SPSS Inc., IBM Corporation, Armonk, NY, USA). ANOVA was performed to determine the significant difference in multiple groups. Student’s t-test was used to compare the significant difference between two groups. Statistical significance was represented as: *P<0.05 or **P<0.01.
Results and discussion
Preparation and characterization of PTXL
PTX is a well-known chemotherapeutic drug with anti-fibrotic function. Encapsulation in liposomes is a method to improve the water solubility and bioavailability of PTX, but the poor stability of PTX liposomes leads to challenges in its clinical application. Lipid nanoemulsion loaded PTX (LDE-PTX) has been confirmed to have tumor-targeting effects, but it is stable for only 8 days at 4°C, which makes it less promising for clinical application.34 Its instability might be attributable to the poor lipophilicity of PTX.35 Therefore, increasing the lipophilicity of PTX may be a way to increase the stability of PTX in drug delivery systems. According to research, the lipid emulsion loaded cholesterol–PTX complex exhibits an ideal particle size, high drug-encapsulation efficiency and excellent stability (12 months at 6°C). Furthermore, cholesterol–PTX emulsion shows better anti-tumor efficacy and better safety profiles compared to the conventional PTX emulsion and Taxol.32,36 Thus, we prepared PTX–cholesterol complexes and loaded them in liposomes to investigate whether PTX is effective in treating keloids. First, we prepared PTX liposomes and cholesterol–PTX liposomes, and investigated their stability after 24 hours at 4°C. After 24 hours, PTX liposomes became cloudy and precipitated, and the EE was only 79.3%. In contrast, the EE of cholesterol–PTX liposomes was 92.1%. The above results suggested that the prepared liposome loading PTX–cholesterol complexes could increase the water solubility of hydrophobic PTX and were more stable.
PTXL showed a spherical shape (Figure 1A) and had an average particle size of 101.43±0.54 nm (Figure 1B) with a PDI of 0.244±0.006; the particle size detection results of two experiments were consistent. The PTXL had a zeta potential of −41.63±0.83 mV (Figure 1C), which ensured the dispersion stability of PTXL through the mutual repulsion effect between identical species charge among liposomes.
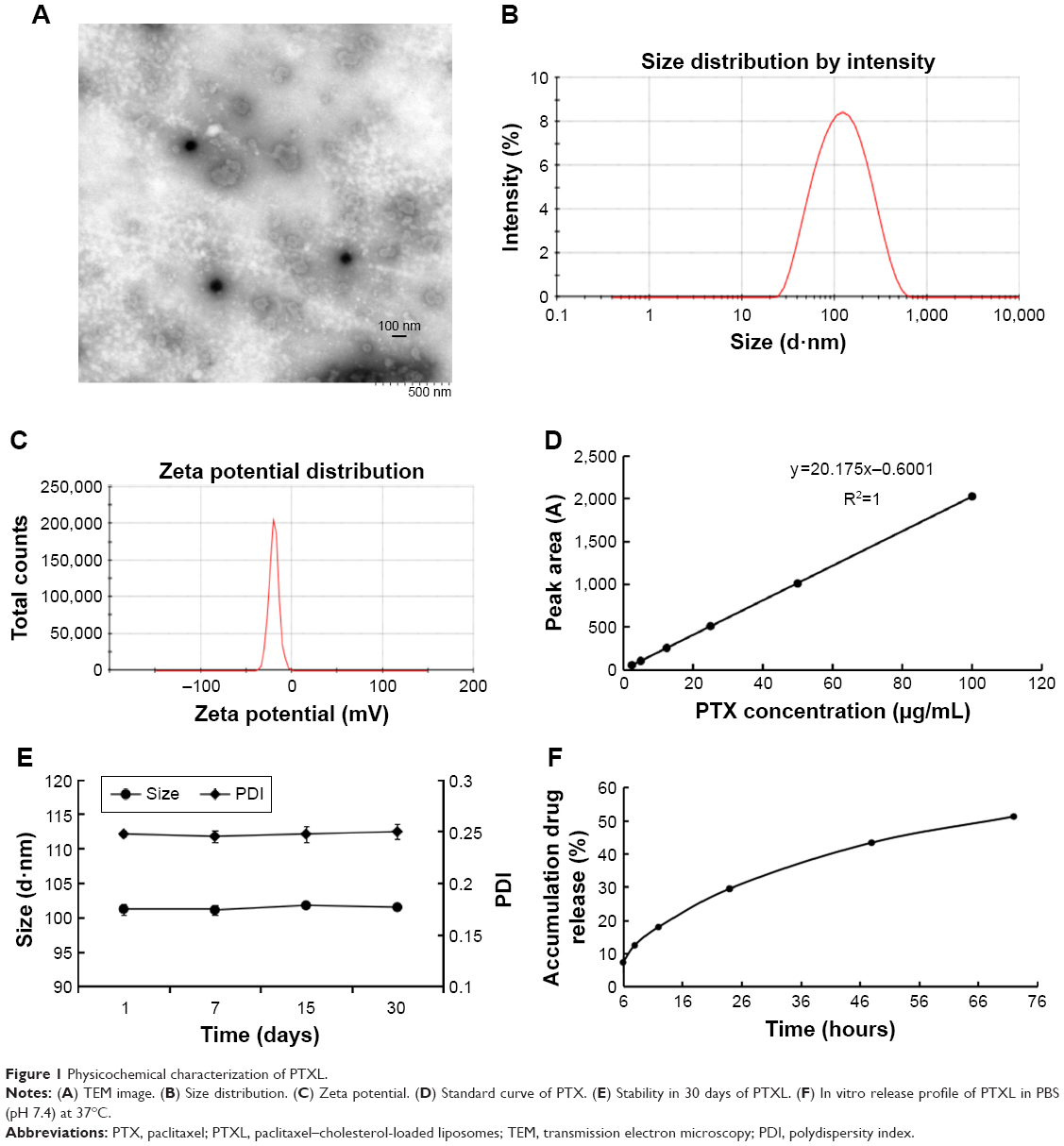 | Figure 1 Physicochemical characterization of PTXL. |
We established an HPLC method to detect the content of PTX. The standard curve between peak area (A) and PTX concentration (C) was: A=20.175 C − 0.6001 (R2=1, n=6) (Figure 1D). It was demonstrated that a good linear relationship exists in the concentration between 2.5 and 100 μg/mL. The EE and LC of PTXL we measured were (95.63±0.70)% and (2.7±0.16)% respectively, indicating that the liposomes have good encapsulation ability of PTX.
EPC, SPC and cholesterol were major components of liposomes, and tended to aggregate because of Van der Waals forces if without other components enhancing stability. mPEG-DSPE2000 inserted into the lipid bilayer of liposomes was capable of diminishing particle aggregation owing to the PEG chain acting like a protective shield.37 The stability assay showed that the size and PDI of PTXL had no obvious alteration when stored at 4°C for 30 days (Figure 1E). The leakage rate in PTXL solution at 1, 7, 15, and 30 days were (0.13±0.05)%, (4.35±1.93)%, (8.74±0.06)%, and (18.23±0.98)%, respectively. These proved that PTXL had good stability for further application.
The accumulative drug release curve obtained by in vitro drug release assay showed that PTX were detected at 6 hours for first time and gently released in following 66 hours, and the accumulative release percent of PTX increased to 51.21% of total drugs after 72 hours. The results suggested that the liposomes could control drug released in a slow and gentle manner (Figure 1F).
Cellular uptake and mechanism
The fluorescence intensity collected by flow cytometry illustrated that the uptake of free C6 and C6-L by HKFs in a time-dependent manner, and the fluorescence intensity, was significantly enhanced by C6-L when compared to C6 within same processing time (P<0.01) (Figure 2A), which was consistent with the result that we observed under a confocal microscopy after incubation for 30 minutes (Figure 3). The fluorescence was mostly distributed in the cytoplasm and the uptake of C6-L by HKFs could be weakened to 40.15% by M-β-CD, but not affected by CPZ both in confocal microscopy and flow cytometry assays (Figures 2B and 3). This suggested that PTXL were partially taken up by cells through caveolin-mediated endocytosis associated with caveolin rather than by clathrin.
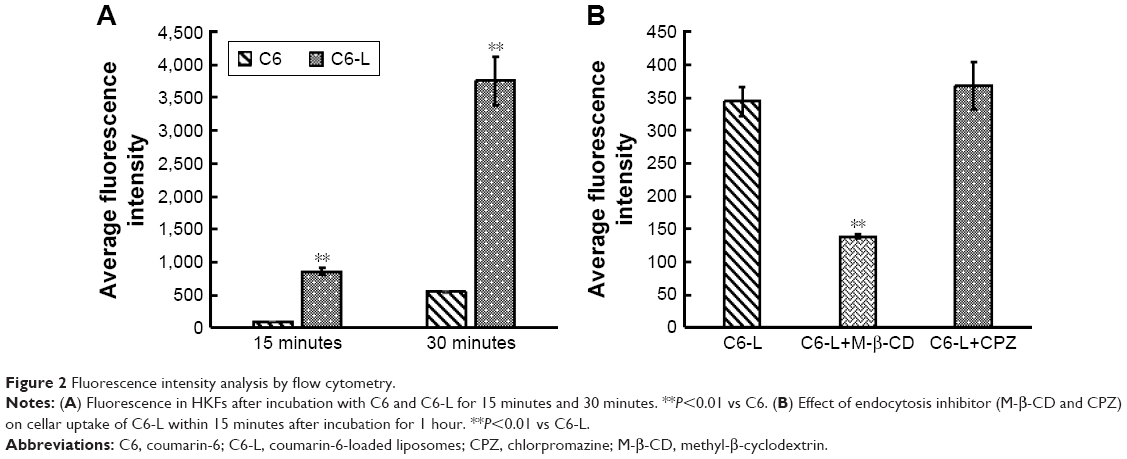 | Figure 2 Fluorescence intensity analysis by flow cytometry. |
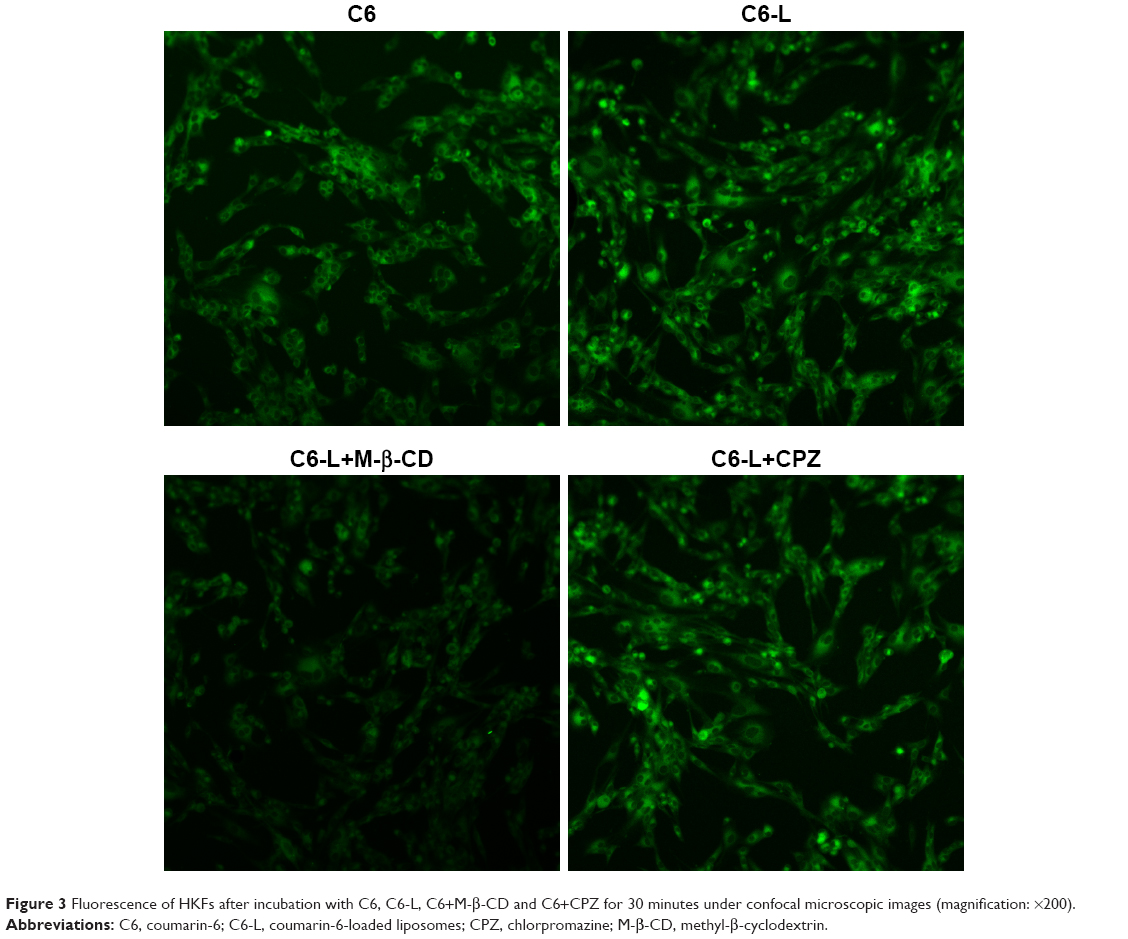 | Figure 3 Fluorescence of HKFs after incubation with C6, C6-L, C6+M-β-CD and C6+CPZ for 30 minutes under confocal microscopic images (magnification: ×200). |
Cell proliferation inhibition
The cytotoxicity of Blank-L and commercial PTX solution (anhydrous ethanol (E)/Kolliphor EL (EL) (E+EL) =1:1) were evaluated by CCK-8 assay. We found that, when the concentration of PTX ranged from 0.05 μg/mL to 1 μg/mL, the inhibition of cell viability caused by Blank-L does not exceed 5% in 48 hours, while blank (E+EL) was <15% in 48 hours. The cytotoxicity of (E+EL) were significantly higher than Blank-L when the concentration was higher than 1 μg/mL (P<0.05 or P<0.01) (Figure 4A). These results proved that liposomal materials exhibited lower toxicity to HKFs.
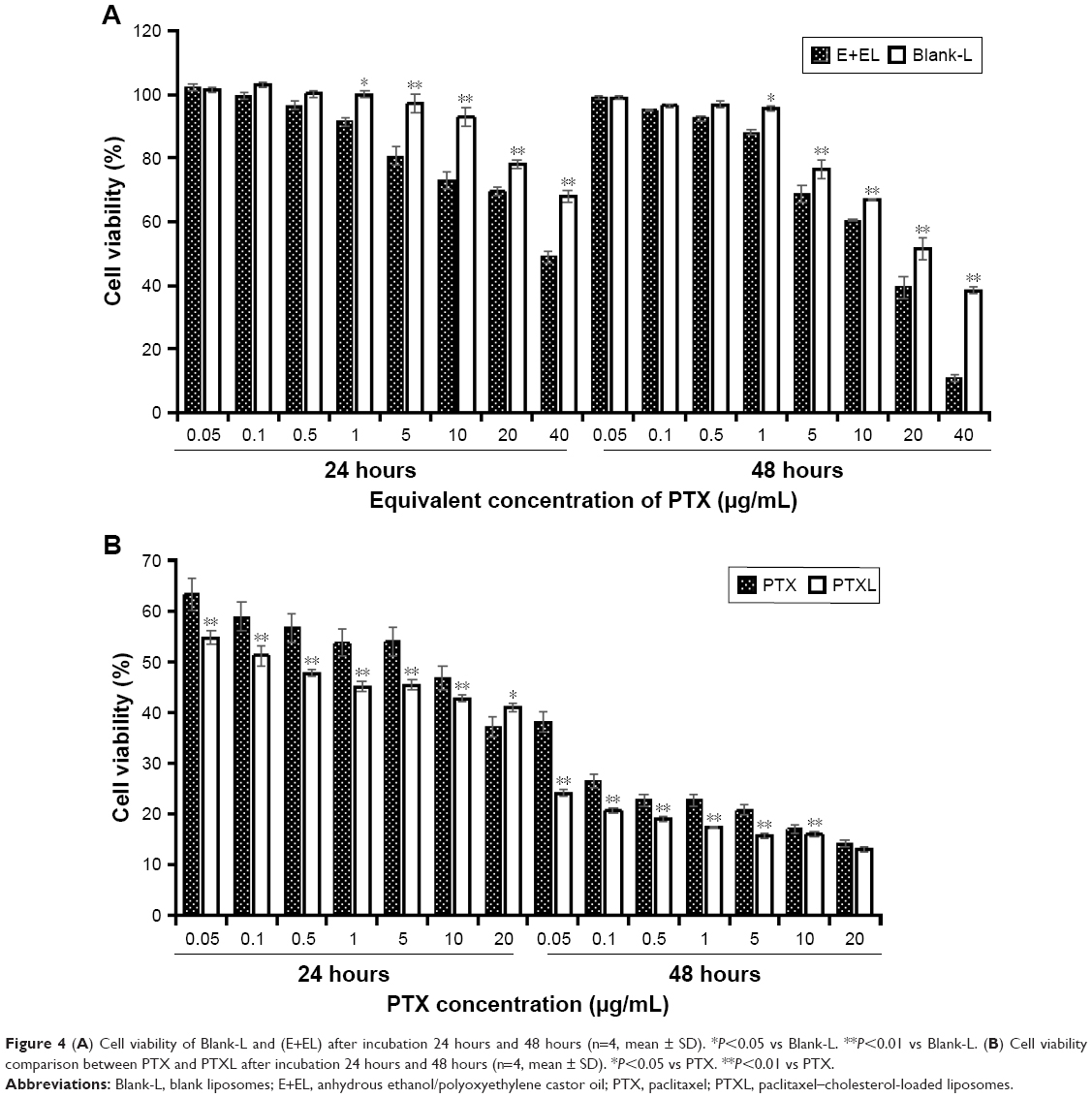 | Figure 4 (A) Cell viability of Blank-L and (E+EL) after incubation 24 hours and 48 hours (n=4, mean ± SD). *P<0.05 vs Blank-L. **P<0.01 vs Blank-L. (B) Cell viability comparison between PTX and PTXL after incubation 24 hours and 48 hours (n=4, mean ± SD). *P<0.05 vs PTX. **P<0.01 vs PTX. |
Subsequently, proliferation inhibition ability of PTXL on HKFs was evaluated and the results showed that PTXL had a strong inhibitory capability on the proliferation of HKFs; the inhibition rate was close to 50% when the concentration of PTX was 0.1 μg/mL. The PTXL inhibited the cell viability in time and concentration dependent manner. PTXL was effective than the PTX on inhibiting cell proliferation under the same concentration (ranges from 0.05 to 10 μg/mL) and time (P<0.01) (Figure 4B).
Influence of PTXL on cell apoptosis and cell cycle
Excessive ECM deposition resulting from insufficient apoptosis of HKFs is known as the primary pathological manifestation of keloids. Therefore, inducing apoptosis of HKFs may be useful for the treatment of keloids. A previous study showed that PTX tends to induce cell apoptosis and arrest cell cycle in multiple tumor cells.13,14 Apoptosis and the cell cycle of HKFs were evaluated. The results illustrated that the proportion of apoptosis cells in the control group was (4.59±0.94)%, which was obviously less than PTX group (P<0.01) and PTXL group (P<0.01) and the proportion of apoptosis cells of the PTXL group was (44.42±1.31)%, which was obviously increased compared to the PTX group (33.92±0.58)% (P<0.01) (Figure 5A and C).
 | Figure 5 Effects of PTXL on cell apoptosis and cell cycle distribution. |
The cell cycle distribution showed that PTX could arrest the HKF cell cycle in G2/M phase after treated for 24 hours. PTX and PTXL increased the proportion of G2/M phase from (14.60±3.48)% in the control group to (78.902±5.32)% and (87.152±1.17)%, separately, statistical difference was existed within any two groups of the above three groups (P<0.01) (Figure 5B and D).
Impact of PTXL on HKF wound healing ability
The observation of wound healing process demonstrated that the wounds of the control group trended to heal after incubation for 24 hours and disappeared at 48 hours. However, the wounds in the PTX group became slightly narrower, accompanied by less cell density than in the control group, while there were no significant changes of the wounds in PTXL group. The morphology of HKFs was changed from a multi-protrusive spindle shape to a more regular triangle or short fusiform shape after drug administration (Figure 6).
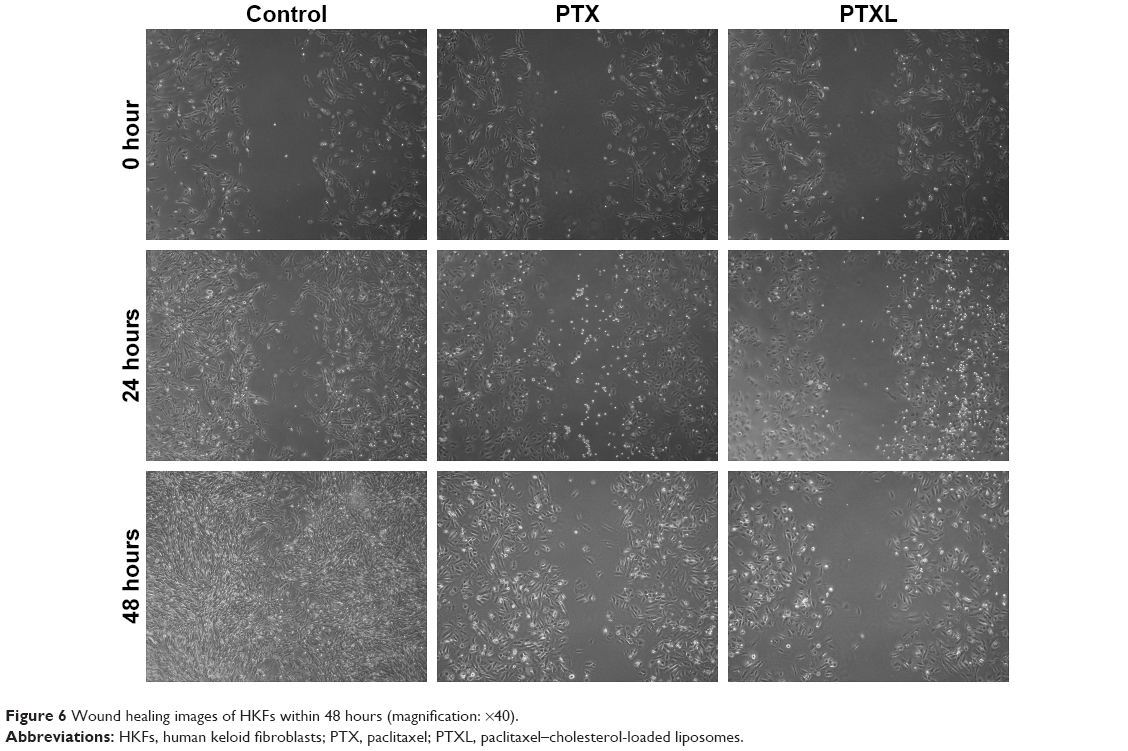 | Figure 6 Wound healing images of HKFs within 48 hours (magnification: ×40). |
Influence of PTXL on cell invasion
Keloids can invade the surrounding normal skin tissue and grow like crab feet. A transwell experiment was performed in which matrigel is used to simulate ECM. The results are displayed in Figure 7. HKFs in the control group showed strong invasiveness, ie, numerous cells invading the matrigel and transwell chamber to the lower layer. However, the invasion rates of PTX and PTXL group were (28.21±7.95)% and (5.77±4.10)%, separately, which showed conspicuous suppression compared to the control group (P<0.01). PTXL showed higher inhibition than PTX (P<0.01). The results suggested that PTXL could significantly inhibit the invasion of HKFs.
 | Figure 7 Crystal violet staining of the invasion cells after incubation for 24 hours. |
In vivo anti-keloid effect of PTXL
The growth curve and weight of keloids in BALB/c nude mice treated by PTXL, PTX and 5% glucose solution showed (Figure 8) that the average weight and volume of keloids were significantly decreased in the PTX and PTXL groups when compared with the control group (P<0.05 and P<0.01, respectively). Meanwhile, PTXL exhibited better inhibition effect compared to the PTX group (P<0.05). H&E staining showed that the density of HKFs in the PTXL group and PTX group was obviously less than the control group (Figure 9A). There were no organic lesions found in nude mice. The results of TUNEL assay showed that cell quantity in keloid tissues were decreased in both PTXL and PTX groups, and more brown-TUNEL-positive cells could be found in PTXL group than in PTX group (Figure 9B). PTXL enhanced the inhibition of PTX on the growth of keloids. This result may be related to the increased PTX uptake of HKFs and the sustained released of PTX in liposomes, which provided an effective and continuous toxicity to keloids.
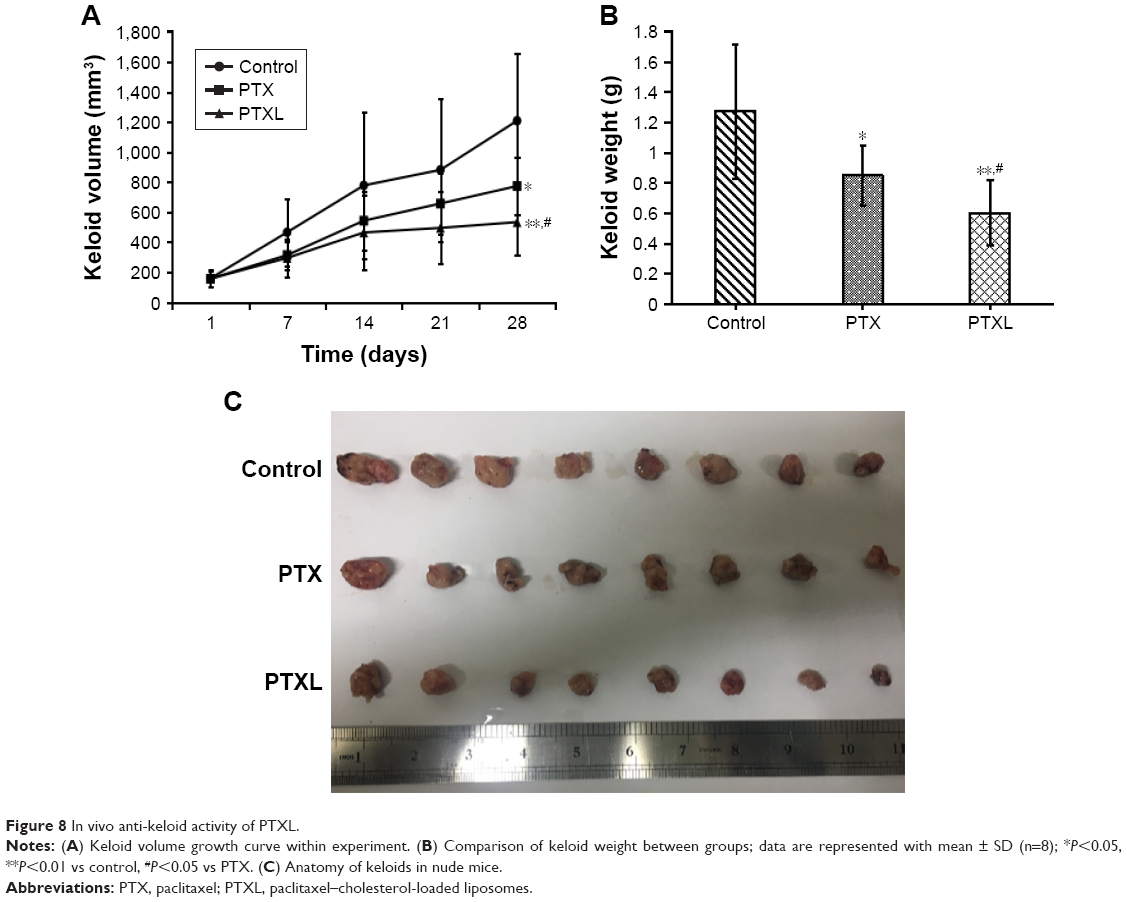 | Figure 8 In vivo anti-keloid activity of PTXL. |
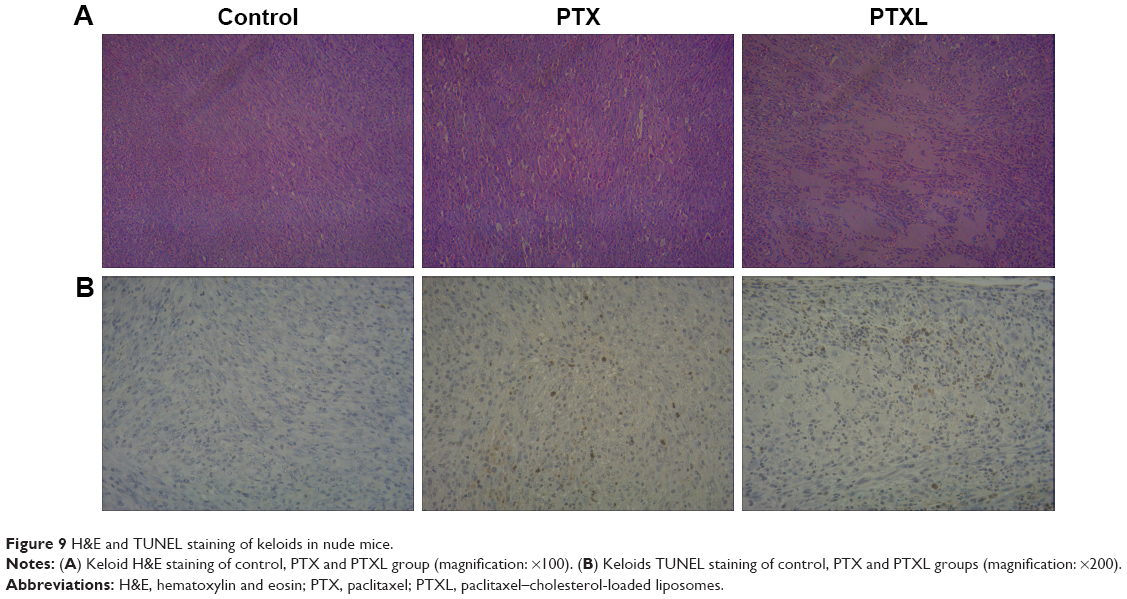 | Figure 9 H&E and TUNEL staining of keloids in nude mice. |
Levels of TNF-α, IL-6 and TGF-β in HKFs
IL6 and TNF-α were proinflammatory factors that could recruit monocytes and T cells to injury sites.38,39 Moreover, TGF-β is necessary to induce fibrosis and cancer. These factors played a critical role in the pathogenesis of various chronic inflammation-related diseases, such as fibrosis and tumor, and could promote cells proliferation and migration.3
The results of an ELISA assay showed that the production of TNF-α, IL-6 and TGF-β was significantly increased in HKFs compared to normal fibroblasts (NFs) (P<0.01), and the expression of above cytokines in HKFs were reduced by PTX (0.1 μg/mL). More interesting, LY294002 (5 μM), a PI3K/AKT inhibitor, also inhibited the expression of the above cytokines (P<0.01) (Figure 10). These results suggested that PTX could suppress the production of TNF-α, IL-6 and TGF-β in keloids, and the inhibition of PI3K/AKT signaling achieved similar effect with PTX in HKFs.
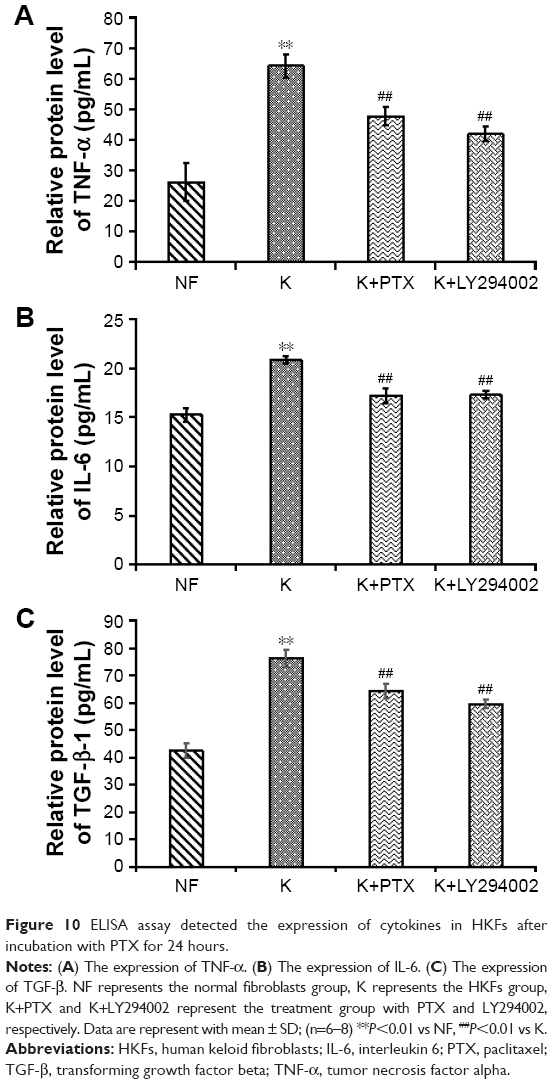 | Figure 10 ELISA assay detected the expression of cytokines in HKFs after incubation with PTX for 24 hours. |
Effect of PTX on AKT/GSK3β pathway and fibrosis
Numerous studies showed that PI3K/AKT signaling is significant for cellular responses to growth factors and for cellular malignant transformation. p-GSK3β is a downstream bioactive product of active PI3K and has been involved in cell proliferation, G2/M phase arrest,40 survival, metabolism and motility.41 It was also reported that AKT/GSK3β modulates the activity of multiple transcription factors involving NF-κB,42 by preventing the degradation of IκBα.43 The excessive activation of the NF-κB signaling pathway is critical in inflammatory responses.44 Previous studies demonstrated that the activation of PI3K/AKT signaling contributed to apoptosis in cancer cells induced by PTX.45
Therefore, we studied the anti-inflammatory and anti-fibrotic effects of PTX in keloids by inhibiting the AKT/GSK3β signaling. HKFs was treated by PTX (0.1 μg/mL) and using LY294002 (5 μM) as a positive control, then Western blot was performed to detect the expression of AKT/GSK3β signaling and the production of α-SMA and collagen I, which were markers in fibrotic remodeling. The results showed that AKT, p-AKT and its downstream molecule GSK3β and p-GSK3β were decreased after PTX treatment, which was consistent with the inhibition of AKT/GSK3β signaling by LY294002 (Figure 11). α-SMA and collagen I were also significantly inhibited by PTX and LY294002. Tissue immunohistochemistry staining results displayed that the brown intensity of AKT/GSK3β signaling and the strength of α-SMA and collagen I were declined after treated with PTX (Figure 12). The results indicated that the inhibition of α-SMA and collagen I by PTX was conducted by the inhibition of AKT/GSK3β signaling in keloids. We speculated that it might be resulted from the reduction of TNF-α, IL-6 and TGF-β through the blocking of AKT/GSK3β signaling pathway.
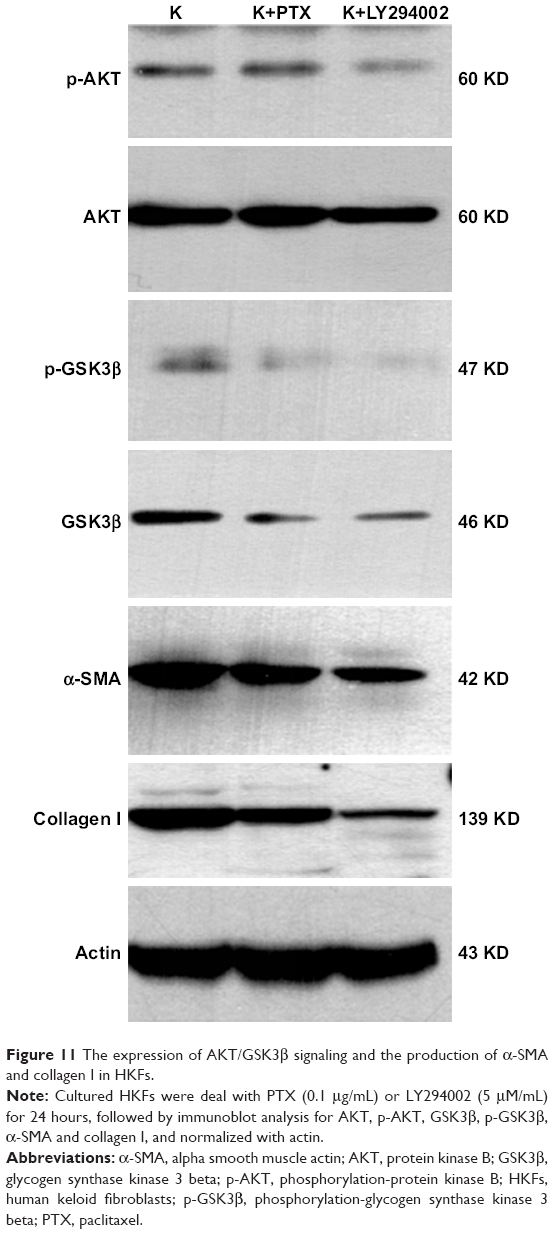 | Figure 11 The expression of AKT/GSK3β signaling and the production of α-SMA and collagen I in HKFs. |
 | Figure 12 PTXL inhibits the activation of AKT/GSK3β signaling and the production of α-SMA and collagen I in keloids bearing in nude mice. |
Conclusion
We prepared PTXL and characterized their physicochemical properties. We also investigated the anti-keloid efficacy of PTXL in vitro and in vivo. It was demonstrated that PTXL could inhibit the growth and invasion of keloids and showed much better effects compared to PTX. Meanwhile, PTXL could also reduce the production of fibrogenic cytokines, including TNF-α, IL-6 and TGF-β, by inhibiting AKT/GSK3β signaling pathway, which consequently led to ameliorating fibrosis progress in keloids. Generally, our results suggested that PTXL could become a promising therapeutic in the field of anti-keloid therapy.
Acknowledgment
This study was supported by CAMS Innovation Fund for Medical Sciences (2017-I2M-1-011), the Drug Innovation Major Project (2018ZX09711001-002-005), the Fundamental Research Funds for the Central Public Welfare Research Institutes (2018PT35002), and the PUMC Youth Fund and the Fundamental Research Funds for the Central Universities (2017350003).
Disclosure
The authors report no conflicts of interest in this work.
References
Wulandari E, Jusman SW, Moenadjat Y, Jusuf AA, Sadikin M, Endah W. Expressions of collagen I and III in hypoxic keloid tissue. Kobe J Med Sci. 2016;62(3):E58–E69. | ||
Andrews JP, Marttala J, Macarak E, Rosenbloom J, Uitto J. Keloids: The paradigm of skin fibrosis – pathomechanisms and treatment. Matrix Biol. 2016;51:37–46. | ||
Bran GM, Goessler UR, Hormann K, Riedel F, Sadick H. Keloids: current concepts of pathogenesis (review). Int J Mol Med. 2009;24:283–293. | ||
Trisliana Perdanasari A, Lazzeri D, Su W, et al. Recent developments in the use of intralesional injections keloid treatment. Arch Plast Surg. 2014;41:620–629. | ||
Gauglitz GG, Korting HC, Pavicic T, Ruzicka T, Jeschke MG. Hypertrophic scarring and keloids: pathomechanisms and current and emerging treatment strategies. Mol Med. 2011;17(1–2):1–25. | ||
Ogawa R. Keloid and hypertrophic scars are the result of chronic inflammation in the reticular dermis. Int J Mol Sci. 2017;18(3):606. | ||
Jiao H, Dong P, Yan L, et al. TGF-β1 induces polypyrimidine tractbinding protein to alter fibroblasts proliferation and fibronectin deposition in keloid. Sci Rep. 2016;6(1):38033. | ||
Sandulache VC, Parekh A, Li-Korotky H, Dohar JE, Hebda PA. Prostaglandin E2 inhibition of keloid fibroblast migration, contraction, and transforming growth factor (TGF)-beta1-induced collagen synthesis. Wound Repair Regen. 2007;15(1):122–133. | ||
Nirodi CS, Devalaraja R, Nanney LB, et al. Chemokine and chemokine receptor expression in keloid and normal fibroblasts. Wound Repair Regen. 2000;8(5):371–382. | ||
Zhang Q, Yamaza T, Kelly AP, et al. Tumor-like stem cells derived from human keloid are governed by the inflammatory niche driven by IL-17/IL-6 axis. PLoS One. 2009;4(11):e7798. | ||
Derynck R, Zhang YE. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature. 2003;425(6958):577–584. | ||
Dong X, Mao S, Wen H, Xianglin D. Upregulation of proinflammatory genes in skin lesions may be the cause of keloid formation (review). Biomed Rep. 2013;1(6):833–836. | ||
Wang C, Wang R, Zhou K, et al. JD enhances the anti-tumour effects of low-dose paclitaxel on gastric cancer MKN45 cells both in vitro and in vivo. Cancer Chemother Pharmacol. 2016;78(5):971–982. | ||
Jin M, Jin G, Kang L, Chen L, Gao Z, Huang W. Smart polymeric nanoparticles with pH-responsive and PEG-detachable properties for co-delivering paclitaxel and survivin siRNA to enhance antitumor outcomes. Int J Nanomedicine. 2018;13(13):2405–2426. | ||
Jeong JY, Kim KS, Moon JS, et al. Targeted inhibition of phosphatidyl inositol-3-kinase p110β, but not p110α, enhances apoptosis and sensitivity to paclitaxel in chemoresistant ovarian cancers. Apoptosis. 2013;18(4):509–520. | ||
Kampan NC, Madondo MT, McNally OM, Quinn M, Plebanski M. Paclitaxel and its evolving role in the management of ovarian cancer. BioMed Res Int. 2015;2015(1):1–21. | ||
Quispe-Soto ET, Calaf GM. Effect of curcumin and paclitaxel on breast carcinogenesis. Int J Oncol. 2016;49(6):2569–2577. | ||
Zhang L, Xu X, Yang R, et al. Paclitaxel attenuates renal interstitial fibroblast activation and interstitial fibrosis by inhibiting Stat3 signaling. Drug Des Devel Ther. 2015;9:2139–2148. | ||
Zhou J. Paclitaxel ameliorates fibrosis in hepatic stellate cells via inhibition of TGF-β/Smad activity. World J Gastroenterol. 2010;16(26):3330–3334. | ||
Tsukada T, Fushida S, Harada S, et al. Low-dose paclitaxel modulates tumour fibrosis in gastric cancer. Int J Oncol. 2013;42(4):1167–1174. | ||
Zhang D, Li Y, Liu Y, Xiang X, Dong Z. Paclitaxel ameliorates lipopolysaccharide-induced kidney injury by binding myeloid differentiation protein-2 to block Toll-like receptor 4-mediated nuclear factor-κB activation and cytokine production. J Pharmacol Exp Ther. 2013;345(1):69–75. | ||
Colombo A, Drzewiecki J, Banning A, et al. Randomized study to assess the effectiveness of slow- and moderate-release polymer-based paclitaxel-eluting stents for coronary artery lesions. Circulation. 2003;108(7):788–794. | ||
Stone GW, Ellis SG, Cox DA, et al. A polymer-based, paclitaxel-eluting stent in patients with coronary artery disease. N Engl J Med. 2004;350(3):221–231. | ||
Chen LQ, Huang W, Gao ZG, Fang WS, Jin MJ. Lx2-32c-loaded polymeric micelles with small size for intravenous drug delivery and their inhibitory effect on tumor growth and metastasis in clinically associated 4T1 murine breast cancer. Int J Nanomedicine. 2016;11:5457–5472. | ||
Hu C, Chen Z, Wu S, et al. Micelle or polymersome formation by PCL-PEG-PCL copolymers as drug delivery systems. Chin Chem Lett. 2017;28(9):1905–1909. | ||
Luo Z, Jiang L, Ding C, et al. Surfactant free delivery of docetaxel by poly[(R)-3-hydroxybutyrate-(R)-3-hydroxyhexanoate]-based polymeric micelles for effective melanoma treatments. Adv Healthc Mater. 2018;7(23):1801221. | ||
Liu X, Li Z, Loh XJ, Chen K, Li Z, Wu YL. Targeted and sustained co-release of chemotherapeutics and gene by injectable supramolecular hydrogel for drug-resistant cancer therapy. Macromol Rapid Commun. 2018;10:e1800117. | ||
Chen X, Qiu YK, Owh C, Loh XJ, Wu YL. Supramolecular cyclodextrin nanocarriers for chemo- and gene therapy towards the effective treatment of drug resistant cancers. Nanoscale. 2016;8(45):18876–18881. | ||
Liu Y, Mei L, Yu Q, et al. Multifunctional tandem peptide modified paclitaxel-loaded liposomes for the treatment of vasculogenic mimicry and cancer stem cells in malignant glioma. ACS Appl Mater Interfaces. 2015;7(30):16792–16801. | ||
Jain RK, Stylianopoulos T. Delivering nanomedicine to solid tumors. Nat Rev Clin Oncol. 2010;7(11):653–664. | ||
Yingchoncharoen P, Kalinowski DS, Richardson DR. Lipid-based drug delivery systems in cancer therapy: what is available and what is yet to come. Pharmacol Rev. 2016;68(3):701–787. | ||
Ye J, Liu Y, Xia X, et al. Improved safety and efficacy of a lipid emulsion loaded with a paclitaxel-cholesterol complex for the treatment of breast tumors. Oncol Rep. 2016;36(1):399–409. | ||
Hong SS, Choi JY, Kim JO, Lee MK, Kim SH, Lim SJ. Development of paclitaxel-loaded liposomal nanocarrier stabilized by triglyceride incorporation. Int J Nanomedicine. 2016;11:4465–4477. | ||
Rodrigues DG, Covolan CC, Coradi ST, Barboza R, Maranhão RC. Use of a cholesterol-rich emulsion that binds to low-density lipoprotein receptors as a vehicle for paclitaxel. J Pharm Pharmacol. 2002;54(6):765–772. | ||
Rodrigues DG, Maria DA, Fernandes DC, et al. Improvement of paclitaxel therapeutic index by derivatization and association to a cholesterol-rich microemulsion: in vitro and in vivo studies. Cancer Chemother Pharmacol. 2005;55(6):565–576. | ||
Xia XJ, Guo RF, Liu YL, et al. Formulation, characterization and hypersensitivity evaluation of an intravenous emulsion loaded with a paclitaxel-cholesterol complex. Chem Pharm Bull. 2011;59(3):321–326. | ||
Wang X, Li ZN, Wang QM, Jin HY, Gao Z, Jin ZH. Lipid nano-bubble combined with ultrasound for anti-keloids therapy. J Liposome Res. 2016;28(1):1–28. | ||
Bollrath J, Phesse TJ, von Burstin VA, et al. Gp130-mediated STAT3 activation in enterocytes regulates cell survival and cell-cycle progression during colitis-associated tumorigenesis. Cancer Cell. 2009;15(2):91–102. | ||
Grivennikov S, Karin E, Terzic J, et al. IL-6 and STAT3 are required for survival of intestinal epithelial cells and development of colitis-associated cancer. Cancer Cell. 2009;15(2):103–113. | ||
Mirlashari MR, Randen I, Kjeldsen-Kragh J. Glycogen synthase kinase-3 (GSK-3) inhibition induces apoptosis in leukemic cells through mitochondria-dependent pathway. Leuk Res. 2012;36(4):499–508. | ||
Manning BD, Toker A. Akt/PKB signaling: navigating the network. Cell. 2017;169(3):381–405. | ||
McQueen J, van Dyk D, Young B, Loewen C, Measday V. The mck1 GSK-3 kinase inhibits the activity of Clb2-Cdk1 post-nuclear division. Cell Cycle. 2012;11(18):3421–3432. | ||
McCubrey JA, Steelman LS, Bertrand FE, et al. GSK-3 as potential target for therapeutic intervention in cancer. Oncotarget. 2014;5(10):2881–2911. | ||
Hong H, Chen F, Qiao Y, et al. GSK-3β activation index is a potential indicator for recurrent inflammation of chronic rhinosinusitis without nasal polyps. J Cell Mol Med. 2017;21(12):3633–3640. | ||
Ren X, Zhao B, Chang H, Xiao M, Wu Y, Liu Y. Paclitaxel suppresses proliferation and induces apoptosis through regulation of ROS and the AKT/MAPK signaling pathway in canine mammary gland tumor cells. Mol Med Rep. 2018;17(6):8289–8299. |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.